

Abstract
During myogenesis, satellite stem cells (SCs) are induced to proliferate and differentiate to myogenic precursors. The role of energy sensors such as the AMP-activated protein kinase (AMPK) and the mammalian Target of Rapamycin (mTOR) in SC activation is unclear. We previously observed that upregulation of ATP through RNA-mediated mitochondrial restoration (MR) accelerates SC activation following skeletal muscle injury. We show here that during regeneration, the AMPK-CRTC2-CREB and Raptor-mTORC-4EBP1 pathways were rapidly activated. The phosho-CRTC2-CREB complex was essential for myogenesis and activated transcription of the critical cell cycle regulator cyclin D1 (Ccnd1). Knockdown (KD) of either mTORC or its subunit Raptor delayed SC activation without influencing the differentiation program. KD of 4EBP1 had no effect on SC activation but enhanced myofiber size. mTORC1 positively regulated Ccnd1 translation but destabilized Ccnd1 mRNA. These antithetical effects of mTORC1 were mediated by two microRNAs (miRs) targeted to the 3′ untranslated region (UTR) of Ccnd1 mRNA: miR-1 was downregulated in mTORC-KD muscle, and depletion of miR-1 resulted in increased levels of mRNA without any effect on Ccnd1 protein. In contrast, miR-26a was upregulated upon mTORC depletion, while anti-miR-26a oligonucleotide specifically stimulated Ccnd1 protein expression. Thus, mTORC may act as a timer of satellite cell proliferation during myogenesis.
INTRODUCTION
Regeneration of skeletal muscle following injury involves formation of new myofibers as well as patch repair of old injured fibers by fusion of satellite cell-derived myoblasts (1). The program of differentiation of satellite cells involves sequential expression of myogenic regulatory factors (MRFs) and their target genes (2–5). Mitochondrial biogenesis occurs at a specific time prior to the formation of myofibers (4, 6), suggesting an energy-requiring step, but the specific role of ATP in differentiation remains unknown. Eukaryotic cells contain a number of enzymes that sense the energy status, particularly, the pools of AMP and ADP (AMP-activated protein kinase [AMPK]) or of ATP (mammalian Target of Rapamycin [mTOR] kinases) (7, 8). It is possible that mitochondrial activity, through modulation of the adenylate pool, impacts these energy sensors, thereby activating or inhibiting downstream pathways.
Although energy sensors such as AMPK and mTORC are known to regulate cellular energy homeostasis and cell growth (9), there are indications that they could also modulate specific developmental processes. It was shown early that the mTORC inhibitor rapamycin inhibits growth of myofibers in regenerating adult muscle (10); such inhibition was overcome by expression of a rapamycin-resistant mTORC gene (11). Genetic ablation of mTORC (12), or of the mTORC1 subunit Raptor (13), or of the mTORC1 target S6K (14) results in severe muscle atrophy. These studies indicate that myofiber growth and maturation are regulated by mTORC1 acting through S6K in vivo but provide no information on the specific effect, if any, of mTORC1 on the early stages of myogenesis. Rapamycin inhibits the proliferation of primary myoblasts in an S6K-independent manner (14), as well as the differentiation of cultured myoblasts to myotubes upon serum deprivation in vitro (15–17), but the inhibition can be rescued by a rapamycin-resistant mTORC through a process that involves insulin-like growth factor II (IGFII) but does not require its kinase activity (17, 18). Knockdown (KD) of mTORC in myoblasts inhibits their differentiation to myotubes in vitro, but unexpectedly, knockdown of Raptor has the opposite effect (19, 20); the role of Rictor, a subunit of the mTORC2 complex, is uncertain, with reports of either inhibition of differentiation (20) or no effect (19) of Rictor KD. From these studies, it is apparent that mTORC can regulate cell growth and division in vitro, that the effect on growth requires S6K whereas that on myoblast division does not, indicating the presence of additional (unknown) mTORC targets, and finally, that mTORC and Raptor regulate the differentiation of cultured myoblasts to myotubes in different ways that are still unexplained. Thus, the precise role of mTORC in early myoblast differentiation is still unclear.
mTORC regulates cellular mRNA translation in different ways. The mTORC1 kinase phosphorylates 4EBP, a repressor of cap-dependent translation initiation, and S6 kinase (S6K), which regulates translation factors, including eukaryotic initiation factor 4A (eIF4A) and eukaryotic elongation factor 2 (eEF2) (21). Additionally, studies in myoblasts have revealed that mTORC up- or downregulates several microRNAs (miRNAs) that regulate translation of their target mRNAs (22). The roles of a number of miRNAs in myoblast differentiation have been elucidated (23–25), but their role in the proliferative step of myogenesis remains unclear.
We have previously observed that a pulse of mitochondrial restoration (MR) in injured rat skeletal muscle through administration of an RNA cocktail targeted to mitochondria results in rapid proliferation of satellite cells and the initiation of a time-resolved sequence of MRF expression, resulting in the deposition of new myofibers by 1 week (4). Activation of satellite cells is evident from the expression of cyclin D1 (Ccnd1), a marker of G1 phase cells, proliferating cell nuclear antigen (PCNA; an indicator of cells in the S phase), and the master myogenic regulator myoD, all at ∼12 h following RNA treatment (4). In the present study, we employed lentiviruses expressing short hairpin RNAs (shRNAs) to selectively knock down various metabolic sensors and their downstream effectors, and we monitored their effects on both early (cell division and differentiation) and late (myofiber biogenesis) myogenic processes. Our results indicate a dual role of mTORC in translational control of myoblast proliferation mediated by miRNAs.
MATERIALS AND METHODS
MR-induced skeletal muscle regeneration.
The quadriceps muscle of male Sprague-Dawley rats (12 to 14 months) was subjected to needle perforation injury followed by RNA-mediated regeneration as described previously (4). All animal experiments were performed according to the guidelines of the IICB Animal Ethics Committee. Polycistronic RNAs (pcRNAs) 1R, 2R, and 3R, encoding various regions of the rat mitochondrial genome (4), and each carrying a 5′ signal tag, were synthesized with T7 RNA polymerase and combined with carrier complex R6 to form an RNP cocktail. At the height of the inflammatory phase (day 6 postinjury), the RNP mixture was injected at the injury site to deliver the RNAs to tissue mitochondria; as a control we used D arm RNA, a 23-nucleotide hairpin constituting the signal tag, which gets delivered to mitochondria but is without any discernible effect (26). Muscle contractile activity was measured ex vivo as isometric contraction force (ICF) in a Radnoti Tissue-Organ Bath (Ad Instruments, Colorado Springs, CO), as described previously (4).
RNAi.
Rats were intramuscularly injected with recombinant lentivirus pLKO.1-CMV-tGFP (where CMV is cytomegalovirus and tGFP is transforming growth factor P; >4.9 × 106 TU/ml; Sigma-Aldrich) containing an expression cassette for short hairpin RNA, either scrambled sequence (nontargeted) or targeted to a specific rat mRNA (sequences available on request). For each mRNA, 2 to 4 different shRNAs were designed, using Block-iT RNA interference (RNAi) designer software and selection of high-probability targets to ensure efficient downregulation. Lentivirus infection (three times daily) was carried out on days 3 and 4 postinjury. pcRNA1-3 was injected on day 6 to initiate regeneration for the indicated times.
miRNA target search.
