

Abstract
Neuroinflammation and endoplasmic reticulum (ER) stress are associated with many neurological diseases. Here, we have examined the interaction between ER stress and JAK/STAT-dependent inflammation in glial cells. We show that ER stress is present in the central nervous system (CNS) concomitant with inflammation and astrogliosis in the multiple sclerosis (MS) mouse model of experimental autoimmune encephalomyelitis (EAE). Astrocytes do not easily succumb to ER stress but rather activate an inflammatory program involving activation of STAT3 in a JAK1-dependent fashion. ER stress-induced activation of the JAK1/STAT3 axis leads to expression of interleukin 6 (IL-6) and several chemokines. Moreover, the activation of STAT3 signaling is dependent on PERK, a central component of the ER stress response, which we show is phosphorylated by JAK1. Disruption of PERK abrogates ER stress-induced activation of STAT3 and subsequent gene expression. Additionally, ER-stressed astrocytes, via paracrine signaling, can stimulate activation of microglia, leading to production of IL-6 and oncostatin M (OSM). These IL-6 cytokines can then synergize with ER stress in astrocytes to drive inflammation. Together, this work describes a new PERK/JAK1/STAT3 signaling pathway that elicits a feed-forward inflammatory loop involving astrocytes and microglia to drive neuroinflammation, which may be relevant in diseases such as MS.
INTRODUCTION
The accumulation of misfolded proteins and the induction of endoplasmic reticulum (ER) stress is prevalent in many neurodegenerative diseases, including Alzheimer's disease (AD), Huntington's disease (HD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS) (1). Excessive accumulation of misfolded proteins can be brought on by protein mutations and polyglutamine expansion, as in the case of neurodegenerative diseases, such as ALS, AD, HD, PD, and others, environmental factors, disruption of ER Ca2+, amino acid deprivation, infection, and inflammation (1–4). ER stress activates the highly conserved unfolded protein response (UPR) that transmits both adaptive and apoptotic signals from the ER to the cytosol and nucleus. This pathway promotes restoration of homeostasis or eliminates the irreparably damaged cell through apoptosis (5). The UPR is initiated by three trans-ER membrane proteins, protein kinase R (PKR)-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1), which are activated in response to misfolded protein accumulation in the ER lumen (2). Following activation of the UPR, protein synthesis is attenuated by the PERK-dependent phosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF2α) to reduce the protein folding burden on the ER (6), and the expression of ER resident molecular chaperones is selectively upregulated as a means to restore homeostasis (2). Recently, a small-molecule kinase inhibitor of PERK, GSK2606414, was shown to attenuate disease in a mouse model of prion infection, suggesting that PERK may be an important target in the treatment of neurodegenerative diseases (7). In the context of neurodegenerative diseases, considerable focus has been placed on the neurotoxic effects of ER stress. However, in neurodegenerative diseases and MS, markers of ER stress are also observed in astrocytes (8–10). Importantly, astrocytes are the most numerous cell type in the central nervous system (CNS) and are immunologically active and able to respond to extracellular cues with the production of numerous cytokines, chemokines, and reactive species (11). While the significance of ER stress in astrocytes has not been fully examined, it is possible that it contributes to the chronic inflammation observed in neurological diseases, including MS.
Not only does the UPR maintain cellular homeostasis, it is also essential for innate and adaptive immunity (12). ER stress induces a systemic acute-phase response involving the expression of serum amyloid P component and C-reactive protein (13), drives the activation of NF-κB (3), and elicits the production of cytokines and chemokines, such as interleukin 6 (IL-6), IL-8, and chemokine (C-C motif) ligand 2 (CCL2) (14). Additionally, ER stress greatly enhances the production of beta interferon (IFN-β), IL-6, IL-1β, and IL-23 in response to the bacterial component lipopolysaccharide (LPS) (15, 16). The interaction between ER stress and inflammation potentially results in a nonresolving inflammatory loop in which ER stress and inflammation drive one another (3). Thus, there is an integral relationship between the ER stress pathway and immunological function.
Janus kinases (JAKs) and signal transducers and activators of transcription (STATs) are critical immunological signaling molecules. Together, the 4 JAKs (JAK1, JAK2, JAK3, and TYK2) and 7 STATs (STATs 1, 2, 3, 4, 5a, 5b, and 6) mediate the biological actions of approximately 60 cytokines and IFNs (17). Among this group is the IL-6 family of cytokines which includes IL-6 itself and oncostatin M (OSM), which are increased in the CNS of MS patients (18, 19). IL-6 and OSM signal via their respective ligand-binding receptor and the common gp130 coreceptor. These membrane-spanning receptors do not have kinase activity but are constitutively associated with JAKs on the cytoplasmic side, juxtaposed to the plasma membrane (20). Following receptor ligation, JAKs tyrosine-phosphorylate gp130, which provides a docking site for STATs, predominantly STAT3. Following recruitment to the receptor complex, STAT3 is phosphorylated by JAKs, leading to transcriptional activation (20). Activated STAT3 then drives the expression of acute-phase proteins as well as a number of cytokines and chemokines, including IL-6, CCL2, and CCL20 (21, 22). Beyond cytokine receptors, JAKs also bind and phosphorylate PKR to promote PKR-mediated eIF2α phosphorylation and translation repression in response to IFNs (23).
Much of the inflammatory gene expression brought on by ER stress has been attributed to NF-κB activation (3). However, recent evidence suggests there may be important interactions between the ER, UPR, and STAT signaling. Deletion of ERp57, an ER resident thiol disulfide oxidoreductase, leads to enhanced STAT3 signaling (24). Additionally, growth hormone-induced STAT5 activation is prolonged by ER stress (25). The STAT3-activating cytokine OSM increases the expression of glucose-regulated protein 78 (GRP78), a key regulator of UPR activation, but does not induce ER stress (26). The STAT1-activating cytokine IFN-γ stimulates ER stress and apoptosis in myelin-producing oligodendrocytes, and activation of PERK in this context provides protection in the MS mouse model of experimental autoimmune encephalomyelitis (EAE) (27, 28), indicating that manipulation of the ER stress response may have therapeutic potential for treatment of MS (29). In glial cells, ER stress enhances IL-6 expression induced by IFN-γ and prostaglandin E2 (PGE2) (30). Moreover, ER stress activates STAT3, which is important in the induction of secretory machinery (31). How STAT3 is activated in response to ER stress and the functional significance in relation to inflammation have not been addressed. In this study, we demonstrate that STAT3 is activated through a novel PERK-JAK1 pathway and has an essential role in ER stress-induced inflammation.
MATERIALS AND METHODS
Reagents.
PERK-deficient mouse embryonic fibroblasts (MEFs) were from Ronald Wek (University of Indiana), and INS832/13 cells were obtained from Chris Newgard (Duke University). PERK-myc vector was from David Ron via Addgene (Addgene plasmid 21814). TaqMan primers and Silencer predesigned select small interfering RNA (siRNA) were purchased from Life Technologies. WST-1 {4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate} and Fugene 6 were purchased from Roche. Enzyme-linked immunosorbent assays (ELISAs) were from BioLegend. Mouse cytokine/chemokine multiplex assay was from EMD Millipore. Antibodies (Abs) for phosphorylated JAK1, phosphorylated and total STAT3, eIF2α, total caspase 3, and PERK were from Cell Signaling Technologies. JAK1 antibody was from Santa Cruz Biotechnology. GAPDH antibody was from Abcam. Total phosphotyrosine antibody was from EMD Millipore. Thapsigargin (Thaps), tunicamycin, and pyridone 6 (P6; JAK inhibitor I) were from Calbiochem. Culture media, fetal bovine serum (FBS), HEPES, nonessential amino acids, l-glutamine, and penicillin-streptomycin were from Cellgro. Nonessential amino acids and gentamicin were from Lonza. Mouse IL-6 and OSM were from R&D Systems. Epidermal growth factor (EGF) and fibroblast growth factor (FGF) were from Miltenyi Biotech. Heparin was from Stemcell Technologies. GSK2606414 was from EMD Millipore. AZD1480, a JAK1/2 inhibitor (32, 33), and AZ-JAK1, a JAK1 inhibitor, were provided by AstraZeneca.
Mice and primary cell preparations.
C57BL/6 mice were bred and housed in the animal facility at the University of Alabama in Birmingham under the care of the animal resources program. Primary murine astrocyte or microglial cultures were prepared as previously described (34). Astrocytes or microglia were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS, 16 mM HEPES, 1× nonessential amino acids, 2 mM l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 50 μg/ml gentamicin. Astrocytes were separated from microglia by shaking at 400 rpm for 2 h, and astrocyte cultures contained >90% glial fibrillary acidic protein (GFAP)-positive cells, as determined by immunofluorescence microscopy. Following shaking, microglia-containing supernatants were centrifuged at 1,000 rpm for 5 min, and microglia were resuspended in fresh medium and plated.
