

Abstract
Glucose-dependent insulinotropic polypeptide (GIP), an incretin hormone secreted from gastrointestinal K cells in response to food intake, has an important role in the control of whole-body metabolism. GIP signals through activation of the GIP receptor (GIPR), a G-protein-coupled receptor (GPCR). Dysregulation of this pathway has been implicated in the development of metabolic disease. Here we demonstrate that GIPR is constitutively trafficked between the plasma membrane and intracellular compartments of both GIP-stimulated and unstimulated adipocytes. GIP induces a downregulation of plasma membrane GIPR by slowing GIPR recycling without affecting internalization kinetics. This transient reduction in the expression of GIPR in the plasma membrane correlates with desensitization to the effects of GIP. A naturally occurring variant of GIPR (E354Q) associated with an increased incidence of insulin resistance, type 2 diabetes, and cardiovascular disease in humans responds to GIP stimulation with an exaggerated downregulation from the plasma membrane and a delayed recovery of GIP sensitivity following cessation of GIP stimulation. This perturbation in the desensitization-resensitization cycle of the GIPR variant, revealed in studies of cultured adipocytes, may contribute to the link of the E354Q variant to metabolic disease.
INTRODUCTION
Glucose-dependent insulinotropic polypeptide (GIP) is secreted by K cells of the gastrointestinal tract in response to food (1, 2). GIP together with the other incretin hormone, glucagon-like peptide 1, have prominent roles in the control of whole-body energy metabolism. A primary function of these hormones is to stimulate glucose-dependent insulin release from pancreatic beta cells (3–5). In addition to its effect on the pancreas, GIP functions to regulate several aspects of adipocyte metabolism, including increasing the sensitivity of adipocytes to insulin, thereby setting the tone for an optimal insulin response (e.g., see references 6–13). GIP signals through the GIP receptor (GIPR), a G-protein-coupled receptor (GPCR) coupled to the stimulatory G alpha subunit and elevated cyclic AMP (cAMP) levels (4, 14, 15).
In individuals with type 2 diabetes mellitus (T2DM), GIP-mediated insulinotropic effects are attenuated despite normal to elevated levels of blood GIP (16–18). This GIP resistance potentially contributes to the pathophysiology of T2DM. The importance of GIP function in metabolic homeostasis is highlighted by the discovery in genome-wide association studies of a number of single nucleotide polymorphisms in the GIPR gene linked to an increased risk of metabolic diseases, including insulin resistance, T2DM, and cardiovascular diseases (19–21). One of these variants results in the substitution of glutamine for glutamic acid at position 354 (E354Q) of GIPR, which has been shown in various studies to be associated with insulin resistance (22), cardiovascular disease (21), and defects in beta-cell function (23).
Despite extensive characterization of GIP's effects on metabolism, little is known about the behavior of GIPR. Because the trafficking behaviors of GPCRs are critical for their signal transduction, we embarked on a study of GIPR trafficking. Here we report that GIPR constitutively cycles between the trans-Golgi network (TGN) and the plasma membrane in both GIP-stimulated and unstimulated adipocytes. GIP induces a rapid, reversible downregulation of plasma membrane GIPR by promoting a slowing of GIPR recycling without an effect on internalization kinetics. The E354Q substitution results in more pronounced GIP-stimulated downregulation and a prolonged desensitization period. Our data suggest that the link between the E354Q substitution and metabolic disease might result from a disruption of the GIPR desensitization-resensitization cycle.
MATERIALS AND METHODS
Reagents and antibodies.
GIP (1-30) a peptide of amino acids 1 to 30 of full-length 42-amino-acid GIP, was purchased from Bachem, Inc. (Torrance, CA); formaldehyde and saponin were obtained from Sigma-Aldrich; antihemagglutinin (anti-HA) antibodies were obtained from Covance (Berkley, CA); rabbit polyclonal anti-LAMP1 antibodies and rabbit anti-TGN46 antibodies were obtained from Abcam (Beverly, MA); Cy3-conjugated anti-mouse IgG, Cy3-conjugated anti-rabbit IgG, and Cy5-conjugated anti-rabbit IgG were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA); Cy3-conjugated bungarotoxin and Cy5-conjugated bungarotoxin were purchased from Life Technologies; and DNA and small interfering RNA (siRNA) oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA).
Cell culture, transfection, and electroporation.
3T3-L1 fibroblasts were cultured, differentiated into adipocytes, and electroporated as described previously (24, 25). Experiments were performed 24 h after electroporation, except for the siRNA experiments. For siRNA experiments, cells were electroporated together with HA-GLUT4-green fluorescent protein (GFP) and siRNA, and experiments were performed 48 h after electroporation. HEK293 cells and F293 packaging cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum and penicillin-streptomycin. Cells were transiently transfected by using Lipofectamine 2000 according to the manufacturer's instructions. Experiments were carried out 48 h after transfection unless otherwise indicated.
DNA constructs.
Human GIPR cDNA in the pCDNA3 vector was a kind gift from Bernaud Thorens (University of Lausanne, Switzerland). HA epitope (YPYDVPDYA)-tagged GIPR was generated by PCR amplification using Pfu Turbo polymerase (Invitrogen), using the GIPR-pCDNA3 plasmid as the template. The purified PCR product was digested and ligated into pEGFP-N1 to generate a plasmid, HA-GIPR-GFP, which contains the HA-GIPR sequence upstream of the GFP coding sequence to express the HA-GIPR-GFP fusion protein. To generate bungarotoxin (BTX)-GIPR-GFP, PCR amplification was done to generate BTX (WRYYESSLEPYPD)-tagged GIPR, and amplified PCR was ligated into pEGF-N1 to express the BTX-GIPR-GFP fusion protein. E354Q HA-GIPR-GFP and E354Q BTX-GIPR were generated by using a QuikChange site-directed mutagenesis kit (Stratagene). Mutation was confirmed by DNA sequencing.
siRNA and quantitative PCR.
GIPR knockdown was determined by using quantitative PCR. 3T3-L1 adipocytes were electroporated with GIPR siRNAs directed against the mouse GIPR sequence (CTAGGACAATCAACTGGAAGGC). After 24 h and 48 h of electroporation, cells were harvested, and RNA was extracted by using the RNeasy kit from Qiagen (Germantown, MD). cDNA was made from extracted RNA by using the Sprint RT Complete oligo(dT) kit from Clontech (Mountain View, CA), and quantitative PCR was performed by using a qSTAR primer pair obtained from OriGene (Rockville, MD).
cAMP assay.
Cells were washed and incubated with serum-free medium for 2 h and then stimulated with GIP for 15 min. The medium was aspirated, and cells were lysed in 0.1 N HCl. Intracellular cAMP was measured by using the direct immunoassay kit from Assay Designs (Plymouth Meeting, PA).
Quantification of cell surface GIPR.
3T3-L1 adipocytes electroporated with HA-GIPR-GFP were serum starved, treated with GIP, and then fixed under nonpermeabilizing conditions. Cells were then stained with anti-HA antibodies, followed by secondary staining with anti-mouse Cy3-conjugated antibodies. Cells were imaged, and the Cy3/GFP ratio was determined for each cell, indicating the ratio of surface/total HA-GIPR-GFP. An average of ∼50 cells/experiment was used to measure the amount of GIPR at the cell surface. Alternatively, to determine the fraction of GIPR at the cell surface after fixing of the cells, one set of cells was permeabilized and stained with anti-HA and anti-mouse Cy3 antibodies to label the entire pool of GIPR. The fraction of GIPR at the cell surface was then determined by taking the ratio of Cy3/GFP staining in nonpermeabilized cells to that in permeabilized cells.
Stable expression of HA-GLUT4-GFP and BTX-GIPR in adipocytes.
HA-GLUT4-GFP stable cells were generated by using the ViraPower lentiviral expression system (Life Technologies). Briefly, HA-GLUT4-GFP was PCR amplified and cloned into a pLenti6.3/V5-TOPO vector by using TA TOPO cloning. 293FT packaging cells were cotransfected with a pLenti6.3/V5-TOPO vector containing HA-GLUT4-GFP and ViraPower packaging mix to produce a lentiviral stock. This lentiviral stock was filtered and used to transduce 3T3-L1 fibroblasts. GFP-positive cells were sorted by flow cytometry, and selected cells were cultured in DMEM–10% calf serum containing blasticidin (250 mg/ml). Expression of HA-GLUT4-GFP was confirmed by fluorescence microscopy.
HA-GLUT4-GFP-expressing 3T3-L1 cells were then engineered to stably express wild-type (WT) BTX-GIPR or E354Q BTX-GIPR by using the pLVX-IRES-tdTomato lentiviral expression system (Clontech). The vector expresses the two proteins from a bicistronic mRNA transcript, allowing tdTomato to be used as an indicator of transduction efficiency and a marker for selection by flow cytometry. Briefly, WT BTX-GIPR and E354Q BTX-GIPR were PCR amplified, digested, and cloned into the pLVX-IRES-tdTomato vector. 293FT packaging cells were cotransfected with the pLVX-IRES-tdTomato vector and packaging mix to produce a lentiviral stock that was used to transduce 3T3-L1 fibroblasts stably expressing HA-GLUT4-GFP. Positive cells were selected by using flow cytometry, and expression of BTX-GIPR was confirmed by uptake of Cy5-conjugated bungarotoxin.
Internalization of GIPR.
Adipocytes electroporated with HA-GIPR-GFP were serum starved for 2 h and incubated with or without 100 nM GIP for 1 h. Cells were then incubated with anti-HA antibodies (100 μg/ml) in the absence or presence of 100 nM GIP for various times so that every receptor that went to the cell surface during these times were labeled with anti-HA antibody. Cells were fixed with 3.7% formaldehyde and incubated with a saturating concentration of anti-mouse Cy5 antibody to bind all the anti-HA antibody at the cell surface. Cells were fixed again, permeabilized with 250 μg/ml saponin, and then stained with anti-mouse Cy3 to label the internal pool of anti-HA. Cells were imaged, and the ratio of internalized GIPR (Cy3) to total GIPR (GFP) was determined and plotted over time. The rate of internalization was determined by measuring the slope of the curve.