Rat miRNA sequences were obtained from miRBASE (www.mirbase.org). The 3′ untranslated region (UTR) of rat Ccnd1 (NCBI accession no. NM_171992) was scanned for miRNA seed sequences in the site RNA Hybrid (http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/welcome.html) or manually, using the criterion of 7 or more contiguous base pairs starting at positions +1 to +4 of the miRNA (27).
Anti-miRNA treatment.
miRCURY locked nucleic acid (LNA) microRNA inhibitors (Exiqon) targeted against rno-miR-1-3p, rno-miR26a-5p, rno-miR-214-5p, or a negative-control (no significant hit to any sequence) oligonucleotide (sequences available on request) were injected into rat quadriceps muscle at 2.5 nmol per injection 3 times a day for 2 consecutive days starting at day 4 postinjury before pcRNA administration, as above.
Confocal imaging.
Muscle sections (10 μm) were incubated with appropriate combinations of primary antibody and Alexa Fluor (AF) 488 (green)- or AF 633 (red)-labeled secondary antibody (details available on request); nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Sections were imaged in an A1R confocal microscope (Nikon) using 401-, 488-, and 627-nm lasers and NIS Elements software. Angiogenesis was visualized with fluorescein isothiocyanate (FITC)-conjugated isolectin B4, an endothelial cell marker.
Histochemistry.
Hematoxylin and eosin (H&E)-stained sections were imaged at a magnification of ×400, and the numbers of intact fibers, or nuclei, per field were determined. Fiber area was estimated using Image J software from 10 random fields.
Promoter analysis.
The rat Ccnd1 5′ upstream promoter region (28) was scanned for putative transcription factor binding sites in the EpiTect ChIP qPCR Primers portal of SA Biosciences.
Northern blot analysis.
Blots of muscle total RNA (10 μg) were probed with 32P-labeled gene-specific primers (sequences available on request). Band intensities were quantified on the inverted image as the average intensity value (a.i.) in the Histogram option of Adobe Photoshop 7 after background subtraction.
Estimation of mRNA stability.
Actinomycin D (2.5 μg/g of body weight) was injected at the injury site 6 h after pcRNA treatment, and RNA was prepared 0, 6, and 12 h thereafter. The average intensities (a.i.) of the bands were plotted versus time, and the best-fit exponential decay curve was obtained in Microsoft Excel. The half-life (t1/2) was computed from the equation Nt = N0 · e−kt, where Nt is the a.i. at time t and k is the decay constant; t1/2 = 0.693/k.
ChIP assays.
For chromatin immunoprecipitation (ChIP) assays, the mononuclear cell fraction was isolated from collagenase-treated muscle as described previously (4). The cells in serum-free culture medium were incubated with 1% formaldehyde for 10 min at room temperature to cross-link proteins to DNA. Cells were homogenized in 10 mM Tris-HCl (pH 8.0), 1.5 mM MgCl2, and 10 mM KCl, and the nuclei were sedimented by centrifugation at 1,500 × g for 10 min at 4°C. The pellet was resuspended in nuclear lysis buffer (50 mM Tris-HCl [pH 8], 10 mM EDTA, 1% SDS). The chromatin was sonicated 10 times for 10 s at 50 Hz to generate DNA fragments of 250 to 3,000 bp. Lysates (10 mg DNA) were diluted with ChIP dilution buffer (20 mM Tris-HCl [pH 8.1], 150 mM NaCl, 2 mM EDTA, 0.01% SDS, 1% Triton X-100). Antibodies (1:25 or 1:50 dilution) were added, and after rotation overnight at 4°C, the immune complexes were collected by the addition of 50 μl of protein G-agarose bead-salmon sperm DNA slurry (Millipore). After extensive washes, immune complexes were eluted with 1% SDS–0.1 M NaHCO3, and cross-linking was reversed by the addition of 190 mM NaCl overnight at 65°C. DNA was purified using a PCR purification kit (Qiagen) and amplified by PCR for 30 cycles, with annealing at 55°C, using primers flanking the respective factor-binding sites within the rat Ccnd1 promoter (sequences available on request). The product was visualized by 0.8% agarose gel electrophoresis. For quantification, SYBR green real-time PCR was performed with appropriate primer pairs, and the signals were normalized to input chromatin signals and expressed as percent recovery as follows: % recovery = 100 × 2(CT[100% input] − CT[antibody]), where CT[100% input] is equal to CT [10% input] − log2 10 and CT is the critical threshold.
Real-time RT-PCR.
RNA was isolated from muscle (∼50 mg) using 0.5 ml of Tri reagent (Sigma), after homogenization and centrifugation at 12,000 rpm at 4°C for 10 min (to remove insoluble material) following the manufacturer's instructions, and suspended in 50 μl water. cDNA was synthesized from 1 μg RNA using Moloney murine leukemia virus (M-MuLV) reverse transcriptase (RT; New England BioLabs) and rat Ccnd1 antisense primer and diluted to 100 μl with water. PCRs (25-μl reaction mixtures) contained cDNA (2 μl), 12.5 μl of SYBR green Supermix (Fermentas), and 1.5 μl each of sense and antisense Ccnd1 primers (sequences available on request). PCR was performed in triplicate in a thermal cycler (model Step One Plus; Applied Biosystems) for 40 cycles with a 45-s annealing step at 55°C. SYBR green signals were recorded by Step One Plus software and expressed as critical threshold (CT) values. β-Actin was used as an internal reference. Gene expression was quantified as the fold change between pcRNA-treated samples and controls [2−ΔΔCT, where ΔCT = CT(target gene) − CT(β-actin)].
Protein analysis.
Clarified muscle homogenates (100 μg protein) were subjected to Western blot analysis using specific primary antibodies (details available on request) and detection by chemiluminescence.
RESULTS
Activation of AMPK and mTORC pathways.
In the presence of AMP or ADP bound to the regulatory γ subunit of AMPK, the catalytic α subunit is activated via phosphorylation at Ser172 (to form phospho-AMPKα) by an upstream kinase (29). In pcRNA-treated regenerating muscle, the level of phospho-AMPKα fell sharply (∼3-fold) between 3 and 6 h posttreatment (Fig. 1A). The level of total AMPKα (detected with a different antibody) was unchanged during this time (Fig. 1A), indicating dephosphorylation of preexisting AMPK; reprobing the same blot with the phospho-AMPKα-specific antibody indicated that the phosphorylation level fell from 76% to 7% of total between 3 and 6 h. In injured muscle treated with control RNA, a high level of phospho-AMPKα was maintained for at least 48 h (Fig. 1A). In normal muscle, most fiber-attached (i.e., quiescent) Pax7+ satellite cells were negative for phospho-AMPKα (Fig. 1B, upper row, and E). In contrast, intense cytoplasmic phospho-AMPK staining of ∼70% of the satellite cells was observed after injury (Fig. 1B, middle row, and E), indicating ATP deficiency. The energy crisis was relieved by pcRNA treatment, as evidenced by low or absent phospho-AMPK in the satellite cells (Fig. 1B, bottom row, and E).
FIG 1.