Neural precursor cell (NPC) isolation and propagation were adapted from reference 35. Briefly, E15 embryos were collected and the telencephalon was isolated. The tissue was then triturated in cold medium and allowed to settle by gravity for 2 min. The cell-containing supernatant was then passed through a 100-μm-pore-size cell strainer and centrifuged at 110 × g for 5 min. The cells were resuspended at a density of 2 × 105 cells/ml and grown on nontreated culture plates. Cells were grown as free-floating neurospheres and passaged as needed by gentle trituration. For experiments, NPCs were cultured as adherent cells on poly-d-lysine- and laminin-coated tissue culture plates. NPC medium contained DMEM–F-12 (1:1), 2% B27 without vitamin A (Neurobrew; Miltenyi Biotech), 2 mM l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, 20 ng/ml EGF, 10 ng/ml FGF, and 0.0002% heparin.
EAE induction and assessment.
Active EAE was induced as previously described (36). Eight- to 12-week-old C57BL/6 mice were immunized subcutaneously with 200 μg of myelin oligodendrocyte glycoprotein peptide (MOG35-55) emulsified in Complete Freund's adjuvant (supplemented with 2 mg/ml of Mycobacterium tuberculosis) and injected intraperitoneally (i.p.) on days 0 and 2 with 500 ng pertussis toxin. Assessment of classical EAE was as follows: 0, no disease; 1, decreased tail tone; 2, hind limb weakness or partial paralysis; 3, complete hind limb paralysis; 4, front and hind limb paralysis; and 5, moribund state (37).
Immunoblotting.
Cells were washed twice with phosphate-buffered saline (PBS) and lysed with immunoprecipitation (IP) lysis buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.5% NP-40, 1 mM phenylmethanesulfonyl fluoride, 25 μg/ml leupeptin, 25 μg/ml aprotinin, and 1× phosphatase inhibitor cocktail [Pierce, Rockford, IL]), as previously described (38). Protein concentrations were determined using the BCA assay (Pierce, Rockford, IL). Equal amounts of protein from each sample were solubilized in Laemmli sample buffer (2% SDS) and heated for 5 min at 95°C. Proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose, and the membranes were blocked in 5% milk followed by an overnight incubation at 4°C with primary Ab diluted in 5% bovine serum albumin (BSA) or milk, according to the manufacturer's recommendation. Horseradish peroxidase-conjugated donkey anti-rabbit or donkey anti-mouse (1:4,000 dilution) secondary Ab was incubated for 1 h at room temperature, followed by detection with enhanced chemiluminescence. Films were digitized with an Epson Perfection V300 photo scanner and quantified using ImageJ.
qRT-PCR.
RNA was isolated using TRIzol (Sigma-Aldrich) as previously described (39). RNA was quantified using a NanoDrop system (NanoDrop Technologies). One microgram of RNA was used for cDNA synthesis using Moloney murine leukemia virus (MMLV) reverse transcriptase (Promega). The cDNA was analyzed by quantitative PCR performed using TaqMan gene expression assays according to the manufacturer's instructions in an ABI Prism 7500 system (Applied Biosystems). Reactions were carried out in 20 μl and analyzed using the threshold cycle (ΔΔCT) method.
JAK1 kinase assay.
Active recombinant JAK1 (Life Technologies) was incubated in kinase buffer (25 mM Tris [pH 7.5], 10 mM MgCl2, 0.5 mM EGTA, 0.5 mM Na3VO4, 5 mM β-glycerophosphate, 2.5 mM dithiothreitol [DTT], 0.01% Triton X-100, 100 μM ATP, and 3 μCi [γ-32P]ATP/reaction), with 2 μg of recombinant PERK (residues 536 to 1116) (Enzo Life Sciences) as a substrate, for 20 min at 30°C. Reactions were carried out in a final volume of 30 μl. Reactions were terminated by the addition of Laemmli sample buffer and heating at 95°C for 5 min. Reactions were resolved by SDS-PAGE, and gels were Coomassie stained. Gels were dried and exposed to a phosphor screen for 1 h. The screens were then imaged on a Typhoon scanner.
Nano-HPLC electrospray ionization multistage tandem mass spectrometry.
Enriched proteins were resolved by 4 to 12% SDS Bis-Tris PAGE and stained with colloidal blue (Invitrogen). For in-gel digestion, the band of interest was excised, equilibrated in 100 mM ammonium bicarbonate, reduced with 10 mM DTT, carbidomethylated with 55 mM iodoacetamide, dehydrated, and digested with trypsin gold (Promega). The digested peptides were concentrated under vacuum, resolubilized in 0.1% formic acid, and injected onto a Surveyor Plus high-performance liquid chromatography (HPLC) system (Thermo Scientific) using a split flow configuration on the back end of a 100-μm-internal-diameter by 13-cm pulled-tip C18 column (Jupiter C18, 300 Å, 5 μm; Phenomenex). Peptide fractions were then directly sprayed into a Thermo Orbitrap Velos Pro hybrid mass spectrometer equipped with a nanoelectrospray source over a 1-h gradient set to increase over 60 min from 0% to 50% acetonitrile in distilled H2O containing 0.1% formic acid with a flow rate of 0.3 μl/min. Following each parent ion scan, fragmentation data were collected on the top most intense 10 ions, in CID mode, with the following instrument settings: spray voltage of 1.9 kV; capillary temperature of 170°C; 1 microscan, with the scan window set at 300 to 2,000 m/z; and maximum inject times of 500 ms and 150 ms for the parent ion and fragmentation scans, respectively. The parent ion scans were obtained at 60-K resolution in the Orbitrap with a minimum signal threshold of 2,000 counts. The activation settings were as follows: charge state, +2; isolation width, 2.0 m/z; normalized collision energy, 35.0; activation Q, 0.250; and activation time, 25 ms. For data-dependent settings, monoisotopic precursor selection was enabled, in addition to charge state screening with rejection of 1+ ions and a dynamic exclusion with a repeat count of 2, a repeat duration of 30 s, an exclusion list size of 500, and an exclusion duration of 90 s.
The XCalibur RAW files collected in profile mode were centroided and converted to MzXML using ReADW v. 3.5.1. The .mgf files were created using MzXML2Search for all scans with a precursor mass between 300 Da and 1,200 Da. The data were searched using SEQUEST set for three maximum missed cleavages, a precursor mass window of 20 ppm, trypsin digestion, a fixed modification at C (57.0293), and variable modifications at M (15.9949), S, T, and Y (79.96633). For the fragment-ion mass tolerance, 0.0 Da was used. Searches were performed with a human subset of the UniRef100 database with protein sequences specific to these experiments. Peptide identifiers were filtered using Scaffold (Proteome Software), with cutoff values set at a peptide length of >5 amino acids, peptide probability of >90% confidence interval (CI), the number of peptides/protein greater than or equal to 2, and protein probability of >99% CI, resulting in protein identifiers with >99.9% confidence. Posttranslational modifications were further analyzed using an “Ascore” algorithm (40), where localization probability and the Ascore were assessed. Each spectra representing the posttranslational modification (PTM) of interest that passed each screen mentioned was then manually assessed for accuracy, and the best representative spectra for each PTM was then manually annotated.
ELISA.
Culture supernatants (100 μl, undiluted) were collected and assayed by ELISA for murine IL-6 and CCL2 or by multiplex ELISA according to the manufacturer's protocol.
Transfections.
Primary astrocytes were transfected with the indicated siRNA (100 to 150 pmol per 35-mm well) using Lipofectamine RNA interference (RNAi) Max (Life Technologies) according to the manufacturer's protocol. Cells were used for experiments 48 to 72 h posttransfection. Primary astrocytes were transfected with 2 μg plasmid DNA per 35-mm well using Fugene 6 according to the manufacturer's protocol. Cells were used for experiments 24 h posttransfection.
WST-1 assay.
WST-1 assay (Roche) was carried out according to the manufacturer's instructions and as previously described (41). Briefly, cells were plated in triplicate in a 96-well plate and treated as indicated. Following treatments, 10 μl of the WST-1 reagent was added, and the cells were incubated for 2 h. Viable, metabolically active cells cleave WST-1, a tetrazolium salt, resulting in a soluble formazan dye (42). The amount of formazan dye was measured by absorbance at 450 nm with a subtraction wavelength of 655 nm.
Statistics.
When comparing more than two data sets, significance was determined by one-way analysis of variance (ANOVA) with post hoc analysis. For two data sets, Student's t test was used. P values of <0.05 were considered statistically significant.
RESULTS
ER stress and inflammatory signaling are increased in the CNS during EAE.