For BTX-GIPR internalization, cells expressing BTX-GIPR-GFP were treated with GIP as described above and incubated with BTX-Cy3 for various times. Cells were fixed and imaged to determine the Cy3-to-GFP ratio over time.
Kinetics of GIPR trafficking.
Adipocytes electroporated with HA-GIPR-GFP were serum starved for 2 h and incubated with or without 100 nM GIP for 1 h. Cells were then incubated with anti-HA antibodies (100 μg/ml) in the absence or presence of 100 nM GIP for various times. Cells were fixed with 3.7% formaldehyde, permeabilized with 250 μg/ml saponin, and then stained with anti-mouse Cy3 to label the entire pool of antibody-labeled HA-GIPR-GFP. Cells were imaged, and the Cy3-to-GFP (total GIPR) ratio was determined and plotted over time. The rate of exocytosis was determined by measuring the slope of the curve.
Desensitization and recovery of GIPR.
Cells transiently expressing HA-GIPR-GFP were serum starved for 2 h and stimulated with various doses of GIP (1 nM to 100 nM) or with 100 nM GIP for various times (15 to 60 min). Surface GIPR was measured as described above.
For resensitization, cells were treated with or without 100 nM GIP for 1 h, washed extensively, and incubated without GIP for the indicated times. The cell surface GIPR level was determined at each time point, as described above. GFP intensity was used to measure total GIPR.
HA-GLUT4-GFP translocation assay.
HA-GLUT4-GFP translocation was measured as described previously (26–28).
Data acquisition and processing.
Fluorescent images were collected on a DMIRB inverted microscope (Leica Microsystems, Deerfield, IL), using a 20× objective. Fluorescence quantifications were done by using MetaMorph image processing software (Molecular Devices, Sunnyvale, CA), as described previously (26, 28, 29).
Statistical analysis.
Statistical significance was calculated by Student's t test.
RESULTS
Functional validation of GIP receptor reporter constructs.
To study the trafficking behavior of GIPR, we generated a reporter with an HA epitope at the extracellular N terminus and GFP at the intracellular C terminus (HA-GIPR-GFP) (Fig. 1A). We made a second construct with a bungarotoxin binding domain (30, 31) at the extracellular N terminus and a GFP tag at the intracellular C terminus (Fig. 1A). GIPR is a Gs-coupled receptor, and its activation results in elevated cAMP levels (14, 32, 33). We validated that the reporters are functional in transient-expression studies of HEK293 cells, which do not express endogenous GIPR. Incubation of mock-transfected HEK293 cells with GIP did not induce formation of cAMP. Stimulation of HEK293 cells transiently expressing either of the tagged receptor constructs or untagged GIPR resulted in a robust increase of the cAMP level (Fig. 1B). The stimulation of cAMP production by the tagged constructs was similar to that by the untagged construct, indicating that the tags do not affect activation of GIPR. Binding of anti-HA antibody or bungarotoxin to the reporters did not affect receptor activity, neither activating the receptors nor affecting GIP activation (Fig. 1C and D). These data establish that the GIPR reporters are functional and that the binding of neither anti-HA to HA-GIPR nor bungarotoxin to BTX-GIPR affects GIP activation of the receptors.
FIG 1.
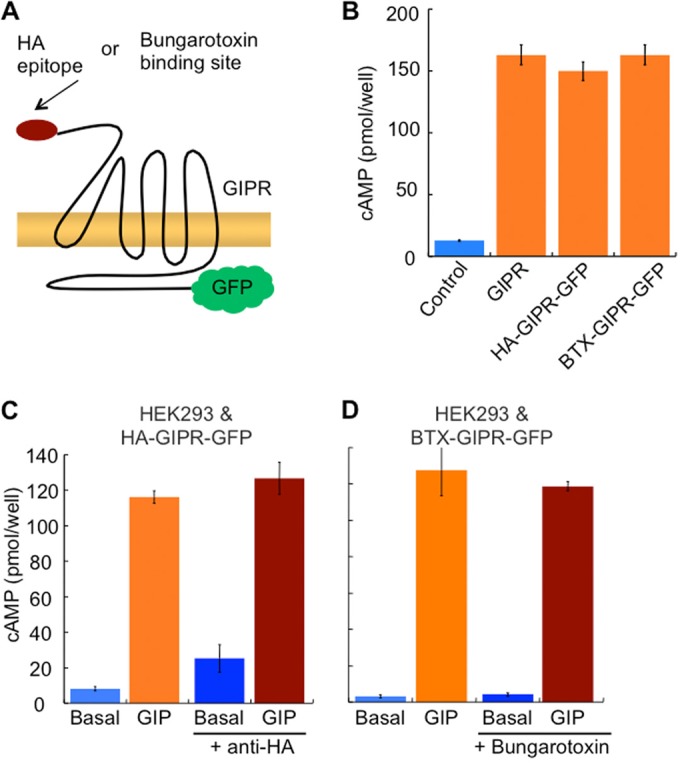
Dually tagged GIPR reporter constructs. (A) Schematic of GIPR reporters HA-GIPR-GFP and BTX-GIPR-GFP. An HA epitope in the former and a bungarotoxin binding site in the latter are inserted at the amino terminus of GIPR. (B) cAMP production in HEK293 cells stimulated with 100 nM GIP ectopically expressing GIPR (untagged), HA-GIPR-GFP, or BTX-GIPR-GFP. The bar labeled Control shows data from mock-transfected HEK293 cells (no GIPR expression). (C) cAMP production in HEK293 cells expressing HA-GIPR-GFP stimulated with GIP, with or without anti-HA in the incubation medium. (D) cAMP production in HEK293 cells expressing BTX-GIPR-GFP stimulated with GIP, with or without bungarotoxin in the medium. A total of 100 nM GIP was used for the stimulations. Each bar represents the average ± standard error of the mean of data from 3 independent experiments.
GIPR is constitutively internalized in adipocytes.
The behaviors of GPCRs are cell context dependent (34, 35). We next studied the behaviors of the GIPR reporters expressed in adipocytes, cells that express endogenous GIPR. In transiently transfected adipocytes, HA-GIPR-GFP, revealed by GFP fluorescence, was distributed between the plasma membrane and a perinuclear compartment (Fig. 2A). To quantify the fraction of GIPR in the plasma membrane, we determined anti-HA binding to nonpermeabilized and permeabilized adipocytes; the former is a measure of the amount of GIPR in the plasma membrane, and the latter is a measure of the total amount of GIPR expressed in cells. This quantification revealed that 44.5% ± 3.5% (average of data from 3 experiments ± standard deviations [SD]; >30 cells per experiment) of the HA-GIPR-GFP was in the plasma membrane of basal adipocytes (that is, without GIP stimulation). When analyzed at the single-cell level, the distribution of tagged GIPR expressed on the plasma membrane was not affected by the amount of the reporter expressed, thereby demonstrating that trafficking steps are not saturated within the range of expression of the tagged GIPR that is achieved by electroporation of adipocytes (Fig. 2B).
FIG 2.

GIPR is internalized in a constitutive manner in the absence of GIP in adipocytes. (A) Epifluorescence images of adipocytes with HA-GIPR-GFP. GFP fluorescence is shown in the left panels, and Cy3 fluorescence is shown in the right panels. Cells were stained by indirect anti-HA immunofluorescence (Cy3) under nonpermeabilizing conditions to measure surface GIPR or with permeabilization to reveal total GIPR. Representative cells are shown. Bar, 10 μm. (B) GIPR trafficking is not altered within the range of expression of WT and E354Q GIPRs in 3T3-L1 cells. To establish that the level of GIPR expression achieved by electroporation does not lead to aberrant trafficking due to saturation of endocytosis and/or recycling, we determined, on an individual-cell basis, the steady-state amount of HA-GIPR-GFP on the plasma membrane in fixed cells by anti-HA epitope indirect immunofluorescence correlated to the total amount of HA-GIPR-GFP expressed in the cell. These data are from a representative experiment, and each point indicates the Cy3 and GFP fluorescence from a single cell. AU, arbitrary units. (C) Cartoon of the internalization experiment protocol. (D) Cells expressing HA-GIPR-GFP were incubated with anti-HA antibodies for the indicated times, fixed, and stained with saturating concentrations of anti-mouse Cy5 secondary antibodies to block the surface. Cells were refixed, permeabilized, and stained with anti-mouse Cy3-conjugated secondary antibodies to label intracellular anti-HA. Representative cells are shown. Bar, 10 μm. (E) Quantification of internalized anti-HA plotted as a function of incubation time. (F) HA-GIPR-GFP trafficking is not altered by incubation of living cells with anti-HA antibody. Adipocytes transiently expressing HA-GIPR-GFP were incubated at 37°C for 10 min in serum-free DMEM or in serum-free DMEM supplemented with anti-HA antibody. The cells were fixed, and the amount of HA-GIPR-GFP in the plasma membrane was determined by incubating the cells with anti-HA antibody, followed by incubation with Cy3-labeled goat anti-mouse secondary antibody. The ratio of the surface distribution to the total distribution of HA-GIPR-GFP was determined by quantitative fluorescence microscopy. ns, not significant. (G) Adipocytes expressing BTX-GIPR-GFP were incubated with BTX-Cy3 for the indicated times. Cells were fixed and imaged. Representative cells are shown. (H) Quantification of cells treated as described above for panel E. The Cy3/GFP fluorescence ratio per cell ratio is plotted as a function of incubation time. The data points are the averages ± standard errors of the means from 3 independent experiments. The data from each experiment were normalized to the Cy3/GFP value at the 30-min time point. Asterisks indicate the nucleus.
