

Abstract
Methylation of histone H3 on lysine 9 or 27 is crucial for heterochromatin formation. Previously considered hallmarks of, respectively, constitutive and facultative heterochromatin, recent evidence has accumulated in favor of coexistence of these two marks and their cooperation in gene silencing maintenance. H3K9me2/3 ensures anchorage at chromatin of heterochromatin protein 1α (HP1α), a main component of heterochromatin. HP1α chromoshadow domain, involved in dimerization and interaction with partners, has additional but still unclear roles in HP1α recruitment to chromatin. Because of previously suggested links between polycomb repressive complex 2 (PRC2), which catalyzes H3K27 methylation, and HP1α, we tested whether PRC2 may regulate HP1α abundance at chromatin. We found that the EZH2 and SUZ12 subunits of PRC2 are required for HP1α stability, as knockdown of either protein led to HP1α degradation. Similar results were obtained upon overexpression of H3K27me2/3 demethylases. We further showed that binding of HP1α/β/γ to H3K9me3 peptides is greatly increased in the presence of H3K27me3, and this is dependent on PRC2. These data fit with recent proteomic studies identifying PRC2 as an indirect H3K9me3 binder in mouse tissues and suggest the existence of a cooperative mechanism of HP1α anchorage at chromatin involving H3 methylation on both K9 and K27 residues.
INTRODUCTION
N-terminal tails of histones protrude from nucleosome core structures and are subject to various posttranslational modifications that regulate chromatin properties (1). Methylation of residues Lys 9 (H3K9me2/3) and Lys 27 (H3K27me2/3) of H3 are largely associated with silent heterochromatic regions. Although H3K9me2/3 and H3K27me2/3 are generally defined as hallmarks of constitutive and facultative heterochromatin (HC), respectively, it recently became evident that these marks frequently colocalize in the genome. For instance, chromatin immunoprecipitation experiments revealed enrichment of H3K9me3 and H3K27me3 modifications at 4q subtelomeric D4Z4 repeats (2) and at telomeres (3), and a significant enrichment of H3K9 and H3K27 trimethylation was also detected at the FXN locus of Friedreich ataxia-derived fibroblasts (4) and at the Deleted in Colon Cancer gene (DCC) promoter (5). Supporting these observations, genome-wide analyses revealed that H3K9me3, apart from heterochromatic regions, is frequently detected at silent gene promoters, one-half of which are cooccupied by H3K27me3 and belong to the family of so-called bivalent gene promoters (6, 7). Further reinforcing of these findings came from mass spectrometry analysis of H3K27me2/3 nucleosomes purified from embryonic stem cells, mouse embryonic fibroblasts, and HeLa cells showing frequent cooccurrence of H3K27me2/3 with H3K9me2/3- and H4K20me2-repressive marks (8). A quantitative mass spectrometry analysis of histone posttranslational modifications performed on mononucleosomes bound by the chromo domain (CD)-containing heterochromatin protein 1 (HP1) further revealed concomitant enrichment of H3K9me2/3 and H3K27me2/3 (9). Finally, two recent studies provided additional evidences of a cross talk between H3K9 and H3K27 methylation either on the inactive X (Xi) chromosome (10) or at the level of developmentally regulated gene promoters (11).
Di/trimethylation of H3K9 is mainly catalyzed by the conserved SUV39H1/2 histone methyltransferases, while the polycomb repressive complex 2 (PRC2) ensures di/trimethylation of H3K27 (12, 13). PRC2 is conserved from Drosophila to mammals and comprises the EZH1/2 catalytic subunit, SUZ12, EED, and RBBP7/4 (13). Recently, several other proteins were identified as PRC2 components—AEBP2, PCL, and JARID—which are not required for PRC2 enzymatic activity in vitro but were shown to enhance the complex activity in vivo and to regulate chromatin recruitment (13). In proliferating cells, transmission of H3K27me3 and H3K9me2/3 epigenetic marks appears to be mediated by direct binding to these marks of, respectively, the EED subunit of PRC2 (14, 15) and HP1, which in turn recruits SUV39H1/2 (16–18). HP1 is a key structural adaptor for macromolecular complex assembly required for constitutive HC formation and maintenance. It is conserved from Schizosaccharomyces pombe (Swi6) to mammals, in which three HP1 isoforms exist: HP1α, -β, and -γ (19). HP1 binding to H3K9me2/3 occurs through direct interactions of the methyl groups with the aromatic cage of HP1 located in the conserved N-terminal CD (20, 21). The evolutionarily conserved chromoshadow domain (CSD) at the C-terminal end defines a second critical region of HP1 required for silencing functions. HP1 CSD is involved both in homo/heterodimerization and in interactions with various partners, the latter playing crucial roles in HP1 association with chromatin, as binding of HP1 to H3K9me2/3, on its own, is fairly weak (19, 22–25).
Previous studies have reported the ability of SUZ12 to interact with HP1α and showed that depletion of SUZ12 alters HP1α distribution in embryonic stem (ES) cells (26, 27). However, how PRC2 regulates HP1α abundance at chromatin remains unknown. Interestingly, large-scale proteomic screens recently identified several PRC2 members as readers of the H3K9me3 mark, but the direct binder for this complex to the K9me3 mark has not been identified (28, 29). With these findings and the growing evidence of cross talks between H3K9me2/3 and H3K27me3 pathways of gene silencing, we tested whether PRC2 may regulate HP1α abundance at chromatin of human cells.
MATERIALS AND METHODS
Cell culture and transfections.
HT1080 is a human fibrosarcoma cell line, and HCA2 cells are primary human foreskin fibroblasts. Small interfering RNAs (siRNAs; Eurogentec) were transfected with Lipofectamine 2000 (Invitrogen). The following siRNAs were used: EZH2-A (30), EZH2-B (31), SUZ12 (32), and luciferase (3) siRNAs. Transfections with plasmids were also performed with Lipofectamine 2000. pGEX-4T1-GST-HP1α (33), pCMV-HA-JMJD3 (34), and pCMV-HA-UTX (34) plasmids 24074, 24167, and 24168 were obtained from Addgene. The plasmid carrying the GFP-HP1α gene, cloned into pIE-NHA-Gw (Invitrogen) (35), was kindly provided by P.-O. Angrand (Institut de Recherche Interdisciplinaire/CNRS, Villeneuve d'Ascq, France). In experiments with proteasome inhibitor, 30 h after transfection with siRNAs, cells were treated with 3 μM MG132 (Calbiochem) for 18 h.
HA-HP1α-V21M and HA-HP1α-I165K plasmid constructs.
HA-HP1α-V21M and HA-HP1α-I165K constructs (where HA is hemagglutinin) were obtained by site-directed mutagenesis using HA-HP1α vector as the template (cloned into pIE-NHA-Gw [Invitrogen] and kindly provided by P.-O. Angrand, Institut de Recherche Interdisciplinaire/CNRS, Villeneuve d'Ascq, France), Pfu enzyme (Thermo Scientific), and the following primers: V21M_forward, GGAGGAGTATGTTATGGAGAAGGTGCTAGAC; V21M_reverse, GTCTAGCACCTTCTCCATAACATACTCCTCC; I165K_forward, GTGAAATGTCCACAAATTGTGAAAGCATTTTATGAAGAGAG; and I165K_reverse, CTCTCTTCATAAAATGCTTTCACAATTTGTGGACATTTCAC, where boldface indicates mutated codons. Plasmids were checked by sequencing.
qRT-PCR.
Total RNA was isolated with TriPure reagent (Roche), and quantitative reverse transcription-PCRs (qRT-PCRs) were performed as described previously (3) using HP1α and ACTB primers (3, 36).
Cell fractionation.
