

Abstract
The 3β-hydroxysteroid dehydrogenase (3β-HSD) is an enzyme crucial for steroid synthesis. Two different 3β-HSD isoforms exist in humans. Classically, HSD3B2 was considered the principal isoform present in the adrenal. However, we recently showed that the alternative isoform, HSD3B1, is expressed specifically within the adrenal zona glomerulosa (ZG), where aldosterone is produced, raising the question of why this isozyme needs to be expressed in this cell type. Here we show that in both human and mouse, expression of the ZG isoform 3β-HSD is rapidly induced upon angiotensin II (AngII) stimulation. AngII is the key peptide hormone regulating the capacity of aldosterone synthesis. Using the human adrenocortical H295R cells as a model system, we show that the ZG isoform HSD3B1 differs from HSD3B2 in the ability to respond to AngII. Mechanistically, the induction of HSD3B1 involves de novo protein synthesis of the nuclear orphan receptors NGFIB and NURR1. The HSD3B1 promoter contains a functional NGFIB/NURR1-responsive element to which these proteins bind in response to AngII. Knockdown of these proteins and overexpression of a dominant negative NGFIB both reduce the AngII responsiveness of HSD3B1. Thus, the AngII-NGFIB/NURR1 pathway controls HSD3B1. Our work reveals HSD3B1 as a new regulatory target of AngII.
INTRODUCTION
The enzyme 3β-hydroxysteroid dehydrogenase/Δ5-Δ4-isomerase (3β-HSD) is essential for the biosynthesis of all active steroid hormones, including those secreted from the adrenal gland (1–4). Whereas two distinct 3β-HSD isoforms (type I 3β-HSD, which is encoded by HSD3B1, and type II 3β-HSD, which is encoded by HSD3B2) exist in humans, it has long been thought that type II 3β-HSD was the only isoform expressed in the human adrenal glands (1). However, this canonical view was recently revised due to the observation that the alternative isoform, HSD3B1, is expressed within zona glomerulosa (ZG) cells (5, 6), where aldosterone is produced. Interestingly, the mouse also has two isoforms in the adrenal: one (Hsd3b1) is ubiquitous in the cortex, but the other (Hsd3b6) is ZG specific (6–9). Thus, in both species, the adrenals possess a ZG-specific isoform, in addition to the ubiquitous one. However, it remains unknown why these different enzymes are expressed simultaneously in ZG cells. Since these isozymes catalyze the same enzymatic reaction, the question remains open why the newly identified ZG-specific isoform needs to be expressed in ZG cells.
Angiotensin II (AngII) is the key peptide hormone in the renin-angiotensin-aldosterone system (RAAS). AngII is a potent secretagogue of the mineralocorticoid aldosterone, which is principally synthesized within the adrenal gland ZG cells through a series of enzymatic reactions that involve multiple enzymes, including 3β-HSDs. The actions of AngII on ZG cells are often divided into two temporally different phases (10–12): (i) an early (within minutes after stimulation) upregulation of aldosterone synthesis through posttranslational activation of steroidogenic acute regulatory (StAR) protein that facilitates the transfer of cholesterol (steroid precursor) to the mitochondria and (ii) a relatively late (hours after stimulation) enhancement of aldosterone synthesis by increasing the capacity to produce aldosterone through increased expression of relevant enzymes in ZG cells. It has been well documented that AngII triggers expression of CYP11B2 (aldosterone synthase), an enzyme also known to be expressed specifically within ZG cells. CYP11B2 and HSD3B1 are the only known steroidogenic enzymes whose expression is limited to ZG cells in the adrenal gland. However, virtually nothing is known about whether the expression of HSD3B1 is under the control of AngII (13, 14). This paucity of knowledge is partly because of the high sequence similarity between HSD3B1 and HSD3B2 (93.6% identity, including the 5′ and 3′ untranslated regions [UTRs]) (5, 6), which made it difficult to achieve isoform-selective quantification of their transcripts without the recent TaqMan MGB probe technology that was devised for single nucleotide discrimination between target genes (6, 15).
A better understanding of HSD3B1 and HSD3B2 is also critical for the comprehension of adrenal disorders. The results of pathological investigations of human idiopathic hyperaldosteronism (5) and its animal model (circadian clock-deficient Cry-null mice) (6) revealed that abnormally increased expression of the ZG isoform (HSD3B1 for human, Hsd3b6 for mouse) is closely associated with the enhanced aldosterone production of this disease. On the other hand, decreased expression of HSD3B1 and HSD3B2 was found in ZG cells located in a nontumor portion of an aldosterone-producing adenoma (APA)-containing adrenal gland (5, 16, 17), suggesting a feedback regulation of 3β-HSDs as a compensatory response to excess aldosterone from the APA. Interestingly, aldosterone synthase CYP11B2 showed sustained expression in APA-associated ZG cells (5, 18). Thus, it appears that the expression of 3β-HSDs and CYP11B2 is differently regulated and that the levels of 3β-HSDs might be an additional regulatory element for determining the capacity of steroid production in ZG cells. However, it is currently unknown how the expression levels of HSD3B1 are regulated.
The present study was undertaken to elucidate whether AngII controls HSD3B1 and/or HSD3B2 and, if so, by what molecular mechanism(s). Using human adrenocortical H295R cells as a model system, we found that AngII is able to induce HSD3B1 but not HSD3B2. Similarly, mouse adrenal glands showed selective induction of Hsd3b6 (ZG isoform) but not Hsd3b1 (ubiquitous isoform) after AngII treatment. Our data also provide evidence that the acute induction of HSD3B1 is fully dependent on AngII-stimulated de novo protein synthesis of the orphan nuclear receptors NGFIB and NURR1. Interestingly, the NGFIB protein family has been implicated in the regulation of CYP11B2 as well (19–22). However, we found that the regulation of this gene is not entirely dependent on de novo protein synthesis. A mechanistic difference between HSD3B1 and CYP11B2 is discussed.
MATERIALS AND METHODS
Animals.
All animal studies were performed with protocols approved by the animal experimentation committee of Kyoto University. C57BL/6 male mice aged 8 weeks were purchased from local suppliers and housed in 12-h light/12-h dark cycle (lights on at 08:00, lights off at 20:00) for 7 days. Then, the animals were transferred to a low-sodium diet (0.001% elemental Na+ diet; CREA, Japan) at 08:00 (zeitgeber time zero), and the adrenal glands were removed by surgery after 24 or 48 h of sodium restriction. Since all samples were collected at 08:00, there is no confounding factor derived from circadian time on the expression of target genes. For AngII treatment, AngII (Peptide Institute, Japan) was injected into the mice at 08:00 intraperitoneally at a dose of 1 mg per kg body weight, and the adrenals were removed from the animals at 4 and 24 h after the injection. For the enucleation of the adrenal gland, the adrenals freed of adherent fat were mechanically separated into the capsular portions according to a conventional method (6, 23). The adrenal samples were harvested in either TRIzol reagent (Invitrogen) for subsequent RNA analysis or 4% paraformaldehyde-containing fixative solution for in situ hybridization (see below).
Cell culture and treatments.
Human adrenocortical H295R cells (ATCC CRL-2128) were cultured in Dulbecco modified Eagle medium (DMEM)–F-12 medium (Invitrogen) supplemented with 2.5% Nu serum (BD Biosciences) and 1% ITS premix (BD Biosciences). H295R cells are one of the best-characterized cellular models for the analysis of adrenal cell biology (24), since this is the human adrenal cell line that preserves the ability to secrete aldosterone in respond to AngII (25). For AngII stimulation, an aliquot of freshly reconstituted AngII (10 μM) was added to the culture medium at a final concentration of 100 nM. To specify the type of AngII receptors involved, we also added either the AT1R blocker CV11974 (final concentration, 100 nM; a generous gift from Takeda Pharmaceutical) or the AT2R blocker PD123319 (final concentration, 1 μM; Sigma) to the culture medium 1 h before AngII treatment. Pharmacological inhibition of de novo protein synthesis was also carried out by adding cycloheximide (CHX; final concentration, 10 μg/ml) to the medium 15 min before AngII treatment. At 1, 2, 4, 6, 8, and 12 h after AngII treatment, the cells were harvested in TRIzol reagent (Invitrogen) for subsequent RNA analysis.
RNA extraction and quantitative reverse transcription-PCR (qRT-PCR).
RNA was extracted using an RNeasy kit (Qiagen) according to the manufacturer's protocol. Total RNA was converted to cDNA with random hexamer primers using SuperScript III first-strand synthesis SuperMix (Invitrogen), and quantitative PCR (qPCR) was run in duplicate with the primers and probes shown below. For the analysis of human HSD3B1 and HSD3B2, qPCR was done with the TaqMan Universal master mix (Applied Biosystems) using gene-specific TaqMan MGB probes. As we previously reported (6), these probes distinguish a few nucleotide differences at the region corresponding to the dehydrogenase catalytic Y-X-X-X-K motif (1) of the human 3β-HSDs. On the other hand, SYBR green-based qPCR was done for the other genes with the aid of a Platinum SYBR green qPCR SuperMix-UDG kit (Invitrogen). As a qPCR device, we used a StepOnePlus real-time PCR monitoring system (Applied Biosystems), and the quantification of target cDNAs was achieved with a standard curve method as described previously (7). The data were normalized to those for Rplp0. The sequences for the primers and probes are as follows: for mouse Hsd3b1, forward primer 5′-AGC ATC CAG ACA CTC TCA TC-3′ and reverse primer 5′-GGA GCT GGT ATG ATA TAG GGT A-3′; for mouse Hsd3b6, forward primer 5′-TGA TGG GAA GAG GGT GGA G-3′ and reverse primer 5′-AGG TGC TGA GAG GCT TGG A-3′; for mouse Rplp0, forward primer 5′-CTC ACT GAG ATT CGG GAT ATG-3′ and reverse primer 5′-CTC CCA CCT TGT CTC CAG TC-3′; for human StAR, forward primer 5′-CCT GAG CAG AAG GGT GTT CA-3′ and reverse primer 5′-CCA ACG GGT GAA GCA CCA T-3′; for human CYP11A1, forward primer 5′-AGC CAG CAT CAA GGA GAC ACT A-3′ and reverse primer 5′-ACC AGT GTC TTG GCA GGA ATC A-3′; for human CYP21A2, forward primer 5′-CAC TGA GAC CAC AGC AAA CAC-3′ and reverse primer 5′-CTG CAG TCG CTG CTG AAT C-3′; for human CYP11B1, forward primer 5′-GCC ATC AAC TAA TCA CGA CT-3′ and reverse primer 5′-TGA TCT TAG CCT TCT AAG CCT T-3′; for human CYP11B2, forward primer 5′-ACT CGC TGG GTC GCA ATG-3′ and reverse primer 5′-GTC TCC ACC AGG AAG TGC-3′; for human NGFIB, forward primer 5′-GCC TCC TGG AGG CTC TTC ATC-3′ and reverse primer 5′-GAG AAG GCC AGG ATA CTG TCA ATC-3′; for human NURR1, forward primer 5′-GGC TCC CAG AGG GAA CTG-3′ and reverse primer 5′-GAG TCC AGC CTG TCC AAT CTC-3′; for human NOR1, forward primer 5′-TCC GCT CCT CCT ACA CTC TC-3′ and reverse primer 5′-GGT GTA TTC CGA GCT GTA TGT CTG-3′; for human RPLP0, forward primer 5′-ATG CAG CAG ATC CGC ATG T-3′ and reverse primer 5′-TTG CGC ATC ATG GTG TTC TT-3′; for human HSD3B1, forward primer 5′-AGA AGA GCC TCT GGA AAA CAC ATG-3′, reverse primer 5′-TAA GGC ACA AGT GTA CAG GGT GC-3′, and probe 5′-FAM-CCA TAC CCA CAC AGC-MGB-3′ (where FAM is 6-carboxyfluorescein); and for human HSD3B2, forward primer 5′-AGA AGA GCC TCT GGA AAA CAC ATG-3′, reverse primer 5′-CGC ACA AGT GTA CAA GGT ATC ACC A-3′, and probe 5′-VIC-TCC ATA CCC GTA CAG CA-MGB-3′.