To assess the relationship between the plasma membrane and intracellular pools of GIPR, we incubated living adipocytes transiently expressing HA-GIPR-GFP with anti-HA antibody. This antibody binds the HA-GIPR-GFP on the plasma membrane and remains bound as the receptor traffics into the cell (Fig. 2C). After various incubation times, the cells were fixed, and plasma membrane anti-HA was revealed by staining with a Cy5-labeled secondary antibody. Anti-HA internalized during the incubation period was revealed with Cy3-labeled secondary antibody staining of permeabilized cells. At 2 min of incubation, most of the fluorescence was restricted to the plasma membrane; however, after 30 min, the intracellular compartments containing GIPR (GFP positive) were labeled with Cy3, demonstrating trafficking of GIPR from the plasma membrane to the perinuclear compartment in unstimulated adipocytes (Fig. 2D). The accumulation of intracellular anti-HA increased exponentially, reaching a plateau level at 15 min, establishing that GIPR is constitutively internalized and recycled in the absence of GIP stimulation (Fig. 2E).
A potential caveat regarding the use of the anti-HA antibody is that its binding to HA-GIPR-GFP might alter receptor behavior. We confirmed that antibody was not inducing internalization of the receptor by establishing that the amount of HA-GIPR-GFP in the plasma membrane was not altered by incubation of living cells with anti-HA antibody (Fig. 2F). In addition, to ensure that anti-HA-induced cross-linking of HA-GIPR-GFP did not alter GIPR trafficking, we studied the behavior of the BTX-GIPR-GFP reporter. Bungarotoxin is monovalent and therefore will not cross-link the receptor reporter (36). Similar to anti-HA uptake, Cy3-labeled bungarotoxin is internalized by BTX-GIPR-GFP to the perinuclear compartment, with Cy3 fluorescence reaching a plateau at 15 min (Fig. 2G and H). Thus, the constitutive internalization of GIPR is not due to effects of anti-HA binding the HA-GIPR-GFP reporter.
GIP induces desensitization of GIPR in adipocytes.
GPCR signal transduction is often characterized by ligand-induced desensitization that results in an attenuation of the response to a second challenge with the agonist (37). The principal mechanism for desensitization is a depletion of receptors in the plasma membrane resulting in a reduced response to the ligand (37–40). Desensitization is a key regulator of the signaling tone of GPCR signaling pathways (41–43).
To determine whether endogenous GIPR undergoes desensitization in adipocytes, we determined the response of adipocytes to sequential challenges with GIP (Fig. 3A). A single 1-h GIP stimulation induced an increase in the amount of cAMP compared to that in nonstimulated adipocytes, whereas the response of these cells to a second challenge with GIP was significantly attenuated (Fig. 3B). Loss of responsiveness after the first challenge with GIP is a demonstration of the desensitization of endogenous GIPR in adipocytes.
FIG 3.

GIPR undergoes homologous desensitization in adipocytes. (A) Scheme of the desensitization experiment. (B) Adipocytes were incubated with or without 100 nM GIP for 60 min, followed by stimulation with 100 nM fresh GIP for 15 min. Cells were lysed, and the amount of cAMP was measured. (C) Epifluorescence images of adipocytes expressing HA-GIPR-GFP. Cells were incubated with 100 nM GIP for the times indicated. Representative cells are shown. (D) Quantification of cells treated as described above for panel C. The plasma membrane GIPR level was determined by indirect immunofluorescence of fixed, nonpermeabilized cells. The data from each experiment were normalized to the Cy3/GFP value at the 0-min time point. (E) Cells were incubated with the indicated concentrations of GIP for the times indicated. Representative cells are shown. Bar, 10 μm. (F) Quantification of cells treated as described above for panel E. The plasma membrane GIPR level was determined by indirect immunofluorescence of fixed, nonpermeabilized cells. Data from each experiment were normalized to the Cy3/GFP value for the no-GIP treatment. Each bar is the average ± standard error of the mean from 3 to 6 independent experiments (*, P < 0.05; **, P < 0.01). Asterisks indicate the nucleus.
To explore the mechanism of GIP-induced desensitization of GIPR signaling, we determined the effect of GIP stimulation on the trafficking of HA-GIPR-GFP in adipocytes. Receptor desensitization (e.g., see Fig. 3B) was coupled with a time-dependent redistribution of the GIPR from the plasma membrane to the intracellular perinuclear compartment (Fig. 3C), resulting in an approximate 40% reduction of GIPR on the cell surface (Fig. 3D). These results suggest that the attenuated cAMP response to the second GIP stimulation is due to a reduction of GIPR on the surface of adipocytes.
Downregulation of GIPR is dependent on the dose of GIP, with 100 nM GIP promoting maximal downregulation (Fig. 3E and F). GIP at a dose of 100 nM has the maximum biological effect on cultured adipocytes (9); thus, GIPR downregulation occurs within the range of GIP concentrations that mediate GIP effects in cultured adipocytes.
Increased GIP-induced desensitization of a natural GIPR variant.
A naturally occurring missense mutation in human GIPR that results in a glutamic acid-to-glutamine substitution at position 354 (E354Q) within the sixth transmembrane domain is associated with an increased risk of insulin resistance and cardiovascular disease (21, 22). It is not known how this mutation interferes with the behavior of GIPR.
We first determined if the E354Q mutation affects GIP-stimulated adenylate cyclase by expressing E354Q GIPR in HEK293 cells (Fig. 4A). Increased cAMP as a function of the GIP dose in cells expressing WT GIPR was identical to that in cells expressing E354Q GIPR, demonstrating that the E354Q substitution does not affect GIP binding or GIP activation of adenylate cyclase.
FIG 4.
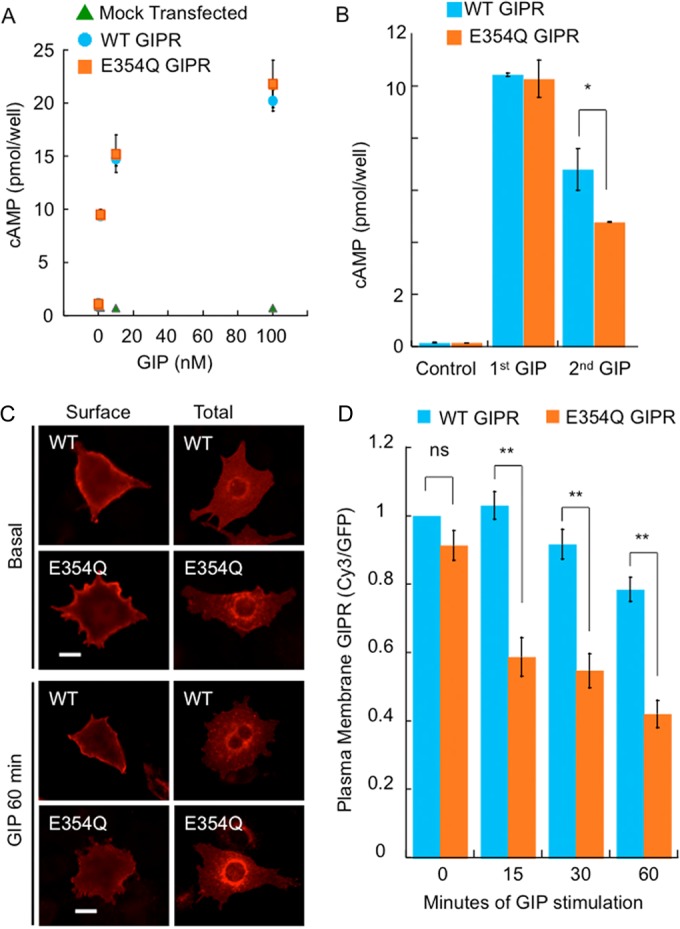
A natural variant of GIPR undergoes enhanced desensitization in response to GIP. (A) HEK293 cells transfected with either the empty vector, WT HA-GIPR-GFP, or E354Q HA-GIPR-GFP were stimulated with the indicated doses of GIP, and the amount of cAMP was measured. (B) HEK293 cells expressing either WT or E354Q HA-GIPR-GFP were incubated with or without 100 nM GIP for 1 h, followed by a second incubation with fresh GIP for 10 min. Cells were lysed, and the amount of cAMP was measured. (C) Adipocytes expressing WT or E354Q HA-GIPR-GFP were stimulated with 100 nM GIP for the indicated times, and anti-HA was revealed by indirect immunofluorescence of nonpermeabilized (Surface) and permeabilized (Total) cells. Representative cells are shown. Bar, 10 μm. (D) Quantification of cells treated as described above for panel C. The plasma membrane GIPR level was determined by indirect immunofluorescence of fixed, nonpermeabilized cells. The data from each experiment were normalized to the Cy3/GFP value for WT GIPR with no GIP treatment. Each bar is the average ± standard error of the mean from 3 to 5 independent experiments (**, P < 0.01; *, P < 0.05).
We next determined whether the substitution affected receptor desensitization by measuring cAMP formation after two consecutive challenges with GIP. The amount of cAMP formation after the first challenge with GIP in cells expressing the E354Q mutant was identical to that in cells expressing the WT receptor; however, the amount of cAMP formation in response to the second challenge in E354Q GIPR-expressing cells was significantly reduced (Fig. 4C). These data demonstrate a normal first response and an enhanced desensitization of the E354Q GIPR variant when expressed in HEK293 cells.