Cells were incubated for 5 min at 4°C in CEB (10 mM HEPES, pH 7.9, 10 mM KCl, 0.34 M sucrose, 1.5 mM MgCl2, 0.5 mM dithiothreitol [DTT], 1% NP-40), and centrifuged at 1,300 × g. Supernatant was collected as the cytoplasm fraction. The pellet was washed with CEB lacking NP-40 and resuspended in 50 mM Tris-Cl (pH 7.4), 150 mM NaCl, 1% NP-40, and 0.25% sodium deoxycholate. Lysates were incubated for 20 min on ice and centrifuged at 10,000 × g. Supernatant was collected as the nucleoplasm and the pellet as the chromatin fraction. The pellet was resuspended in Laemmli sample buffer and boiled at 98°C for 5 min before sonication.
Western blotting.
Western blotting was performed according to standard procedures using the following antibodies (manufacturer, catalog no., and dilution are given in parentheses): H3 (Abcam, ab1791, 1:2,500), H3K9me3 (Millipore, 17-442, 1:1,000), H3K27me3 (Millipore, 17-622, 1:2,000), HP1α (Millipore, 05-689, 1:2,500), HP1β (Active Motif, 39979, 1:1,000), HP1γ (Active Motif, 39981, 1:1,000), β-actin (Sigma, A5441, 1:10,000), EZH2 (Diagenode, 004-050, 1:1,000), SUZ12 (Millipore, 04-46, 1:1,000), Rb (Santa Cruz, sc-50, 1:1,000), and HA (Sigma, H6908, 1:2,000). Streptavidin-horseradish peroxidase (HRP; 1:5,000) was from Invitrogen. Signal quantifications were done using either GelEval or LI-COR Image Studio software.
Immunofluorescence.
Cells were treated with permeabilization buffer (20 mM Tris-HCl, pH 8.0, 50 mM NaCl, 3 mM MgCl2, 300 mM sucrose, 0.5% Triton X-100) for 5 min at room temperature, fixed with 3% formaldehyde for 15 min, and incubated again in permeabilization buffer for 10 min. Cells were then blocked with 3% bovine serum albumin (BSA) for 30 min at 37°C and incubated with primary antibodies for 1 h at 37°C, followed by phosphate-buffered saline (PBS) washes and addition of a second primary antibody before 1 h of incubation with a mix of secondary antibodies. The following antibodies were used: H3K9me3 (Millipore, 17-442, 1:100), H3K27me3 (Millipore, 17-622, 1:200), HP1α (Millipore, 05-689, 1:50), HP1β (Active Motif, 39979, 1:400), HP1γ (Active Motif, 39981, 1:200), and HA (Sigma, H6908, 1:50; or Roche, 3F10, 1:200). Samples were mounted in 2.33% DABCO (1,4-diazabicyclo[2.2.2]octane), 90% glycerol, 20 mM Tris (pH 8.0), and 0.6 μg/ml DAPI (4′,6-diamidino-2-phenylindole). Images were acquired with a Cell Observer spinning disc (Zeiss) confocal microscope with a 100× objective and analyzed using ImageJ software (National Institutes of Health).
Bacterial protein purification.
GST-HP1α recombinant protein was expressed in Escherichia coli strain BL21 incubated with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 4 h. Glutathione S-transferase (GST)–HP1α was then purified using a standard protocol and a Pierce glutathione chromatography cartridge (Thermo Scientific). The N-terminal GST tag was cleaved using biotinylated thrombin (Novagen), and HP1α was then purified by incubation with avidin-agarose (Thermo Scientific) and glutathione-Sepharose (GE Healthcare) beads for 30 min at room temperature under gentle agitation.
Pulldown experiments with H3 N-terminal tail peptides.
H3 biotinylated peptides comprising the 40 N-terminal amino acids of H3, followed by the YCK sequence added for coupling purposes (ARTKQTARKSTGGKAPRKQLATKAARKSAPATGGVKKPHR-YCK [biotin]; boldface indicates the positions of K9 and K27 residues) (14) were synthesized on the solid phase using conventional fluorenylmethoxycarbonyl chemistry, purified by reverse-phase high-pressure liquid chromatography (HPLC), and characterized by mass spectrometry. Pulldown assays with HT1080 cell extracts were performed as described previously (14). For pulldown assays with bacterially purified protein, 6 μg of H3 peptides were incubated with 1.5 μg of purified HP1α and 50 μl avidin-agarose beads (Thermo Scientific). Pulldown assays were carried out overnight at 4°C in 1 ml total volume of 0.1% Triton X-100–2 mM DTT and protease inhibitors in PBS. Beads were washed 3 times with 150 mM NaCl and 0.1% Triton X-100 in PBS before resuspension in Laemmli sample buffer.
ChIP.
Chromatin immunoprecipitation (ChIP) was performed according to standard procedures. Chromatin was sonicated with a BioRuptor sonicator (Diagenode) to obtain fragments between 200 and 600 bp. Samples were incubated overnight with antibodies and immunoprecipitated using the OneDay ChIP kit (Diagenode) or, for siRNA-treated samples, the LowCell# ChIP kit (Diagenode) according to the manufacturer's instructions. The following antibodies (manufacturer, catalog no.) were used: H3K9me3 (Abcam ab8898), H3K27me3 (Millipore 17-622), HP1γ (Active Motif 39981), and green fluorescent protein (GFP; Abcam ab290). Nonspecific total rabbit IgGs were from Millipore. Immunoprecipitated DNA was analyzed by qPCR using the Kapa SYBR Fast qPCR kit (Kapa Biosystems) and the following primers: 4qHOX (2), CHR7q (5′-CCTCGCTTTGACACGACTCGG and 5′-GCACAGGATTCAGACGGGCTTT), TSH2B (5′-GCAGCACTGCCTGAATGTTA and 5′-TGTATTTGGCGGCAGTGTTA), MB exon2 (LowCell# ChIP kit; Diagenode), c-FOS promoter (37), and GAPDH TSS (3). Results were presented either as the percentage of starting material (% input) or as relative occupancy of specific signal over c-FOS promoter control locus [% input (specific locus)/% input (control locus)].
Statistical analyses.
Error bars in graphs represent standard deviations from independent experiments. Student's t tests were applied to compare differences between means.
RESULTS
EZH2 or SUZ12 depletion leads to HP1α loss from chromatin and degradation.
Despite a previous report establishing that HP1α interacts with SUZ12 (26), a direct role for PRC2 in regulating HP1α abundance at chromatin has not been demonstrated. Here, we wished to test whether PRC2 impacts HP1α binding to chromatin. As EZH2, SUZ12, EED, and RBBP7/4 have been established as the components of the minimum functional PRC2 core complex (13), we first depleted EZH2 histone-lysine N-methyltransferase in HT1080 human fibrosarcoma cancer cells. Cells were treated with either siRNA against EZH2 (siEZH2-A) or control siRNA (siLuci), and lysates were collected every 24 h over a 4-day period. An ∼40% reduction in EZH2 protein levels was noticeable already 24 h after transfection, reaching 80% after 96 h (Fig. 1A and B). In agreement with previous findings, knockdown of EZH2 led to depletion of another protein of the PRC2 complex, SUZ12 (32) (Fig. 1A). As anticipated, loss of EZH2/SUZ12 was associated with a reduction in K27 trimethylation of histone H3 (Fig. 1A). Interestingly, EZH2 depletion resulted in a strong decrease of total cellular levels of HP1α, an effect that was already observed 24 h after treatment and that mirrored EZH2 depletion in time course experiments (Fig. 1A and B). We also observed a slight reduction of H3K9me3 levels at later time points of siEZH2-A treatment, which is likely to result from a reduced ability to recruit SUV39H1/2 H3K9 methyltransferase to chromatin in response to HP1α loss (16–18). To confirm the Western blot data, we also monitored HP1α levels after siEZH2 treatment using microscopy coupled with immunofluorescence in Triton-extracted cells. In comparison to control cells (siLuci), HP1α levels were reduced by 45% after treatment with siEZH2-A, and this occurred without any noticeable reduction of H3K9me3 levels (Fig. 1C and D). The observed phenomenon was not a consequence of siRNA transfection, as siLuci transfection showed protein levels similar to those of mock control (Fig. 1E). Degradation of HP1α upon EZH2 knockdown was also observed with a different siRNA against EZH2 (siEZH2-B), as well as in normal HCA2 fibroblasts (Fig. 1F and G), further supporting our findings.