Regulation of energy sensors in regenerating muscle. (A) Injured rat muscle was treated with pcRNA cocktail (left) or control D arm RNA (right) bound to carrier complex R6 for the indicated times. Expression levels of the indicated proteins at the injury site were analyzed by Western blotting. An asterisk indicates an antibody specific for the phosphorylated form of the protein. The percentage of the phosphorylated form (P-form, upper band) of 4EBP1 is shown below the lanes. (B to D) Confocal micrographs of sections of normal muscle, injured untreated (inj) muscle, or injured muscle treated with pcRNAs for 6 h, showing Pax7+ (green) satellite cells stained (red) for phospho-AMPKα (B), phospho-Raptor (C), or phospho-4EBP1 (D); scale bar, 20 μm. (E to G) Quantification of Pax7+ cells that are positive for phosphoforms of AMPKα (E), Raptor (F), or 4EBP1 (G).
One of the known targets of AMPK is the transcriptional coactivator CRTC2, which in association with CREB activates the transcription of many cellular genes with cyclic AMP response element (CRE) regulatory elements; phosphorylation of CRTC2 by AMPK in liver leads to its retention in the cytosol and thus to downregulation of the target promoter (30). In pcRNA-treated muscle, the level of phosphorylated CRTC2 was sharply reduced between 3 and 6 h, disappearing subsequently (Fig. 1A) and mirroring the early deactivation of AMPK.
AMPK phosphorylates Raptor, an mTORC1 complex subunit (31), thereby leading to inactivation of mTORC. In pcRNA-treated muscle, the total amount of Raptor was not appreciable altered, but the phospho form was reduced from 91% of total to 44% between 3 and 6 h after pcRNA treatment (Fig. 1A), with a reduction in the number of phospho-Raptor+ cells (Fig. 1C and F). In parallel, phosphorylation of the mTORC substrate 4EBP1 increased from 21% in injured muscle to ∼80% by 12 h following pcRNA treatment, gradually declining thereafter to a steady level of ∼50% (Fig. 1A); there was an increase in the number of phospho-4EBP1 cells in the regenerating tissue (Fig. 1D and G). These results show that AMPK is inactivated and mTORC1 is activated in satellite cells within 6 h of regeneration, i.e., before the onset of the activation program, which occurs at ∼12 h.
Effects of KD of energy sensors on myogenesis.
To determine the effect of specifically depleting energy sensors or their effectors on regeneration, injured rat quadriceps muscle was infected with lentiviruses expressing shRNAs targeting individual mRNAs and then treated with pcRNAs for MR. Expression of nontargeted (NT) shRNA in normal muscle had no effect on the level of any of the targets (Fig. 2A), indicating the lack of a general inhibition due to lentivirus infection. Expression of target-specific shRNA resulted in downregulation of the target by 80 to 95% (Fig. 2A). Downregulation was maintained in regenerating muscle and was target specific; KD of AMPKα resulted in loss of phosphorylation of the AMPK targets CRTC2 and Raptor, as expected (data not shown). Knockdown was stable until at least 2 weeks (see Fig. 4A). We monitored the effects of RNA interference in terms of the following: (i) levels of PCNA, a marker for cells in the S phase; (ii) expression of myogenic regulatory factors (MRFs); (iii) number and size of newly formed myofibers after 2 weeks; and (iv) formation of the microcapillary network (MCN) as an indicator of angiogenesis. Functional recovery was assayed as the isometric contraction force (ICF) of the regenerated muscle ex vivo.
FIG 2.

Effects of lentivirus-mediated KD of energy-sensing pathway components on myogenesis. (A) Effects of KD on expression of the total levels of the indicated targets in normal muscle infected with lentivirus expressing the indicated shRNA for 3 days. (B to G) Injured rat quadriceps muscle was infected with lentivirus expressing shRNA targeting the specified mRNA or nontargeted (NT) shRNA on day 3 and then treated with pcRNA1-3 on day 6. (B) Hematoxylin and eosin (H&E) stain of injury site 2 weeks after treatment with pcRNA; scale bar, 100 μm. (C) Isolectin B4 staining of endothelial cells (green) at injury site at 2 weeks, merged with the differential interference contrast (DIC) image (gray) and nuclear DAPI stain (blue); scale bar, 10 μm. (D to G) Quantification (means ± standard deviations [SD]) of the number of myofibers (D), myofiber cross-sectional area (E), number of nuclei (F), and isometric contraction force (ICF, in milliNewtons) (G) of muscle infected with lentivirus targeting the indicated mRNA after treatment with pcRNAs for 2 weeks.
FIG 4.

Effects of KD of energy-sensing pathway components on the satellite cell differentiation program. Injury site muscle infected with lentivirus targeting the indicated mRNA or expressing nontargeted (NT) shRNA (top of each panel), at the indicated number of days following pcRNA treatment, was analyzed by Western blotting using antibodies specific to the proteins indicated on the right side. (A) Levels of the indicated proteins in KD muscle at various times, compared to the NT controls. Time zero represents injured muscle prior to pcRNA treatment; n, normal muscle. (B) Time course of expression of PCNA and MRFs in control or KD muscle at various times. (C) Levels of total or phospho-4EBP1 or-S6K1 in control and KD muscle at indicated times following pcRNA treatment.
In control regenerating muscle expressing nontargeted shRNA, formation of myofibers was complete by 2 weeks (Fig. 2B), though the fiber density was slightly higher (29.6 ± 6.8 versus 23.3 ± 2.5 fibers/field) (Fig. 2D), and the fiber size lower (0.018 ± 0.004 versus 0.030 ± 0.006 mm2) (Fig. 2E), than in normal muscle; formation of the MCN around the new myofibers was complete by this time (Fig. 2B). In contrast to normal, mature muscle, NT-virus-infected muscle is actually regenerating muscle after 2 weeks, when the fibers have been formed but are yet to fully grow and mature; thus, a larger number of smaller fibers are packed in the same area. In AMPKα-KD muscle, myofibers formed normally after 2 weeks, though there was a slight increase in the number and a decrease in the average size (Fig. 2B, D, and E); the ex vivo contractile activity of the regenerated muscle was slightly higher than normal (Fig. 2G). Thus, genetic and enzymatic deactivations of AMPK (which occur in KD and control muscle, respectively) have similar effects on myogenesis. Although myofibers were normally formed in AMPKα-KD regenerating muscle, the microcapillary network around them was absent, and there was significant inhibition of the numbers of myofiber-attached isolectin B4-positive endothelial cells (Fig. 2C). Thus, AMPK has a positive role in the angiogenesis program.
KD of CRTC2 or of CREB (Fig. 2A) had profound effects on myogenesis. Sections of the injury site 2 weeks after MR revealed only torn myofibers and the general lack of mononuclear cells, including myoblasts (Fig. 2B and D), and failure of recovery of muscle function (Fig. 2G). Angiogenesis was also affected (Fig. 2C). Over a period of 2 weeks, the lentivirus-mediated downregulation was stable, and there was no evidence of myoblast proliferation (PCNA expression) or MRF expression (see Fig. 4A). Thus, the activation of satellite cells is critically dependent upon the presence of the CRTC2/CREB pair.