To test whether the increased desensitization of E354Q GIPR is coupled to an alteration in the trafficking of the receptor, we created this substitution in the HA-GIPR-GFP reporter. In unstimulated adipocytes, E354Q GIPR was distributed between the plasma membrane and the perinuclear compartment (Fig. 4C). Quantification of this distribution revealed that the E354Q substitution did not affect the distribution: 44.5% ± 3.00% and 42.0% ± 7.6% of WT GIPR and E354Q GIPR were on the plasma membrane, respectively (mean ± SD; n = 3). These data indicate that the substitution does not alter trafficking of GIPR through the biosynthetic pathway, its delivery to the plasma membrane, or its behavior in unstimulated adipocytes.
Upon GIP stimulation, E354Q GIPR, like WT GIPR, was redistributed to the perinuclear compartment (Fig. 4C). Quantification of the effect of GIP on E354Q GIPR reveled that downregulation of E354Q GIPR occurred more rapidly and to a greater extent than for WT GIPR (Fig. 4D). These studies demonstrate that with GIP stimulation, E354Q GIPR undergoes enhanced desensitization, resulting in a greater reduction of GIPR at the cell surface available for subsequent activation in response to a second challenge with GIP than for WT GIPR studied in the same system (Fig. 4B).
Enhanced desensitization of E354Q GIPR is due to impaired exocytosis.
To investigate the mechanism underlying the enhanced desensitization, we determined the effect of the E354Q substitution on the trafficking kinetics of the HA-GIPR-GFP reporter. The amount of GIPR at the cell surface of adipocytes is dynamically maintained, reflecting the balance between internalization and recycling (Fig. 2). In unstimulated adipocytes, WT and E354Q GIPRs were internalized at identical rates (Fig. 5A and B). A 60-min incubation with GIP had no effect on the internalization kinetics of WT GIPR or E354Q GIPR (slope), despite the larger decrease of E354Q GIPR on the cell surface compared to the decrease in the amount of WT GIPR (y intercept) (Fig. 5A and B). These data demonstrate that modulation of internalization kinetics does not account for GIP-induced desensitization of either WT or E354Q GIPR, nor does it explain the more pronounced downregulation of the E354Q GIPR mutant.
FIG 5.

Enhanced desensitization of E354Q GIPR is due to impaired exocytosis after exposure to GIP. (A and B) Adipocytes electroporated with WT or E354Q GIPR were serum starved and incubated without or with 100 nM GIP for 1 h. The cells were then incubated with anti-HA antibodies for the indicated time points, fixed, permeabilized, and stained with anti-mouse Cy3 antibodies. Cell-associated anti-HA (Cy3 fluorescence) normalized to GFP is plotted as a function of incubation time. The slope is proportional to the rate of GIPR internalization. (C and D) Adipocytes expressing WT or E354Q GIPR were incubated with or without 100 nM GIP for 1 h, followed by incubation with anti-HA antibodies for the indicated times. GIP was included in the incubation of cells that were stimulated with GIP during the first 60-min incubation. Cells were fixed, permeabilized, and stained with anti-mouse Cy3. Cell-associated anti-HA (Cy3 fluorescence) normalized to GFP is plotted as a function of incubation time. The data were fit to an exponential rise, with k being the exocytosis rate constant. Each graph represents the averages ± standard errors of the means of data from 3 to 4 independent experiments (**, P < 0.01; *, P < 0.05).
We next compared the recycling of WT GIPR and E354Q GIPR. In unstimulated adipocytes, there was no difference in the recycling kinetics between WT and E354Q GIPRs (Fig. 5C and D). However, a 60-min stimulation with GIP induced a 30% decrease of WT GIPR recycling. This reduced recycling accounts for the decreased expression of WT GIPR on the plasma membrane of GIP-stimulated adipocytes. The effect of GIP on E354Q GIPR recycling was more pronounced, with an ∼60% reduction in the recycling rate constant, thereby accounting for the enhanced desensitization of E354Q GIPR (Fig. 5C and D). These data demonstrate that modulation of exocytosis kinetics accounts for GIP-induced desensitization of WT GIPR as well as the more pronounced downregulation of the E354Q GIPR mutant.
Recycled intracellular GIPR repopulates the plasma membrane after GIP stimulation.
We next explored the resensitization of adipocytes following GIP stimulation. Cells were stimulated with GIP for 60 min, washed, and incubated in medium without GIP. At various times during the recovery period, the amount of GIPR in the plasma membrane was measured (Fig. 6A). WT GIPR repopulates the plasma membrane to prestimulation levels within 60 min, whereas it takes about 240 min for the E354Q GIPR mutant to achieve prestimulation levels. It takes longer for the E354Q mutant to repopulate the plasma membrane, in large part because more of the E354Q mutant is redistributed intracellularly during GIP stimulation.
FIG 6.

WT GIPR and E354Q GIPR recycle back to the cell surface after GIP challenge. (A and B) Adipocytes were treated without or with 100 nM GIP for 1 h, washed and fixed, or washed and further incubated in serum-free medium for the times indicated. Plasma membrane GIPR (A) and total GIPR (B) levels were determined. Each graph represents averages ± standard errors of the means of data from 3 to 4 independent experiments (**, P < 0.01; *, P < 0.05). (C and D) Epifluorescence images of adipocytes expressing WT or E354Q HA-GIPR-GFP, stained for TGN46. Cells were incubated without or with 100 nM GIP for 1 h. One set of GIP-treated cells was washed and incubated in serum-free medium for an additional 1 h. The cells were fixed, permeabilized, and stained with anti-TGN46 antibodies, followed by secondary staining with anti-rabbit Cy3 antibodies. Bar, 10 μm. (E) Confocal optical sections of WT HA-GIPR-GFP and E354Q HA-GIPR-GFP colocalization with TGN46 in adipocytes. Arrows indicate colocalization of GIPR with TGN46 in the perinuclear region of cells.
During the course of GIP stimulation and recovery, we monitored the total expression of HA-GIPR-GFP by quantifying the amount of GFP fluorescence per cell. The total GFP signal did not significantly change during the GIP stimulation or recovery period, indicating that the depletion and repopulation of the plasma membrane GIPR resulted from a redistribution of GIPR rather than degradation and new synthesis (Fig. 6B).
GIPR accumulates in a TGN46-positive intracellular compartment in basal and GIP-stimulated cells.
GIPR perinuclear accumulation in basal adipocytes colocalizes with TGN46, a marker of the trans-Golgi network, and this colocalization is maintained in GIP-stimulated adipocytes (Fig. 6C). These data suggest that the reduced recycling of GIPR that determines plasma membrane downregulation during desensitization occurs by regulated (slowed) release from this compartment, rather than altered recycling kinetics being due to GIPR trafficking through distinct compartments under unstimulated and GIP-stimulated conditions. The E354Q GIPR mutant also colocalizes to the TGN46 compartment in unstimulated adipocytes and stimulated adipocytes and remains colocalized with TGN46 during the slowed recovery from desensitization, suggesting that the E354Q mutant traffics to the same compartments as the WT albeit with altered kinetics (Fig. 6D). Examination of WT and E354Q GIPRs by confocal microscopy supports the conclusion that intracellular GIPR is localized to TGN46-positive compartments upon GIP stimulation (Fig. 6E). Additional studies are required to reveal the details of the intracellular trafficking itinerary of GIPR.
Both WT and E354Q GIPRs partially colocalized with transferrin internalized from the medium, indicating that GIPR traffics via the transferrin-positive endosomal system during transit to and from the TGN46-positive compartment (Fig. 7A). Neither WT nor E354Q GIPR colocalizes with LAMP1, a marker of late endosomes/lysosomes, during or after recovery from desensitization (Fig. 7B), consistent with our results that there is little degradation of GIPR during desensitization (Fig. 6B).
FIG 7.
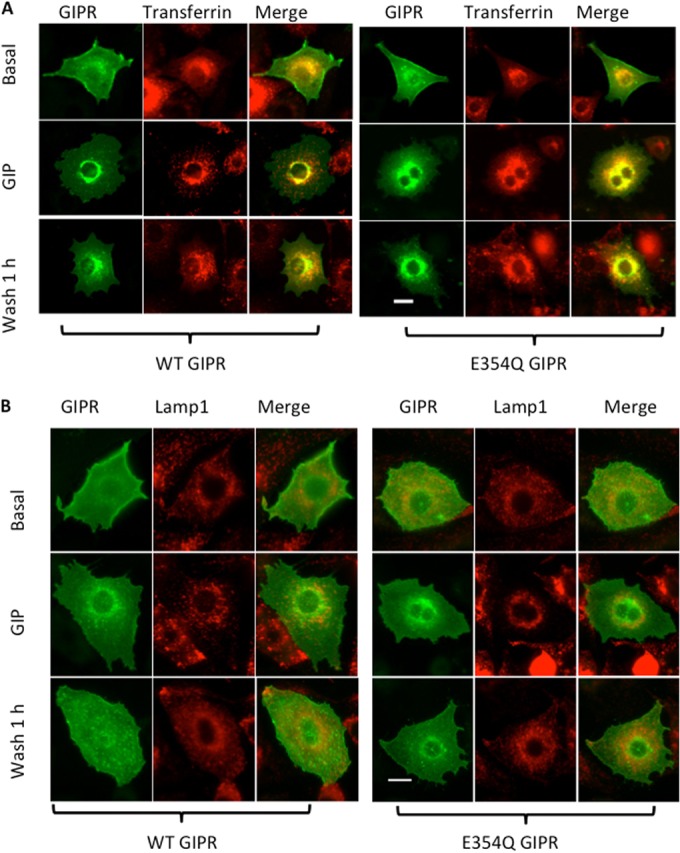
(A) Epifluorescence images of 3T3-L1 adipocytes electroporated with WT or E354Q HA-GIPR-GFP and stained. (A) Cells were serum starved, incubated with rhodamine-labeled transferrin (2 mg/ml), and stimulated without or with 100 nM GIP for 1 h. One set of GIP-treated cells was washed and left in serum-free medium for 1 h. The cells were fixed and imaged. Bar, 10 μm. (B) Epifluorescence images of 3T3-L1 adipocytes electroporated with WT or E354Q HA-GIPR-GFP and stained. Cells were serum starved and incubated without or with 100 nM GIP for 1 h. One set of GIP-treated cells was washed and left in serum-free medium for 1 h. The cells were fixed, permeabilized, and stained with anti-LAMP1 antibodies, followed by secondary staining with anti-rabbit Cy3 antibodies. Bar, 10 μm.