FIG 1.

HP1α dissociates from chromatin and is degraded upon EZH2 depletion. (A) HT1080 cells were treated with siEZH2-A for the indicated time lengths or with siLuciferase (siLuci) for 48 h, and whole-cell extracts were analyzed by Western blotting with the specified antibodies. (B) Quantification from three independent time course experiments of EZH2 and HP1α protein levels is shown after normalization, first to β-actin and then to siLuci. (C) Immunofluorescence analyses of H3K27me3 and H3K9me3 (red, left and right panels, respectively) and HP1α (green) in Triton-extracted HT1080 cells transfected with siEZH2-A or siLuci for 48 h. Scale bars, 10 μm. (D) Quantification of immunofluorescence shown in panel C on at least 20 nuclei from three independent experiments. Mean fluorescence intensity of nuclei in siEZH2-A-treated cells was compared to that of siLuci control cells. (E) Control for siLuci treatment. HT1080 cells were either mock transfected or treated with siEZH2-A or siLuci for 72 h, and whole-cell extracts were analyzed by Western blotting with the specified antibodies. (F) Knockdown efficiency of EZH2 in HT1080 (upper panel) or HCA2 (lower panel) cells treated with either siEZH2-A or siEZH2-B. β-Actin is shown as a loading control. (G) Western blot analysis of the nucleoplasm and chromatin fractions from HT1080 (upper panel) or HCA2 (lower panel) cells collected 72 h after transfection with two different siRNAs against EZH2. Membranes were analyzed separately for the presence of HP1α, H3K27me3, and H3K9me3. H3 and Rb levels were used as loading controls for the chromatin and nucleoplasm fractions, respectively. Control cells were transfected with siLuci.
HP1α loss upon EZH2 depletion is not a consequence of cell cycle perturbation or reduced HP1α transcription but results from proteasomal degradation.
Having found that HP1α levels are drastically downregulated upon EZH2 depletion, we checked that this was not a consequence of cell cycle perturbation, as depletion or inhibition of EZH2 was previously reported to increase the number of cells in either G2/M (38, 39) or G1/S (40). DNA content analysis of HT1080 cells treated with siEZH2-A for 72 h did not reveal major changes in cell cycle distribution; however, there was a slight increase in G2/M cells (Fig. 2A). As HP1 proteins naturally detach from chromatin upon phosphorylation of H3 on serine 10 residue during metaphase (41), we checked whether siEZH2 treatment of cells increases the level of H3Ser10P. As seen in Fig. 2B, there was no difference in H3 phosphorylation between control and siEZH2-treated cells; thus, the effect of HP1α loss upon EZH2 depletion was not caused by increased H3Ser10 phosphorylation. Based on the above control experiments and on the fact that reduction in total HP1α levels was seen as early as 24 h after siEZH2 treatment, we concluded that cell cycle perturbations could not account for the loss of HP1α under our experimental conditions.
FIG 2.
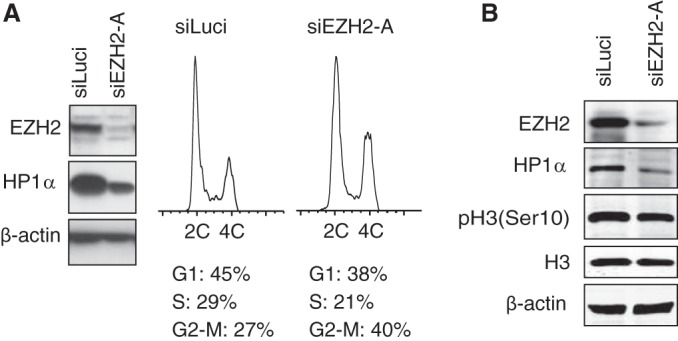
EZH2 depletion is not associated with major cell cycle defects. (A) Cell cycle distribution analyses of HT1080 cells treated with siRNAs against EZH2 or luciferase for 72 h. siEZH2-A-treated cells displayed minor enrichment in G2/M cells. (B) HT1080 cells were treated with siEZH2-A or with siLuci for 72 h, and cell extracts were analyzed by Western blotting to monitor the phosphorylation levels of H3 on Ser10 [pH 3(Ser10)]. Total H3 and β-actin levels are shown as loading controls, and EZH2 is shown as a control for siRNA knockdown efficiency.
Disruption of HP1α interaction with H3K9me3 marks results in its detachment from chromatin and subsequent recovery of the protein in the soluble fraction, leaving total cellular levels of HP1α unaltered (16). Conversely, our results indicated a strong reduction of total HP1α cellular levels upon EZH2 knockdown with no recovery of the detached protein in the soluble fraction (nucleoplasm or cytoplasm) (Fig. 3A). As we did not detect any downregulation of HP1α transcript levels upon siEZH2-A/-B treatment (Fig. 3B), this suggested that it is the protein, and not the transcript level of HP1α, that is affected. It has been reported that HP1α undergoes proteasomal degradation upon overexpression of lamin A mutants (42, 43). Hence, we tested whether EZH2 depletion-induced loss of HP1α was due to proteasomal degradation. Accordingly, we observed that treatment with MG132 proteasome inhibitor mostly suppressed HP1α degradation upon EZH2 depletion (Fig. 3C and D). Note that due to high toxicity of MG132, siEZH2 treatment was applied for 48 h instead of 72 h, explaining why the decrease in HP1α levels was less drastic in these experiments.
FIG 3.

HP1α loss upon EZH2 depletion does not result from reduced HP1α transcription but is due to proteasomal degradation. (A) HT1080 cells were transfected with either siLuci or siEZH2-A and collected 72 h after transfection. Cell extracts were subjected to subfractionation, and total, nucleoplasm, chromatin, and cytoplasm fractions were analyzed by Western blotting using the indicated antibodies. (B) Transcript levels of HP1α gene were measured by qRT-PCR and normalized to β-actin (ACTB) transcripts in HT1080 cells treated for 72 h with two different siRNAs against EZH2 or with siLuci (n = 3 independent experiments). (C) MG132 proteasome inhibitor suppresses HP1α degradation in siEZH2-treated cells. Thirty hours after siRNA transfection, HT1080 cells were incubated with either 3 μM MG132 (+) or dimethyl sulfoxide (DMSO) (−) for 18 h. Total protein levels for HP1α, EZH2, and β-actin were analyzed. (D) Quantification of HP1α protein levels from panel C (n = 3 independent experiments). (E) HP1α levels are also decreased in SUZ12-depleted cells. HT1080 cells were treated with siSUZ12 or siLuci for 6 days and processed as described for panel A.
SUZ12 depletion is also associated with loss of PRC2 integrity (44, 45). Therefore, we tested whether similar effects on HP1α can be observed when cells are treated with siRNA against SUZ12. As detachment of HP1α from chromatin upon knockdown of SUZ12 was similarly associated with degradation of HP1α and lack of recovery in the cytoplasmic fraction (Fig. 3E), we concluded that loss of PRC2 integrity induces proteasomal degradation of HP1α.
Removal of H3K27me3 marks is sufficient to detach HP1α from chromatin.
It is well established that HP1 proteins detect and bind to H3K9me3 (46). How then can we explain the role of PRC2 in HP1α abundance at chromatin? We reasoned that PRC2 might stabilize the interaction between HP1α and H3K9me3. As PRC2 binds to its own product, H3K27me3 (14, 15), this raised the interesting hypothesis that HP1α anchorage at chromatin may be stabilized through interactions with H3K27me3-bound PRC2. Alternatively, HP1α binding efficiency to the H3K9me3 mark may be increased through direct interactions with K27me3, as purified HP1α protein was reported to display low affinity for the K27me3 mark in pulldown experiments using H3 tails (47). In both cases (indirect or direct interaction of HP1α with H3K27me3), we expected the H3K27me3 mark to be involved in the stabilization of HP1α at chromatin. To test this, we globally depleted cells of H3K27me3 by transiently overexpressing JMJD3, a potent H3K27me2/3 demethylase (34). We transfected HT1080 cells with either a control HA or an HA-JMJD3 vector and analyzed, by immunofluorescence, the residual levels of H3K27me3, H3K9me3, and HP1α. Twenty-four hours after transfection with HA-JMJD3, H3K27me3 marks were reduced to 32% of the control levels, while H3K9me3 marks were unaltered (Fig. 4A, B, and D). In agreement with a role played by H3K27me3 in PRC2-dependent stabilization of HP1α, we found that chromatin-bound HP1α had dropped to 15% of the control levels in JMJD3-overexpressing cells (Fig. 4C and D). Similar results were observed when we transiently overexpressed HA-UTX H3K27me2/3 demethylase (34) using the same experimental settings (Fig. 4E and F).