KD of mTORC, or of the mTORC1 subunit Raptor, resulted in a significant slowing down of myogenesis. After 2 weeks, few new myofibers were detectable at the injury site (Fig. 2B and D), which was populated with myoblasts (Fig. 3A). The total number of nuclei per field was higher in the regenerating muscle expressing nontargeted shRNA than in normal, uninjured controls (Fig. 2F) but was not significantly different from that in mTOR- or Raptor-KD muscle (Fig. 2F), indicating that the mTORC1 complex does not affect the quantum of myoblast proliferation.
FIG 3.
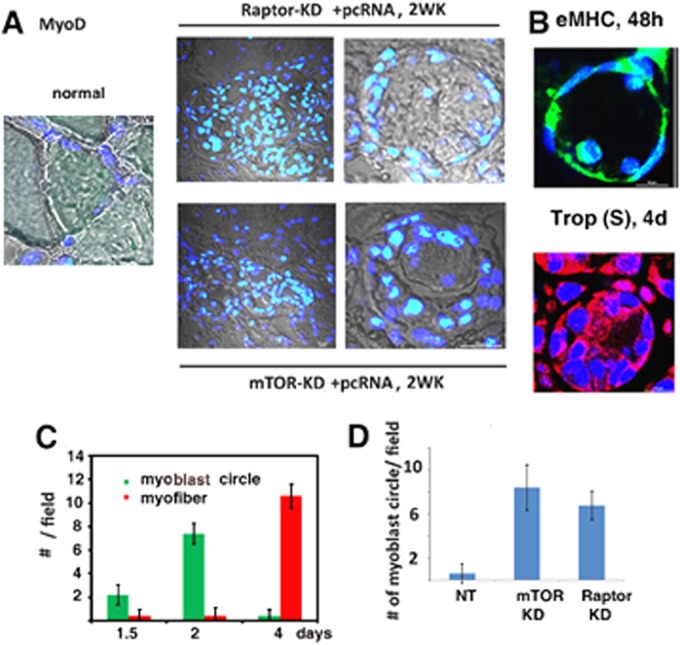
Myoblast rings in regenerating muscle. (A) DIC confocal images (gray) of sections of normal (left), Raptor-KD (upper right), or mTORC-KD (lower right) muscle after 2 weeks of pcRNA treatment, superimposed on merged DAPI (blue)- and anti-MyoD antibody (green)-stained nuclei. Left and right panels show lower and higher magnifications, respectively. (B) Ring structures in normal regenerating muscle after 2 days, stained for eMHC (green) or, after 4 days, stained for troponin (Trop) slow form (S) (red). Nuclei were DAPI stained. (C) Numbers of myoblast circles or myofibers per field (means ± SD) at various times in normal regenerating muscle. (D) Number of myoblast circles after 2 weeks in muscle expressing nontargeted (NT), mTORC-KD, or Raptor-KD muscle (means ± SD).
The mononuclear cells in sections from mTOR or raptor KD muscle were predominantly myoD-positive myoblasts; no such cells were observed in normal muscle (Fig. 3A). Many of the myoblasts formed novel ring structures enclosing one or a few cells (Fig. 3A). Similar myoblast rings were observed at earlier times in normal regenerating muscle stained for intermediate (eMHC) or late (troponin) markers (Fig. 3B). The number of such circles in normal MR induced myogenesis and peaked at ∼2 days, while new myofibers were apparent only after 4 days (Fig. 3C); moreover, the number of such ring structures after 2 weeks was considerably higher in mTOR- or Raptor-KD muscle than in the nontargeted control (Fig. 3D). Thus, depletion of either component of the mTORC1 complex led to the accumulation of putative intermediates in myofiber maturation.
One of the direct targets of the mTORC1 complex is 4EBP1, which in the dephosphorylated form binds to and inactivates translation initiation factor eIF4E (21). In 4EBP1-KD muscle, the myofibers formed were fewer but larger than those formed in nontargeted muscle or even than normal myofibers (Fig. 2B to E), indicating a repressive effect of 4EBP1 on cytoplasmic growth during myogenesis.
Deficiency of mTORC1 causes a delay in the onset of satellite cell activation.
Satellite cell activation during MR-induced myogenesis is characterized by an early burst of cellular proliferation (as indicated by expression of Ccnd1 and PCNA) and upregulation of the myogenic factors MyoD and Myf5; this is followed by the differentiation program, i.e., sequential expression of Myog and MRF4 (4). In view of the incomplete myogenesis observed in mTORC- or Raptor-deficient muscle (Fig. 2 and 3), we queried how this sequence of events was altered by analyzing KD muscle at various times of regeneration for the expression of these cell cycle and differentiation markers.
The level of each lentiviral target was reduced by 80 to 90% in injured muscle prior to the onset of pcRNA-induced regeneration (time zero) and maintained at this low level until at least 2 weeks after pcRNA treatment (Fig. 4A). In control muscle expressing nontargeted shRNA, the cells passed into the S phase between 6 and 24 h, as previously observed (4) (Fig. 4B). However, PCNA expression was evident only after 1 week in Raptor- or mTOR-KD muscle (Fig. 4B). This was accompanied by the expression of myoD and myogenin, as in the case of control muscle (Fig. 4B). Thus, deficiency of mTORC1 slowed down satellite cell activation without significantly altering the differentiation program.
The timing of myoblast proliferation and the program of MRF expression in 4EBP1-depleted muscle were not detectably different from those in nontargeted muscle, except that expression levels were generally higher and the timing of myogenin expression was advanced relative to that of myoD (Fig. 4B). This indicates that myogenin mRNA translation is subject to regulation by 4EBP1.
The mTORC1 complex phosphorylates 4EBP1, a translation repressor, and S6K1, a translation activator (32); phosphorylation of these proteins is therefore an indicator of translation upregulation and could constitute the actual mechanism regulating the timing of differentiation. In mTORC or Raptor-KD muscle, the targeted proteins were maintained at a low level up to at least 2 weeks following MR (Fig. 4A). However, phosphorylation of both of the mTORC1 substrates, 4EBP1 and S6K1, although at low levels initially, was stimulated at 1 to 2 weeks; in the case of 4EBP1, the fraction of total protein phosphorylated went up from near 0 at 1 day to ∼50% after 1 week (Fig. 4C). The timing of this second mTORC1-independent phosphorylation event coincided with the onset of the delayed differentiation program (Fig. 4B), suggesting either a causal relationship between the two or activation of a parallel pathway for cytoplasmic growth.
Transcriptional regulation of cyclin D1 by CRTC2-CREB.
Expression of Ccnd1 is an accurate indicator of the passage of cells through the G1 phase of the cell cycle. In normal regenerating muscle, Ccnd1 transcripts peaked at 6 h and declined thereafter (Fig. 5A). In AMPKα-KD muscle, the transcription of the Ccnd1 gene was marginally stimulated compared to the nontargeted control (Fig. 5B). In CRTC2-or CREB-depleted muscle, the level of Ccnd1 at 6 h was inhibited ∼6-fold (Fig. 5B), suggesting a role of CRTC2 and CREB in the activation of the Ccnd1 promoter. The rat Ccnd1 promoter region contains a cyclic AMP response element (CRE) at position −44 with respect to the transcription start site, which is known to bind CREB (Fig. 5C). The association of CREB and CRTC2 with the Ccnd1 promoter during regeneration was profiled using quantitative chromatin immunoprecipitation (Q ChIP) assays. Binding of phospho-CREB and of its coactivator CRTC2 to the Ccnd1 promoter was initiated at 6 h and peaked at 12 h (Fig. 5D). These experiments show that Ccnd1 transcription is activated during regeneration by the CRTC2-CREB complex.