Impaired recovery of E354Q GIPR after desensitization affects receptor function.
We have previously shown that GIP acutely increases the insulin sensitivity of adipocytes (9). In those studies, we used insulin-stimulated translocation of GLUT4 to the plasma membrane of adipocytes as a measure of insulin action. To study the effect of the E354Q substitution of GIPR on GIP-induced insulin sensitivity, we generated adipocytes stably expressing either the WT or E354Q BTX-GIPR reporters. The GIP dose-responses (elevated cAMP levels) were identical in cells expressing the WT and the E354Q mutant, as were the timings of the responses, with maximum levels of cAMP being achieved within 15 min of GIP stimulation (Fig. 8A and B). These data are consistent with results showing that the signaling response of transiently expressed E354Q HA-GIPR-GFP to a single challenge with GIP is identical to that of WT HA-GIPR-GFP (Fig. 4A).
FIG 8.

Impaired recovery of E354Q GIPR after desensitization affects receptor function. (A) Dose-response of WT and E354Q BTX-GIPR stably expressed in adipocytes. The data are averages of data from 3 experiments ± standard errors of the means. (B) Elevated cAMP levels in adipocytes stably expressing WT or E354Q BTX-GIPR as a function of time of GIP stimulation. Data are averages of data from 2 experiments ± SD. (C) The amount of HA-GLUT4-GFP in the plasma membrane of adipocytes stably expressing WT BTX-GIPR or E354Q BTX-GIPR after a 30-min stimulation with 0.2 nM insulin or 0.2 nM insulin and 100 nM GIP or with no stimulation (basal) was determined. (D) In parallel, the amount of HA-GLUT4-GFP in the plasma membrane of adipocytes preincubated with 100 nM GIP for 60 min, washed free of GIP, and allowed to recover for either 60 min or 240 min before stimulation with 0.2 nM insulin or 0.2 nM insulin and 100 nM GIP was determined. Each bar represents the average ± standard error of the mean from 3 independent experiments (*, P < 0.02; ns, not significant).
We next determined whether the enhanced desensitization and impaired recovery of E354Q GIPR (Fig. 4 and 6) impair GIPR control of insulin sensitivity. In previous studies, we have shown that GIP acutely sensitizes adipocytes to insulin, as measured by insulin-stimulated translocation of GLUT4 to the plasma membrane (9). Insulin-stimulated translocation of GLUT4 to the plasma membrane of adipocytes and muscle cells is a key mechanism underlying the disposal of dietary glucose and is a convenient quantitative functional measure of insulin action. Adipocytes were incubated with GIP for 60 min, washed, and incubated for an additional 60 or 240 min without GIP to allow for the repopulation of the plasma membrane before a second GIP stimulation. In the first challenge, GIP promoted a similar increase in GLUT4 translation over the effect of insulin alone in both adipocytes expressing WT GIPR and adipocytes expressing E354Q GIPR (Fig. 8C). These data establish that E354Q GIPR, similar to WT GIPR, functions to promote insulin sensitivity in response to a single challenge of GIPR. Cells expressing WT BTX-GIPR were responsive to a second GIP challenge after a 60-min recovery, with GIP promoting a significant increase in insulin-stimulated GLUT4 translocation to the plasma membrane (Fig. 8D). These data are consistent with WT GIPR repopulating the plasma membrane within 60 min (Fig. 6A). However, cells expressing E354Q BTX-GIPR were refractory to a GIP challenge after only a 60-min recovery but regained responsiveness to GIP (i.e., increased translocation of GLUT4 to the plasma membrane) after a 240-min recovery (Fig. 8D). The recovery of GIP sensitivity is consistent with E354Q GIPR repopulating the plasma membrane within 240 min (Fig. 6A). These results demonstrate that the enhanced desensitization and impaired recovery of E354Q GIPR affect its function as an insulin sensitizer.
In the above-described functional studies, the activities of the ectopic WT and E354Q GIPRs were assayed in the background of the endogenous 3T3-L1 GIPR. To control for any effects of endogenous GIPR, we transiently knocked down mouse GIPR using an siRNA that does not target the ectopically expressed human BTX-GIPR (Fig. 9A). Knockdown of endogenous GIPR in 3T3-L1 adipocytes completely blocked the effect of GIP on insulin action, demonstrating that GIPR is required for GIP-promoted insulin sensitivity (Fig. 9B). The knockdown of endogenous GIPR in cells stably expressing WT or E354Q BTX-GIPR did not affect GIP action. Cells expressing the WT receptor responded similarly to primary and secondary challenges with GIP whether or not endogenous GIPR was transiently knocked down by siRNA (Fig. 9C). In addition, transient knockdown of endogenous GIPR in cells expressing E354Q BTX-GIPR did not affect the GIP response (Fig. 9C). These cells responded to the primary GIP challenge but failed to respond to a second challenge 60 min after the first. These data demonstrate that the stable ectopically expressed BTX-GIPR constructs functionally replace endogenous GIPR.
FIG 9.
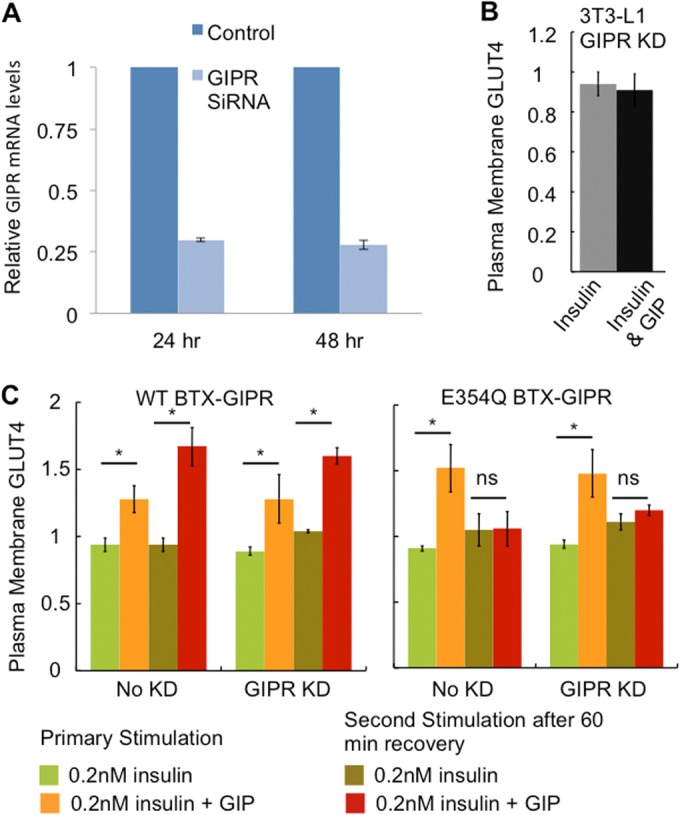
(A) Adipocytes were electroporated with GIPR siRNAs directed against the mouse GIPR sequence (CTAGGACAATCAACTGGAAGGC). After 24 and 48 h of electroporation, cells were harvested, and RNA was extracted. Quantitative PCR was performed as described in Materials and Methods. Each graph represents averages ± SD of data from 2 independent experiments. (B) The effects of 0.2 nM insulin and 0.2 nM insulin plus 100 nM GIP on the expression of HA-GLUT4-GFP in the plasma membrane were determined in 3T3-L1 adipocytes in which endogenous GIPR was transiently knocked down (KD). The knockdown of GIPR eliminated the effect of GIP on insulin-stimulated GLUT4 translocation. The data are averages ± SD of data from 2 experiments. (C) The amount of HA-GLUT4-GFP in the plasma membrane of adipocytes stably expressing WT BTX-GIPR or E354Q BTX-GIPR in which endogenous GIPR was transiently knocked down with an siRNA targeting mouse GIPR compared to cells in which endogenous GIPR was not knocked down was determined after a 30-min stimulation with 0.2 nM insulin or 0.2 nM insulin and 100 nM GIP (primary stimulation). In parallel, the amount of HA-GLUT4-GFP in the plasma membrane of adipocytes preincubated with 100 nM GIP for 60 min, washed free of GIP, and allowed to recover for 60 min before stimulation with either 0.2 nM insulin or 0.2 nM insulin and 100 nM GIP was determined (second stimulation). Each bar represents the average ± standard error of the mean of data from 3 independent experiments (*, P < 0.05; ns, not significant).
DISCUSSION
Here we show that GIPR undergoes constitutive internalization and recycling in the absence of GIP, demonstrating that in unstimulated cultured adipocytes, the functional complement of plasma membrane GIPR is actively maintained. Stimulation of adipocytes with GIP results in a downregulation of plasma membrane GIPR and a consequent desensitization to further GIP stimulation. The reduction of plasma membrane GIPR is achieved by a GIP-induced slowing of receptor recycling without an effect on internalization kinetics. As is the case in unstimulated adipocytes, in GIP-stimulated cells, the intracellular and plasma membrane pools of GIPR are in dynamic equilibrium. Upon cessation of GIP stimulation, the clamp on recycling is released, and the plasma membrane receptor pool is dynamically repopulated with GIPR from inside the cells, resulting in a resensitization to GIP stimulation.