FIG 4.

Overexpressed H3K27me2/3 demethylases remove HP1α from chromatin. Immunofluorescence analyses of H3K27me3 (A), H3K9me3 (B), or HP1α (C) (green) in HT1080 cells transiently transfected (24 h) with either the HA-JMJD3 construct or the HA plasmid. Transfections were monitored with anti-HA (red). Scale bars, 10 μm. (D) Quantification of immunofluorescence analyses from at least 15 nuclei and from three independent experiments. (E) Immunofluorescence analyses of HP1α (green) in HT1080 cells transiently transfected with the HA-UTX construct. Transfections were monitored with anti-HA (red). (F) Quantification of immunofluorescence signals from three independent experiments.
Hence, whether resulting from knockdown of the H3K27 histone methyltransferase or from overexpression of the corresponding demethylase, these data supported an important role of H3K27me3 in regulating HP1α abundance at chromatin.
HP1α binding to H3K9me3 peptide tails is increased in the presence of H3K27me3.
The above data strongly suggested that the H3K27me3 mark is involved in HP1α anchorage at chromatin. To directly test the impact of H3K27me3 on HP1α affinity for the H3K9me3 mark, we performed pulldown experiments using HT1080 cell lysates and H3 tail peptides with either K9me3, K27me3, or both modifications. As expected, HP1α binding to H3 tails was strictly dependent on the presence of K9me3 (Fig. 5A). Strikingly, the binding of HP1α was significantly increased more than 4-fold (P < 0.001) when H3K9me3 peptide was also carrying the K27me3 modification (Fig. 5A and B). HP1α proteins contain two highly conserved domains: (i) the CD, which recognizes and directly binds to H3K9me3; and (ii) the CSD, involved in homo/heterodimerization and interactions with various proteins, including SUV39H1, KAP-1, CAF1p150, SUZ12, lamin B receptor, and TAFII130 (19, 33). Mutational analyses showed that both domains are required for correct binding of HP1 isoforms to the H3K9me3 mark (16, 17, 24). To test whether these domains are also needed for binding onto peptides bearing the dual methyl mark, we performed pulldown experiments using lysates from cells expressing HA-tagged HP1α constructs with mutations either in the CD (V21M) or the CSD (I165K) (Fig. 5C). As seen in Fig. 5D, similarly to H3K9me3 histone tails, HP1α binding to H3K9me3K27me3 was completely abolished in the presence of a mutation in either of the domains.
FIG 5.

Binding of human cell extract-derived HP1α to H3 histone tails is increased in the presence of both K9me3 and K27me3 marks. (A) Pulldown experiments with biotinylated H3 tail peptides (H3, unmodified; K9, H3K9me3; K27, H3K27me3; K9;K27, H3K9me3K27me3) and HT1080 cell lysates. After elution, bound HP1α proteins were analyzed by Western blotting and H3 peptides were monitored with HRP-conjugated streptavidin. (B) Quantification of data from panel A from four independent pulldown experiments (n = 4) by normalizing first to the H3 peptide amount and then to HP1α binding to H3K9me3 peptide. (C and D) Both CD and CSD are required for HP1α binding to H3K9me3K27me3 tails. Extracts from HT1080 cells overexpressing either wild-type (WT), V21M, or I165K HA-HP1α proteins (C) were incubated in the presence of immobilized H3 tails as indicated. Bound HA-HP1α and H3 peptides were visualized by, respectively, anti-HA antibodies and Coomassie blue staining (D).
Bacterially expressed HP1α protein does not display increased affinity for H3 peptide tails with both K9me3 and K27me3 modifications.
One possible explanation to account for the increased binding of human cell-derived HP1α to H3K9me3 in the presence of a nearby H3K27me3 mark would be that the binding of one HP1α monomer to H3K9me3 increases the affinity of the second HP1α molecule for H3K27me3. Alternatively, HP1α may not be directly bound to H3K27me3, but the latter mark may recruit other proteins, such as PRC2, which through interactions with HP1α dimers would stabilize their binding to H3K9me3. Since mutation of the I165 residue of HP1α not only abolishes dimerization of the protein but also prevents the CSD-mediated interactions with other factors (47), it was difficult to deduce which of the two, dimerization or interactions with other factors, is important for the enhanced binding of HP1α proteins to chromatin. To assess whether auxiliary factors may be required, we repeated pulldown assays using bacterially expressed and purified HP1α (Fig. 6A). As shown previously (24), the recombinant HP1α protein spontaneously dimerizes in solution (Fig. 6B). When this recombinant protein was used instead of human cell extracts, we detected similar binding efficiencies of HP1α to H3K9me3 and to H3K9me3K27me3 (Fig. 6C and D). Using serial dilutions of purified HP1α protein in the presence of steady amounts of H3 peptides, we confirmed that the loss of enhanced binding to peptides bearing both marks was not due to saturation of H3 peptides (Fig. 6E). These results suggested that the increased binding of HP1α to H3K9me3K27me3 that we observed when human cell extracts were used in pulldown experiments is not accomplished exclusively by HP1α but requires additional cofactors.
FIG 6.

Bacterially produced HP1α does not show increased binding to H3 tails combining K9me3 and K27me3 marks. (A) GST-HP1α protein was purified from bacteria (lane 1) and cleaved with thrombin protease (lane 2) before affinity purification on glutathione-agarose beads (lanes 3 and 4). (B) Purified recombinant HP1α spontaneously forms dimers in solution with an approximate molecular mass of 54 kDa. Fractions 5 to 13 recovered from a Superdex 200 gel filtration column were analyzed by Western blotting. Molecular mass markers are shown on the left, and the calculated molecular masses for fractions 8 to 10 are indicated. (C) Purified recombinant HP1α protein was incubated in the presence of immobilized biotinylated H3 peptides modified as indicated (H3, unmodified; K9, H3K9me3; K27, H3K27me3; K9;K27, H3K9me3K27me3). After elution, bound HP1α proteins, H3K9me3, and H3K27me3 were analyzed by Western blotting, and H3 peptides were monitored by Coomassie blue staining. (D) Quantification of data from panel C by normalizing to the binding to H3K9me3. Quantifications were obtained from three independent experiments (n = 3). (E) Serial dilutions (2.4, 1.2, 0.6, and 0.3 μg) of bacterially produced HP1α were incubated in the presence of 6 μg of either H3K9me3 (K9) or H3K9me3K27me3 (K9;K27) peptide, and HP1α binding was monitored by Western blotting. HP1α levels were normalized first to H3K9me3 and then to the ratios obtained with 2.4 μg of recombinant HP1α.
EZH2 is required for increased binding of HP1α to H3 tails displaying both K9me3 and K27me3 marks.