FIG 5.

Regulation of cyclin D1 expression during regeneration. (A) Q PCR analysis of CcnD1 RNA in injured muscle treated with pcRNA1-3 for the indicated times, showing fold changes with respect to the corresponding controls treated with D arm RNA. (B) Injured rat quadriceps muscle was infected with lentivirus targeting the specified mRNA and then treated with pcRNA1-3 for 6 h. Bars represent the fold changes of Ccnd1 transcripts measured by Q PCR in pcRNA-treated muscle knocked down at the specified locus versus nontargeted-shRNA-expressing control. (C) Map of transcription factor binding sites in the 5′ upstream region of the rat Ccnd1 gene. Numbers indicate distances in base pairs from the ATG codon. (D) ChIP signals from mononuclear cells in injured muscle treated with pcRNA1-3 for the indicated times using antibody against the specified protein and amplified using Ccnd1 promoter primers and 10% of PCR product visualized by gel electrophoresis. In10, PCR product from 10% of input DNA. Numbers indicate the percent recovery from SYBR Green Q-PCR data. (E, F) Ccnd1 mRNA (E) or protein (F) levels in control (NT) or KD muscle at various times following pcRNA treatment. (G) Northern blot analysis of Ccnd1 mRNA in NT or mTORC-KD regenerating muscle in the absence or presence of actinomycin D added at 6 h following pcRNA treatment.
Dual posttranscriptional regulation of cyclin D1 by mTORC1.
KD of mTORC or of the mTORC1 subunit Raptor did not have a significant effect on the level of Ccnd1 mRNA in regenerating muscle at the time of the onset of Ccnd1 transcription, i.e., 6 h after MR (Fig. 5B and E). However, while in normal muscle Ccnd1 RNA was sharply reduced by 24 h, it rose at this time in mTORC or Raptor KD muscle and persisted up to 1 to 2 weeks (Fig. 5E). This indicates stabilization of the mRNA in the absence of a functional mTORC1 complex. To estimate the half-life (t1/2) of Ccnd1 mRNA, regenerating muscle was treated with actinomycin D at 6 h to stop further transcription and the CCnd1 mRNA levels were quantified at different times thereafter. Analysis of the decay curves revealed a t1/2 of 3.6 h in control muscle expressing nontargeted shRNA, which went up to 69 h upon KD of mTORC (Fig. 5F).
Expression of Ccnd1 protein was not directly related to the mRNA level but increased after the mRNA peak in control regenerating muscle (Fig. 5E). In mTORC- or Raptor-depleted muscle, the lag was more pronounced, with protein expression occurring several days after the mRNA peak (Fig. 5E). This suggests that, independent of its role in mRNA degradation, the mTORC1 complex times the translation of Ccnd1 mRNA. In 4EBP1-KD muscle, there was no significant effect on the level of Ccnd1 mRNA (Fig. 5B and E) or on the timing of Ccnd1 translation (Fig. 5F), indicating a 4EBP1-independent mechanism of translation control of this gene.
mTORC-mediated early regulation of microRNAs.
The absence of any significant influence of 4EBP1, which regulates cap-dependent translation, on Ccnd1 expression prompted us to examine other regulatory mechanisms operating during myogenesis. Several mouse miRNAs are regulated during myoblast differentiation in vitro; many of these are up- or downregulated in the presence of rapamycin, indicating a regulatory role of the mTORC1 complex (22). The expression of a few of the corresponding rat miRNAs after muscle injury and regeneration was examined. Subsequent to injury, there was a significant upregulation of miRNA-1 (miR-1) and miR-26a, increasing between 1 and 7 days, but the levels of miR-214 and miR-199 were unaffected (Fig. 6). During MR-induced regeneration in controls expressing nontargeted shRNA, there was a further increase of all 4 miRNAs at 1 day, but the effect was transient, with the levels decreasing during the following week (Fig. 6). Particularly, miR-1 and miR-26a were induced ∼8-fold in 1-day regenerating muscle relative to the injured-state muscle. In mTORC-KD muscle, miR-1, miR-214, and miR-199 were downregulated, whereas the level of miR-26a was further increased compared to nontargeted control (Fig. 6). Thus, mTORC positively regulates miR-1 but partially represses miR-26a.
FIG 6.

Role of miRNAs in cyclin D1 expression during regeneration. (A) Putative seed sequences for the indicated miRNAs in the rat Ccnd1 mRNA 3′-UTR. Numbers indicate the start positions of the seed sequences relative to the AUG codon (+1) of the Ccnd1 gene. (B to F) Injured rat quadriceps muscle infected with nontargeted (NT) or anti-mTORC lentivirus was further injected with negative control (nt) or the indicated anti-miR LNA oligonucleotide and then treated with pcRNAs for the indicated times. (B, C) Northern blots probed with the indicated anti-miR (B) or anti-Ccnd1 (C) primer, with U6 snRNA or 28S rRNA as the respective loading control. (D) Quantification of the Ccnd1mRNA levels at 1 or 7 days (d) in regenerating muscle that had been treated with the indicated lentivirus and negative-control or anti-miR1 LNA oligonucleotide. (E) Western blots probed with anti-Ccnd1 antibody. (F) Quantification of the Western blot signals in muscle treated with the indicated lentivirus and negative-control or anti-miR26a LNA oligonucleotide.
miRNA mediated posttranscriptional control of cyclin D1 by mTORC-regulated miRNAs.
We hypothesized that the mTORC1 complex regulates Ccnd1 expression through one or more of these miRNAs. Examination of the 3′ untranslated region of rat Ccnd1 mRNA revealed single putative miRNA seed sequences (a contiguous target sequence of 7 or more nucleotides complementary to the 5′-terminal region of the miRNA starting at +1 to + 4 [27]) for miR-1, miR-214-5p, and miR-26a-3p and two such sequences each for miR-26a-5p and miR214-3p (Fig. 6A). Antisense LNA oligonucleotides targeted against individual miRs or a nontargeted negative control was administered to injured rat quadriceps muscle expressing lentivirus-encoded nontargeted or mTORC-targeted shRNA, which was then allowed to regenerate in the presence of pcRNAs. At day 1, there was complete downregulation of the targeted miRNA specifically, but the effect was transient, as the levels at 7 days were not significantly different from those in the nontargeted control (Fig. 6B). In control oligonucleotide-treated muscle, there was a significant decline (by ∼95%) in Ccnd1 mRNA between 1 and 7 days following pcRNA treatment (Fig. 6C and D). Anti-miR-26a or anti-miR-214a had no significant effect on either the level or the degradation rate of Ccnd1 mRNA in this time interval (Fig. 6C). However, anti-miR-1 upregulated Ccnd1 mRNA ∼2.5-fold over control at 1 day, and a significant level persisted after 1 week (Fig. 6C and D). Thus, of the 3 miRNAs examined, only miR-1 regulates the level of Ccnd1 mRNA. In mTORC-KD muscle, there was upregulation of Ccnd1 mRNA, and anti-miR-1 additionally increased the mRNA level by ∼25% (Fig. 6C and D). KD of mTORC resulted in an ∼98% reduction in miR-1 (Fig. 7); the marginal stimulation of Ccnd1 mRNA by anti-miR-1 is therefore a result of inhibition of the residual miR-1. These results indicate that mTORC1 regulates the stability of Ccnd1 mRNA by upregulating miR-1, which targets the 3′-UTR of the mRNA, leading to its degradation.