Most GPCRs reside at the plasma membrane in unstimulated cells, undergoing internalization only in response to an agonist. This agonist-stimulated internalization leads to a depletion of the receptor in the plasma membrane and desensitization to further agonist stimulation, a process critical for sculpting GPCR signal transduction (42, 44, 45). In these cases, the internalized GPCRs are either sorted to lysosomes for degradation (and prolonged desensitization) or targeted to a recycling pathway that returns the receptor to the plasma membrane (and rapid resensitization) (46–48). Nonetheless, the triggering event for downregulation is agonist-induced internalization. Hence, GIPR does not conform to the typical GPCR behavior. The constitutive cycling of GIPR is not unique; other GPCRs are known to constitutively cycle (49–53). However, to our knowledge, only in the case of melanocortin receptor 4 (MC4R) has it been shown that desensitization of a constitutively cycling GPCR occurs by impairment of recycling back to the plasma membrane (49).
Because it is the most common mechanism of GPCR desensitization, downregulation due to regulated internalization has been intensively studied (54). Much less is known at the molecular level about downregulation that occurs via a slowing of recycling. Regardless of the specific molecular mechanism underlying the regulation of GIPR recycling, our data support a model in which GIP-induced slowing of GIPR recycling is achieved by regulation of receptor traffic through the same compartments traversed in unstimulated adipocytes, in contrast to a mechanism involving activated receptor recycling by a distinct pathway. In both unstimulated and GIP-stimulated adipocytes, GIPR partially accumulates in a TGN46-positive intracellular compartment, indicating that traffic from this compartment is the rate-limiting step in GIPR recycling in both GIP-stimulated and unstimulated cells. TGN46 is a TGN-resident protein; therefore, our data are consistent with the site of GIPR regulation being its trafficking from the TGN (or a TGN subdomain) to the plasma membrane. However, additional studies are required to identify the intracellular itinerary of the GIPR receptor and the molecular mechanism responsible for the differential control of GIPR trafficking in unstimulated and GIP-stimulated cells.
K cells of the gastrointestinal tract secrete GIP in response to the lipid and carbohydrate content of the meal. Blood GIP levels increase in the postprandial state, returning to prefeeding levels within 3 to 4 h (55–57). Thus, cells are exposed to various GIP levels throughout the day, resulting in multiple rounds of response, desensitization, and resensitization. We propose that GIPR desensitization that occurs by the dynamic redistribution of a constitutively cycling pool of receptors provides a means for cells to be rapidly desensitized and resensitized to various blood GIP levels. It is important to note that in studies of cultured cells, including those presented here, pharmacological amounts of GIP are used. Postprandial blood GIP levels are on the order of 100 pM, whereas 10 to 100 nM GIP is used for studies of cultured and primary adipocytes, including those presented here (e.g., see references 58 and 59). Thus, although previous studies suggest that the activities of GIP assayed in cultured cells reflect GIP activities in vivo, our conclusions on the behavior of GIPR based on studies of cultured adipocytes need to be tested in primary cells and tissues. Nonetheless, our results provide the foundation for these future studies of GIPR function and regulation.
The response to GIP is blunted in insulin resistance, obesity, and T2DM (60–63). The underlying cause of this GIP resistance is not known. Our data suggest that one possible mechanism of resistance is that persistent elevations of GIP levels due to overstimulation of K cells, as a consequence of overnutrition, induce prolonged agonist-induced desensitization (downregulation) of the GIP receptor (60). Since the downregulation of the receptor occurs in a dose-dependent fashion, even small but persistent elevations of GIP levels could result in desensitization over time.
Genome-wide association studies have revealed that genetic variations in GIPR (rs1800437), which results in a glutamine-for-glutamic acid substitution at position 354 of the sixth transmembrane of GIPR, are associated with an increased risk of insulin resistance and cardiovascular diseases (21, 22). It is not known how the E354Q substitution affects GIPR function. The results of studies of E354Q GIPR ectopically expressed in Chinese hamster ovary cells or HEK cells are conflicting. In one study, the GIP-stimulated increases in cAMP formation downstream of E354Q GIPR activation were found to be blunted due to reduced localization on the plasma membrane (64), whereas in a second study, no differences in signaling or expression on the cell surface were observed (23). A limitation of those studies is that GIPR is not normally expressed in either of the cell types used. There is ample evidence that GPCR behaviors are cell context dependent (34, 35).
Here, in studies of GIPR in an appropriate cell type, we show that the E354Q substitution affects GIP-induced desensitization/resensitization of the GIPR. The E354Q GIPR mutant responds normally to a single challenge with GIP, as determined by measurements of cAMP formation after stimulation and effects on adipocyte insulin sensitivity. However, GIP-induced downregulation of E354Q GIPR from the plasma membrane is pronounced and the repopulation of the plasma membrane following cessation of GIP stimulation is prolonged compared to WT GIPR. This increased recovery time from GIP stimulation results in an extension of the time that cells are refractory to a second challenge of GIP, which disrupts GIP control of the insulin response. The impact of this effect of the resensitization period on the biological activity of GIP will be amplified over time because every GIPR desensitization/resensitization cycle will be affected. The impaired recovery of the E354Q mutant after GIP challenge may contribute to the association of this variant with an increased risk of insulin resistance and cardiovascular disease.
Mutations in GPCRs often lead to retention in the biosynthetic system due to misfolding and a consequent reduction in expression in the plasma membrane or to impaired agonist potency due to an alteration in agonist binding and/or receptor activation (65–69). Our study reveals how a specific and subtle alteration in the trafficking of a GPCR can profoundly affect the biological function of the receptor. Our data suggest that the effect of the E354Q substitution is to increase the affinity or efficiency of GIPR interactions with the cellular machinery that regulates the trafficking of WT GIPR, rather than E354Q inducing aberrant trafficking of GIPR, since increased intracellular retention is achieved by a more pronounced slowing of recycling of E354Q GIPR from the TGN46. Future studies are required to achieve a mechanistic understanding of the factors involved in the intracellular retention of E354Q GIPR that lead to enhanced desensitization and slowed resensitization after exposure to the agonist. The discoveries that we have made regarding the behavior of GIPR through studies of a cultured cell model system will serve as a foundation for work to link the characteristics of GIPR behavior, both the WT and the E354Q variant, to the role of the GIP/GIPR in the control of whole-body metabolism.
Here we have focused on a detailed cell-biological analysis of the behavior of WT and E354Q variant GIPRs in adipocytes as a model cell type. GIP also has an important role in promoting glucose-stimulated insulin secretion from beta cells (4). It will be of interest in future studies to determine whether GIPR behaves in a similar fashion in beta cells as it does in adipocytes. For example, previous studies in beta cells suggest that GIP desensitization occurs downstream of GIPR, although detailed behavior of GIPR has not been reported for beta cells (14).
ACKNOWLEDGMENTS
We thank Bernaud Thorens, University of Lausanne, for providing human GIPR cDNA and Eva Gonzalez, Reema Vazirani, and Salihah Dick for helpful discussions and critically reading the manuscript.
This work was supported by National Institutes of Health grant DK096925 (T.E.M.). J.B. was supported by a Medical Scientist Training Program grant from the National Institute of General Medical Sciences of the National Institutes of Health (T32GM07739) to the Weill Cornell/Rockefeller/Sloan-Kettering Tri-Institutional MD-PhD Program.