In situ hybridization.
Radioisotopic in situ hybridization was performed as described previously (6). Briefly, paraformaldehyde-fixed adrenal glands were frozen and sectioned at a thickness of 30 μm. The free-floating sections were then transferred sequentially through 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) buffer for 10 min, 1 μg/ml proteinase K in 0.1 M Tris buffer (pH 8.0) with 50 mM EDTA for 15 min at 37°C, 0.25% acetic anhydride in 0.1 M triethanolamine for 10 min, and 2× SSC buffer for 10 min. Then, the sections were transferred to hybridization buffer (55% formamide, 10% dextran sulfate, 10 mM Tris-HCl [pH 8.0], 1 mM EDTA, 0.6 M NaCl, 0.2% N-laurylsarcosine, 0.5 g/ml tRNA, 1× Denhardt's solution, 0.25% SDS, 10 mM dithiothreitol [DTT]) containing radiolabeled riboprobes (4 × 106 dpm) and incubated at 60°C for 16 h. Following a high-stringency posthybridization wash, the sections were treated with RNase A. Air-dried sections were exposed to X-ray films.
Western blotting.
The H295R cells that had been treated with 100 nM AngII for periods ranging from 1 to 12 h were harvested in Laemmli buffer, and the lysates were subjected to Western blot analysis with antibodies to NGFIB (1:500 dilution; M-210 antibody; Santa Cruz Biotechnology), NURR1 (1:1,000 dilution; N1404 antibody; Perseus Proteomics), NOR1 (1:500 dilution; H7833 antibody; Perseus Proteomics), and β-actin (1:1,000 dilution; AC-15 antibody; Sigma) or α-tubulin (1:1,000 dilution; T6199 antibody; Sigma). For the detection of the dominant negative (DN) form of NGFIB (DN-NGFIB; which corresponds to the C-terminal 249-amino-acid portion of NGFIB), we employed an alternative antibody (1:500 dilution; E-20 antibody; Santa Cruz Biotechnology) that was raised against the C-terminal region of NGFIB. Immunoblotting was done according to our standard protocol (26).
Electrophoretic mobility shift assay (EMSA).
Nuclear proteins extracted from H295R cells were subjected to a gel shift assay as described previously (27), with certain modifications. The sequence of the oligonucleotide probe (5′-TAA CCC AAA GGT CAC TAT TTT-3′) was designed to encompass the NGFIB-responsive element (NBRE) site of the human HSD3B1 promoter (underlined). Radioactive probe was generated by end labeling the annealed oligonucleotide pairs with T4 polynucleotide kinase and [γ-32P]ATP. The binding reactions were performed on ice for 30 min in a 20-μl reaction mixture that contained 4 μg of the nuclear extract proteins together with 4 × 105 dpm of the radiolabeled oligonucleotide probe in 10% glycerol, 250 mM NaCl, 25 mM Tris-HCl (pH 7.9), 0.5 mM DTT, 0.5 mM EDTA, 6.25 mM MgCl2, 1 μg/μl bovine serum albumin (BSA), and 0.2 μg/μl poly(dG-dC). For antibody supershift assays, 1 μg of specific antibodies that recognize NGFIB (anti-NGFIB; M-210 antibody), NURR1 (anti-NURR1; N-20 antibody), or NOR1 (anti-NOR1; A-20 antibody) or those recognizing both NGFIB and NURR1 (anti-NGFIB/NURR1; E-20 antibody) (all antibodies were from Santa Cruz Biotechnology) were preincubated with the nuclear extract on ice for 30 min before the addition of the radioactive probe. In control experiments, the antibody was replaced with nonimmune antibodies (IgG). For competition assay, a 100-fold molar excess of unlabeled probe was added to the reaction mixture simultaneously with the radiolabeled probe. The mutant oligonucleotide probe used for the competition assay was as follows: 5′-TAA CCC AGA ATT CAC TAT TTT-3′ (underlining indicates mutated NBRE). After the binding reactions, the resultant DNA/protein complexes were separated on a 4% native polyacrylamide gel in 1× Tris-glycine buffer containing 1% glycerol. The gel was dried and exposed to an X-ray film.
Chromatin immunoprecipitation (ChIP).
H295R cells grown to confluence on a 10-cm dish (∼1 × 107 cells) were treated with either AngII (100 nM) or vehicle for 3 or 12 h. Then, the cells were homogenized in 1.2 ml (per dish) of phosphate-buffered saline (PBS) containing 2 mM disuccinimidyl glutarate (Pierce), and the homogenates were kept for 20 min at room temperature. Then, methanol-free formaldehyde (final concentration, 1%; Thermo) was added for 5 min at room temperature, and 125 μl of 1.5 M glycine (final concentration, 150 mM) was added to stop the reaction on ice. The homogenates were centrifuged at 700 × g, and the resultant nuclear pellets were washed twice with ice-cold PBS. The nuclei were resuspended in 2.5 ml of immunoprecipitation (IP) assay buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride [PMSF], protease inhibitor cocktail) and sonicated 10 times for 30 s each time at 4°C using a Bioruptor UCW-201TM apparatus (Tosho Denki, Yokohama, Japan). Approximately 1.5 μg of fragmented chromatin was precleared by incubating with 40 μl of protein A-agarose (Roche) for 2 h at 4°C on a rotating wheel. Precleared chromatin was then incubated with 1 μg of anti-NGFIB/NURR1 antibody (E-20 antibody; catalog number sc-990; Santa Cruz Biotechnology) or control normal rabbit IgG (catalog number sc-2027; Santa Cruz Biotechnology) overnight at 4°C on a rotating wheel, 10 μl of protein A/G Plus-agarose (Santa Cruz Biotechnology) was added to each sample, and the mixture was incubated for 2 h at 4°C. Beads were then washed once with IP assay buffer, once with high-salt wash buffer (20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 2 mM EDTA, 1% Triton X-100, 1 mM PMSF), once with LiCl wash buffer (20 mM Tris-HCl, pH 8.0, 250 mM LiCl, 2 mM EDTA, 0.5% Nonidet P-40, 1% sodium deoxycholate, 1 mM PMSF), and twice with TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA). Coimmunoprecipitated DNA fragments were eluted with 100 μl of elution buffer (20 mM Tris-HCl, pH 8.0, 5 mM EDTA, 0.5% sodium dodecyl sulfate) and then reverse cross-linked at 65°C overnight, incubated with 10 μg of RNase A for 30 min at 37°C and 50 μg of proteinase K for 90 min at 55°C, and then purified using a QIAquick PCR purification kit (Qiagen). Immunoprecipitated DNA fragments were quantified by TaqMan qPCR for the NBRE site of HSD3B1 with the following primers and probe: forward primer 5′-CCT GTT AAG GCT AAA CCC AAG AC-3′, reverse primer 5′-CAT TGC TCT CTC CTC CTA TGG G-3′, and TaqMan probe 5′-VIC-TGC CAC ACT GCA GCA TTA GGA TGG G-NFQ-MGB-3′.
Reporter assays.
Expression vectors for human NGFIB (hNGFIB/pEZ; catalog number EX-A0227-M02) and NURR1 (hNURR1/pEZ; catalog number EX-I0539-M02) were purchased from GeneCopoeia. The following reporter plasmids were used in this study: (i) pGL4.10 HSD3B1 promoter-Luc(1332), in which a 1,332-bp genomic DNA fragment upstream of the translation start site (position +274) of the human HSD3B1 was cloned into the pGL4.10(luc2) vector (Promega); (ii) pGL4.10 HSD3B1 promoter-Luc(599), in which a 599-bp sequence upstream of the transcription start site of HSD3B1 was cloned into pGL4.10(luc2); (iii) pGL4.10 HSD3B1 promoter-Luc(200) and pGL4.10 HSD3B1 promoter-Luc(100), which are deletion constructs of pGL4.10 HSD3B1 promoter-Luc(599) containing either 200 or 100 bp of the 5′-flanking sequence; (iv) pGL4.10 HSD3B1 mut NBRE promoter-Luc (599), in which the NBRE sequence of pGL4.10 HSD3B1 promoter-Luc(599) was mutated to 5′-AGAATTCA-3′ with a standard sequential PCR method (28); (v) pGL4.23 NBRE-Luc, in which a DNA fragment containing nine tandem copies of the sequence corresponding to the HSD3B1 NBRE with its flanking sequences (positions −141 to −72) was inserted into the pGL4.23(luc2/minP) vector (Promega); and (vi) pGL4.23 mut NBRE-Luc, which is the same as pGL4.23 NBRE-Luc, except that every NGFIB-binding site was mutated to 5′-AGAATTCA-3′. Cell culture and transfection assays were carried out as previously described (6), except that the Lipofectamine LTX/Plus reagent (Invitrogen) was used as the transfection reagent according to the manufacturer's protocol.
siRNA transfection.