FIG 7.
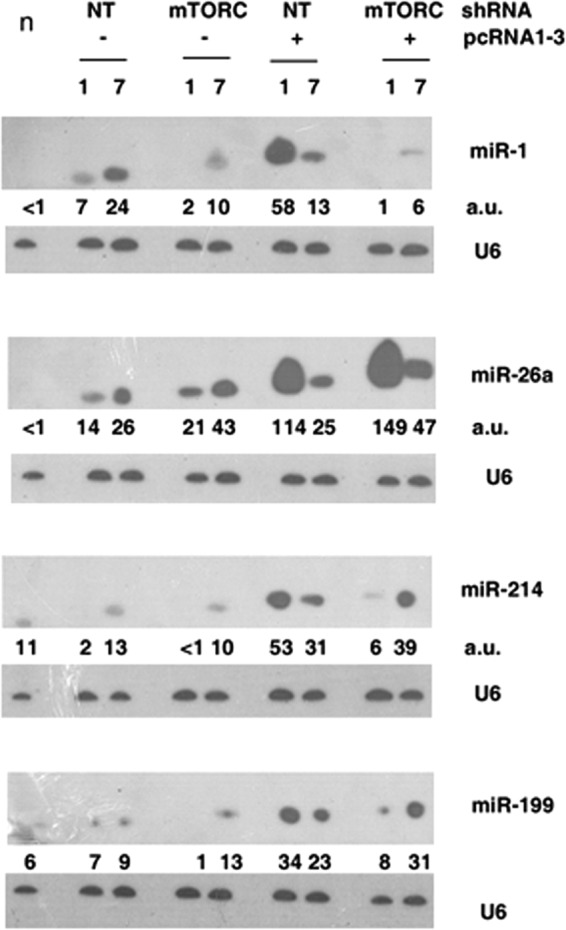
Northern blot analysis of RNA from normal (n), injured (−pcRNA) or regenerating (+pcRNA) muscle infected with nontargeted (NT) or anti-mTORC lentivirus at 1 or 7 days, probed with the indicated antisense oligonucleotide. Numbers below each panel represent the relative RNA levels in terms of average intensity units (a.u.).
In the presence of anti-miR-1 or anti-miR-214, the levels of Ccnd1 protein in regenerating muscle at 1 and 7 days were indistinguishable from those in the control muscle (Fig. 6E), indicating the lack of a specific effect of these miRs on Ccnd1 translation, even though in the case of anti-miR-1, the amount of Ccnd1 mRNA increased 2.5-fold (Fig. 6D). In the presence of anti-miR-26a, the Ccnd1 protein level was upregulated ∼33% at 1 day and ∼3-fold at 7 days (Fig. 6E and F), indicating prolonged translation of Ccnd1 mRNA. In mTORC-KD muscle, Ccnd1 translation was delayed relative to nontargeted lentivirus infected muscle (Fig. 6E), as previously observed (Fig. 5). The timing was not affected by anti-miR-1 or anti-miR-214, but in the presence of anti-miR26a, there was significant Ccnd1 protein after 1 day, increasing further up to 7 days (Fig. 6F). These results indicate that mTORC mediates the timing of Ccnd1 translation through miR-26a.
DISCUSSION
A number of alternative mechanisms involving growth factors or inhibitory proteins for maintaining satellite cell quiescence or initiating activation have been proposed. Our results highlight the previously unrecognized involvement of metabolic sensors (AMPK and mTORC1) in influencing the activity of satellite cells in vivo. We found that whereas the quiescent satellite cell is low in the active form of the AMP/ADP sensor AMPK, high cytoplasmic staining for phospho-AMPK and phospho-Raptor occurred in satellite cells postinjury (Fig. 1), reflecting an energy crisis (in terms of high AMPK and low mTORC activities) during the inflammatory phase; the latter cells presumably represent a postquiescent, repressed state. Restoration of the adenylate pool (through mitochondrial or glycolytic activity) is necessary to deactivate AMPK and activate satellite cell proliferation.
We have carried out a time course study of the effects of lentivirus-mediated knockdown of several components of the energy-sensing pathways on muscle regeneration. This differs from previous tissue-specific gene knockout studies that focused on the endpoint, i.e., myofiber number and size, consequently failing to monitor the early steps of myogenesis. Thus, the atrophy that is a common feature of skeletal muscle in which mTORC (12) or Raptor (13) has been knocked out could be a consequence of either reduced cellular growth or delayed differentiation of myogenic precursors. Studies of the effect of rapamycin on muscle regeneration (10, 33) have similarly focused on myofiber maturation. In the present study, lentivirus-mediated RNA interference just prior to the onset of synchronized MR-induced regeneration, combined with monitoring of the early steps of myogenesis, revealed stable as well as transient phenotypic effects: whereas KD of CRTC2 or CREB (as well as of components of the Notch signaling pathway [S. Jash and S. Adhya, unpublished data]) resulted in a permanent failure of satellite cell activation, KD of Raptor or mTORC produced a similar delay in activation without substantially altering the differentiation program in terms of the sequence of MRF expression (Fig. 4). This illustrates the importance of monitoring the time-dependent effects of target depletion on the differentiation program.
We found that inactivation of AMPK caused by mitochondrial activation is necessary for the proliferation of SCs at the injury site and that the active CRTC2-CREB transcription factor regulated by AMPK is critical for this process (Fig. 2). AMPK is known to upregulate mitochondrial biogenesis through the transcriptional coactivator PGC1α (29); thus, AMPK inactivation would result in lower overall mitochondrial activity. This feedback loop would ensure a proper balance between mitochondrial and AMPK activity during the passage of SCs through the cell cycle. Interestingly, in AMPKα-depleted muscle, myogenesis occurred normally but formation of the MCN was affected (Fig. 2); this could reflect a failure of the endothelial cells to attach to the AMPK-deficient myofibers.
The delayed differentiation program in mTORC- or Raptor-deficient muscle resulted in the accumulation of novel MyoD+ myoblast ring structures of about the same diameter as mature myotubes, which were also observed at earlier times in normal regenerating muscle, being replaced by myotubes at later times (Fig. 3), indicating that they might be intermediates in the biogenesis of new fibers. Recent work in mice and Drosophila has revealed adhesion, migration, and signaling components involved in myoblast fusion, but it is unclear how myoblasts assemble to form the cylindrical structures of new myotubes during regeneration of adult mammalian muscle (34, 35). The observed rings, enclosing one or more myoblasts, bear a distant resemblance to the myoblast aggregates formed during Drosophila myogenesis, consisting of founder cells surrounded by fusion-competent myoblasts (34), but whether the internal myoblasts observed here behave as founder cells remains to be seen.