Footnotes
Published ahead of print 21 July 2014
REFERENCES
- 1.Dupre J, Ross SA, Watson D, Brown JC. 1973. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J. Clin. Endocrinol. Metab. 37:826–828. 10.1210/jcem-37-5-826 [DOI] [PubMed] [Google Scholar]
- 2.Ross SA, Dupre J. 1978. Effects of ingestion of triglyceride or galactose on secretion of gastric inhibitory polypeptide and on responses to intravenous glucose in normal and diabetic subjects. Diabetes 27:327–333. 10.2337/diab.27.3.327 [DOI] [PubMed] [Google Scholar]
- 3.Vilsboll T, Krarup T, Madsbad S, Holst JJ. 2003. Both GLP-1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul. Pept. 114:115–121. 10.1016/S0167-0115(03)00111-3 [DOI] [PubMed] [Google Scholar]
- 4.Drucker DJ. 2006. The biology of incretin hormones. Cell Metab. 3:153–165. 10.1016/j.cmet.2006.01.004 [DOI] [PubMed] [Google Scholar]
- 5.McIntyre N, Holdsworth CD, Turner DS. 1965. Intestinal factors in the control of insulin secretion. J. Clin. Endocrinol. Metab. 25:1317–1324. 10.1210/jcem-25-10-1317 [DOI] [PubMed] [Google Scholar]
- 6.Miyawaki K, Yamada Y, Ban N, Ihara Y, Tsukiyama K, Zhou H, Fujimoto S, Oku A, Tsuda K, Toyokuni S, Hiai H, Mizunoya W, Fushiki T, Holst JJ, Makino M, Tashita A, Kobara Y, Tsubamoto Y, Jinnouchi T, Jomori T, Seino Y. 2002. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat. Med. 8:738–742. 10.1038/nm727 [DOI] [PubMed] [Google Scholar]
- 7.Asmar M, Simonsen L, Madsbad S, Stallknecht B, Holst JJ, Bulow J. 2010. Glucose-dependent insulinotropic polypeptide may enhance fatty acid re-esterification in subcutaneous abdominal adipose tissue in lean humans. Diabetes 59:2160–2163. 10.2337/db10-0098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asmar M, Simonsen L, Arngrim N, Holst JJ, Dela F, Bulow J. 2014. Glucose-dependent insulinotropic polypeptide has impaired effect on abdominal, subcutaneous adipose tissue metabolism in obese subjects. Int. J. Obes. 38:259–265. 10.1038/ijo.2013.73 [DOI] [PubMed] [Google Scholar]
- 9.Mohammad S, Ramos LS, Buck J, Levin LR, Rubino F, McGraw TE. 2011. Gastric inhibitory peptide controls adipose insulin sensitivity via activation of cAMP-response element-binding protein and p110beta isoform of phosphatidylinositol 3-kinase. J. Biol. Chem. 286:43062–43070. 10.1074/jbc.M111.289009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yip RG, Boylan MO, Kieffer TJ, Wolfe MM. 1998. Functional GIP receptors are present on adipocytes. Endocrinology 139:4004–4007. 10.1210/endo.139.9.6288 [DOI] [PubMed] [Google Scholar]
- 11.Starich GH, Bar RS, Mazzaferri EL. 1985. GIP increases insulin receptor affinity and cellular sensitivity in adipocytes. Am. J. Physiol. 249:E603–E607 [DOI] [PubMed] [Google Scholar]
- 12.Getty-Kaushik L, Song DH, Boylan MO, Corkey BE, Wolfe MM. 2006. Glucose-dependent insulinotropic polypeptide modulates adipocyte lipolysis and reesterification. Obesity 14:1124–1131. 10.1038/oby.2006.129 [DOI] [PubMed] [Google Scholar]
- 13.Zhou H, Yamada Y, Tsukiyama K, Miyawaki K, Hosokawa M, Nagashima K, Toyoda K, Naitoh R, Mizunoya W, Fushiki T, Kadowaki T, Seino Y. 2005. Gastric inhibitory polypeptide modulates adiposity and fat oxidation under diminished insulin action. Biochem. Biophys. Res. Commun. 335:937–942. 10.1016/j.bbrc.2005.07.164 [DOI] [PubMed] [Google Scholar]
- 14.Tseng CC, Zhang XY. 2000. Role of G protein-coupled receptor kinases in glucose-dependent insulinotropic polypeptide receptor signaling. Endocrinology 141:947–952. 10.1210/endo.141.3.7365 [DOI] [PubMed] [Google Scholar]
- 15.Kim W, Egan JM. 2008. The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol. Rev. 60:470–512. 10.1124/pr.108.000604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. 1993. Preserved incretin activity of glucagon-like peptide-1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J. Clin. Invest. 91:301–307. 10.1172/JCI116186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elahi D, Mcaloondyke M, Fukagawa NK, Meneilly GS, Sclater AL, Minaker KL, Habener JF, Andersen DK. 1994. The insulinotropic actions of glucose-dependent insulinotropic polypeptide (Gip) and glucagon-like peptide-1(7-37) in normal and diabetic subjects. Regul. Pept. 51:63–74. 10.1016/0167-0115(94)90136-8 [DOI] [PubMed] [Google Scholar]
- 18.Fritsche A, Stefan N, Hardt E, Haring H, Stumvoll M. 2000. Characterisation of beta-cell dysfunction of impaired glucose tolerance: evidence for impairment of incretin-induced insulin secretion. Diabetologia 43:852–858. 10.1007/s001250051461 [DOI] [PubMed] [Google Scholar]
- 19.Saxena R, Hivert MF, Langenberg C, Tanaka T, Pankow JS, Vollenweider P, Lyssenko V, Bouatia-Naji N, Dupuis J, Jackson AU, Kao WH, Li M, Glazer NL, Manning AK, Luan J, Stringham HM, Prokopenko I, Johnson T, Grarup N, Boesgaard TW, Lecoeur C, Shrader P, O'Connell J, Ingelsson E, Couper DJ, Rice K, Song K, Andreasen CH, Dina C, Kottgen A, Le Bacquer O, Pattou F, Taneera J, Steinthorsdottir V, Rybin D, Ardlie K, Sampson M, Qi L, van Hoek M, Weedon MN, Aulchenko YS, Voight BF, Grallert H, Balkau B, Bergman RN, Bielinski SJ, Bonnefond A, Bonnycastle LL, Borch-Johnsen K, Bottcher Y, et al. 2010. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat. Genet. 42:142–148. 10.1038/ng.521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu C, Zhang R, Wang C, Wang J, Ma X, Hou X, Lu J, Yu W, Jiang F, Bao Y, Xiang K, Jia W. 2010. Variants from GIPR, TCF7L2, DGKB, MADD, CRY2, GLIS3, PROX1, SLC30A8 and IGF1 are associated with glucose metabolism in the Chinese. PLoS One 5:e15542. 10.1371/journal.pone.0015542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nitz I, Fisher E, Weikert C, Burwinkel B, Li Y, Mohlig M, Boeing H, Schreiber S, Schrezenmeir J, Doring F. 2007. Association analyses of GIP and GIPR polymorphisms with traits of the metabolic syndrome. Mol. Nutr. Food Res. 51:1046–1052. 10.1002/mnfr.200700048 [DOI] [PubMed] [Google Scholar]
- 22.Sauber J, Grothe J, Behm M, Scherag A, Grallert H, Illig T, Hinney A, Hebebrand J, Wiegand S, Gruters A, Krude H, Biebermann H. 2010. Association of variants in gastric inhibitory polypeptide receptor gene with impaired glucose homeostasis in obese children and adolescents from Berlin. Eur. J. Endocrinol. 163:259–264. 10.1530/EJE-10-0444 [DOI] [PubMed] [Google Scholar]
- 23.Almind K, Ambye L, Urhammer SA, Hansen T, Echwald SM, Holst JJ, Gromada J, Thorens B, Pedersen O. 1998. Discovery of amino acid variants in the human glucose-dependent insulinotropic polypeptide (GIP) receptor: the impact on the pancreatic beta cell responses and functional expression studies in Chinese hamster fibroblast cells. Diabetologia 41:1194–1198 [DOI] [PubMed] [Google Scholar]
- 24.Karylowski O, Zeigerer A, Cohen A, McGraw TE. 2004. GLUT4 is retained by an intracellular cycle of vesicle formation and fusion with endosomes. Mol. Biol. Cell 15:870–882. 10.1091/mbc.E03-07-0517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeigerer A, McBrayer MK, McGraw TE. 2004. Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mol. Biol. Cell 15:4406–4415. 10.1091/mbc.E04-04-0333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lampson MA, Racz A, Cushman SW, McGraw TE. 2000. Demonstration of insulin-responsive trafficking of GLUT4 and vpTR in fibroblasts. J. Cell Sci. 113(Part 22):4065–4076 [DOI] [PubMed] [Google Scholar]
- 27.Martin OJ, Lee A, McGraw TE. 2006. GLUT4 distribution between the plasma membrane and the intracellular compartments is maintained by an insulin-modulated bipartite dynamic mechanism. J. Biol. Chem. 281:484–490. 10.1074/jbc.M505944200 [DOI] [PubMed] [Google Scholar]
- 28.Zeigerer A, Lampson MA, Karylowski O, Sabatini DD, Adesnik M, Ren M, McGraw TE. 2002. GLUT4 retention in adipocytes requires two intracellular insulin-regulated transport steps. Mol. Biol. Cell 13:2421–2435. 10.1091/mbc.E02-02-0071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lampson MA, Schmoranzer J, Zeigerer A, Simon SM, McGraw TE. 2001. Insulin-regulated release from the endosomal recycling compartment is regulated by budding of specialized vesicles. Mol. Biol. Cell 12:3489–3501. 10.1091/mbc.12.11.3489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hannan S, Wilkins ME, Dehghani-Tafti E, Thomas P, Baddeley SM, Smart TG. 2011. Gamma-aminobutyric acid type B (GABA(B)) receptor internalization is regulated by the R2 subunit. J. Biol. Chem. 286:24324–24335. 10.1074/jbc.M110.220814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hannan S, Wilkins ME, Thomas P, Smart TG. 2013. Tracking cell surface mobility of GPCRs using alpha-bungarotoxin-linked fluorophores. Methods Enzymol. 521:109–129. 10.1016/B978-0-12-391862-8.00006-5 [DOI] [PubMed] [Google Scholar]
- 32.Gault VA, Flatt PR, Bailey CJ, Harriott P, Greer B, Mooney MH, O'Harte FP. 2002. Enhanced cAMP generation and insulin-releasing potency of two novel Tyr1-modified enzyme-resistant forms of glucose-dependent insulinotropic polypeptide is associated with significant antihyperglycaemic activity in spontaneous obesity-diabetes. Biochem. J. 367:913–920. 10.1042/BJ20020319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gespach C, Emami S, Rosselin G. 1984. Gastric inhibitory peptide (GIP), pancreatic glucagon and vasoactive intestinal peptide (VIP) are cAMP-inducing hormones in the human gastric cancer cell line HGT-1. Homologous desensitization of VIP receptor activity. Biochem. Biophys. Res. Commun. 120:641–649 [DOI] [PubMed] [Google Scholar]