ER stress has been observed in the CNS of MS patients and in mice during EAE (43–45). To confirm these previous reports, mice were immunized with MOG peptide to induce EAE (Fig. 1A), and markers of ER stress were examined over time (Fig. 1B to D). We observed PERK activation, as demonstrated by increased mobility shift and eIF2α phosphorylation (Fig. 1B), and increased expression of ER stress-responsive genes, including the astrocyte-selective transcription factor old astrocyte specifically induced substance (OASIS) (46) (Fig. 1C and D). These data confirm the presence of ER stress in the CNS during EAE. In addition to ER stress, inflammatory signaling was also apparent during the same time course. This included increased activation-associated phosphorylation of STAT3 (Fig. 1B), increased expression of IL-6 and CCL2, and increased expression of the astrogliosis marker GFAP in the brain and spinal cord (Fig. 1C and D). The upregulation of GFAP and OASIS suggested that astrocytes were experiencing ER stress, consistent with observations of MS brain lesions in which ER stress is observed in multiple cell types, including astrocytes (44). As astrocytes have a key role in regulating inflammation in the CNS, we next examined the vulnerability and inflammatory reaction of astrocytes in response to ER stress.
FIG 1.

STAT3 activation, ER stress, and inflammation are present in the CNS during EAE. (A) Mice were immunized with MOG peptide to induce EAE, and disease was scored over time. Brain tissue was collected from EAE mice at the indicated time points and analyzed by immunoblotting (B) or by reverse transcription-quantitative PCR (qRT-PCR) (C). (D) Spinal cord tissue was collected from EAE mice at the indicated time points and analyzed by qRT-PCR.
Astrocytes are highly resistant to ER stress-induced cell death.
Multiple investigations have shown that neurons and related cell lines are vulnerable to ER stress-induced apoptosis (47–50). We now find that primary murine glial cells (astrocytes and microglia) are remarkably resistant to ER stress-induced cell death. Astrocytes, microglia, and NPCs were treated with the ER stress-inducing agents thapsigargin (Thaps) or tunicamycin (Tunic), and cell viability was assessed by WST-1 assay (Fig. 2A). Astrocytes were unaffected by Thaps at all concentrations tested and only modestly sensitive to high concentrations of Tunic, while microglia were slightly more sensitive. Consistent with this, there was no detectable caspase-3 cleavage in astrocytes following a 24-h exposure to relatively high concentrations of Thaps or Tunic (Fig. 2B). Osmotic stress (OS) was included as a positive control. NPCs were highly susceptible to ER stress-induced cell death (Fig. 2A) and displayed increased caspase-3 cleavage (see Fig. 4B). Despite the absence of cell death, astrocytes activate a robust UPR, as evident by PERK activation (increased mobility shift), increased phosphorylation of eIF2α (Fig. 2C), and the rapid induction of the ER stress-induced transcription factor CHOP (Fig. 2D). Concomitant with UPR activation, we observed an inflammatory reaction in astrocytes in response to ER stress, consistent with that of other cell types (51–53). Treatment of astrocytes with Thaps increased mRNA expression of IL-6 and the chemokines CCL2 and CCL20 (Fig. 3A). We also confirmed that ER stress increased IL-6 and CCL2 protein levels. For the experiments shown in Fig. 3B and C, cells were treated transiently (2 h) with Thaps or Tunic, respectively, followed by washing and incubation in fresh medium for 24 h to allow proper protein folding and secretion. ER stress significantly increased IL-6 and CCL2 protein levels (Fig. 3B and C). The transient ER stress paradigm is not strictly necessary, as we also observed similar levels of IL-6 protein even in the continued presence of Thaps for 16 h (Fig. 3D). Neither prolonged nor transient Thaps exposure resulted in caspase-3 cleavage (Fig. 3D). Together, these data indicate that ER stress does not easily impair astrocytes but elicits an inflammatory reaction that may contribute to a neuroinflammatory environment.
FIG 2.

Astrocytes are resistant to ER stress-induced cell death. (A) Primary astrocytes, NPCs, and microglia were treated with Thaps (0.03 to 3 μM) or Tunic (0.1 to 10 μM) for 24 h followed by assessment of cell viability by WST-1 assay. (B) Astrocytes were treated with Thaps or Tunic for 24 h followed by immunoblotting for cleaved caspase 3. Osmotic stress (OS; 500 mM sucrose for 4 h) was included as a positive control. Astrocytes were treated with Thaps (1 μM, 2 h) followed by immunoblotting (C), and CHOP mRNA was measured by qRT-PCR at the indicated times (D).
FIG 4.

ER stress stimulates JAK1-dependent STAT3 activation. Astrocytes (A) or the pancreatic β-cell line INS832/13 (C) was treated with Thaps (1 μM) for the indicated times followed by immunoblotting. (B) NPCs were treated with the indicated concentrations of Thaps for 2 h. (D) Astrocytes were treated with Thaps (1 μM, 1 h) in the absence or presence of the pan-JAK inhibitor P6 (0.5 μM) or the JAK1/2 inhibitor AZD1480 (1.0 μM), followed by immunoblotting for P-STAT3, STAT3, P-JAK1, and JAK1. (E) Astrocytes were treated with OSM (0.5 ng/ml) for 15 min in the presence of the indicated concentrations of the JAK1 inhibitor AZ-JAK1, followed by immunoblotting. (F) Astrocytes were treated with Thaps (1 μM) for the indicated times in the absence or presence of AZ-JAK1 (1.0 μM) followed by immunoblotting for P-STAT3 and STAT3. (G) Astrocytes were transfected with control or p65 siRNA (50 pmol/ml) for 72 h. Cells were then treated with Thaps (1 μM) for the indicated times followed by immunoblotting. (H) Astrocytes were pretreated (1 h) with cycloheximide (20 μg/ml) and then treated with Thaps (1 μM) for the indicated times, followed by immunoblotting and ELISA. (I) Astrocytes were treated with OSM (1 ng/ml) for the indicated times in the absence or presence of Thaps (1 μM, 30-min pretreatment) followed by immunoblotting. Data are the averages ± SD from 2 independent experiments.
FIG 3.
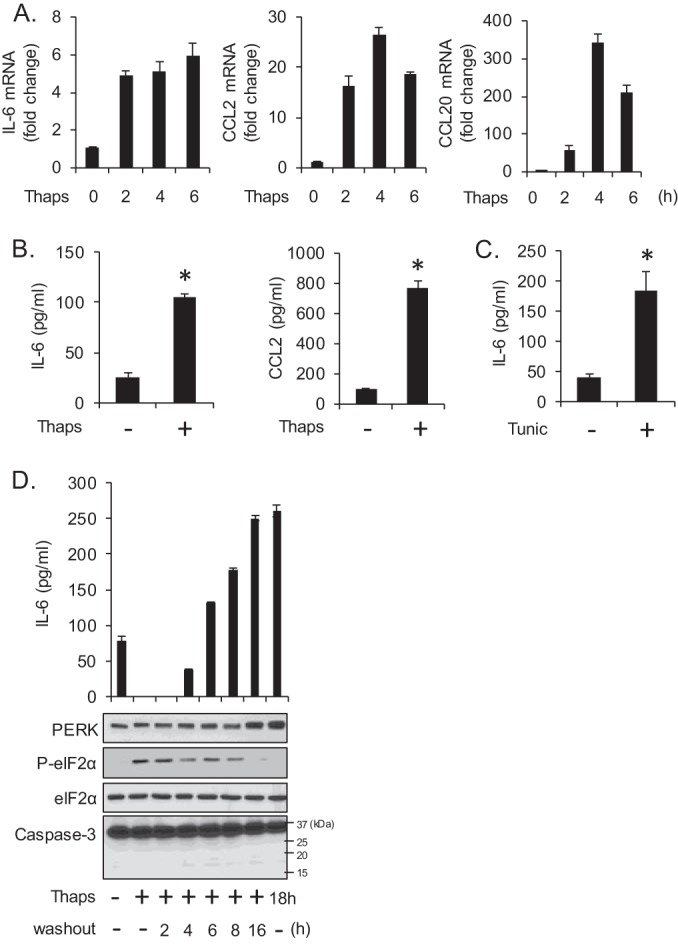
ER stress stimulates an inflammatory response. (A) Astrocytes were treated with Thaps (1 μM) for the indicated times followed by analysis of IL-6, CCL2, and CCL20 mRNA by qRT-PCR. (B) Astrocytes were exposed transiently to Thaps (1 μM, 2 h); the cells were then washed and incubated in fresh medium for 24 h. IL-6 and CCL2 in the cell culture supernatants were measured by ELISA. (C) Astrocytes were exposed transiently to Tunic (5 μM, 2 h); the cells were then washed and incubated in fresh medium for 24 h. IL-6 in the cell culture supernatants was measured by ELISA. (D) Astrocytes were exposed transiently to Thaps (1 μM, 2 h); the cells were then washed and incubated in fresh medium for the indicated times (washout). Alternatively, astrocytes were cultured in the continued presence of Thaps (last lane). Medium and cells were then collected, and IL-6 in the medium was measured by ELISA, and cell lysate was examined by immunoblotting. Data are means ± standard deviations (SD) from 3 independent experiments or representative of independent experiments analyzed in duplicate or triplicate. n = 3; *, P < 0.05.