Interactions with SUZ12 were previously detected for HP1α and -γ (26, 27). Since our data indicated that depletion of either PRC2 subunit, EZH2 or SUZ12, resulted in substantial loss of HP1α binding to chromatin, we anticipated that PRC2 binding to H3K27me3 might stabilize HP1α interactions with the H3K9me3 mark. In support of our hypothesis and as expected (14), EZH2 binds to H3 peptides containing the K27me3 mark (Fig. 7A). Hence, to test whether EZH2 has any impact on the increased binding of human cell-derived HP1α proteins to K9me3/K27me3-containing H3 peptides, we chose to repeat pulldown experiments with extracts coming from HT1080 cells treated with siEZH2-A. However, as shown above, HP1α protein is almost completely degraded upon EZH2 knockdown, and the amount of HP1α that is recovered is thus too low for pulldowns. In light of the previously reported formation of heterodimers between HP1α and other HP1 isoforms (48), we reasoned that we may probe for the binding of HP1β and -γ, instead of that of HP1α, as we showed the following: (i) treatment of cells with siEZH2-A, although reducing the protein levels of HP1β and -γ, does not lead to complete loss of the proteins (Fig. 7B, left panel); (ii) HP1β and -γ are released from chromatin upon treatment with siEZH2-A (Fig. 7B and C); and (iii) binding of human cell-derived HP1β and -γ to the H3K9me3 tail is also increased in the presence of K27me3 (Fig. 7D). Assuming that chromatin-bound HP1 proteins are more susceptible to degradation upon EZH2 knockdown, higher residual levels of HP1β and -γ than of HP1α are likely due to the high abundance of these two isoforms in the nucleoplasm (Fig. 7B). Interestingly, we also found that the stimulating effect of H3K27me3 mark on HP1α/β/γ binding to H3K9me3 occurred whether the marks were present on the same H3 tail (K9;K27) or on different peptides (K9 + K27) (Fig. 7D), suggesting that K27me3 may also act in trans to stabilize HP1 binding to K9me3 H3 tails.
FIG 7.

EZH2 is required for increased binding of human cell extract-derived HP1β/γ to H3 histone tails with both K9me3 and K27me3 marks. (A) EZH2 binding is detected on K27me3-containing H3 tails. HT1080 cell lysates were incubated in the presence of immobilized biotinylated H3 peptides modified as indicated, and bound EZH2 was detected by Western blotting. The total amount of H3 peptides was monitored with Ponceau S. (B) Residual HP1α/β/γ levels upon EZH2 depletion. HT1080 cells were transfected with either siLuci or siEZH2-A and collected 72 h after transfection. Total cell extracts and chromatin and nucleoplasm fractions were analyzed by Western blotting using the indicated antibodies. (C) Immunofluorescence analyses of HP1β and HP1γ (green) in HT1080 cells transiently transfected with HA-JMJD3 construct and performed as described in legend to Fig. 4A to C. (D) Pulldown experiments with HT1080 cell lysates and biotinylated H3 tail peptides (H3, unmodified; K9, H3K9me3; K27, H3K27me3; K9;K27, H3K9me3K27me3; K9 + K27, equimolar mix of H3K9me3 and H3K27me3). (E to G) Binding of HP1β and γ to H3 tails with K9me3 and K27me3 is reduced in the absence of EZH2. (E) Western blot of cell extracts after HT1080 treatment with siLuci or siEZH2-A used as inputs for pulldown experiment shown in panel F. (F) H3K9me3 or a mixture of H3K9me3 and H3K27me3 peptides was used for the pulldown experiment. Bound HP1β/γ and H3K9me3 mark levels were monitored by Western blotting. Loading of H3 peptides was monitored by Coomassie blue staining. (G) Quantification of data from panel F by normalizing to H3K9me3 binding.
Since all the conditions were met to monitor binding of HP1β and -γ instead of HP1α, we repeated the pulldown experiments with extracts from cells treated with either siLuci or siEZH2-A using a mixture of K9me3 and K27me3 H3 tails. In agreement with our hypothesis, we observed an increased binding of HP1β and -γ in the presence of both K9me3 and K27me3 marks in siLuci control samples, but this increase was no longer detected upon EZH2 depletion (Fig. 7E to G).
In conclusion, the evidence collected so far clearly suggests that EZH2 influences binding of HP1 proteins to chromatin and that its presence is essential for the observed enhanced binding to H3 tails carrying both methyl marks.
EZH2 depletion reduces HP1γ abundance at H3K27me3-enriched loci bound by EZH2.
Our data indicate that the stability of all three chromatin-bound HP1 isoforms is drastically reduced upon EZH2 knockdown. Because of the well-established requirement for the H3K9me2/3 mark in maintaining HP1 at chromatin, our observations would suggest that most H3K9me2/3-enriched loci are in close proximity to H3K27me2/3 marks. Indeed, recently published papers provide evidence in favor of this. First, Voigt et al. data suggested that about 55% of H3K27me2/3-marked mononucleosomes further display either di- or trimethylation on H3K9 residue (8). The fact that H3K27me2/3 marks are very abundant and detected in a total of about 50% of HEK293 nucleosomes genomewide (9) would imply that one-fourth of all nucleosomes display di- or trimethylation on both H3K9 and H3K27 residues. Furthermore, quantitative mass spectrometry analysis of histone posttranslational modifications revealed concomitant and similar enrichment of H3K9me2/3 and H3K27me2/3 marks on HP1α- or -β-bound mononucleosomes (9) (Fig. 8A).
FIG 8.

EZH2 depletion induces loss of HP1γ from chromatin at H3K27me3-enriched loci. (A) Analysis of “Additional file 2” data from LeRoy et al. (9) The graph shows relative H3 or H4 posttranslational modification enrichment determined by quantitative mass spectrometry in chromatin samples immunoprecipitated with FLAG antibodies and obtained from HEK293 cells expressing either FLAG-HP1α or FLAG-HP1β. Data were normalized to genomic levels of these marks. (B) qPCR analysis of ChIP DNA using primers specific for the indicated genomic regions. HT1080 chromatin was immunoprecipitated with antibodies against H3K9me3 or H3K27me3. Nonspecific total rabbit IgGs were used as a control. Genomic regions for qPCR analysis were chosen on the basis of the presence of H3K9me3 and H3K27me3. For heterochromatic/transcriptionally silent loci, we used primers amplifying the subtelomeric region of the D4Z4 repeat sequences on chromosome 4q (4qHOX), the chromosome 7 subtelomeric region, ∼200 bp away from the telomeric TTAGGG repeats (CHR7q), the promoter region of testis-specific histone 2B variant (TSH2B), and the exon 2 of myoglobin gene (MB exon2). The GAPDH TSS (transcription start site) region, extending from −46 to +119 relative to the TSS, was chosen as a locus positive for HP1γ binding but negative for H3K9me3, H3K27me3, and EZH2. The c-FOS promoter region was chosen as a negative control for all heterochromatic proteins. Data are shown as percent enrichment relative to input. (C) HT1080 cells were treated for 72 h with siRNA against EZH2 (siEZH2-A) or luciferase (siLuci), and chromatin was used for immunoprecipitation with specific antibodies against HP1γ (top panel), EZH2 (middle panel), H3K27me3 (bottom panel), or a nonspecific total rabbit IgG. Data are shown as enrichment over the negative control (the c-FOS promoter region). Quantifications were obtained from three independent experiments (n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.005. (D) HT1080 cells stably expressing GFP-HP1α protein were treated for 4 days with siEZH2-A or siLuci and reductions in either endogenous or GFP-tagged HP1α levels were monitored by Western blotting. H3K27me3 levels are shown as an indication of EZH2 knockdown and β-actin as a loading control. (E) qPCR analysis of ChIP samples using chromatin samples from panel D. ChIP was carried out with antibodies against GFP or nonspecific total rabbit IgG. Data are shown as percent enrichment relative to input. Quantifications were obtained from three independent experiments (n = 3). ***, P < 0.005; NS, not significant.