- 34.Tobin AB, Butcher AJ, Kong KC. 2008. Location, location, location. Site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol. Sci. 29:413–420. 10.1016/j.tips.2008.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butcher AJ, Prihandoko R, Kong KC, McWilliams P, Edwards JM, Bottrill A, Mistry S, Tobin AB. 2011. Differential G-protein-coupled receptor phosphorylation provides evidence for a signaling bar code. J. Biol. Chem. 286:11506–11518. 10.1074/jbc.M110.154526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sekine-Aizawa Y, Huganir RL. 2004. Imaging of receptor trafficking by using alpha-bungarotoxin-binding-site-tagged receptors. Proc. Natl. Acad. Sci. U. S. A. 101:17114–17119. 10.1073/pnas.0407563101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hausdorff WP, Caron MG, Lefkowitz RJ. 1990. Turning off the signal: desensitization of beta-adrenergic receptor function. FASEB J. 4:2881–2889 [PubMed] [Google Scholar]
- 38.Lefkowitz RJ, Wessels MR, Stadel JM. 1980. Hormones, receptors, and cyclic AMP: their role in target cell refractoriness. Curr. Top. Cell. Regul. 17:205–230. 10.1016/B978-0-12-152817-1.50011-0 [DOI] [PubMed] [Google Scholar]
- 39.Stadel JM, Lefkowitz RJ. 1980. Molecular mechanisms of beta-adrenergic receptor desensitization. Proc. West. Pharmacol. Soc. 23:469–475 [PubMed] [Google Scholar]
- 40.Luttrell LM, Lefkowitz RJ. 2002. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 115:455–465 [DOI] [PubMed] [Google Scholar]
- 41.Shankaran H, Wiley HS, Resat H. 2007. Receptor downregulation and desensitization enhance the information processing ability of signalling receptors. BMC Syst. Biol. 1:48. 10.1186/1752-0509-1-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferguson SS, Barak LS, Zhang J, Caron MG. 1996. G-protein-coupled receptor regulation: role of G-protein-coupled receptor kinases and arrestins. Can. J. Physiol. Pharmacol. 74:1095–1110. 10.1139/y96-124 [DOI] [PubMed] [Google Scholar]
- 43.Penn RB, Pronin AN, Benovic JL. 2000. Regulation of G protein-coupled receptor kinases. Trends Cardiovasc. Med. 10:81–89. 10.1016/S1050-1738(00)00053-0 [DOI] [PubMed] [Google Scholar]
- 44.Aramori I, Ferguson SS, Bieniasz PD, Zhang J, Cullen B, Cullen MG. 1997. Molecular mechanism of desensitization of the chemokine receptor CCR-5: receptor signaling and internalization are dissociable from its role as an HIV-1 co-receptor. EMBO J. 16:4606–4616. 10.1093/emboj/16.15.4606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelly E, Bailey CP, Henderson G. 2008. Agonist-selective mechanisms of GPCR desensitization. Br. J. Pharmacol. 153(Suppl 1):S379–S388. 10.1038/sj.bjp.0707604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Magalhaes AC, Dunn H, Ferguson SS. 2012. Regulation of GPCR activity, trafficking and localization by GPCR-interacting proteins. Br. J. Pharmacol. 165:1717–1736. 10.1111/j.1476-5381.2011.01552.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ritter SL, Hall RA. 2009. Fine-tuning of GPCR activity by receptor-interacting proteins. Nat. Rev. Mol. Cell Biol. 10:819–830. 10.1038/nrm2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galet C, Hirakawa T, Ascoli M. 2004. The postendocytotic trafficking of the human lutropin receptor is mediated by a transferable motif consisting of the C-terminal cysteine and an upstream leucine. Mol. Endocrinol. 18:434–446. 10.1210/me.2003-0293 [DOI] [PubMed] [Google Scholar]
- 49.Mohammad S, Baldini G, Granell S, Narducci P, Martelli AM, Baldini G. 2007. Constitutive traffic of melanocortin-4 receptor in Neuro2A cells and immortalized hypothalamic neurons. J. Biol. Chem. 282:4963–4974. 10.1074/jbc.M608283200 [DOI] [PubMed] [Google Scholar]
- 50.Gilliland CT, Salanga CL, Kawamura T, Trejo J, Handel TM. 2013. The chemokine receptor CCR1 is constitutively active, which leads to G protein-independent, beta-arrestin-mediated internalization. J. Biol. Chem. 288:32194–32210. 10.1074/jbc.M113.503797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grimsey NL, Goodfellow CE, Dragunow M, Glass M. 2011. Cannabinoid receptor 2 undergoes Rab5-mediated internalization and recycles via a Rab11-dependent pathway. Biochim. Biophys. Acta 1813:1554–1560. 10.1016/j.bbamcr.2011.05.010 [DOI] [PubMed] [Google Scholar]
- 52.Snyder JC, Rochelle LK, Lyerly HK, Caron MG, Barak LS. 2013. Constitutive internalization of the leucine-rich G protein-coupled receptor-5 (LGR5) to the trans-Golgi network. J. Biol. Chem. 288:10286–10297. 10.1074/jbc.M112.447540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trivedi RR, Bhattacharyya S. 2012. Constitutive internalization and recycling of metabotropic glutamate receptor 5 (mGluR5). Biochem. Biophys. Res. Commun. 427:185–190. 10.1016/j.bbrc.2012.09.040 [DOI] [PubMed] [Google Scholar]
- 54.von Zastrow M. 2001. Endocytosis and downregulation of G protein-coupled receptors. Parkinsonism Relat. Disord. 7:265–271. 10.1016/S1353-8020(00)00069-9 [DOI] [PubMed] [Google Scholar]
- 55.Tseng CC, Kieffer TJ, Jarboe LA, Usdin TB, Wolfe MM. 1996. Postprandial stimulation of insulin release by glucose-dependent insulinotropic polypeptide (GIP). Effect of a specific glucose-dependent insulinotropic polypeptide receptor antagonist in the rat. J. Clin. Invest. 98:2440–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lyssenko V, Eliasson L, Kotova O, Pilgaard K, Wierup N, Salehi A, Wendt A, Jonsson A, De Marinis YZ, Berglund LM, Taneera J, Balhuizen A, Hansson O, Osmark P, Duner P, Brons C, Stancakova A, Kuusisto J, Bugliani M, Saxena R, Ahlqvist E, Kieffer TJ, Tuomi T, Isomaa B, Melander O, Sonestedt E, Orho-Melander M, Nilsson P, Bonetti S, Bonadonna R, Miccoli R, Delprato S, Marchetti P, Madsbad S, Poulsen P, Vaag A, Laakso M, Gomez MF, Groop L. 2011. Pleiotropic effects of GIP on islet function involve osteopontin. Diabetes 60:2424–2433. 10.2337/db10-1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jorde R, Schulz TB, Burhol PG, Waldum HL. 1982. Effect of gastrin on fasting and postprandial plasma GIP release in man. Digestion 25:81–87. 10.1159/000198815 [DOI] [PubMed] [Google Scholar]
- 58.Ahlqvist E, Osmark P, Kuulasmaa T, Pilgaard K, Omar B, Brons C, Kotova O, Zetterqvist AV, Stancakova A, Jonsson A, Hansson O, Kuusisto J, Kieffer TJ, Tuomi T, Isomaa B, Madsbad S, Gomez MF, Poulsen P, Laakso M, Degerman E, Pihlajamaki J, Wierup N, Vaag A, Groop L, Lyssenko V. 2013. Link between GIP and osteopontin in adipose tissue and insulin resistance. Diabetes 62:2088–2094. 10.2337/db12-0976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim SJ, Nian C, McIntosh CH. 2010. GIP increases human adipocyte LPL expression through CREB and TORC2-mediated trans-activation of the LPL gene. J. Lipid Res. 51:3145–3157. 10.1194/jlr.M006841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tseng CC, Boylan MO, Jarboe LA, Usdin TB, Wolfe MM. 1996. Chronic desensitization of the glucose-dependent insulinotropic polypeptide receptor in diabetic rats. Am. J. Physiol. 270:E661–E666 [DOI] [PubMed] [Google Scholar]
- 61.Vilsboll T, Krarup T, Madsbad S, Holst JJ. 2002. Defective amplification of the late phase insulin response to glucose by GIP in obese type II diabetic patients. Diabetologia 45:1111–1119. 10.1007/s00125-002-0878-6 [DOI] [PubMed] [Google Scholar]
- 62.Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. 1993. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J. Clin. Invest. 91:301–307. 10.1172/JCI116186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou J, Livak MF, Bernier M, Muller DC, Carlson OD, Elahi D, Maudsley S, Egan JM. 2007. Ubiquitination is involved in glucose-mediated downregulation of GIP receptors in islets. Am. J. Physiol. Endocrinol. Metab. 293:E538–E547. 10.1152/ajpendo.00070.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fortin JP, Schroeder JC, Zhu Y, Beinborn M, Kopin AS. 2010. Pharmacological characterization of human incretin receptor missense variants. J. Pharmacol. Exp. Ther. 332:274–280. 10.1124/jpet.109.160531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luo R, Jin Z, Deng Y, Strokes N, Piao X. 2012. Disease-associated mutations prevent GPR56-collagen III interaction. PLoS One 7:e29818. 10.1371/journal.pone.0029818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chiang NY, Hsiao CC, Huang YS, Chen HY, Hsieh IJ, Chang GW, Lin HH. 2011. Disease-associated GPR56 mutations cause bilateral frontoparietal polymicrogyria via multiple mechanisms. J. Biol. Chem. 286:14215–14225. 10.1074/jbc.M110.183830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sanchez-Laorden BL, Herraiz C, Valencia JC, Hearing VJ, Jimenez-Cervantes C, Garcia-Borron JC. 2009. Aberrant trafficking of human melanocortin 1 receptor variants associated with red hair and skin cancer: steady-state retention of mutant forms in the proximal Golgi. J. Cell. Physiol. 220:640–654. 10.1002/jcp.21804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xiang Z, Proneth B, Dirain ML, Litherland SA, Haskell-Luevano C. 2010. Pharmacological characterization of 30 human melanocortin-4 receptor polymorphisms with the endogenous proopiomelanocortin-derived agonists, synthetic agonists, and the endogenous agouti-related protein antagonist. Biochemistry 49:4583–4600. 10.1021/bi100068u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Granell S, Mohammad S, Ramanagoudr-Bhojappa R, Baldini G. 2010. Obesity-linked variants of melanocortin-4 receptor are misfolded in the endoplasmic reticulum and can be rescued to the cell surface by a chemical chaperone. Mol. Endocrinol. 24:1805–1821. 10.1210/me.2010-0071 [DOI] [PMC free article] [PubMed] [Google Scholar]