H295R cells were transfected using Nucleofector technology (Amaxa Biosystems). Three million log-phase cells were resuspended in 100 μl Nucleofector solution R containing 2 μM (final concentration) small interfering RNA (siRNA) mixtures for NGFIB (siRNAs s6978 and s6979), NURR1 (siRNAs s9785 and s9786), and NOR1 (siRNAs s15541 and s15543) or negative-control siRNA (catalog number 12935112) (all siRNAs were from Life Technologies) and electroporated using the proprietary program P-20. Cells were allowed to recover for 15 min in RPMI 1640 medium at 37°C and then plated in 48-well plates with 0.5 ml H295R cell complete medium at a density of 5 × 105 cells/well. One day after the electroporation, dead cell debris was removed by refreshing the medium, and the cells were cultured for 48 h. Then, the culture medium was removed and the cells were incubated in serum-free medium (DMEM–F-12 medium) for 24 h, followed by a treatment with or without AngII for 4 h. At the end of the treatment, the cells were washed with PBS and immediately lysed in TRIzol reagent for RNA extraction or boiled in Laemmli buffer for Western blotting.
Isolation of DN-NGFIB-transfected H295R cells by the MACSelect system.
DN-NGFIB (the C-terminal 249-amino acid portion of NGFIB) was produced by deleting the N-terminal transactivation domain of human NGFIB. This protein is defective in transactivation function but shares the same DNA sequence binding specificity with the NGFIB family. Thus, the DN-NGFIB protein acts as an inhibitor for all three subfamily members: NGFIB, NURR1, and NOR1 (29, 30). For coexpression of DN-NGFIB with a cell surface marker protein, H-2Kk (Miltenyi Biotec), we used the pMACS Kk.II expression vector (Miltenyi Biotec), which allows the simultaneous expression of H-2Kk and the gene of interest from a single vector. Since in H295R cells the simian virus 40 (SV40) promoter shows lower levels of activity than the cytomegalovirus (CMV) promoter, the SV40 promoter region of this vector was replaced by the CMV promoter. Then, the modified vector (pMACS Kk.II CMV) was used for cloning and expression of DN-NGFIB. Transient transfection of H295R cells was done by lipofection using the Lipofectamine LTX/Plus reagent (Invitrogen), for which we routinely observed an ∼25% transfection efficiency (an estimation from green fluorescent protein-transfected H295R cells). The cells seeded in 10-cm dishes (5 × 106 cells/dish) were transfected with 24 μg of the DN-NGFIB/pMACS Kk.II CMV plasmid. After transfection, the cells were allowed to recover for 72 h prior to AngII treatment. Following 3 h treatment with AngII (100 nM) or vehicle, the cells were washed and removed with Versene solution (Invitrogen) and then dissociated by gentle pipetting and passed through a 40-μm-pore-size cell strainer (BD Falcon). The resultant cell suspensions were resuspended in ice-cold PBS (150 μl) containing 0.5% BSA and 1 mM EDTA and mixed with MACSelect Kk microbeads (1:375 dilution; Miltenyi Biotec). After incubation on ice for 15 min, the cells were isolated on a magnetic separation column (Miltenyi Biotec) under a magnetic field according to the manufacturer's protocol. The cells underwent two consecutive rounds of purification. Then, the second round of eluted cells and the first round of flowthrough cells were washed twice with ice-cold PBS, and these cell samples were either lysed in TRIzol reagent for qRT-PCR or boiled in Laemmli sample buffer for Western blotting.
Statistical analysis.
We used Student's t test for the comparison of two test groups (see Fig. 1B and D). For the experiments in which three or more test groups were compared, we used one-way analysis of variance (ANOVA) with Bonferroni's post hoc test (see Fig. 1A). The differences arising from two independent parameters (see Fig. 1E, 4G, 6A, and 7C) were assessed through two-way ANOVA, followed by Bonferroni's multiple-comparison test.
FIG 1.

The mouse adrenal Hsd3b6 increases in response to AngII treatment and a low-salt diet. (A to C) Adrenal Hsd3b6 and Hsd3b1 mRNA levels in mice after a change to a low-salt (LS) diet. The adrenals were removed from animals at 0, 24, and 48 h after sodium restriction. Total RNA was prepared from either whole adrenal (A) or the ZG cell layer-containing capsular portion of the adrenal gland (B). The mRNA levels of Hsd3b6 and Hsd3b1 were determined by gene-selective qRT-PCR. All the values (means ± SEMs, n = 3) were normalized to the levels of Rplp0, and the values at time zero were set equal to 1. *, P < 0.01. Autoradiographs (C) show mouse adrenal sections hybridized with a radioisotopic hybridization probe for Hsd3b6 and Hsd3b1 before and after 48 h of sodium restriction. Bar, 0.5 mm. (D to F) Adrenal Hsd3b6 and Hsd3b1 mRNA levels in mice after a single intraperitoneal administration of AngII (1 mg/kg) or the vehicle (Veh) control (saline). Adrenals were removed from the animals at 4 and 24 h after administration, and total RNAs prepared from either whole adrenal (D) or the capsule portion (E) were analyzed by qRT-PCR, as described in the legends to panels A and B. *, P < 0.01. Autoradiographs (F) show the sections of the adrenal collected at 4 h after the administration of AngII or vehicle. Bar, 0.5 mm.
FIG 4.

NGFIB and NURR1 protein levels increase upon AngII stimulation in a pattern similar to that of HSD3B1 mRNA. (A) Genomic structure upstream of the translation start site of human HSD3B1. Numbers indicate the positions relative to the transcription initiation site (position +1). The sequence of the potential NBRE site (positions −129 to −113) is compared with that of the consensus NGFIB-binding motif (W, A or T). *, sequences that conform to the consensus motif; arrows, positions of primers used in the ChIP assay. (B to D) Gene expression profiles of NGFIB (B), NURR1 (C), and NOR1 (D) after AngII (100 nM) stimulation in H295R cells with or without CHX (10 μg/ml) pretreatment. Relative mRNA levels (mean ± SEM, n = 3) were determined by qRT-PCR and normalized to those of RPLP0. (E to G) Immunoblots showing the protein expression profiles of NGFIB (E), NURR1 (F), and NOR1 (G) after AngII (100 nM) stimulation in H295R cells. Relative band intensities were determined by densitometry and expressed as the means ± variation from two independent experiments. For comparison, HSD3B1 mRNA expression profiles (replots of Fig. 2B) are displayed in parallel. (H) Western blots showing the protein expression profiles of NGFIB, NURR1, and NOR1 after AngII treatment in the presence or absence of CHX in H295R cells.
FIG 6.
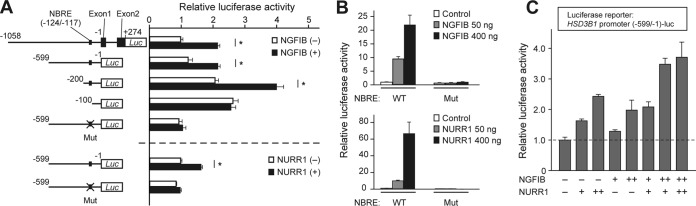
NGFIB and NURR1 possess the ability to enhance HSD3B1 promoter activity. (A) Luciferase reporter assays of NGFIB and NURR1 for the HSD3B1 promoter. Reporter constructs containing serial deletions of the HSD3B1 promoter (positions −1059 to +274, −599 to −1, −200 to −1, and −100 to −1) as well as the mutant derivative for the NBRE (Mut; positions −124 to −117) were transiently introduced (20 ng plasmid/well for each) into H295R cells with either an empty pEZ expression vector (500 ng) or an expression vector containing the coding sequence of human NGFIB or NURR1 (500 ng). After recovery for 24 h, cells were lysed, and luciferase activity was measured. Deletion constructs are numbered relative to the transcription initiation site. Shown are representative data from replicate experiments with similar results. Values represent the means ± SEMs (n = 4). *, P < 0.01, Bonferroni test. (B) Luciferase reporter assays of NGFIB and NURR1 for the isolated NBRE. A luciferase reporter construct (20 ng) that contains either nine copies of the NBRE sequence of HSD3B1 (wild type) or those of the mutated sequence was transiently introduced into H295R cells with increasing doses of the expression vector for NGFIB or NURR1 (50, 400 ng). Values are the means ± SEMs (n = 4). (C) Luciferase reporter assays examining the effect of coexpression of NGFIB and NURR1 on the promoter activities of HSD3B1. The reporter plasmid containing the HSD3B1 promoter (positions −599 to −1) (20 ng) was transiently introduced into H295R cells with the indicated combinations of expression plasmids for NGFIB and NURR1. +, 200 ng, ++, 400 ng. Values are the means ± SEMs (n = 4).
FIG 7.

Knockdown of NGFIB and NURR1 attenuates AngII-stimulated HSD3B1 induction. (A) Gene-selective knockdown by electroporation of siRNAs against NGFIB and NURR1. H295R cells were transfected by electroporation using the indicated siRNA mixtures, each directed against NGFIB (siNGFIB) or NURR1 (siNURR1), and treated with 100 nM AngII or vehicle for 4 h. Total RNA was isolated, and expression of NGFIB and NURR1 was determined by qRT-PCR. All values (means ± SEMs, n = 3) were normalized to the levels of RPLP0. As a control, the electroporation was also performed with a negative-control siRNA (NC). (B) Reduction of protein expression of NGFIB and NURR1 by siRNA-mediated knockdown. H295R cells transfected with the indicated siRNAs were stimulated by 4 h AngII treatment (10 nM or 100 nM) and subjected to Western blotting with antibodies against NGFIB and NURR1. α-Tubulin was used as a loading control. (C) Attenuated AngII response of HSD3B1 and CYP11B2 by knockdown of NGFIB and NURR1. H295R cells were transfected with the indicated siRNAs and then stimulated with either 100 nM or 10 nM AngII or vehicle for 4 h. The levels of HSD3B1 and CYP11B2 mRNA were determined by qRT-PCR (n = 3 for each), and the means of vehicle treatment were set equal to 1 after normalization to the level of RPLP0. Error bars indicate SEMs. *, P < 0.05 (versus 10 nM AngII induction of negative-control siRNA-transfected cells); #, P < 0.05 (versus 100 nM AngII induction of negative-control siRNA-transfected cells). (D) Unimpaired AngII response of HSD3B1 by electroporation of siRNAs targeting NOR1 (siNOR1). H295R cells transfected with the indicated siRNAs (s15541, s15543, or the negative-control siRNA) were treated with 100 nM AngII or vehicle for 4 h. Total RNA was isolated, and expression of NGFIB, NURR1, NOR1, and HSD3B1 was determined by qRT-PCR. All values (means ± SEMs, n = 3) were normalized to the level of RPLP0.