Activation of STAT3 signaling is JAK1 dependent and a primary response to ER stress.
We have previously demonstrated that IL-6, CCL2, and CCL20 expression can be driven by JAK/STAT3 signaling (34, 41). Therefore, we tested if ER stress could also stimulate STAT3 activation. As shown in Fig. 4A, Thaps treatment of primary murine astrocytes leads to a rapid and prolonged increase in the activation-associated phosphorylation of tyrosine 705 of STAT3. Interestingly, this prolonged STAT3 phosphorylation is different from the transient phosphorylation induced by cytokines. Similarly, ER stress stimulated STAT3 activation in NPCs, along with caspase-3 cleavage (Fig. 4B), consistent with the sensitivity of these cells to ER stress. ER stress-induced STAT3 activation was also observed in the pancreatic β-cell line INS832/13, indicating that ER stress leads to STAT3 activation in a variety of cell types (Fig. 4C). In response to cytokine stimulation, STAT3 phosphorylation is dependent on upstream kinases, namely, JAKs. Therefore, we tested if ER stress-induced STAT3 activation was also JAK dependent. Astrocytes were treated with Thaps, which increased phosphorylation of STAT3 and JAK1 (Fig. 4D). The addition of the pan-JAK inhibitor pyridone 6 (P6) or the JAK1/2 inhibitor AZD1480 blocked ER stress-induced STAT3 phosphorylation (Fig. 4D). To determine if JAK1 was essential for this response, we used the JAK1 selective inhibitor AZ-JAK1. We first confirmed that AZ-JAK1, in a concentration-dependent fashion, could block canonical JAK/STAT signaling stimulated by OSM (Fig. 4E). We then treated astrocytes with Thaps in the absence or presence of AZ-JAK1. As shown in Fig. 4F, AZ-JAK1 blocked STAT3 phosphorylation in response to ER stress. These results indicate that ER stress activates STAT3 in a JAK1-dependent fashion. However, it is possible that this activation is indirect via NF-κB-dependent cytokine production, leading to autocrine activation of JAK/STAT signaling. To test this, p65, the transactivation domain-containing subunit of NF-κB, was knocked down and ER stress-induced activation of STAT3 was examined. p65 knockdown did not prevent Thaps-induced phosphorylation of STAT3 (Fig. 4G). To test if STAT3 activation is caused by autocrine signaling, de novo protein synthesis was inhibited with cycloheximide in order to block autocrine signaling. Alone, both Thaps and cycloheximide increased STAT3 phosphorylation, and together this effect was additive (Fig. 4H), indicating that inhibition of protein synthesis did not inhibit Thaps-induced STAT3 activation. Moreover, the activation of STAT3 is temporally disconnected from autocrine signaling, as shown in Fig. 4H. STAT3 activation preceded IL-6 production at these early time points, as measured by ELISA. An alternative possibility is that rather than stimulating STAT3 phosphorylation, ER stress blocks dephosphorylation. To test this, astrocytes were stimulated with OSM to induce STAT3 phosphorylation in the absence or presence of Thaps. As shown in Fig. 4I, OSM induced strong STAT3 phosphorylation at 15 min that is reduced by dephosphorylation at 90 min. The addition of Thaps did not prevent dephosphorylation following OSM stimulation (Fig. 4I). These data indicate that STAT3 activation is a JAK1-dependent primary response to ER stress and is not due to autocrine production of STAT-activating cytokines, such as IL-6, or blockade of STAT3 dephosphorylation.
ER stress-induced inflammatory gene expression is JAK/STAT and NF-κB dependent.
We next sought to determine if ER stress-induced inflammatory gene expression was dependent on JAK/STAT signaling. Astrocytes were treated with Thaps in the absence or presence of JAK inhibitor P6 or AZD1480. As shown in Fig. 5A, JAK inhibition significantly attenuated Thaps-induced IL-6 and CCL2 but had no effect on CHOP expression. To further examine which ER stress-induced inflammatory molecules were dependent on JAK/STAT signaling, we analyzed astrocyte supernatants by multiplex ELISA. Cells were treated transiently with Thaps, as described for Fig. 3B, in the absence or presence of JAK inhibitors. The JAK inhibitors were present for the duration of the experiment. Interestingly, we observed that ER stress induced IL-6 and macrophage colony-stimulating factor (M-CSF), but other cytokines were not detected, while several chemokines and vascular endothelial growth factor (VEGF) were markedly increased. Inhibition of JAK signaling significantly attenuated Thaps-induced CCL11, IL-6, and CCL2. CXCL10, CCL3, and CCL4 levels were also reduced but did not reach statistical significance in this assay. VEGF was greatly enhanced by ER stress and was unaffected by JAK inhibition (Fig. 5B). A traditional ELISA was used to measure IL-6 in the same samples analyzed by multiplex ELISA to validate the results (Fig. 5C). These data demonstrate that JAK/STAT signaling is essential for a robust ER stress-induced inflammatory response.
FIG 5.

ER stress drives JAK-dependent gene expression. (A) Astrocytes were treated with Thaps (1 μM, 4 h) in the absence or presence of P6 (0.5 μM) or AZD1480 (1.0 μM), followed by analysis of IL-6, CCL2, and CHOP mRNA by qRT-PCR. (B) Astrocytes were exposed transiently to Thaps (1 μM, 2 h) in the absence or presence of P6 or AZD1480; the cells were then washed and incubated in fresh medium without or with JAK inhibitors for 24 h. The cell culture supernatants were then measured by multiplex ELISA. (C) To validate multiplex data, the same supernatants as those described for panel B were analyzed by traditional IL-6 ELISA. Data are means ± SD from independent experiments. n = 3 or 4; *, P < 0.05.
To confirm the importance of JAK1 in ER stress-induced STAT3 activation and inflammatory gene expression, we used JAK1 siRNA. Astrocytes were transfected with control or JAK1 siRNA followed by treatment with Thaps. JAK1 siRNA effectively reduced JAK1 protein levels and blocked ER stress-induced STAT3 phosphorylation (Fig. 6A). Consistent with reduced STAT3 activation, JAK1 siRNA attenuated ER stress-induced IL-6, CCL2, and CCL20 mRNA expression but had no effect on CHOP (Fig. 6B). This further verifies the involvement of JAK1/STAT3 signaling in the inflammatory response to ER stress.
FIG 6.

JAK1 and NF-κB contribute to inflammatory gene expression. (A) Astrocytes were transfected with control or JAK1 siRNA (50 pmol/ml) for 48 h. Cells were then treated with Thaps (1 μM) for the indicated times followed by immunoblotting for JAK1, P-STAT3, STAT3, P-eIF2α, and eIF2α. (B) Astrocytes were transfected with control or JAK1 siRNA (50 pmol/ml) for 48 h. Cells were then treated with Thaps (1 μM, 4 h), and gene expression was measured by qRT-PCR. (C) Astrocytes were transfected with control or p65 siRNA (50 pmol/ml) for 72 h. Cells were then treated with Thaps (1 μM) in the absence or presence of AZ-JAK1, and gene expression was measured by qRT-PCR. Data are means ± SD and are representative of results from 3 independent experiments analyzed in duplicate.
In light of the importance of both NF-κB and JAK/STAT signaling in mediating the inflammatory response to ER stress, we examined the relative contribution of each. Astrocytes were transfected with p65 siRNA followed by treatment with Thaps in the absence or presence of AZ-JAK1. Knockdown of p65 modestly attenuated Thaps-induced IL-6 mRNA expression, while AZ-JAK1 ablated IL-6 expression (Fig. 6C), indicating that JAK1 is predominantly responsible for IL-6 expression, in agreement with the ELISA data in Fig. 5. CCL2 expression was attenuated by p65 knockdown and further reduced by JAK1 inhibition, suggesting that both NF-κB and JAK1 are involved in ER stress-induced CCL2 expression. CHOP was unaffected by either p65 knockdown or JAK inhibition (Fig. 6C). These data indicate that both NF-κB and JAK/STAT are involved in the inflammatory response to ER stress.
ER stress-induced STAT3 activation is PERK dependent.