We next wished to provide additional evidence that EZH2 depletion from a given locus leads to detachment of HP1 proteins from the same locus. To do this, we performed ChIP experiments in HT1080 cells treated with siEZH2-A, and since assessing chromatin-associated HP1α by ChIP is reportedly difficult and usually requires the use of tagged proteins, we monitored the residual abundance, at various loci, of HP1γ, EZH2, and H3K27me3. Interestingly, HP1γ has been reported to localize within both H3K27me3-enriched heterochromatin and H3K27me3-free euchromatic loci, including the highly transcribed GAPDH gene (49, 50), thus offering the possibility to control for residual HP1γ levels at loci that are not bound by EZH2. As shown in Fig. 8B, 4qHOX and CHR7q subtelomeric loci on one hand and testis-specific histone 2B variant TSH2B promoter or myoglobin exon 2 on the other hand are enriched in both H3K9me3 and H3K27me3 repressive marks. Conversely, and as expected, the c-FOS promoter did not display any of these two marks (Fig. 8B). Agreeing with a previous report (49), we found that HP1γ is recruited around the transcription start site (TSS) of the highly transcribed GAPDH gene in the absence of either H3K9me3 or H3K27me3 (Fig. 8B and C). The mechanisms of HP1γ recruitment to highly transcribed loci are not completely elucidated yet but may depend on the direct interaction of HP1γ with the activated form of RNA polymerase II (49).
Supporting a role played by the H3K27me3 mark in stabilizing HP1 proteins at chromatin, we found that depletion of EZH2 and the subsequent loss of the H3K27me3 mark were associated with reduced abundance of HP1γ at the four repressed loci that we analyzed (4qHOX, Chr7q, TSH2B, and myoglobin) (Fig. 8C). Conversely, and in agreement with the absence of EZH2 and H3K27me3, EZH2 knockdown did not affect HP1γ enrichment around the TSS of GAPDH (Fig. 8C).
Finally, since we failed to get ChIP to work with endogenous HP1α proteins, we established an HT1080 cell line stably expressing GFP-HP1α (Fig. 8D). The expression level of GFP-HP1α protein was slightly higher than the one of endogenous HP1α, and the impact of siEZH2 treatment was less drastic than the one observed on endogenous HP1α (Fig. 8D). We nevertheless observed a reduction of GFP-HP1α stability upon siEZH2 treatment (Fig. 8D). Monitoring GFP-HP1α abundance in siEZH2-A-treated cells revealed a significant reduction at both 4qHOX and subtelomeric CHR7q loci (Fig. 8E), further emphasizing the important role of EZH2 in ensuring HP1α stability at heterochromatin.
Collectively, these data clearly suggest that genomic regions associated with H3K9me3 and H3K27me3 repressive marks also display binding of the HP1 and EZH2 proteins and that knockdown of EZH2 leads to loss of HP1 proteins from these loci.
DISCUSSION
Until recently, it was assumed that H3K9me2/3 and H3K27me2/3 marks had distinct functions and that their presence in the genome was mutually exclusive. However, recent studies demonstrated the cooccupancy of these repressive marks at various genomic loci (8, 9) and, interestingly, suggested the existence of cooperative mechanisms between these methylation marks in maintaining silencing. Cooperation was proposed to be mediated either by the combined impact of H3K9me2 and H3K27me3 marks on the recruitment, to Xi, of CDYL chromodomain-containing protein, itself interacting with H3K9 histone methyltransferase (10), or through direct interaction between H3K9 and H3K27 histone methyltransferases (11). Further supporting an interplay between the two marks was the observation that direct recruitment of EZH2 and SUZ12 to the proximal promoter of the metastasis suppressor RKIP gene was accompanied by transcriptional repression mediated by both H3K27me3 and H3K9me3 modifications (51).
In this study, we provide additional evidence in favor of a cooperative mechanism between the two histone lysine methylation pathways by showing that proximity of the H3K27me3 mark increases the binding of HP1α to H3K9me3 and that it is dependent on the PRC2 complex. Although the interplay between HP1 and the PRC2 complex has been suggested before (26, 27), this is the first report showing that HP1α dissociates from chromatin and is degraded by the proteasome upon disruption of the PRC2 complex by siRNA-mediated knockdown. Based on our findings, we thus propose that thanks to PRC2 binding, H3K27me3 marks increase the stability of H3K9me2/3-bound HP1α (Fig. 9). It is still unclear, however, whether PRC2 directly interacts with HP1α or if it is mediated through yet another factor, as we were unable to detect interactions between endogenous HP1α and EZH2/SUZ12 PRC2 subunits. Interestingly, while investigating the maintenance of H3K27me3 by PRC2 complex, Hansen et al. (14) similarly detected increased HP1α and -γ binding to H3 peptides in the presence of both K9me3 and K27me3 marks; however, this observation was not addressed at all in the study. In light of the recently proposed cooperation between HP1α domains to bridge nearby H3K9 methylated nucleosomes (52), the fact that we observed the same effect on HP1α binding to H3 tails when the K9me3 and K27me3 marks were in cis or in trans suggests that PRC2 may also contribute to HP1α-mediated bridging activity between adjacent nucleosomes (Fig. 9).
FIG 9.
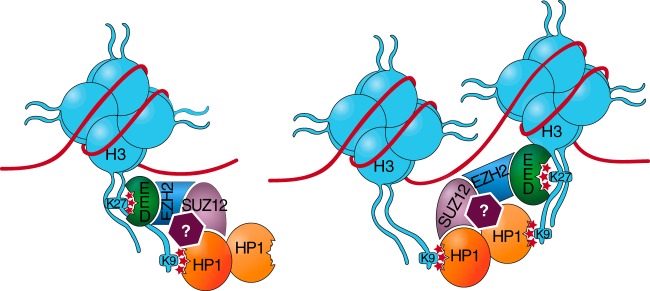
The H3K27me3-H3K9me3 cooperation model. We propose that H3K27me3-bound PRC2 stabilizes H3K9me3-anchored HP1α, either directly (interactions with SUZ12) or indirectly (interactions with another factor illustrated by a question mark). In this model, the second CD of each HP1α dimer remains free for binding to a nearby methylated H3K9 residue to bridge adjacent nucleosomes, as recently suggested (52).
How can we explain the destabilization of HP1 isoforms upon EZH2 or SUZ12 knockdown? A number of studies have shown that HC preferentially associates with nuclear periphery and that the lamin A/B network plays an important role in this localization (53). HP1α was shown to directly interact with the lamin B receptor, and three-dimensional (3D) microscopy analysis revealed a high degree of H3K9me3 and H3K27me3 overlapping signals at the nuclear periphery (54, 55). Interestingly, analyses of cells containing a lamin A mutation causing the Hutchinson-Gilford progeria syndrome showed that expression of the mutated protein decreases EZH2 and H3K27me3 levels, and this was later accompanied by a reduction in HP1α and H3K9me3 levels (56). In addition, overexpression of lamin A mutants caused proteasomal degradation of HP1α/β (42, 43). The precise molecular mechanism linking lamins with histone methylation and HP1 stability is still unknown, but it has been hypothesized that the lamin A/B network acts as a 3D scaffold hub for HC complexes assembly (57). Hence, changes in nuclear lamina structure and/or depletion of the PRC2 complex may lead to disruption of this HC network and proteasome-mediated degradation of chromatin-bound HP1α proteins. In this scenario, and knowing that HP1α is subject to various posttranslational modifications, including phosphorylation (58), it is possible that HC network disruption activates a kinase that phosphorylates chromatin-bound HP1α to trigger its ubiquitin-dependent proteolysis. Because of the connections that have also been established between the PRC2 and PRC1 polycomb group complexes (59), we also cannot exclude a possible role for PRC1 in contributing to overall heterochromatin formation and HP1 stabilization.
Altogether, we propose that cross talks exist between H3K27 and H3K9 methylation marks, which likely contribute to both the maintenance of constitutive heterochromatin and the stable silencing of developmentally regulated genes. Cooperation between these two marks is also likely involved in embryonic stem (ES) cell state maintenance, as loss of either polycomb proteins or SetDB1 H3K9 methyltransferase upregulates bivalent developmental regulators that are normally kept silent in ES cells (6). Given that HP1 helps in recruitment of DNA methyltransferases (60), this may also provide an explanation as to why bivalent promoters are more prone to DNA hypermethylation during aging and cancer (61). Supporting this hypothesis, epigenetic silencing of DCC tumor suppressor gene is mediated, on one hand, by CpG island hypermethylation and, on the other hand, by the presence of H3K9me3 and H3K27me3 marks at the promoter (5). In the near future, we expect more evidence in favor of cooperative mechanisms of heterochromatin formation/spreading to be uncovered.