RESULTS
AngII triggers expression of Hsd3b6 but not Hsd3b1 in the mouse adrenal gland.
The mouse Hsd3b6 gene is the specific 3β-HSD isoform whose adrenal expression is confined to ZG cells (6, 7). To study whether the activation of the renin-angiotensin system influences this gene's expression profile in the adrenal, mice were placed on a low-sodium diet (0.001% elemental Na+) for 1 or 2 days, and the levels of adrenal expression of Hsd3b6 and Hsd3b1 mRNA were examined by qRT-PCR with gene-specific primers (Fig. 1A). We found that sodium restriction significantly increased Hsd3b6 mRNA levels in the whole adrenal gland within 24 h after treatment (Fig. 1A). In contrast, Hsd3b1 mRNA levels did not show any appreciable increase even after 48 h of sodium restriction (Fig. 1A). Analogous results were also observed for the enucleated adrenal gland sample (i.e., the capsular portions of the adrenal gland), which consists largely of ZG cells (Fig. 1B): the capsular mRNA levels of the ZG-specific isoform, Hsd3b6, were significantly increased by more than 3-fold after 2 days of sodium restriction, whereas those of the canonical isoform, Hsd3b1, remained unchanged. In situ hybridization of the whole adrenal sections with a radiolabeled probe for Hsd3b6 (Fig. 1C) confirmed selective induction of this gene in ZG cells, which constitute the outer layer of the cortex.
In order to determine whether the expression of Hsd3b6 is directly regulated by AngII, mice were then treated with AngII (Fig. 1D to F). We found that a single intraperitoneal administration of AngII (1 mg/kg) caused a rapid and transient induction of Hsd3b6 (Fig. 1D): the expression of Hsd3b6 mRNA in the whole adrenal gland increased approximately 3-fold over the basal levels (vehicle treatment) after 4 h of administration, but it returned to near basal levels within 24 h after AngII treatment (Fig. 1E). No significant increase was observed for Hsd3b1 (Fig. 1D and E). Selective induction of Hsd3b6 was also observed in the qPCR analysis of the enucleated adrenal gland (Fig. 1E), as well as with the isoform-selective in situ hybridization of adrenal gland sections (Fig. 1F).
AngII triggers expression of HSD3B1 but not that of HSD3B2 in human adrenocortical H295R cells.
We recently showed that HSD3B1 is exclusively expressed in ZG cells in the human adrenal gland (5, 6). This identification raises the question of whether the expression of this gene is under the regulation of AngII. Using adrenocortical H295R cells as a model system, we next sought to investigate AngII responsiveness of the human 3β-HSD isoform genes (Fig. 2). We treated H295R cells with AngII (100 nM) over a range of times (1, 2, 4, 8, and 12 h), and the gene expression profiles of human HSD3B1 and HSD3B2 were examined by qRT-PCR with the aid of isoform-specific TaqMan MGB probes and primers that we developed previously (6) (see also Materials and Methods) (Fig. 2). Because of the high degree of cDNA sequence similarity between the two isoforms (93.6% identity, including the 5′ and 3′ UTRs) (5, 6), studies aimed at isoform-selective comparison between HSD3B1 and HSD3B2 have not been available in the past for H295R cells (13, 14). Interestingly, we observed that AngII caused a marked transient induction of HSD3B1 (Fig. 2): after 4 h AngII treatment, the levels of HSD3B1 mRNA were dramatically increased up to about 10-fold over basal levels. On the other hand, HSD3B2 was not responsive to AngII (Fig. 2): no appreciable increase was observed for this gene over 12 h of AngII treatment. Thus, these results indicate that the ZG-specific isoform HSD3B1 differs from HSD3B2 in the ability to respond to AngII stimulation.
FIG 2.

AngII triggers HSD3B1 induction through the AT1 receptor in human adrenocortical H295R cells. (A) Schematic of the human aldosterone biosynthesis pathway. (B) Expression profiles of the genes involved in aldosterone production in H295R cells before and after AngII treatment. H295R cells were pretreated with the vehicle control, CV11974 (CV; 100 nM), or PD123319 (PD; 1 μM) for 1 h and then stimulated with AngII (100 nM). After the indicated periods of time, total RNA was isolated from the cells and the levels of mRNA for HSD3B1 and HSD3B2 as well as StAR, CYP11A1, CYP21A2, CYP11B1, and CYP11B2 were determined by qRT-PCR. All the values (means ± SEM, n = 3) were normalized to the levels of RPLP0, and the values for vehicle treatment for each gene at time zero (the time immediately before AngII stimulation) were set equal to 1.
There are two different AngII receptor subtypes, AT1 and AT2. Previous studies have shown that AT1 is the principal AngII receptor for the regulation of aldosterone biosynthesis in the human adrenal gland as well as in H295R cells (24, 31). Consistent with this, AngII-induced induction of HSD3B1 mRNA in H295R cells was completely blocked by pharmacological treatment with the AT1 receptor antagonist CV11974, whereas no inhibition was observed in response to treatment with the AT2 receptor antagonist PD123319 (Fig. 2). These results indicate that the AT1 receptor is responsible for the AngII-induced HSD3B1 mRNA expression.
As reported, AngII stimulation-dependent acute mRNA induction was observed for StAR, CYP21A, CYP11B1, and CYP11B2 but not for CYP11A1 (Fig. 2) (14, 32–35). Not all steroidogenic enzymes were increased upon AngII stimulation (11). Moreover, the expression profiles were time dependent. For example, HSD3B1 showed the maximum mRNA expression at 4 h after AngII stimulation, while expression of the other genes, such as CYP11B2, increased continuously for 8 h after AngII stimulation (Fig. 2). These results indicate that AngII controls different steroidogenic genes with different kinetics.
AngII-driven transcriptional activation of HSD3B1 relies on de novo protein synthesis.
In the process of the gene expression time course studies (Fig. 3), we noticed that HSD3B1 did not show any increase during the first hour of AngII stimulation. The levels of HSD3B1 mRNA began to increase 2 h after AngII treatment. This late onset of HSD3B1 induction might be ascribed to a need for de novo protein synthesis of the relevant trans-acting regulator(s) of this gene. We therefore studied whether cycloheximide (CHX) treatment affects the AngII-responsive mRNA expression profiles of HSD3B1 in H295R cells (Fig. 3). Notably, pharmacological blockade of new protein synthesis by CHX treatment completely blocked the AngII response of HSD3B1, while the induction profiles of StAR, CYP11B1, and CYP11B2 were essentially unimpaired by this treatment (Fig. 3). These data clearly indicate that HSD3B1 differs from StAR, CYP11B1, and CYP11B2 in requiring de novo protein synthesis for the response to AngII stimulation.
FIG 3.

AngII-driven transcriptional activation of HSD3B1 relies on de novo protein synthesis. H295R cells were treated with either CHX (10 μg/ml) or the vehicle control for 15 min and then stimulated with AngII (100 nM). After the indicated periods of time, total RNA was isolated from the cells and the levels of HSD3B1 and HSD3B2 mRNA as well as StAR, CYP11A1, CYP21A2, CYP11B1, and CYP11B2 mRNA were determined by qRT-PCR. All values (means ± SEM, n = 3) were normalized to the levels of RPLP0, and the values for vehicle treatment at time zero were set equal to 1.
We observed that HSD3B2 was again AngII stimulation insensitive, regardless of the CHX treatment. Also, our results reproduced a previously reported CHX-sensitive AngII response of CYP21A (34), while this gene's induction was relatively modest compared to that of HSD3B1 (Fig. 3).
Upon AngII stimulation, NGFIB and NURR1 protein levels increase synchronously with HSD3B1 mRNA levels.
The 5′-flanking genomic sequence of HSD3B1 contains a consensus NGFIB-binding motif in the vicinity of the transcription initiation site (Fig. 4A). Specifically, the element at position −117 contains 9 nucleotides that closely resemble the consensus NGFIB-responsive element [NBRE; (A/T)AAAGGTCA] (36). This potential NBRE site attracted our attention, since all NGFIB nuclear receptor gene family members, which include NGFIB, NURR1, and NOR1, are known to be AngII stimulation responsive in H295R cells (21, 37, 38). Importantly, the induction of these genes occurred rapidly, within 1 h, after stimulation even in the presence of CHX (Fig. 4B to D). Such features fulfill the criteria for the potential regulators of HSD3B1. In contrast, while previous studies (21, 37, 38) showed that AngII can also induce the expression of FOS, JUNB, and EGR, neither a potential AP-1 site (to which FOS and JUNB bind) nor the consensus EGR binding motif was found in the sequence of the 5′-flanking region of HSD3B1 (1).
Western blot analysis of NGFIB (Fig. 4E) further demonstrated that the levels of the NGFIB protein in H295R cells were increased rapidly, within 2 h, after AngII stimulation. Following the peak of expression at about 4 h, the levels of NGFIB expression began to decrease by 8 h and returned to near basal levels after 12 h of AngII treatment. These profiles temporally correlate with the kinetics of the rise and fall of the levels of the HSD3B1 transcript (Fig. 4E). These data support the hypothesis that the regulated expression of HSD3B1 mRNA involves the rapid de novo protein synthesis of the short-lived transcription factor NGFIB.
We confirmed that the CHX treatment that blocked HSD3B1 induction indeed blocked AngII-stimulated induction of the NGFIB protein (Fig. 4H), reinforcing the hypothesis that we raised above. We also examined the other members of the NGFIB protein family: NURR1 was increased upon AngII stimulation in a pattern similar to that of NGFIB (Fig. 4F and H). In comparison, the NOR1 protein showed a relatively slow accumulation (Fig. 4G and H): NOR1 peaked at 8 h and remained expressed even 12 h after AngII treatment. These profiles of the NOR1 protein do not account for the transient expression of HSD3B1 mRNA.
AngII-induced NGFIB and NURR1 bind to the NBRE site on the HSD3B1 promoter.
There are many studies describing the AngII response of NGFIB, NURR1, and NOR1 in H295R cells (19, 39, 40), but it remains unexplored whether the corresponding proteins bind to the cognate DNA element in an AngII stimulation-dependent manner. We therefore performed an electrophoretic mobility shift assay (EMSA) to determine whether the AngII-induced NGFIB family proteins bind to the potential NBRE site of the HSD3B1 promoter (Fig. 5A). Nuclear extracts were prepared from H295R cells after 3 h of incubation with AngII or vehicle (3 h treatment was chosen to see the proteins bound to NBRE during the phase of the increase in HSD3B1). We observed that the H295R cell nuclear extracts were able to form two separate complexes, C1 and C2, with the radiolabeled HSD3B1 NBRE oligonucleotide probe (Fig. 5A). Competition assays with nonlabeled probes demonstrated that the complexes C1 and C2 both compete with the wild-type but not the mutant NBRE oligonucleotide probes (see lanes 9 to 12), indicating that both complexes are formed through the sequence of the NBRE.