Next, we examined which arm(s) of the UPR was responsible for STAT3 activation. As shown in Fig. 5 and 6, ER stress-induced CCL2 and IL-6 are dependent on JAK/STAT activation; therefore, we used these genes as readouts in screening for relevant UPR components. Astrocytes were transfected with control, PERK, IRE1, or ATF6 siRNA (mRNA of target genes is shown in Fig. 7A, right) followed by treatment with Thaps and analysis of IL-6 and CCL2 mRNA. Knockdown of IRE1 or ATF6 had no effect on ER stress-induced CCL2 or IL-6. However, knockdown of PERK attenuated both CCL2 and IL-6, suggesting that PERK is the relevant signal transducer (Fig. 7A). To verify this, astrocytes were transfected with control or PERK siRNA followed by treatment with Thaps. PERK knockdown effectively reduced the level of PERK protein and the PERK-dependent phosphorylation of eIF2α in response to Thaps. Importantly, PERK knockdown reduced Thaps-stimulated phosphorylation of JAK1 and STAT3 (Fig. 7B). We also observed that knockdown of PERK increased the basal levels of total JAK1, phosphorylated JAK1 (P-JAK1), and P-STAT3 (Fig. 7B, lane 4), possibly a compensatory mechanism. PERK knockdown did not reduce IL-6 or OSM-induced STAT3 phosphorylation (Fig. 7C), indicating that cytokine-induced JAK/STAT signaling remains intact in the absence of PERK. To confirm the role of PERK, we examined STAT3 activation in PERK−/− MEFs. As shown in Fig. 7D, wild-type MEFs respond to ER stress with increased P-STAT3, while there is no increase in phosphorylation of STAT3 in PERK−/− MEFs. Consistent with these data, PERK−/− MEFs have greatly attenuated CCL2 and CCL20 expression in response to Thaps compared to that of wild-type MEFs (Fig. 7E). CHOP expression is also reduced (Fig. 7E), consistent with loss of PERK signaling (54). These data demonstrate that ER stress-induced JAK/STAT signaling is PERK dependent.
FIG 7.

ER stress-induced JAK/STAT signaling is PERK dependent. (A) Astrocytes were transfected with control, PERK, IRE1, or ATF6 siRNA (50 pmol/ml) for 48 h. Cells were then treated with Thaps (1 μM, 4 h), and the levels of CCL2 and IL-6 mRNA were measured by qRT-PCR. The untreated levels of UPR signaling molecules targeted by siRNA are shown to the right. (B) Astrocytes were transfected with control or PERK siRNA (50 pmol/ml) for 48 h. Cells were then treated with Thaps (1 μM) for the indicated times. Cell lysates were analyzed by immunoblotting. (C) Cells were transfected as described for panel B and then treated with IL-6 (10 ng/ml, 30 min) or OSM (1 ng/ml, 30 min), followed by immunoblotting. Wild-type or PERK−/− MEFs were treated with Thaps (1 μM) for the indicated times followed by immunoblotting (D) or qRT-PCR analysis (E). Data are means ± SD and representative of results from 3 independent experiments analyzed in duplicate.
JAK1 interacts with and phosphorylates PERK.
Previous work from Su and colleagues has demonstrated that JAK1 and TYK2 interact with and phosphorylate PKR (23). Considering the homology and functional similarities between PKR and PERK (55, 56), we examined if PERK and JAK1 could also interact. To test this, primary astrocytes were transfected with myc-tagged PERK followed by immunoprecipitation of endogenous JAK1 and immunoblotting of PERK. As shown in Fig. 8A, PERK coimmunoprecipitated with JAK1. PERK is heavily autophosphorylated on serine and threonine in response to ER stress (6) in addition to functionally important tyrosine phosphorylation (57). Therefore, we tested if JAK1 could phosphorylate PERK in an in vitro kinase assay. JAK1 phosphorylated PERK in a concentration-dependent fashion, and this could be blocked by the addition of a JAK1 inhibitor (Fig. 8B). To extend these findings, PERK tyrosine phosphorylation was examined in astrocytes. ER stress increased PERK tyrosine phosphorylation, and this was attenuated by the addition of AZD1480 (Fig. 8C), indicating that JAK(s) (likely JAK1) could phosphorylate PERK. We next used liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) to identify the sites on PERK phosphorylated by JAK1 in vitro. We identified the phosphorylation sites on PERK as Y585 and Y619, with Ascores of 47.7 and 1,000, respectively, indicating high confidence in these sites (40) (Fig. 8D). Both tyrosines are highly conserved. We hypothesize that this phosphorylation may be similar to the JAK-dependent phosphorylation of cytokine receptors that produces a phosphotyrosine motif to recruit STAT3 via the SH2 domain. To test if SH2-mediated phosphotyrosine binding of STAT3 is required for ER stress-induced activation, astrocytes were treated with Thaps without or with STATTIC. STATTIC is a small molecule that selectively blocks SH2 domain interaction of STAT3 (58) and was effective at blocking ER stress-induced STAT3 activation (Fig. 8E). These data alone do not directly show that STAT3 binds phosphotyrosine residues on PERK. Moreover, if STAT3 and PERK interact, it is likely transient, as we could not detect the interaction (not shown), consistent with previous work, which also failed to detect an interaction (59). Next, we examined if the kinase activity of PERK was essential for STAT3 activation. Astrocytes were treated with the ATP-competitive PERK kinase inhibitor GSK2606414, followed by treatment with Thaps. PERK inhibition reduced eIF2α phosphorylation, confirming PERK inhibition, and attenuated STAT3 phosphorylation in a concentration-dependent fashion, indicating PERK kinase activity is essential for ER stress-induced STAT3 activation (Fig. 8F). Taken together, these data suggest a model in which ER stress stimulates the activation and dimerization of PERK, bringing the interacting JAK(s) into proximity to allow transphosphorylation and activation as well as phosphorylation of the cytoplasmic domain of PERK at Y585 and Y619. STAT3 may then be recruited through SH2 domain-dependent interactions to the PERK/JAK complex, where it is phosphorylated by JAK1, becoming activated and driving inflammatory gene expression. One possible model supported by these data is shown in Fig. 8G.
FIG 8.

JAK1 interacts with and phosphorylates PERK. (A) Astrocytes were transfected with empty vector (−) or PERK-myc (+) followed by immunoprecipitation of JAK1 and immunoblotting for PERK and JAK1. Antibody was omitted in the negative control (neg ctl). (B) Recombinant PERK (2 μg cytoplasmic domain amino acids [aa] 536 to 1116) was used as a substrate in an in vitro kinase assay with increasing amounts of recombinant active JAK1. The JAK1 inhibitor AZ-JAK1 (10 nM) was included in the kinase reaction where indicated. (C) Astrocytes were treated with Thaps (1 μM, 1 h) in the absence or presence of AZD1480 (1 μM). Total phosphotyrosine was immunoprecipitated and immunoblotted for PERK. (D) An in vitro kinase assay was performed as described for panel B with nonradioactive ATP, and PERK was analyzed by LC-ESI-MS/MS. The identified phosphorylation sites, the respective MS/MS spectra, and the homology of each site are shown. (E) Astrocytes were treated with Thaps (1 μM, 1 h) in the absence or presence of the indicated concentrations of STATTIC. (F) Astrocytes were treated with Thaps (1 μM, 2 h) in the absence or presence of increasing concentrations of the PERK kinase inhibitor GSK2606414, followed by immunoblotting. (G) Potential signaling model in which JAK1 phosphorylates PERK and mediates the ER stress-induced activation of STAT3.
ER stress modulates astrocyte-microglia interactions.
Having demonstrated that ER stress drives a novel PERK/JAK1/STAT3 pathway leading to the production of a host of inflammatory mediators in astrocytes, we examined if this would influence microglia, the major CNS-resident immune cell. Astrocytes without or with PERK knockdown were left untreated or treated with Thaps transiently (2 h), followed by washing and incubation in fresh medium for 24 h. The astrocyte-conditioned medium (ACM) was then transferred directly to primary microglia for 4 h followed by mRNA analysis. Conditioned medium from Thaps-treated astrocytes greatly increased IL-1β and IL-6 mRNA expression by microglia, and this effect was attenuated when conditioned medium from PERK knockdown astrocytes was applied (Fig. 9A). These results indicate that ER-stressed astrocytes, in a PERK-dependent fashion, can activate microglia. The IL-6 family of cytokines, which includes IL-6 and OSM, are important regulators of inflammation in the CNS, are elevated in various diseases, and have been shown to induce IL-6 expression in astrocytes (60, 61). As such, we examined if IL-6 could feed-forward to influence ER-stressed astrocytes. Astrocytes were pretreated (1 h) with Thaps, followed by treatment with IL-6. As shown in Fig. 9B, the combination of ER stress plus IL-6 leads to enhanced IL-6 mRNA expression. We also examined if OSM would produce a similar effect. Astrocytes were exposed to ER stress using either Thaps or Tunic and then treated with OSM, resulting in a robust enhancement of IL-6 mRNA and protein expression (Fig. 9C and D). Additionally, the enhanced response was PERK dependent, as knockdown of PERK attenuated IL-6 induced by the combination of OSM and Thaps (Fig. 9E). These data demonstrate that ER stress drives an inflammatory reaction in astrocytes that can subsequently stimulate microglia, potentially leading to a feed-forward, PERK-dependent autocrine/paracrine loop. This signaling pathway may contribute to the excessive and nonresolving neuroinflammation associated with neurological diseases such as MS.