ACKNOWLEDGMENTS
This work was supported by the Fonds National de la Recherche Scientifique, FNRS, the Télévie, and WELBIO.
We are grateful to Asma Boujtat for help with GST-HP1α purification, to A. Van Beneden for GFP-HP1α cells, to P. O. Legrand for sharing reagents, to F. Lagarde for support, and to G. Almouzni, R. O'Sullivan, and C. De Smet for critical comments on the manuscript.
Footnotes
Published ahead of print 21 July 2014
REFERENCES
- 1.Kouzarides T. 2007. Chromatin modifications and their function. Cell 128:693–705. 10.1016/j.cell.2007.02.005 [DOI] [PubMed] [Google Scholar]
- 2.Zeng W, de Greef JC, Chen YY, Chien R, Kong X, Gregson HC, Winokur ST, Pyle A, Robertson KD, Schmiesing JA, Kimonis VE, Balog J, Frants RR, Ball AR, Jr, Lock LF, Donovan PJ, van der Maarel SM, Yokomori K. 2009. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet. 5(7):e1000559. 10.1371/journal.pgen.1000559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnoult N, Van Beneden A, Decottignies A. 2012. Telomere length regulates TERRA levels through increased trimethylation of telomeric H3K9 and HP1α. Nat. Struct. Mol. Biol. 19:948–956. 10.1038/nsmb.2364 [DOI] [PubMed] [Google Scholar]
- 4.De Biase I, Chutake YK, Rindler PM, Bidichandani SI. 2009. Epigenetic silencing in Friedreich ataxia is associated with depletion of CTCF (CCCTC-Binding Factor) and antisense transcription. PLoS One 4(11):e7914. 10.1371/journal.pone.0007914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Derks S, Bosch LJ, Niessen HE, Moerkerk PT, van den Bosch SM, Carvalho B, Mongera S, Voncken JW, Meijer GA, de Bruïne AP, Herman JG, van Engeland M. 2009. Promoter CpG island hypermethylation- and H3K9me3 and H3K27me3-mediated epigenetic silencing targets the deleted in colon cancer (DCC) gene in colorectal carcinogenesis without affecting neighboring genes on chromosomal region 18q21. Carcinogenesis 30:1041–1048. 10.1093/carcin/bgp073 [DOI] [PubMed] [Google Scholar]
- 6.Bilodeau S, Kagey MH, Frampton GM, Rahl PB, Young RA. 2009. SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev. 23:2484–2489. 10.1101/gad.1837309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, Antosiewicz-Bourget J, Ye Z, Espinoza C, Agarwahl S, Shen L, Ruotti V, Wang W, Stewart R, Thomson JA, Ecker JR, Ren B. 2010. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 6:479–491. 10.1016/j.stem.2010.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voigt P, LeRoy G, Drury WJ, III, Zee BM, Son J, Beck DB, Young NL, Garcia BA, Reinberg D. 2012. Asymmetrically modified nucleosomes. Cell 151:181–193. 10.1016/j.cell.2012.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.LeRoy G, Chepelev I, DiMaggio PA, Blanco MA, Zee BM, Zhao K, Garcia BA. 2012. Proteogenomic characterization and mapping of nucleosomes decoded by Brd and HP1 proteins. Genome Biol. 13:R68. 10.1186/gb-2012-13-8-r68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Escamilla-Del-Arenal M, da Rocha ST, Spruijt CG, Masui O, Renaud O, Smits AH, Margueron R, Vermeulen M, Heard E. 2013. Cdyl, a new partner of the inactive X chromosome and potential reader of H3K27me3 and H3K9me2. Mol. Cell. Biol. 33:5005–5020. 10.1128/MCB.00866-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mozzetta C, Pontis J, Fritsch L, Robin P, Portoso M, Proux C, Margueron R, Ait-Si-Ali S. 2014. The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol. Cell 53:277–289. 10.1016/j.molcel.2013.12.005 [DOI] [PubMed] [Google Scholar]
- 12.Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. 2000. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406:593–599. 10.1038/35020506 [DOI] [PubMed] [Google Scholar]
- 13.Margueron R, Reinberg D. 2011. The Polycomb complex PRC2 and its mark in life. Nature 469:343–349. 10.1038/nature09784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, Rappsilber J, Lerdrup M, Helin K. 2008. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 10:1291–1300. 10.1038/ncb1787 [DOI] [PubMed] [Google Scholar]
- 15.Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, III, Voigt P, Martin SR, Taylor WR, De Marco V, Pirrotta V, Reinberg D, Gamblin SJ. 2009. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461:762–767. 10.1038/nature08398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. 2001. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410:120–124. 10.1038/35065138 [DOI] [PubMed] [Google Scholar]
- 17.Lachner M, O'Carroll D, Rea SR, Mechtler K, Jenuwein T. 2001. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410:116–120. 10.1038/35065132 [DOI] [PubMed] [Google Scholar]
- 18.Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. 2001. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292:110–113. 10.1126/science.1060118 [DOI] [PubMed] [Google Scholar]
- 19.Maison C, Almouzni G. 2004. HP1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 5:296–304. 10.1038/nrm1355 [DOI] [PubMed] [Google Scholar]
- 20.Jacobs SA, Khorasanizadeh S. 2002. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science 295:2080–2083. 10.1126/science.1069473 [DOI] [PubMed] [Google Scholar]
- 21.Nielsen PR, Nietlispach D, Mott HR, Callaghan J, Bannister A, Kouzarides T, Murzin AG, Murzina NV, Laue ED. 2002. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature 416:103–107. 10.1038/nature722 [DOI] [PubMed] [Google Scholar]
- 22.Fanti L, Giovinazzo G, Berloco M, Pimpinelli S. 1998. The heterochromatin protein 1 prevents telomere fusions in Drosophila. Mol. Cell 2:527–538. 10.1016/S1097-2765(00)80152-5 [DOI] [PubMed] [Google Scholar]
- 23.Sadaie M, Kawaguchi R, Ohtani Y, Arisaka F, Tanaka K, Shirahige K, Nakayama J. 2008. Balance between distinct HP1 family proteins controls heterochromatin assembly in fission yeast. Mol. Cell. Biol. 28:6973–6988. 10.1128/MCB.00791-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eskeland R, Eberharter A, Imhof A. 2007. HP1 binding to chromatin methylated at H3K9 is enhanced by auxiliary factors. Mol. Cell. Biol. 27:453–465. 10.1128/MCB.01576-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher 3rd FJ. 2002. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromaic genes by KRAB zinc-finger proteins. Genes Dev. 16:919–932. 10.1101/gad.973302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamamoto K, Sonoda M, Inokuchi J, Shirasawa S, Sasazuki T. 2004. Polycomb group suppressor of zeste 12 links heterochromatin protein 1alpha and enhancer of zeste 2. J. Biol. Chem. 279:401–406. 10.1074/jbc.M307344200 [DOI] [PubMed] [Google Scholar]
- 27.de la Cruz CC, Kirmizis A, Simon MD, Isono K, Koseki H, Panning B. 2007. The polycomb group protein SUZ12 regulates histone H3 lysine 9 methylation and HP1 alpha distribution. Chromosome Res. 15:299–314. 10.1007/s10577-007-1126-1 [DOI] [PubMed] [Google Scholar]
- 28.Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, Mann M. 2010. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 142:967–980. 10.1016/j.cell.2010.08.020 [DOI] [PubMed] [Google Scholar]
- 29.Eberl HC, Spruijt CG, Kelstrup CD, Vermeulen M, Mann M. 2013. A map of general and specialized chromatin readers in mouse tissues generated by label-free interaction proteomics. Mol. Cell 49:368–378. 10.1016/j.molcel.2012.10.026 [DOI] [PubMed] [Google Scholar]