FIG 5.

AngII-induced NGFIB and NURR1 bind to the NBRE site of the HSD3B1 promoter. (A and B) EMSA analysis for NGFIB binding at the NBRE. A radiolabeled oligonucleotide probe for the HSD3B1 NBRE (positions −130 to −110) was incubated with the nuclear extracts from AngII-treated or vehicle-treated H295R cells. Supershift assays were performed by preincubating the nuclear extracts with the indicated antibodies. Normal IgG was used as a control. A cold competition assay was performed by adding a 100-fold molar excess of unlabeled wild-type (WT) or mutant (Mut) NBRE probe. (C) ChIP of HSD3B1 promoter. After 3 or 12 h treatment with vehicle or AngII, cross-linked nuclear extracts from H295R cells were subject to ChIP assays with anti-NGFIB/NURR1 antibody and analyzed by qPCR using specific primers and a TaqMan probe targeting the DNA fragments containing the HSD3B1 NBRE. Normal rabbit IgG was used as a control for immunoprecipitation. ChIP values are expressed as a percentage of the input amount of chromatin. The results are the means ± SEMs for three independent samples. *, P < 0.01, Bonferroni test.
Notably, the amount of C1 complex profoundly increased upon AngII stimulation (Fig. 5A, lanes 1 to 4; see the bands indicated by a filled arrowhead). Also, the amount of C2 complex increased upon stimulation, but the increase was modest and appreciably less than the level of induction of C1. Thus, it was crucial to determine whether or not the C1 and C2 complexes contained a member of the NGFIB protein family. To this end, we performed gel mobility supershift assays (Fig. 5B) with antibodies against NGFIB (anti-NGFIB; lanes 7 and 8), NURR1 (anti-NURR1; lanes 9 and 10), and NOR1 (anti-NOR1; lanes 11 and 12). We found that whereas neither the C1 complex nor the C2 complex was diminished by anti-NOR1, the antibodies specific to NGFIB (anti-NGFIB) and NURR1 (anti-NURR1) both induced a selective reduction of the C1 complex (Fig. 5B, lanes 7 to 10). Moreover, we found that EMSAs with the antibody recognizing both NGFIB and NURR1 (anti-NGFIB/NURR1) led to a complete loss of the C1 complex with the concomitant generation of a slowly migrating supershifted band (Fig. 5A and B, lanes 5 and 6). Control IgG did not induce any noticeable shifting or reduction of the complexes in the EMSAs (Fig. 5A, lanes 7 and 8). The antibody shift assays thus strongly suggest that the AngII stimulation-induced protein complexes formed on the NBRE site of HSD3B1 are yielded primarily from NGFIB and NURR1.
The recruitment of the NGFIB/NURR1 protein to the promoter region of HSD3B1 was further analyzed by chromatin immunoprecipitation (ChIP) assays (Fig. 5C). H295R cells were incubated with or without AngII. Then, cross-linked, sheared chromatin fragments were immunoprecipitated with anti-NGFIB/NURR1. DNA fragments from the immunoprecipitates were examined by qPCR with a sequence-specific TaqMan probe. We found that 3 h AngII treatment significantly increased the levels of NGFIB/NURR1 binding to the promoter region of HSD3B1 (Fig. 5C). We observed that this AngII-induced binding was decreased to nearly basal levels after 12 h AngII treatment (Fig. 5C). This transient binding property is in parallel with the kinetics of HSD3B1 gene expression. These data indicate that AngII-induced NGFIB/NURR1 protein binds to the HSD3B1 promoter at the time of activation of this gene.
NGFIB and NURR1 have the ability to enhance transcription of HSD3B1.
Next, we performed reporter gene expression assays in H295R cells to know whether NGFIB and NURR1 have the ability to increase the promoter activity of HSD3B1 (Fig. 6A). We found that the reporter activities under the control of the HSD3B1 promoter (a 1,332-bp DNA fragment upstream of the translation start site of HSD3B1) were significantly increased by ectopically expressed NGFIB. Deletion and mutational analyses of the 5′-flanking region of HSD3B1 further demonstrated that the DNA sequence to which NGFIB and NURR1 proteins bound in the EMSA as well as in the ChIP assay (Fig. 5) was indeed responsible for the NGFIB-mediated transactivation of HSD3B1 (Fig. 6A). Importantly, similar to NGFIB, NURR1 increased the activity of the HSD3B1 promoter in a manner that depended on the NBRE sequence (Fig. 6A). In addition, we also found that both NGFIB and NURR1 can activate transcription from an artificial promoter that contains multiple copies of the isolated NBRE sequence of HSD3B1 (Fig. 6B). These activities of NGFIB and NURR1 were entirely abolished when the NBRE sequences were mutated, indicating that this NBRE site is essential and sufficient to induce the transactivation function of NGFIB and NURR1.
The potential cooperation between NGFIB and NURR1 was next examined (Fig. 6C), since these two proteins synchronously accumulate after AngII stimulation and bind to the same NBRE site of the HSD3B1 promoter (Fig. 5). Interestingly, reporter luciferase assays with the HSD3B1 promoter (Fig. 6C) demonstrated that while both NGFIB and NURR1 could independently activate the promoter activities of HSD3B1 in a dose-dependent fashion by approximately 2-fold, concomitant expression of NGFIB and NURR1 resulted in a further enhancement of the promoter activities to approximately less than 4-fold. It seems likely that NGFIB and NURR1 can additively activate HSD3B1 transcription in a dose-dependent manner.
Inactivation of NGFIB and NURR1 attenuates AngII-stimulated HSD3B1 induction.
In the promoter assays, luciferase reporter plasmids were recombinant and episomal. Thus, it remains uncertain whether NGFIB and NURR1 are indeed involved in the regulation of the endogenous native promoter of HSD3B1. In an attempt to clarify this point and further explore a potential contribution of NGFIB and NURR1 to the AngII-responsive induction of HSD3B1, we finally investigated the expression of the endogenous HSD3B1 mRNA under the conditions where NGFIB and NURR1 were inactivated. For this analysis, we used two different experimental tools, small interfering RNAs (siRNAs) targeting NGFIB and NURR1 (Fig. 7) and a dominant negative NGFIB (DN-NGFIB) that is known to competitively inhibit all members of the NGFIB family (29, 30) (Fig. 8).
FIG 8.

DN-NGFIB interferes with AngII-induced HSD3B1 expression in H295R cells. (A) Cell separation scheme for DN-NGFIB-transfected cells (eluate) and control nontransfected cells (flowthrough). The cells were treated with AngII or vehicle and separated with a cell surface marker antibody-based immunomagnetic microbead system. (B) Western blots of the separated cell samples with antibodies against endogenous NGFIB, β-actin, and DN-NGFIB. *, a nonspecific band. (C) Immunoblot of DN-NGFIB-transfected cells (eluate) with anti-C-terminus NGFIB antibody that allows the simultaneous detection of endogenous NGFIB and N-terminally truncated DN-NGFIB. Note that in the transfected cells DN-NGFIB predominates over the endogenously induced NGFIB. WB, Western blotting. (D) Levels of HSD3B1, HSD3B2, StAR, CYP11B2, and NGFIB mRNA in the separated cell samples. All the values (mean ± SEMs, n = 3) were determined by qRT-PCR (the primers used for NGFIB do not cross-react with DN-NGFIB), and the value for vehicle treatment in eluate cells for each gene was set equal to 1 after normalization to the levels of RPLP0.
Because H295R is a very difficult cell line to transfect (41, 42), electroporation was performed with the Nucleofector system (Amaxa Biosystems) for efficient introduction of siRNAs (43, 44). We observed that electroporation of siRNA mixtures, each directed against NGFIB or NURR1, led to a specific knockdown of the target gene without any apparent off-target cross-reactions between the members of the NGFIB family (Fig. 7A and B for mRNA and protein, respectively). In agreement with the previously described role for the NGFIB family in CYP11B2 expression (19–22), knockdown of NGFIB and NURR1 decreased AngII-stimulated induction of CYP11B2 (Fig. 7C, right). Importantly, we also observed that knockdown of NGFIB and NURR1, either each alone or both, led to a significant attenuation of HSD3B1 mRNA induction at any of the dosages of AngII used for stimulation (10 nM and 100 nM) (Fig. 7C, left). These data demonstrate that NGFIB and NURR1 can each play a role in increasing the expression of HSD3B1 in response to AngII. We observed that NOR1 knockdown did not attenuate AngII-induced HSD3B1 expression in H295R cells (Fig. 7D).
In order to further confirm the roles of NGFIB and NURR1, we used DN-NGFIB (Fig. 8). To see the effect of DN-NGFIB on the endogenous gene expression profiles in H295R cells, we used a magnetic cell separation system (MACSelect H-2Kk) that allows selective enrichment of transfected cells and thereby circumvents the inherent problems associated with the low-level transfection efficiency observed for H295R cells. As shown in Fig. 8A, 3 days after cotransfection of DN-NGFIB with a cell surface selection marker (H-2Kk), the cells were treated with either AngII or vehicle for 3 h. Then, immediately after the treatment, the cells were subjected to the MACSelect separation system and isolated into two cell fractions: H-2Kk-positive, transfected cells (eluate) and H-2Kk-negative, nontransfected cells (flowthrough) (Fig. 8A). The preparation of each cell sample was accomplished within 1 h to minimize potential alterations of target gene induction. Importantly, immunoblot analysis of DN-NGFIB confirmed the successful fractionation of the cells (Fig. 8B). Moreover, the expression levels of DN-NGFIB were profoundly higher than those of the endogenously induced NGFIB (Fig. 8C), suggestive of the predominance of DN-NGFIB over the endogenous pathway. Under these conditions, we assessed the influence of AngII stimulation on endogenous HSD3B1 mRNA expression. Notably, qRT-PCR analysis (Fig. 8D) revealed that the induction of this gene was completely blocked by the presence of DN-NGFIB (compare the results for the eluate versus the flowthrough in Fig. 8D). Importantly, the transfected cells (eluate) still possessed the ability to respond to AngII stimulation, as in these cells DN-NGFIB did not inhibit the induction of StAR and endogenous NGFIB mRNA (Fig. 8D). On the other hand, DN-NGFIB caused a suppression of CYP11B2 induction (Fig. 8D), a result congruent with that of a previous study (20) (also see Discussion). Finally, we also confirmed that HSD3B2 was not responsive to AngII, irrespective of the presence of DN-NGFIB.