FIG 9.

ER-stressed astrocytes stimulate a PERK-dependent microglia-astrocyte feed-forward inflammatory loop. (A) Astrocytes were transfected with control or PERK siRNA (50 pmol/ml) for 48 h and then exposed transiently to Thaps (1 μM, 2 h); the cells were then washed and incubated in fresh medium for 24 h. Primary microglia were then exposed to the astrocyte-conditioned media (ACM) for 4 h followed by analysis of gene expression by qRT-PCR. The conditions refer to astrocytes, whereas the data shown are from microglia. (B) Astrocytes were treated with Thaps (1 μM, 1 h pre-IL-6) in the absence or presence of IL-6 (10 ng/ml, 4 h), followed by analysis of IL-6 mRNA by qRT-PCR. (C) Astrocytes were treated with Thaps (1 μM, 1 h pre-OSM) or Tunic (5 μM, 1 h pre-OSM) in the absence or presence of OSM (1 ng/ml, 4 h), followed by analysis of IL-6 mRNA expression by qRT-PCR. (D) Astrocytes were treated with Thaps (1 μM, 1 h pre-OSM) in the absence or presence of OSM (1 ng/ml, 24 h), followed by analysis of IL-6 protein levels by ELISA. (E) Astrocytes were transfected with control or PERK siRNA (50 pmol/ml) for 48 h. Cells were then treated with Thaps (1 μM, 1 h pre-OSM) in the absence or presence of OSM (1 ng/ml, 4 h), followed by analysis of IL-6 mRNA expression by qRT-PCR. Data are means ± SD and representative of results from 3 independent experiments analyzed in duplicate.
DISCUSSION
The well-accepted inflammatory reaction activated by ER stress likely represents a conserved innate immune response (12). Here, we show that cells of the CNS experience ER stress during the neuroinflammatory disease of EAE. We describe a new pathway involved in mediating inflammation in response to ER stress. We have shown that ER stress leads to PERK-dependent activation of JAK/STAT signaling and subsequent inflammatory gene expression. JAK/STAT activation is a primary response to ER stress, as it is independent of NF-κB, temporally precedes increases in the acute-phase cytokine IL-6, and occurs faster (<30 min versus >1.5 h) than the NF-κB-dependent autocrine activation by tumor necrosis factor alpha (TNF-α) we recently reported (62). We speculate and provide evidence that, in activating JAK/STAT signaling, PERK may act analogous to type I and type II cytokine receptors. These cytokine receptors are associated with and phosphorylated by JAKs following receptor ligation and oligomerization, resulting in STAT recruitment and activation (17). Similarly, PERK is associated with and phosphorylated by JAK1 at Y585 and Y619 (and possibly other JAKs) during ER stress, resulting in PERK- and JAK1-dependent activation of STAT3. Y585 is a novel phosphorylation site, while phosphorylation at Y619 has been previously reported and attributed to autophosphorylation (57). The model shown in Fig. 8G is one potential mechanism that the data support. However, additional questions still need to be addressed to fully elucidate this pathway. Does PERK phosphorylate JAK1 and/or STAT3? Does JAK1 phosphorylation of PERK modulate its function? Can STAT3 bind a phosphotyrosine motif on PERK? Additional studies will address these questions. In the current study, we identified a novel PERK/JAK1/STAT3 pathway that is important for ER stress-induced expression of IL-6 and multiple chemokines, suggesting that this pathway is critical to the innate immune reaction produced in response to ER stress.
ER stress and inflammation have been implicated in numerous widespread diseases. ER stress and inflammation are particularly relevant to neurodegenerative diseases, which frequently involve misfolded and aggregated proteins (3). Moreover, ER stress is important in demyelinating and neuroinflammatory diseases such as MS (45). Our finding that astrocytes are highly resistant to ER stress-induced cell death suggests that chronic ER stress, associated with neurodegeneration and neuroinflammation, may promote nonresolving inflammation.
The JAK-dependent inflammatory program activated by ER stress that we have identified includes at least IL-6, CCL2, CCL20, and CCL11. The expression of CCL2 would support the recruitment of phagocytic monocytes (microglia/macrophages), possibly in an effort to promote clearance of protein aggregates (63), while CCL20 would signal for the recruitment of CCR6-expressing lymphocytes, such as Th17 cells, which may be pathogenic in MS (64). Interestingly, CCL11 was recently shown by Villeda and colleagues to be elevated in older individuals and to impair neurogenesis and cognitive function (65). This could indicate that ER-stressed astrocytes may directly impair neurological function through the secretion of soluble mediators such as CCL11. This may be an exacerbating factor associated with the cognitive impairment observed in some MS patients (29). While the chemokines would promote the influx of a variety of cell types, the presence of IL-6 would influence the activation and functional phenotype of the infiltrating cells, particularly T cells. Thus, in the face of ER stress, astrocytes are positioned to control the inflammatory microenvironment. Under acute conditions, this likely facilitates restoration of CNS homeostasis (66); however, chronically, this may facilitate neurodegeneration.
Consistent with a role for astrocytes in influencing inflammatory cell function, we found that ER-stressed astrocytes, via PERK-dependent paracrine signaling, could modulate the inflammatory profile of microglia leading to the expression of IL-6 cytokines. Additionally, we found that OSM and IL-6 could synergistically enhance ER stress-induced IL-6 production in astrocytes, indicating that ER stress in astrocytes may drive a feed-forward inflammatory loop among glial cells. This synergy is also observed in glial cells exposed to ER stress plus IFN-γ/PGE2 (30), suggesting a more widespread interaction between ER stress and STAT-activating cytokines. Our data show that the enhanced IL-6 expression was, at least in part, PERK dependent. We have not yet fully examined which downstream signals may be involved in the cross talk between OSM and ER stress signaling. Both of these stimuli activate similar signaling pathways, including STAT3, NF-κB, and mitogen-activated protein kinases (MAPKs). The combination of OSM and ER stress could result in robust activation of both STAT3 and NF-κB that could functionally interact, which is well known to occur (67), to drive enhanced gene expression. This fits well with recent studies that showed NF-κB and STAT3 work together in starved cancer cells to drive IL-6 expression. Importantly, starved cancer cells undergo ER stress (68). However, our data indicate that in primary astrocytes, ER stress-induced IL-6 expression is mostly dependent on JAK/STAT signaling.
IL-6 appears to be the key cytokine associated with ER stress, as multiple arms of the UPR have been reported to drive IL-6 expression. During plasma cell differentiation, IL-6 is induced through the IRE1/XBP-1 arm of the UPR (69), whereas during glucose deprivation in cancer cells, IL-6 expression is driven by a PERK/ATF4-dependent pathway (70). Our data point toward a PERK/JAK1/STAT3-dependent pathway driving IL-6 expression in primary astrocytes as well as other cell types in response to classic ER stress inducers. While we have shown that JAK1-dependent STAT3 activation is needed for full engagement of the ER stress-induced inflammatory program, it is likely that STAT3 is part of a larger coordinated transcriptional reprograming in response to ER stress. STAT3 may be working in concert with other transcriptional regulators, such as XBP-1, ATF4, and NF-κB, to drive the ER stress-induced inflammatory program. Additional studies will determine how STAT3 interacts with other ER stress-induced transcriptional regulators. Collectively, this suggests that cytokine and chemokine production, particularly IL-6, is an integral part of the ER stress response, and the previously unrecognized PERK/JAK1/STAT3 pathway could be a new therapeutic target to combat neuroinflammation associated with diseases such as MS.
ACKNOWLEDGMENTS
This work was supported in part by the National Institutes of Health grants NS57563 and CA158534 (E.N.B.), a Career Transition Award from the National Multiple Sclerosis Society (TA3050-A-1; G.P.M), and an NMSS Collaborative Award from the National Multiple Sclerosis Society (CA-1059-A-13; E.N.B). The UAB Comprehensive Cancer Center-Mass Spectrometry/Proteomics Shared Facility is supported by NIH grant P30CA13148-38.
We thank Ronald Wek (Indiana University) for the PERK-deficient MEFs, Chris Newgard (Duke University) for the INS832/13 cells, and David Ron (via Addgene) for the PERK-myc vector. We thank AstraZeneca for generously providing AZD1480 (Dennis Huszar) and AZ-JAK1 (Richard Woessner) and Braden McFarland (University of Alabama at Birmingham) for direction in regard to the use of these compounds. We thank Andrew Holdbrooks, Kyle Brawner, and Kavitha Abiraman for technical assistance. We thank Chad Steele (University of Alabama at Birmingham) for assistance with multiplex ELISA.
We declare no conflicts of interest.