The lack of a specific effect of the mTORC1 complex on the satellite cell differentiation program that we observed in vivo is in direct contrast to in vitro studies indicating a role of mTORC in the differentiation of myoblasts to myotubes (15–17, 19, 20). Moreover, in contrast to the opposite effects of mTORC and Raptor on in vitro differentiation (19, 20), we observed that KD of mTORC and Raptor have identical effects on early satellite cell differentiation in vivo, indicating that the mTORC1 kinase, rather than the individual components, mediates the timing of satellite cell activation. One explanation of these discrepancies is that the microenvironment of the myoblasts at the injury site being different from that in culture, energy-sensing mechanisms differ in their contribution to the differentiation program. Alternatively, the differentiation program in vivo may be coupled to the activation of satellite cells but is decoupled in the continuously cultured myoblasts.
The program of satellite cell activation (expression of PCNA, cyclin D1, and myoD) and subsequent myoblast differentiation was identical in the presence or absence of mTORC1 (Fig. 4 and 5), as was the timing of 4EBP1 and S6K1 phosphorylation (Fig. 4). The appearance of activation at late times was not due to loss of RNA interference, since the levels of the targeted proteins remained low up to 2 weeks (Fig. 4), This suggests the presence of an alternative kinase that is activated in the absence of mTORC1 but has a similar or identical range of targets that are rate limiting for the activation process; the default kinase would normally be inhibited by mTORC1, preventing the late expression of activation factors in normal cells. The nature of this default kinase remains unknown at present. Previous studies indicated that non-mTORC kinases could be involved in phosphorylating at least some of the 5 sites in 4EBP1. Thus, two of the sites (Thr37 and Thr46 of the human protein) are rapamycin sensitive and appear to prime the phosphorylation of the remaining sites, which are serum stimulated; moreover, in the absence of serum, Thr37/Thr46 phosphorylation becomes more rapamycin resistant, indicating the involvement of a non-mTORC1 kinase (33). For example, the insulin-activated Akt kinase acts upstream of mTORC and is subject to negative feedback through inhibition of IRS-1 by the mTORC1 substrate S6K (36). Thus, a deficiency of mTORC or raptor would result in hyperactivation of Akt, which could directly or through some other kinase phosphorylate 4EBP1. Similarly, S6K has multiple phosphorylation sites, only one of which is a target of mTORC1, while others are phosphorylated in response to growth factors and mitogens (37).
mTORC1-dependent or -independent phosphorylation of 4EBP1 coincided with the onset of the differentiation program (Fig. 4), but 4EBP1 is unlikely to act as a switch, since KD of 4EBP1, with expected translational derepression from time zero, failed to shift the program initiation to an earlier time (Fig. 4). On the other hand, in common with S6K (14), 4EBP1 had a clear role in the cytoplasmic growth of myofibers (Fig. 2). 4EBP1 regulates a distinct subset of mRNAs with pyrimidine-rich (TOP) motifs in their 5′ untranslated regions (UTR) (38). Thus, the phosphorylation of 4EBP1 and S6K1 signals the upregulation of translation of 5′ TOP and other growth control mRNAs during satellite cell activation. The role of 4EBP1 in the cell cycle is controversial. 4EBP1 has been reported to promote cell proliferation but not cell growth in embryonic fibroblasts (39), but KD of 4EBP1 had no effect on tumor cell proliferation (40). Similarly, the cell cycle regulator Ccnd1 was reported to be a target of 4EBP1 in tumor cells (40) but not in embryonic fibroblasts (38). We found that KD of 4EBP1 failed to influence the timing of translation of Ccnd1 mRNA (Fig. 5). It is possible that Ccnd1 translation is regulated differently in different cellular contexts.
miRNAs are known to inhibit translation of target mRNAs either directly, or indirectly, by inducing mRNA destabilization (41). We found that the muscle-specific miR-1 promotes the degradation of Ccnd1 mRNA but has no effect on its translation (Fig. 6). miR-1 is a muscle-specific miRNA that promotes degradation of a large number of mRNAs that encode inhibitors of myoblast differentiation (23, 25); we show here that, in parallel, miR-1 inhibits progression of the cell cycle through destabilization of Ccnd1 mRNA. In contrast, miR-26a was found to repress translation of Ccnd1 mRNA without affecting its abundance (Fig. 6). In a mouse model, miR-26a was reported to affect both the level and the translation of Smad1 and Smad4 mRNAs, which are components of the transforming growth factor β (TGF-β) signaling pathway, thereby promoting differentiation (24). At present, it is not clear how a specific miRNA directs an mRNA toward degradation or repression. It is possible that the nature of the seed sequence and its surrounding region influences an miRNA-bound mRNA toward either or both fates. Thus, the same miRNA, in this case, miR-26a, may have different effects on different target mRNAs. There are 3 putative seed sequences in the Ccnd1 3′-UTR (Fig. 6), which may have variable affinity for miR-26a. It may be noted that a high level of miR-26a is present in control regenerating muscle 1 day after MR (Fig. 7), at which time there is significant expression of Ccnd1 protein (Fig. 5 and 6), indicating that this level is insufficient to block translation; repression is only achieved after 1 day in mTORC-KD muscle (Fig. 5 and 8), when the miR-26a level is further elevated (Fig. 7). This implies the occurrence of threshold concentrations of the microRNA and cooperative effects between different miRNA-binding sites located close to each other in the 3′-UTR.
FIG 8.
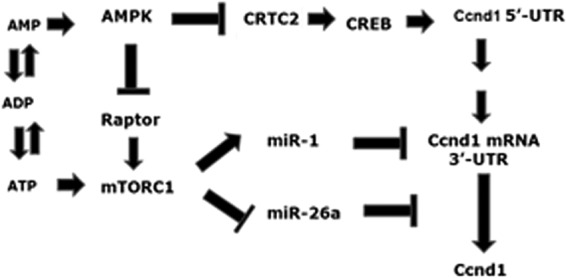
Regulation of cyclin D1 expression by energy sensors during satellite cell activation. Ccnd1 transcription is activated by the AMPK-regulated CRTC2-CREB pair through binding of these factors to the 5-UTR of the Ccnd1 gene. mTORC regulates Ccnd1 mRNA stability through miR-1 and its translation through miR-26a. See the text for details.
We observed that mTORC regulates several miRNAs in the early, proliferative phase of myogenesis (Fig. 7). A previous study of miRNA profiles in cultured mouse myoblasts revealed the presence of groups of miRNAs that are either inhibited or stimulated by rapamycin (22), indicating mTORC1-mediated positive or negative regulation, respectively. In that study, miR-1 was reported to be rapamycin sensitive, whereas miR-26a, miR-199, and miR-214 were upregulated by rapamycin (22). However, we found that miR-199 and miR-214 were both downregulated in mTORC-KD rat muscle (Fig. 7). The reasons for this discrepancy are unknown, but we note that in these two cases there was a partial restoration of the miRNA after 7 days in mTORC-KD muscle, obscuring the early downregulation at 1 day (Fig. 7); thus, the early and late effects of mTORC1 on miRNA levels may be quite different.
mTORC1-regulated miRNAs provide an explanation for the paradoxical observation that mTORC1, while destabilizing Ccnd1 mRNA, also promotes its translation (Fig. 8). Ccnd1 transcription is initiated early, between 6 and 12 h (Fig. 5); during this time, mTORC1 is activated allosterically (by ATP) (Fig. 1), leading to upregulation of miR-1 and initiating the degradation phase of Ccnd1 mRNA (Fig. 5). Active mTORC1 simultaneously downregulates miR-26a to a subthreshold level, resulting in derepression of Ccnd1 mRNA translation; further on, the mRNA is degraded to a negligible level by continued action of miR-1. As a result of the combination of these two effects, Ccnd1 expression is restricted to a narrow time window, as appropriate for the controlled passage of the myoblasts through the G1 to the S phases of the cell cycle.