DISCUSSION
The 3β-HSD enzyme family is comprised of multiple, structurally similar isozymes that are encoded by different genes. The major finding of this study is that the ZG-specific isozyme HSD3B1 (Hsd3b6 for mouse) has a different characteristic from HSD3B2 (Hsd3b1) in terms of the ability to respond to AngII stimulation (see Fig. 9). We showed that AngII stimulation triggers expression of HSD3B1 but not HSD3B2 in human adrenocortical H295R cells. Pharmacological studies demonstrated that the induction of HSD3B1 relies entirely on the proteins that are newly synthesized upon AngII stimulation. The orphan nuclear receptor NGFIB family proteins NGFIB and NURR1 showed a rapid and transient increase in expression in a pattern similar to that of HSD3B1. Moreover, the results from EMSAs, ChIP assays, and reporter-based promoter assays revealed that the HSD3B1 promoter contains a functional NGFIB/NURR1-responsive element to which these proteins bind in response to AngII. Both knockdown of these proteins and overexpression of a dominant negative NGFIB resulted in a reduction of AngII-induced expression of HSD3B1. Taken together, our data demonstrate that HSD3B1 is an AngII-responsive 3β-HSD isoform gene and that the mechanism by which AngII promotes HSD3B1 expression involves the activation of the AT1 receptor downstream pathway, leading to rapid de novo protein synthesis of the nuclear orphan receptors NGFIB and NURR1 (Fig. 9).
FIG 9.
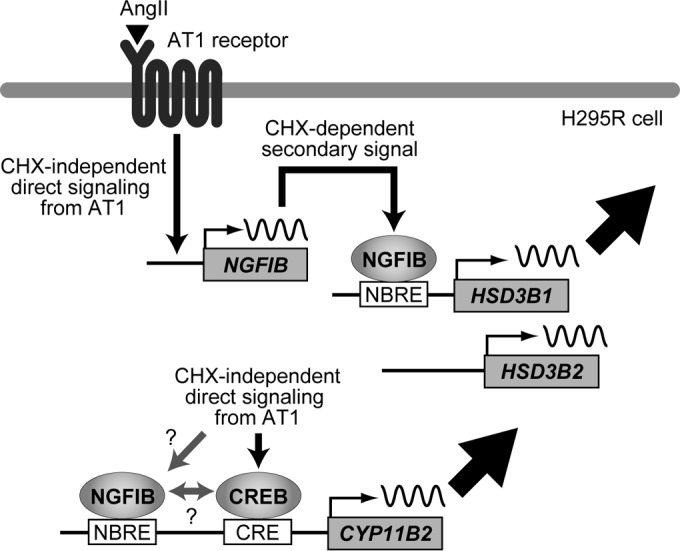
Schematic model showing AngII regulation of HSD3B1 in H295R cells. The AngII-AT1 receptor-NGFIB/NURR1 pathway leads to the upregulation of HSD3B1. CYP11B2 differs from HSD3B1 in relying on a CHX-independent pathway(s) from the AT1 receptor.
Adrenal ZG cells are the principal place where aldosterone is biosynthesized from cholesterol via a series of enzymatic reactions involving 3β-HSD. CYP11B2 (aldosterone synthase) is the last and unique enzyme in the aldosterone biosynthetic pathway exclusively expressed in ZG cells. This spatially limited expression of the enzyme CYP11B2 precludes aldosterone production in the region outside ZG cells (45–47). Moreover, the centripetal blood flow in the adrenal cortex also prevents the precursors of aldosterone in the fasciculata cells from being supplied to ZG cells (which are located in the outer layer of the cortex). Therefore, all the steroid precursors devoted to aldosterone production are synthesized locally in ZG cells. This locality assigns a potential role of importance to the ZG-specific isozyme in the regulation of aldosterone synthesis. We previously showed that in both human and mouse, the adrenals express two distinct 3β-HSD isoform genes with different zone specificities (5–7): one is ubiquitous for ZG and zona fasciculata (ZF) (HSD3B2 for human and Hsd3b1 for mouse), but the other is exclusive to ZG (HSD3B1 for human and Hsd3b6 for mouse). The data in this study therefore extend the difference between the two enzymes from zonal specificity to AngII stimulation reactivity. We showed that HSD3B1 (Hsd3b6) has the ability to respond to AngII. A low-sodium diet also upregulated the expression of the ZG isoform Hsd3b6 in the mouse adrenal gland without affecting the expression of the canonical adrenal isoform Hsd3b1. These data imply that the subtype-specific AngII responses of 3β-HSDs represent a feature that is evolutionally conserved in humans and mice.
At present, it remains unknown whether the enzymes catalyzing the intermediate steps of aldosterone synthesis have a role in changing the capacity of AngII-induced aldosterone production in human adrenal cells. A study based on RNA interference knockdown might be a way to evaluate whether HSD3B1 plays a role. Unfortunately, however, our efforts to achieve 3β-HSD isoform-selective silencing were unsuccessful: all siRNAs that we tested (three independent siRNAs for each subtype) had a severe off-target cross-reaction between the subtypes, a consequence perhaps due to the extremely high sequence similarity between HSD3B1 and HSD3B2 (5, 6). Recently developed subtype-selective monoclonal antibodies to HSD3B1 and HSD3B2 provide clinical evidence that altered expression of the ZG-specific isoform HSD3B1 correlates with the pathology of idiopathic hyperaldosteronism (5). On the basis of the evolutionary conservation, it would be of interest to explore the role of this enzyme by using genetically modified mice specifically lacking Hsd3b6.
The NGFIB nuclear orphan receptor superfamily includes three members, NGFIB (also termed NR4A1), NURR1 (NR4A2), and NOR1 (NR4A3) (48). These transcription factors belong to the nuclear receptor family but have no identified ligand. They regulate transcription through changes in their expression level and phosphorylation (48). Importantly, all three family members are expressed in adrenal ZG cells, while they are also expressed in the ZF with different intensities (19, 49–51). Moreover, previous transcriptome studies demonstrate that all the three NGFIB family members are acutely induced upon AngII stimulation in H295R cells (21, 37, 38) as well as in primary cultures of rat and bovine ZG cells (37, 52). These common responses of NGFIB family members across different species suggest that the AngII induction of these transcription factors may be necessary for the coordinate regulation of steroid synthesis. Consistent with this hypothesis, Nogueira et al. previously demonstrated that overexpression of a dominant negative NGFIB mutant causes a significant reduction of aldosterone synthesis in AngII-stimulated H295R cells (20). However, probably because NGFIB family members have redundant roles, prior studies using targeted disruption of individual family members have not found adrenal phenotypes (53, 54). In the present study, we showed that both NGFIB and NURR1 have the potential to activate HSD3B1 promoter activity. AngII-induced protein induction of NGFIB and NURR1 was indeed accompanied by a concomitant increase of HSD3B1 mRNA expression. In contrast, NOR1 showed a relatively delayed accumulation. Moreover, our in vitro EMSA data further demonstrate that NGFIB and NURR1 are the major components of the protein-DNA complex formed on the NBRE site of the HSD3B1 promoter in H295R cells (note that essentially no residual C1 complex was found after incubation with anti-NGFIB/NURR1 antibody, which does not cross-react with NOR1). Thus, the NOR1 protein in H295R cells appears to target other NBRE sites of the genes under AngII control. It has been shown that subtle changes in the consensus NBRE can affect the transactivation ability of each family member differently (55). The target gene selectivity of NGFIB family members might also be modulated through posttranslational modifications and different heterodimerization abilities (48).
We showed that AngII can induce HSD3B1 but not HSD3B2. Mechanistically, the AngII-NGFIB/NURR1 pathway is integral to the activation of HSD3B1. However, it remains unknown why the same signal pathway does not induce HSD3B2. The HSD3B2 promoter contains NGFIB-responsive elements (1, 13), and these elements have already been implicated in the regulation of this gene's induction upon adrenocorticotropic hormone (ACTH) stimulation (13). Thus, the different responsiveness to AngII that was observed between the two 3β-HSD isoforms cannot be explained by NGFIB alone, suggesting that a yet unknown additional mechanism may be involved in the isoform-selective induction of 3β-HSD. In this respect, it is interesting to note that in the plasmid-based reporter assays, NGFIB could induce only a modest fold increase in the promoter activity of HSD3B1. The same was also true for NURR1. Thus, it is likely that despite the necessity of de novo-synthesized NGFIB and NURR1, the protein accumulation of these transcription factors alone may not be sufficient for inducing the maximal expression of HSD3B1. This probably suggests the requirement for other molecules to be synthesized or posttranslationally modulated upon AngII signaling. Further studies will be needed to understand the isoform-selective AngII regulation of different 3β-HSDs.
We found that the mechanisms controlling HSD3B1 and CYP11B2 are not identical. Notably, the induction of HSD3B1 required rapid de novo protein synthesis, but that of CYP11B2 did not. Moreover, the induction kinetics of HSD3B1 was relatively transient, but CYP11B2 had a continuous increase even after HSD3B1 was decreased to nearly basal levels. These behaviors of CYP11B2 would be somewhat interesting, given a number of studies in the literature showing that the NGFIB family is involved in the mechanisms regulating CYP11B2 (19–22). The CYP11B2 promoter contains two canonical NBRE sites, for which we confirmed anti-NGFIB/NURR1 binding in ChIP assays (T. Ota, M. Doi, and H. Okamura, unpublished results). Moreover, we reproduced a reported reducing effect of DN-NGFIB on AngII-induced CYP11B2 expression in H295R cells (20), indicating that the members of the NGFIB protein family are indeed involved in the upregulation of CYP11B2. However, it is currently difficult to explain how they contribute to the AngII-induced transcription of CYP11B2 that occurs without relying on de novo protein synthesis. Considering that activator transcription factor (ATF)/CREB family members, namely, ATF1, ATF2, CREB, and CREM, have already been shown to activate CYP11B2 transcription via a posttranslational modification-based transactivation mechanism (43, 56, 57), one possible mechanism is that the NGFIB family may interact with AngII-activated ATF/CREB family members to modulate their transactivation function (Fig. 9 depicts this model for discussion): the CYP11B2 promoter contains not only NBRE sites but also a Ca2+/cyclic AMP response element (CRE) to which the ATF/CREB family members bind (43, 57), and a potential interplay between NGFIB and ATF/CREB has been proposed for the regulation of the transcription of CYP11B2 (43) and other genes (58). An alternative explanation is also possible. For example, CHX-independent posttranslational direct modification of the NGFIB family proteins might be involved (Fig. 9). Thus, we would propose that a posttranslational protein-protein interaction(s) and/or modification(s) of the preexisting, basal-level NGFIB family proteins (but not of the newly synthesized ones) might be a part of the mechanisms regulating CYP11B2. Additional experiments will be required to test these hypotheses.