Footnotes
Published ahead of print 11 August 2014
REFERENCES
- 1.Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, Marciniak SJ. 2013. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 12:105–118. 10.1016/S1474-4422(12)70238-7 [DOI] [PubMed] [Google Scholar]
- 2.Walter P, Ron D. 2011. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334:1081–1086. 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- 3.Zhang K, Kaufman RJ. 2008. From endoplasmic-reticulum stress to the inflammatory response. Nature 454:455–462. 10.1038/nature07203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. 2008. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135:61–73. 10.1016/j.cell.2008.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malhotra J, Kaufman R. 2007. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 18:716–731. 10.1016/j.semcdb.2007.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harding H, Zhang Y, Ron D. 1999. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397:271–274. 10.1038/16729 [DOI] [PubMed] [Google Scholar]
- 7.Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, Mallucci GR. 2013. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 5:206ra138. 10.1126/scitranslmed.3006767 [DOI] [PubMed] [Google Scholar]
- 8.Nijholt DAT, van Haastert ES, Rozemuller AJM, Scheper W, Hoozemans JJM. 2012. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J. Pathol. 226:693–702. 10.1002/path.3969 [DOI] [PubMed] [Google Scholar]
- 9.Ito Y, Yamada M, Tanaka H, Aida K, Tsuruma K, Shimazawa M, Hozumi I, Inuzuka T, Takahashi H, Hara H. 2009. Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol. Dis. 36:470–476. 10.1016/j.nbd.2009.08.013 [DOI] [PubMed] [Google Scholar]
- 10.Cunnea P, Mháille AN, McQuaid S, Farrell M, McMahon J, FitzGerald U. 2011. Expression profiles of endoplasmic reticulum stress-related molecules in demyelinating lesions and multiple sclerosis. Mult. Scler. 17:808–818. 10.1177/1352458511399114 [DOI] [PubMed] [Google Scholar]
- 11.Farina C, Aloisi F, Meinl E. 2007. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 28:138–145. 10.1016/j.it.2007.01.005 [DOI] [PubMed] [Google Scholar]
- 12.Martinon F, Glimcher LH. 2011. Regulation of innate immunity by signaling pathways emerging from the endoplasmic reticulum. Curr. Opin. Immunol. 23:35–40. 10.1016/j.coi.2010.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, Kaufman RJ. 2006. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 124:587–599. 10.1016/j.cell.2005.11.040 [DOI] [PubMed] [Google Scholar]
- 14.Gargalovic PS, Gharavi NM, Clark MJ, Pagnon J, Yang W-P, He A, Truong A, Baruch-Oren T, Berliner JA, Kirchgessner TG, Lusis AJ. 2006. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 26:2490–2496. 10.1161/01.ATV.0000242903.41158.a1 [DOI] [PubMed] [Google Scholar]
- 15.Liu Y-P, Zeng L, Tian A, Bomkamp A, Rivera D, Gutman D, Barber GN, Olson JK, Smith JA. 2012. Endoplasmic reticulum stress regulates the innate immunity critical transcription factor IRF3. J. Immunol. 189:4630–4639. 10.4049/jimmunol.1102737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodall JC, Wu C, Zhang Y, McNeill L, Ellis L, Saudek V, Gaston JSH. 2010. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc. Natl. Acad. Sci. U. S. A. 107:17698–17703. 10.1073/pnas.1011736107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Shea JJ, Plenge R. 2012. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 36:542–550. 10.1016/j.immuni.2012.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erta M, Quintana A, Hidalgo J. 2012. Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 8:1254–1266. 10.7150/ijbs.4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruprecht K, Kuhlmann T, Seif F, Hummel V, Kruse N, Brück W, Rieckmann P. 2001. Effects of oncostatin M on human cerebral endothelial cells and expression in inflammatory brain lesions. J. Neuropathol. Exp. Neurol. 60:1087–1098 [DOI] [PubMed] [Google Scholar]
- 20.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. 2003. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 374:1–20. 10.1042/BJ20030407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu H, Pardoll D, Jove R. 2009. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9:798–809. 10.1038/nrc2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meares GP, Ma X, Qin H, Benveniste EN. 2012. Regulation of CCL20 expression in astrocytes by IL-6 and IL-17. Glia 60:771–781. 10.1002/glia.22307 [DOI] [PubMed] [Google Scholar]
- 23.Su Q, Wang S, Baltzis D, Qu L-K, Raven JF, Li S, Wong AH-T, Koromilas AE. 2007. Interferons induce tyrosine phosphorylation of the eIF2alpha kinase PKR through activation of Jak1 and Tyk2. EMBO Rep. 8:265–270. 10.1038/sj.embor.7400891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coe H, Jung J, Groenendyk J, Prins D, Michalak M. 2010. ERp57 modulates STAT3 signaling from the lumen of the endoplasmic reticulum. J. Biol. Chem. 285:6725–6738. 10.1074/jbc.M109.054015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flores-Morales A, Fernández L, Rico-Bautista E, Umana A, Negrín C, Zhang JG, Norstedt G. 2001. Endoplasmic reticulum stress prolongs GH-induced Janus kinase (JAK2)/signal transducer and activator of transcription (STAT5) signaling pathway. Mol. Endocrinol. 15:1471–1483. 10.1210/mend.15.9.0699 [DOI] [PubMed] [Google Scholar]
- 26.Vollmer S, Haan C, Behrmann I. 2010. Oncostatin M up-regulates the ER chaperone Grp78/BiP in liver cells. Biochem. Pharmacol. 80:2066–2073. 10.1016/j.bcp.2010.07.015 [DOI] [PubMed] [Google Scholar]
- 27.Lin W, Lin Y, Li J, Fenstermaker AG, Way SW, Clayton B, Jamison S, Harding HP, Ron D, Popko B. 2013. Oligodendrocyte-specific activation of PERK signaling protects mice against experimental autoimmune encephalomyelitis. J. Neurosci. 33:5980–5991. 10.1523/JNEUROSCI.1636-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin W, Harding HP, Ron D, Popko B. 2005. Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J. Cell Biol. 169:603–612. 10.1083/jcb.200502086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Getts MT, Getts DR, Kohm AP, Miller SD. 2008. Endoplasmic reticulum stress response as a potential therapeutic target in multiple sclerosis. Therapy 5:631–640. 10.2217/14750708.5.5.631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hosoi T, Honda M, Oba T, Ozawa K. 2013. ER stress upregulated PGE2/IFN-γ-induced IL-6 expression and down-regulated iNOS expression in glial cells. Sci. Rep. 3:3388. 10.1038/srep03388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahyi A-NN, Quinton LJ, Jones MR, Ferrari JD, Pepper-Cunningham ZA, Mella JR, Remick DG, Mizgerd JP. 2013. Roles of STAT3 in protein secretion pathways during the acute-phase response. Infect. Immun. 81:1644–1653. 10.1128/IAI.01332-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ioannidis S, Lamb ML, Wang T, Almeida L, Block MH, Davies AM, Peng B, Su M, Zhang H-J, Hoffmann E, Rivard C, Green I, Howard T, Pollard H, Read J, Alimzhanov M, Bebernitz G, Bell K, Ye M, Huszar D, Zinda M. 2011. Discovery of 5-chloro-N2-[(1S)-1-(5-fluoropyrimidin-2-yl)ethyl]-N4-(5-methyl-1H-pyrazol-3-yl)pyrimidine-2,4-diamine (AZD1480) as a novel inhibitor of the Jak/Stat pathway. J. Med. Chem. 54:262–276. 10.1021/jm1011319 [DOI] [PubMed] [Google Scholar]
- 33.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, Buettner R, Proia D, Kowolik CM, Xin H, Armstrong B, Bebernitz G, Weng S, Wang L, Ye M, McEachern K, Chen H, Morosini D, Bell K, Alimzhanov M, Ioannidis S, McCoon P, Cao ZA, Yu H, Jove R, Zinda M. 2009. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell 16:487–497. 10.1016/j.ccr.2009.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meares GP, Qin H, Liu Y, Holdbrooks AT, Benveniste EN. 2013. AMP-activated protein kinase restricts IFN-gamma signaling. J. Immunol. 190:372–380. 10.4049/jimmunol.1202390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rietze RL, Reynolds BA. 2006. Neural stem cell isolation and characterization. Methods Enzymol. 419:3–23. 10.1016/S0076-6879(06)19001-1 [DOI] [PubMed] [Google Scholar]