- 30.Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. 2003. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 22:5323–5335. 10.1093/emboj/cdg542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu ZL, Zheng SS, Li ZM, Qiao YY, Aau MY, Yu Q. 2010. Polycomb protein EZH2 regulates E2F1-dependent apoptosis through epigenetically modulating Bim expression. Cell Death Differ. 17:801–810. 10.1038/cdd.2009.162 [DOI] [PubMed] [Google Scholar]
- 32.Fiskus W, Pranpat M, Balasis M, Herger B, Rao R, Chinnaiyan A, Atadja P, Bhalla K. 2006. Histone deacetylase inhibitors deplete enhancer of zeste 2 and associated polycomb repressive complex 2 proteins in human acute leukemia cells. Mol. Cancer Ther. 5:3096–3104. 10.1158/1535-7163.MCT-06-0418 [DOI] [PubMed] [Google Scholar]
- 33.Vassallo MF, Tanese N. 2002. Isoform-specific interaction of HP1 with human TAFII130. Proc. Natl. Acad. Sci. U. S. A. 99:5919–5924. 10.1073/pnas.092025499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K. 2007. UTX and JMJD3 are H3K27 demethylases involved in HOX gene regulation and development. Nature 449:731–734. 10.1038/nature06145 [DOI] [PubMed] [Google Scholar]
- 35.Rosnoblet C, Vandamme J, Völkel P, Angrand P-O. 2011. Analysis of the human HP1 interactome reveals novel binding partners. Biochem. Biophys. Res. Commun. 413:206–211. 10.1016/j.bbrc.2011.08.059 [DOI] [PubMed] [Google Scholar]
- 36.Lessard J, Baban S, Sauvageau G. 1998. Stage-specific expression of polycomb group genes in human bone marrow cells. Blood 91:1216–1224 [PubMed] [Google Scholar]
- 37.Yang SH, Sharrocks AD. 2005. PIASx acts as an Elk-1 coactivator by facilitating derepression. EMBO J. 24:2161–2171. 10.1038/sj.emboj.7600690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA, Chinnaiyan AM. 2002. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419:624–629. 10.1038/nature01075 [DOI] [PubMed] [Google Scholar]
- 39.Gonzalez ME, Li X, Toy K, DuPrie M, Ventura AC, Banerjee M, Ljungman M, Merajver SD, Kleer CG. 2009. Downregulation of EZH2 decreases growth of estrogen receptor-negative invasive breast carcinoma and requires BRCA1. Oncogene 28:843–853. 10.1038/onc.2008.433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fussbroich B, Wagener N, Macher-Goeppinger S, Benner A, Fälth M, Sültmann H, Holzer A, Hoppe-Seyler K, Hoppe-Seyler F. 2011. EZH2 depletion blocks the proliferation of colon cancer cells. PLoS One 6(7):e21651. 10.1371/journal.pone.0021651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD. 2005. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438:1116–1122. 10.1038/nature04219 [DOI] [PubMed] [Google Scholar]
- 42.Chaturvedi P, Parnaik VK. 2010. Lamin A rod mutants target heterochromatin protein 1alpha and beta for proteasomal degradation by activation of F-box protein, FBXW10. PLoS One 5(5):e10620. 10.1371/journal.pone.0010620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chaturvedi P, Khanna R, Parnaik VK. 2012. Ubiquitin ligase RNF123 mediates degradation of heterochromatin protein 1a and b in lamin A/C knock-down cells. PLoS One 7(10):e47558. 10.1371/journal.pone.0047558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cao R, Zhang Y. 2004. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 15:57–67. 10.1016/j.molcel.2004.06.020 [DOI] [PubMed] [Google Scholar]
- 45.Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K. 2004. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 23:4061–4071. 10.1038/sj.emboj.7600402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. 2003. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 17:1870–1881. 10.1101/gad.1110503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brasher SV, Smith BO, Fogh RH, Nietlispach D, Thiru A, Nielsen PR, Broadhurst RW, Ball LJ, Murzina NV, Laue ED. 2000. The structure of mouse HP1 suggests a unique mode of single peptide recognition by the shadow chromo domain dimer. EMBO J. 19:1587–1597. 10.1093/emboj/19.7.1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nielsen AL, Oulad-Abdelghani M, Ortiz JA, Remboutsika E, Chambon P, Losson R. 2001. Heterochromatin formation in mammalian cells: interaction between histones and HP1 proteins. Mol. Cell 7:729–739. 10.1016/S1097-2765(01)00218-0 [DOI] [PubMed] [Google Scholar]
- 49.Vakoc CR, Mandat SA, Olenchock BA, Blobel GA. 2005. Histone H3 lysine 9 methylation and HP1γ are associated with transcription elongation through mammalian chromatin. Mol. Cell 19:381–391. 10.1016/j.molcel.2005.06.011 [DOI] [PubMed] [Google Scholar]
- 50.Kwon SH, Workman JL. 2011. The changing faces of HP1: from heterochromatin formation and gene silencing to euchromatic gene expression. Bioessays 33:280–289. 10.1002/bies.201000138 [DOI] [PubMed] [Google Scholar]
- 51.Ren G, Baritaki S, Marathe H, Feng J, Park S, Beach S, Bazeley PS, Beshir AB, Fenteany G, Mehra R, Daignault S, Al-Mulla F, Keller E, Bonavida B, de la Serna I, Yeung KC. 2012. Polycomb protein EZH2 regulates tumor invasion via the transcriptional repression of the metastasis suppressor RKIP in breast and prostate cancer. Cancer Res. 72:3091–3104. 10.1158/0008-5472.CAN-11-3546 [DOI] [PubMed] [Google Scholar]
- 52.Canzio D, Chang EY, Shankar S, Kuchenbecker KM, Simon MD, Madhani HD, Narlikar GJ, Al-Sady B. 2011. Chromodomain-mediated oligomerization of HP1 suggests a nucleosome-bridging mechanism for heterochromatin assembly. Mol. Cell 41:67–81. 10.1016/j.molcel.2010.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kind J, van Steensel B. 2010. Genome-nuclear lamina interactions and gene regulation. Curr. Opin. Cell Biol. 22:320–325. 10.1016/j.ceb.2010.04.002 [DOI] [PubMed] [Google Scholar]
- 54.Ye Q, Callebaut I, Pezhman A, Courvalin JC, Howard J, Worman HJ. 1997. Domain-specific interactions of human HP1-type chromodomain proteins and inner nuclear membrane protein LBR. J. Biol. Chem. 272:14983–14989. 10.1074/jbc.272.23.14983 [DOI] [PubMed] [Google Scholar]
- 55.Zinner R, Albiez H, Walter J, Peters AH, Cremer T, Cremer M. 2006. Histone lysine methylation patterns in human cell types are arranged in distinct three-dimensional nuclear zones. Histochem. Cell Biol. 125:3–19. 10.1007/s00418-005-0049-1 [DOI] [PubMed] [Google Scholar]
- 56.Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson MR, Goldman AE, Khuon S, Collins FS, Jenuwein T, Goldman RD. 2006. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. U. S. A. 103:8703–8708. 10.1073/pnas.0602569103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dechat T, Adam SA, Goldman RD. 2009. Nuclear lamins and chromatin: when structure meets function. Adv. Enzyme Regul. 49:157–166. 10.1016/j.advenzreg.2008.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.LeRoy G, Weston JT, Zee BM, Young NL, Plazas-Mayorca MD, Garcia BA. 2009. Heterochromatin protein 1 is extensively decorated with histone code-like post-translational modifications. Mol. Cell. Proteomics 8:2432–2442. 10.1074/mcp.M900160-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sparmann A, van Lohuizen M. 2006. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 6:846–856. 10.1038/nrc1991 [DOI] [PubMed] [Google Scholar]
- 60.Fuks F, Hurd PJ, Deplus R, Kouzarides T. 2003. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 31:2305–2312. 10.1093/nar/gkg332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Decottignies A, d'Adda di Fagagna F. 2011. Epigenetic alterations associated with cellular senescence: a barrier against tumorigenesis or a red carpet for cancer? Semin. Cancer Biol. 21:360–366. 10.1016/j.semcancer.2011.09.003 [DOI] [PubMed] [Google Scholar]