In conclusion, our work revealed that HSD3B1 (Hsd3b6) is an adrenal gland zona glomerulosa-specific 3β-HSD isoform gene that is regulated by AngII. HSD3B1 (Hsd3b6) differs from HSD3B2 (Hsd3b1) in the ability to respond to AngII. We found that the induction of HSD3B1 occurs through a mechanism differing from that of CYP11B2. Notably, the AngII-induced induction of HSD3B1 relies entirely on the de novo protein synthesis of NGFIB and NURR1. Aldosterone synthesis is a complex process potentially subject to many levels of regulation. Future studies aimed at deciphering the mechanism underlying the differential regulation between HSD3B1 and CYP11B2 will help to provide a better understanding of the AngII-dependent dynamic and coordinated regulation of the capacity of aldosterone production by the human adrenal gland.
ACKNOWLEDGMENTS
We thank Masao Matsuoka and Junichiro Yasunaga for technical help and advice for electroporation of siRNA.
T.O. was supported by research fellowships from the Japan Society for the Promotion of Science for Young Scientists. This work was supported in part by a Health Labor Sciences research grant (to H.O.), a Grant-in-Aid for Specially Promoted Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to H.O.), the Funding Program for Next Generation World-Leading Researchers (NEXT program) from the Japan Society for the Promotion of Science (to M.D.), and a grant from the Takeda Science Foundation (to H.O.) and the Mochida Memorial Foundation for Medical and Pharmaceutical Research (to M.D.).
Footnotes
Published ahead of print 4 August 2014
REFERENCES
- 1.Simard J, Ricketts ML, Gingras S, Soucy P, Feltus FA, Melner MH. 2005. Molecular biology of the 3beta-hydroxysteroid dehydrogenase/delta5-delta4 isomerase gene family. Endocr. Rev. 26:525–582. 10.1210/er.2002-0050 [DOI] [PubMed] [Google Scholar]
- 2.Payne AH, Hales DB. 2004. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr. Rev. 25:947–970. 10.1210/er.2003-0030 [DOI] [PubMed] [Google Scholar]
- 3.Luu-The V, Labrie F. 2010. The intracrine sex steroid biosynthesis pathways. Prog. Brain Res. 181:177–192. 10.1016/S0079-6123(08)81010-2 [DOI] [PubMed] [Google Scholar]
- 4.Mason JI, Keeney DS, Bird IM, Rainey WE, Morohashi K, Leers-Sucheta S, Melner MH. 1997. The regulation of 3 beta-hydroxysteroid dehydrogenase expression. Steroids 62:164–168. 10.1016/S0039-128X(96)00176-6 [DOI] [PubMed] [Google Scholar]
- 5.Doi M, Satoh F, Maekawa T, Nakamura Y, Fustin JM, Tainaka M, Hotta Y, Takahashi Y, Morimoto R, Takase K, Ito S, Sasano H, Okamura H. 2014. Isoform-specific monoclonal antibodies against 3beta-hydroxysteroid dehydrogenase/isomerase family provide markers for subclassification of human primary aldosteronism. J. Clin. Endocrinol. Metab. 99:E257-E262. 10.1210/jc.2013-3279 [DOI] [PubMed] [Google Scholar]
- 6.Doi M, Takahashi Y, Komatsu R, Yamazaki F, Yamada H, Haraguchi S, Emoto N, Okuno Y, Tsujimoto G, Kanematsu A, Ogawa O, Todo T, Tsutsui K, van der Horst GTJ, Okamura H. 2010. Salt-sensitive hypertension in circadian clock-deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nat. Med. 16:67-74. 10.1038/nm.2061 [DOI] [PubMed] [Google Scholar]
- 7.Yamamura K, Doi M, Hayashi H, Ota T, Murai I, Hotta Y, Komatsu R, Okamura H. 2014. Immunolocalization of murine type VI 3beta-hydroxysteroid dehydrogenase in the adrenal gland, testis, skin, and placenta. Mol. Cell. Endocrinol. 382:131-138. 10.1016/j.mce.2013.09.014 [DOI] [PubMed] [Google Scholar]
- 8.Doi M. 2012. Circadian clock-deficient mice as a tool for exploring disease etiology. Biol. Pharm. Bull. 35:1385-1391 [DOI] [PubMed] [Google Scholar]
- 9.Ota T, Fustin JM, Yamada H, Doi M, Okamura H. 2012. Circadian clock signals in the adrenal cortex. Mol. Cell. Endocrinol. 349:30-37. 10.1016/j.mce.2011.08.010 [DOI] [PubMed] [Google Scholar]
- 10.Hattangady NG, Olala LO, Bollag WB, Rainey WE. 2012. Acute and chronic regulation of aldosterone production. Mol. Cell. Endocrinol. 350:151–162. 10.1016/j.mce.2011.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nogueira EF, Bollag WB, Rainey WE. 2009. Angiotensin II regulation of adrenocortical gene transcription. Mol. Cell. Endocrinol. 302:230–236. 10.1016/j.mce.2008.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spat A, Hunyady L. 2004. Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol. Rev. 84:489–539. 10.1152/physrev.00030.2003 [DOI] [PubMed] [Google Scholar]
- 13.Bassett MH, Suzuki T, Sasano H, De Vries CJ, Jimenez PT, Carr BR, Rainey WE. 2004. The orphan nuclear receptor NGFIB regulates transcription of 3beta-hydroxysteroid dehydrogenase. Implications for the control of adrenal functional zonation. J. Biol. Chem. 279:37622–37630. 10.1074/jbc.M405431200 [DOI] [PubMed] [Google Scholar]
- 14.Bird IM, Pasquarette MM, Rainey WE, Mason JI. 1996. Differential control of 17 alpha-hydroxylase and 3 beta-hydroxysteroid dehydrogenase expression in human adrenocortical H295R cells. J. Clin. Endocrinol. Metab. 81:2171–2178. 10.1210/jc.81.6.2171 [DOI] [PubMed] [Google Scholar]
- 15.Kutyavin IV, Afonina IA, Mills A, Gorn VV, Lukhtanov EA, Belousov ES, Singer MJ, Walburger DK, Lokhov SG, Gall AA, Dempcy R, Reed MW, Meyer RB, Hedgpeth J. 2000. 3′-Minor groove binder-DNA probes increase sequence specificity at PCR extension temperatures. Nucleic Acids Res. 28:655–661. 10.1093/nar/28.2.655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamura Y, Satoh F, Morimoto R, Kudo M, Takase K, Gomez-Sanchez CE, Honma S, Okuyama M, Yamashita K, Rainey WE, Sasano H, Ito S. 2011. 18-Oxocortisol measurement in adrenal vein sampling as a biomarker for subclassifying primary aldosteronism. J. Clin. Endocrinol. Metab. 96:E1272–E1278. 10.1210/jc.2010-2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sasano H. 2000. The adrenal cortex, p 221–252 Stefaneanu L, Sasano H, Kovacs K. (ed), Molecular and cellular endocrine pathology. Arnold, London, United Kingdom [Google Scholar]
- 18.Nishimoto K, Nakagawa K, Li D, Kosaka T, Oya M, Mikami S, Shibata H, Itoh H, Mitani F, Yamazaki T, Ogishima T, Suematsu M, Mukai K. 2010. Adrenocortical zonation in humans under normal and pathological conditions. J. Clin. Endocrinol. Metab. 95:2296–2305. 10.1210/jc.2009-2010 [DOI] [PubMed] [Google Scholar]
- 19.Bassett MH, Suzuki T, Sasano H, White PC, Rainey WE. 2004. The orphan nuclear receptors NURR1 and NGFIB regulate adrenal aldosterone production. Mol. Endocrinol. 18:279–290. 10.1210/me.2003-0005 [DOI] [PubMed] [Google Scholar]
- 20.Nogueira EF, Xing Y, Morris CA, Rainey WE. 2009. Role of angiotensin II-induced rapid response genes in the regulation of enzymes needed for aldosterone synthesis. J. Mol. Endocrinol. 42:319–330. 10.1677/JME-08-0112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Romero DG, Rilli S, Plonczynski MW, Yanes LL, Zhou MY, Gomez-Sanchez EP, Gomez-Sanchez CE. 2007. Adrenal transcription regulatory genes modulated by angiotensin II and their role in steroidogenesis. Physiol. Genomics 30:26–34. 10.1152/physiolgenomics.00187.2006 [DOI] [PubMed] [Google Scholar]
- 22.Romero DG, Gomez-Sanchez EP, Gomez-Sanchez CE. 2010. Angiotensin II-regulated transcription regulatory genes in adrenal steroidogenesis. Physiol. Genomics 42A:259–266. 10.1152/physiolgenomics.00098.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giroud CJ, Stachenko J, Venning EH. 1956. Secretion of aldosterone by the zona glomerulosa of rat adrenal glands incubated in vitro. Proc. Soc. Exp. Biol. Med. 92:154–158. 10.3181/00379727-92-22416 [DOI] [PubMed] [Google Scholar]
- 24.Bird IM, Hanley NA, Word RA, Mathis JM, McCarthy JL, Mason JI, Rainey WE. 1993. Human NCI-H295 adrenocortical carcinoma cells: a model for angiotensin-II-responsive aldosterone secretion. Endocrinology 133:1555–1561. 10.1210/en.133.4.1555 [DOI] [PubMed] [Google Scholar]
- 25.Rainey WE, Saner K, Schimmer BP. 2004. Adrenocortical cell lines. Mol. Cell. Endocrinol. 228:23–38. 10.1016/j.mce.2003.12.020 [DOI] [PubMed] [Google Scholar]
- 26.Doi M, Ishida A, Miyake A, Sato M, Komatsu R, Yamazaki F, Kimura I, Tsuchiya S, Kori H, Seo K, Yamaguchi Y, Matsuo M, Fustin J-M, Tanaka R, Santo Y, Yamada H, Takahashi Y, Araki M, Nakao K, Aizawa S, Kobayashi M, Obrietan K, Tsujimoto G, Okamura H. 2011. Circadian regulation of intracellular G-protein signalling mediates intercellular synchrony and rhythmicity in the suprachiasmatic nucleus. Nat. Commun. 2:327. 10.1038/ncomms1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doi M, Cho S, Yujnovsky I, Hirayama J, Cermakian N, Cato AC, Sassone-Corsi P. 2007. Light-inducible and clock-controlled expression of MAP kinase phosphatase 1 in mouse central pacemaker neurons. J. Biol. Rhythms 22:127-139. 10.1177/0748730406298332 [DOI] [PubMed] [Google Scholar]