- 36.Qin H, Yeh WI, De Sarno P, Holdbrooks AT, Liu Y, Muldowney MT, Reynolds SL, Yanagisawa LL, Fox TH, Park K, Harrington LE, Raman C, Benveniste EN. 2012. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc. Natl. Acad. Sci. U. S. A. 109:5004–5009. 10.1073/pnas.1117218109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stromnes IM, Goverman JM. 2006. Active induction of experimental allergic encephalomyelitis. Nat. Protoc. 1:1810–1819. 10.1038/nprot.2006.285 [DOI] [PubMed] [Google Scholar]
- 38.Meares GP, Zmijewska AA, Jope RS. 2004. Heat shock protein-90 dampens and directs signaling stimulated by insulin-like growth factor-1 and insulin. FEBS Lett. 574:181–186. 10.1016/j.febslet.2004.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma X, Reynolds SL, Baker BJ, Li X, Benveniste EN, Qin H. 2010. IL-17 enhancement of the IL-6 signaling cascade in astrocytes. J. Immunol. 184:4898–4906. 10.4049/jimmunol.1000142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beausoleil SA, Villén J, Gerber SA, Rush J, Gygi SP. 2006. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24:1285–1292. 10.1038/nbt1240 [DOI] [PubMed] [Google Scholar]
- 41.McFarland BC, Ma J-Y, Langford CP, Gillespie GY, Yu H, Zheng Y, Nozell SE, Huszar D, Benveniste EN. 2011. Therapeutic potential of AZD1480 for the treatment of human glioblastoma. Mol. Cancer Ther. 10:2384–2393. 10.1158/1535-7163.MCT-11-0480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berridge MV, Herst PM, Tan AS. 2005. Tetrazolium dyes as tools in cell biology: new insights into their cellular reduction. Biotechnol. Annu. Rev. 11:127–152. 10.1016/S1387-2656(05)11004-7 [DOI] [PubMed] [Google Scholar]
- 43.Lin W, Bailey SL, Ho H, Harding HP, Ron D, Miller SD, Popko B. 2007. The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J. Clin. Invest. 117:448–456. 10.1172/JCI29571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mháille AN, McQuaid S, Windebank A, Cunnea P, McMahon J, Samali A, FitzGerald U. 2008. Increased expression of endoplasmic reticulum stress-related signaling pathway molecules in multiple sclerosis lesions. J. Neuropathol. Exp. Neurol. 67:200–211. 10.1097/NEN.0b013e318165b239 [DOI] [PubMed] [Google Scholar]
- 45.Deslauriers AM, Afkhami-Goli A, Paul AM, Bhat RK, Acharjee S, Ellestad KK, Noorbakhsh F, Michalak M, Power C. 2011. Neuroinflammation and endoplasmic reticulum stress are coregulated by crocin to prevent demyelination and neurodegeneration. J. Immunol. 187:4788–4799. 10.4049/jimmunol.1004111 [DOI] [PubMed] [Google Scholar]
- 46.Kondo S, Murakami T, Tatsumi K, Ogata M, Kanemoto S, Otori K, Iseki K, Wanaka A, Imaizumi K. 2005. OASIS, a CREB/ATF-family member, modulates UPR signalling in astrocytes. Nat. Cell Biol. 7:186–194. 10.1038/ncb1213 [DOI] [PubMed] [Google Scholar]
- 47.Meares GP, Mines MA, Beurel E, Eom TY, Song L, Zmijewska AA, Jope RS. 2011. Glycogen synthase kinase-3 regulates endoplasmic reticulum (ER) stress-induced CHOP expression in neuronal cells. Exp. Cell Res. 317:1621–1628. 10.1016/j.yexcr.2011.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stefani IC, Wright D, Polizzi KM, Kontoravdi C. 2012. The role of ER stress-induced apoptosis in neurodegeneration. Curr. Alzheimer Res. 9:373–387. 10.2174/156720512800107618 [DOI] [PubMed] [Google Scholar]
- 49.Higo T, Hamada K, Hisatsune C, Nukina N, Hashikawa T, Hattori M, Nakamura T, Mikoshiba K. 2010. Mechanism of ER stress-induced brain damage by IP(3) receptor. Neuron 68:865–878. 10.1016/j.neuron.2010.11.010 [DOI] [PubMed] [Google Scholar]
- 50.Smith MI, Deshmukh M. 2007. Endoplasmic reticulum stress-induced apoptosis requires bax for commitment and Apaf-1 for execution in primary neurons. Cell Death Differ. 14:1011–1019. 10.1038/sj.cdd.4402089 [DOI] [PubMed] [Google Scholar]
- 51.Wheeler MC, Rizzi M, Sasik R, Almanza G, Hardiman G, Zanetti M. 2008. KDEL-retained antigen in B lymphocytes induces a proinflammatory response: a possible role for endoplasmic reticulum stress in adaptive T cell immunity. J. Immunol. 181:256–264. 10.4049/jimmunol.181.1.256 [DOI] [PubMed] [Google Scholar]
- 52.Mahadevan NR, Fernandez A, Rodvold JJ, Almanza G, Zanetti M. 2010. Prostate cancer cells undergoing ER stress in vitro and in vivo activate transcription of pro-inflammatory cytokines. J. Inflamm. Res. 3:99–103. 10.2147/JIR.S11190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iwasaki Y, Suganami T, Hachiya R, Shirakawa I, Kim-Saijo M, Tanaka M, Hamaguchi M, Takai-Igarashi T, Nakai M, Miyamoto Y, Ogawa Y. 2013. Activating transcription factor 4 links metabolic stress to interleukin-6 expression in macrophages. Diabetes 63:152–161. 10.2337/db13-0757 [DOI] [PubMed] [Google Scholar]
- 54.Harding H, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. 2000. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6:1099–1108. 10.1016/S1097-2765(00)00108-8 [DOI] [PubMed] [Google Scholar]
- 55.Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC. 1998. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 18:7499–7509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wek R, Jiang H, Anthony T. 2006. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 34:7–11. 10.1042/BST0340007 [DOI] [PubMed] [Google Scholar]
- 57.Su Q, Wang S, Gao HQ, Kazemi S, Harding HP, Ron D, Koromilas AE. 2008. Modulation of the eukaryotic initiation factor 2 alpha-subunit kinase PERK by tyrosine phosphorylation. J. Biol. Chem. 283:469–475. 10.1074/jbc.M704612200 [DOI] [PubMed] [Google Scholar]
- 58.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. 2006. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 13:1235–1242. 10.1016/j.chembiol.2006.09.018 [DOI] [PubMed] [Google Scholar]
- 59.Shen S, Niso-Santano M, Adjemian S, Takehara T, Malik SA, Minoux H, Souquere S, Mariño G, Lachkar S, Senovilla L, Galluzzi L, Kepp O, Pierron G, Maiuri MC, Hikita H, Kroemer R, Kroemer G. 2012. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol. Cell 48:667–680. 10.1016/j.molcel.2012.09.013 [DOI] [PubMed] [Google Scholar]
- 60.Van Wagoner NJ, Choi C, Repovic P, Benveniste EN. 2000. Oncostatin M regulation of interleukin-6 expression in astrocytes: biphasic regulation involving the mitogen-activated protein kinases ERK1/2 and p38. J. Neurochem. 75:563–575 [DOI] [PubMed] [Google Scholar]
- 61.Chen S-H, Benveniste EN. 2004. Oncostatin M: a pleiotropic cytokine in the central nervous system. Cytokine Growth Factor Rev. 15:379–391. 10.1016/j.cytogfr.2004.06.002 [DOI] [PubMed] [Google Scholar]
- 62.McFarland BC, Hong SW, Rajbhandari R, Twitty GB, Gray GK, Yu H, Benveniste EN, Nozell SE. 2013. NF-κB-induced IL-6 ensures STAT3 activation and tumor aggressiveness in glioblastoma. PLoS One 8:e78728. 10.1371/journal.pone.0078728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. 2007. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 13:432–438. 10.1038/nm1555 [DOI] [PubMed] [Google Scholar]
- 64.Becher B, Segal BM. 2011. T(H)17 cytokines in autoimmune neuro-inflammation. Curr. Opin. Immunol. 23:707–712. 10.1016/j.coi.2011.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A, Lucin KM, Czirr E, Park J-S, Couillard-Després S, Aigner L, Li G, Peskind ER, Kaye JA, Quinn JF, Galasko DR, Xie XS, Rando TA, Wyss-Coray T. 2011. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477:90–94. 10.1038/nature10357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sofroniew MV. 2005. Reactive astrocytes in neural repair and protection. Neuroscientist 11:400–407. 10.1177/1073858405278321 [DOI] [PubMed] [Google Scholar]
- 67.Grivennikov SI, Karin M. 2010. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 21:11–19. 10.1016/j.cytogfr.2009.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yoon S, Woo SU, Kang JH, Kim K, Shin H-J, Gwak H-S, Park S, Chwae Y-J. 2012. NF-κB and STAT3 cooperatively induce IL6 in starved cancer cells. Oncogene 31:3467–3481. 10.1038/onc.2011.517 [DOI] [PubMed] [Google Scholar]
- 69.Iwakoshi NN, Lee A-H, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. 2003. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 4:321–329. 10.1038/ni907 [DOI] [PubMed] [Google Scholar]
- 70.Wang Y, Alam GN, Ning Y, Visioli F, Dong Z, Nör JE, Polverini PJ. 2012. The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res. 72:5396–5406. 10.1158/0008-5472.CAN-12-0474 [DOI] [PMC free article] [PubMed] [Google Scholar]