- 28.Cormack B. 2001. Directed mutagenesis using the polymerase chain reaction. Curr. Protoc. Mol. Biol. Chapter 8:Unit 8.5. 10.1002/0471142727.mb0805s37 [DOI] [PubMed] [Google Scholar]
- 29.Cheng LE, Chan FK, Cado D, Winoto A. 1997. Functional redundancy of the Nur77 and Nor-1 orphan steroid receptors in T-cell apoptosis. EMBO J. 16:1865–1875. 10.1093/emboj/16.8.1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song KH, Park JI, Lee MO, Soh J, Lee K, Choi HS. 2001. LH induces orphan nuclear receptor Nur77 gene expression in testicular Leydig cells. Endocrinology 142:5116–5123. 10.1210/endo.142.12.8525 [DOI] [PubMed] [Google Scholar]
- 31.Bird IM, Mason JI, Rainey WE. 1994. Regulation of type 1 angiotensin II receptor messenger ribonucleic acid expression in human adrenocortical carcinoma H295 cells. Endocrinology 134:2468–2474. 10.1210/endo.134.6.8194473 [DOI] [PubMed] [Google Scholar]
- 32.Clark BJ, Pezzi V, Stocco DM, Rainey WE. 1995. The steroidogenic acute regulatory protein is induced by angiotensin II and K+ in H295R adrenocortical cells. Mol. Cell. Endocrinol. 115:215–219. 10.1016/0303-7207(95)03683-0 [DOI] [PubMed] [Google Scholar]
- 33.Denner K, Rainey WE, Pezzi V, Bird IM, Bernhardt R, Mathis JM. 1996. Differential regulation of 11 beta-hydroxylase and aldosterone synthase in human adrenocortical H295R cells. Mol. Cell. Endocrinol. 121:87–91. 10.1016/0303-7207(96)03853-1 [DOI] [PubMed] [Google Scholar]
- 34.Bird IM, Mason JI, Rainey WE. 1998. Protein kinase A, protein kinase C, and Ca(2+)-regulated expression of 21-hydroxylase cytochrome P450 in H295R human adrenocortical cells. J. Clin. Endocrinol. Metab. 83:1592–1597. 10.1210/jc.83.5.1592 [DOI] [PubMed] [Google Scholar]
- 35.Holland OB, Mathis JM, Bird IM, Rainey WE. 1993. Angiotensin increases aldosterone synthase mRNA levels in human NCI-H295 cells. Mol. Cell. Endocrinol. 94:R9–R13. 10.1016/0303-7207(93)90175-J [DOI] [PubMed] [Google Scholar]
- 36.Wilson TE, Fahrner TJ, Johnston M, Milbrandt J. 1991. Identification of the DNA binding site for NGFI-B by genetic selection in yeast. Science 252:1296–1300. 10.1126/science.1925541 [DOI] [PubMed] [Google Scholar]
- 37.Nogueira EF, Vargas CA, Otis M, Gallo-Payet N, Bollag WB, Rainey WE. 2007. Angiotensin-II acute regulation of rapid response genes in human, bovine, and rat adrenocortical cells. J. Mol. Endocrinol. 39:365–374. 10.1677/JME-07-0094 [DOI] [PubMed] [Google Scholar]
- 38.Szekeres M, Nadasy GL, Turu G, Supeki K, Szidonya L, Buday L, Chaplin T, Clark AJ, Hunyady L. 2010. Angiotensin II-induced expression of brain-derived neurotrophic factor in human and rat adrenocortical cells. Endocrinology 151:1695–1703. 10.1210/en.2009-1060 [DOI] [PubMed] [Google Scholar]
- 39.Szekeres M, Turu G, Orient A, Szalai B, Supeki K, Cserzo M, Varnai P, Hunyady L. 2009. Mechanisms of angiotensin II-mediated regulation of aldosterone synthase expression in H295R human adrenocortical and rat adrenal glomerulosa cells. Mol. Cell. Endocrinol. 302:244–253. 10.1016/j.mce.2008.12.015 [DOI] [PubMed] [Google Scholar]
- 40.Romero DG, Plonczynski M, Vergara GR, Gomez-Sanchez EP, Gomez-Sanchez CE. 2004. Angiotensin II early regulated genes in H295R human adrenocortical cells. Physiol. Genomics 19:106–116. 10.1152/physiolgenomics.00097.2004 [DOI] [PubMed] [Google Scholar]
- 41.Ye P, Nakamura Y, Lalli E, Rainey WE. 2009. Differential effects of high and low steroidogenic factor-1 expression on CYP11B2 expression and aldosterone production in adrenocortical cells. Endocrinology 150:1303–1309. 10.1210/en.2008-0667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Romero DG, Welsh BL, Gomez-Sanchez EP, Yanes LL, Rilli S, Gomez-Sanchez CE. 2006. Angiotensin II-mediated protein kinase D activation stimulates aldosterone and cortisol secretion in H295R human adrenocortical cells. Endocrinology 147:6046–6055. 10.1210/en.2006-0794 [DOI] [PubMed] [Google Scholar]
- 43.Nogueira EF, Rainey WE. 2010. Regulation of aldosterone synthase by activator transcription factor/cAMP response element-binding protein family members. Endocrinology 151:1060–1070. 10.1210/en.2009-0977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Romero DG, Yanes LL, de Rodriguez AF, Plonczynski MW, Welsh BL, Reckelhoff JF, Gomez-Sanchez EP, Gomez-Sanchez CE. 2007. Disabled-2 is expressed in adrenal zona glomerulosa and is involved in aldosterone secretion. Endocrinology 148:2644–2652. 10.1210/en.2006-1509 [DOI] [PubMed] [Google Scholar]
- 45.Domalik LJ, Chaplin DD, Kirkman MS, Wu RC, Liu WW, Howard TA, Seldin MF, Parker KL. 1991. Different isozymes of mouse 11 beta-hydroxylase produce mineralocorticoids and glucocorticoids. Mol. Endocrinol. 5:1853–1861. 10.1210/mend-5-12-1853 [DOI] [PubMed] [Google Scholar]
- 46.Ogishima T, Suzuki H, Hata J, Mitani F, Ishimura Y. 1992. Zone-specific expression of aldosterone synthase cytochrome P-450 and cytochrome P-45011 beta in rat adrenal cortex: histochemical basis for the functional zonation. Endocrinology 130:2971–2977. 10.1210/en.130.5.2971 [DOI] [PubMed] [Google Scholar]
- 47.Pascoe L, Jeunemaitre X, Lebrethon MC, Curnow KM, Gomez-Sanchez CE, Gasc JM, Saez JM, Corvol P. 1995. Glucocorticoid-suppressible hyperaldosteronism and adrenal tumors occurring in a single French pedigree. J. Clin. Invest. 96:2236–2246. 10.1172/JCI118279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maxwell MA, Muscat GE. 2006. The NR4A subgroup: immediate early response genes with pleiotropic physiological roles. Nucl. Recept. Signal. 4:e002. 10.1621/nrs.04002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson TE, Mouw AR, Weaver CA, Milbrandt J, Parker KL. 1993. The orphan nuclear receptor NGFI-B regulates expression of the gene encoding steroid 21-hydroxylase. Mol. Cell. Biol. 13:861–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davis IJ, Lau LF. 1994. Endocrine and neurogenic regulation of the orphan nuclear receptors Nur77 and Nurr-1 in the adrenal glands. Mol. Cell. Biol. 14:3469–3483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernandez PM, Brunel F, Jimenez MA, Saez JM, Cereghini S, Zakin MM. 2000. Nuclear receptors Nor1 and NGFI-B/Nur77 play similar, albeit distinct, roles in the hypothalamo-pituitary-adrenal axis. Endocrinology 141:2392–2400. 10.1210/endo.141.7.7562 [DOI] [PubMed] [Google Scholar]
- 52.Romero DG, Plonczynski MW, Welsh BL, Gomez-Sanchez CE, Zhou MY, Gomez-Sanchez EP. 2007. Gene expression profile in rat adrenal zona glomerulosa cells stimulated with aldosterone secretagogues. Physiol. Genomics 32:117–127. 10.1152/physiolgenomics.00145.2007 [DOI] [PubMed] [Google Scholar]
- 53.Crawford PA, Sadovsky Y, Woodson K, Lee SL, Milbrandt J. 1995. Adrenocortical function and regulation of the steroid 21-hydroxylase gene in NGFI-B-deficient mice. Mol. Cell. Biol. 15:4331–4316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zetterstrom RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. 1997. Dopamine neuron agenesis in Nurr1-deficient mice. Science 276:248–250. 10.1126/science.276.5310.248 [DOI] [PubMed] [Google Scholar]
- 55.Murphy EP, Dobson AD, Keller C, Conneely OM. 1996. Differential regulation of transcription by the NURR1/NUR77 subfamily of nuclear transcription factors. Gene Expr. 5:169–179 [PMC free article] [PubMed] [Google Scholar]
- 56.Bassett MH, Zhang Y, White PC, Rainey WE. 2000. Regulation of human CYP11B2 and CYP11B1: comparing the role of the common CRE/Ad1 element. Endocr. Res. 26:941–951. 10.3109/07435800009048620 [DOI] [PubMed] [Google Scholar]
- 57.Clyne CD, Zhang Y, Slutsker L, Mathis JM, White PC, Rainey WE. 1997. Angiotensin II and potassium regulate human CYP11B2 transcription through common cis-elements. Mol. Endocrinol. 11:638–649. 10.1210/mend.11.5.9920 [DOI] [PubMed] [Google Scholar]
- 58.Mynard V, Latchoumanin O, Guignat L, Devin-Leclerc J, Bertagna X, Barre B, Fagart J, Coqueret O, Catelli MG. 2004. Synergistic signaling by corticotropin-releasing hormone and leukemia inhibitory factor bridged by phosphorylated 3′,5′-cyclic adenosine monophosphate response element binding protein at the Nur response element (NurRE)-signal transducers and activators of transcription (STAT) element of the proopiomelanocortin promoter. Mol. Endocrinol. 18:2997–3010. 10.1210/me.2003-0417 [DOI] [PubMed] [Google Scholar]