ACKNOWLEDGMENTS
This work was supported by the XII Plan CSIR-IICB Suprainstitutional Project (BenD, BSC0206). S.J. and U.G. were Senior Research Fellows of CSIR and UGC, respectively; G.D. was a Research Associate of CSIR.
We thank Tapas Chowdury and Tanay Roy for technical help, Anupam Banerjee for confocal microscopy, Uday Bandyopadhyay for oxygraph measurements, and K. P. Mohanakumar for cryotome sectioning.
Footnotes
Published ahead of print 21 July 2014
REFERENCES
- 1.Le Grand F, Rudnicki MA. 2007. Skeletal muscle satellite cells and adult myogenesis. Curr. Opin. Cell Biol. 19:628–633. 10.1016/j.ceb.2007.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blais A, Tsikitis M, Acosta-Alvear D, Sharan R, Kluger Y, Dynlach BD. 2005. An initial blueprint for myogenic differentiation. Genes Dev. 19:553–569. 10.1101/gad.1281105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brack AS, Conboy IM, Conboy MJ, Shen J, Rando TA. 2008. A temporal switch from Notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell 2:50–59. 10.1016/j.stem.2007.10.006 [DOI] [PubMed] [Google Scholar]
- 4.Jash S, Adhya S. 2012. Induction of muscle regeneration by RNA-mediated mitochondrial restoration. FASEB J. 26:4187–4197. 10.1096/fj.11-203232 [DOI] [PubMed] [Google Scholar]
- 5.Vasyutina E, Lenhard DC, Birchmeier C. 2007. Notch function in myogenesis. Cell Cycle 6:1451–1454. 10.4161/cc.6.12.4372 [DOI] [PubMed] [Google Scholar]
- 6.Duguez S, Féasson L, Denis C, Freyssenet D. 2002. Mitochondrial biogenesis during skeletal muscle regeneration. Am. J. Physiol. Endocrinol. Metab. 282:E802–E809 [DOI] [PubMed] [Google Scholar]
- 7.Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G. 2001. Mammalian TOR: a homeostatic ATP sensor. Science 294:1102–1105. 10.1126/science.1063518 [DOI] [PubMed] [Google Scholar]
- 8.Oakhill JS, Steel R, Chen Z-P, Scott JW, Ling N, Tam S, Kemp BE. 2011. AMPK is a direct adenylate charge-regulated protein kinase. Science 332:1433–1435. 10.1126/science.1200094 [DOI] [PubMed] [Google Scholar]
- 9.Sanchez AMJ, Candau RB, Csibi A, Pagano AF, Raibon A, Bernardi H. 2012. The role of AMP-activated protein kinase in the coordination of skeletal muscle turnover and energy homeostasis. Am. J. Physiol. Cell Physiol. 303:C475–C485. 10.1152/ajpcell.00125.2012 [DOI] [PubMed] [Google Scholar]
- 10.Pallafacchina G, Calabria E, Serrano AL, Kalhovde JM, Schiaffino S. 2002. A protein kinase B-dependent and rapamycin sensitive pathway controls skeletal muscle growth but not fiber type specification. Proc. Natl. Acad. Sci. U. S. A. 99:9213–9218. 10.1073/pnas.142166599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ge Y, Wu AL, Warnes C, Liu J, Zhang C, Kawasome H, Terada N, Boppart MD, Schoenherr CJ, Chen J. 2009. mTOR regulates skeletal muscle regeneration in vivo through kinase-dependent and kinase-independent mechanisms. Am. J. Physiol. Cell Physiol. 297:C1434–C1444. 10.1152/ajpcell.00248.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Risson V, Mazelin L, Roceri M, Sanchez H, Moncollin V, Corneloup C, Richard-Bulteau H, Vignaud A, Baas D, Defour A, Freyssenet D, Tanti JF, Le-Marchand-Brustel Y, Ferrier B, Conjard-Duplany A, Romanino K, Bauché S, Hantaï D, Mueller M, Kozma SC, Thomas G, Rüegg MA, Ferry A, Pende M, Bigard X, Koulmann N, Schaeffer L, Gangloff YG. 2009. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J. Cell Biol. 187:859–874. 10.1083/jcb.200903131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bentzinger CF, Romanino K, Cloëtta D, Lin S, Mascarenhas JB, Oliveri F, Xia J, Casanova E, Costa CF, Brink M, Zorzato F, Hall MN, Rüegg MA. 2008. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 8:411–424. 10.1016/j.cmet.2008.10.002 [DOI] [PubMed] [Google Scholar]
- 14.Ohanna M, Sobering AK, Lapointe T, Lorenzo L, Praud C, Petroulakis E, Sonenberg N, Kelly PA, Sotiropoulos A, Pende M. 2005. Atrophy of S6K1-/- skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nat. Cell Biol. 7:286–294. 10.1038/ncb1231 [DOI] [PubMed] [Google Scholar]
- 15.Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR. 1997. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J. Biol. Chem. 272:6653–6662. 10.1074/jbc.272.10.6653 [DOI] [PubMed] [Google Scholar]
- 16.Cuenda A, Cohen P. 1999. Stress-activated protein kinase-2/p38 and a rapamycin-sensitive pathway are required for C2C12 myogenesis. J. Biol. Chem. 274:4341–4346. 10.1074/jbc.274.7.4341 [DOI] [PubMed] [Google Scholar]
- 17.Erbay E, Chen J. 2001. The mammalian target of rapamycin regulates C2C12 myogenesis via a kinase-independent mechanism. J. Biol. Chem. 276:36079–36082. 10.1074/jbc.C100406200 [DOI] [PubMed] [Google Scholar]
- 18.Erbay E, Park IH, Nuzzi PD, Schoenherr CJ, Chen J. 2003. IGF-II transcription in skeletal myogenesis is controlled by mTOR and nutrients. J. Cell Biol. 163:931–936. 10.1083/jcb.200307158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ge Y, Yoon MS, Chen J. 2011. Raptor and Rheb negatively regulate skeletal myogenesis through suppression of insulin receptor substrate 1 (IRS1). J. Biol. Chem. 286:35675–35682. 10.1074/jbc.M111.262881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaafar R, Zeiller C, Pirola L, Di Grazia A, Naro F, Vidal H, Lefai E, Némoz G. 2011. Phospholipase D regulates myogenic differentiation through the activation of both mTORC1 and mTORC2 complexes. J. Biol. Chem. 286:22609–22621. 10.1074/jbc.M110.203885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laplante M, Sabatini DM. 2012. mTOR signaling in growth control and disease. Cell 149:274–293. 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Y, Ge Y, Drnevich J, Zhao Y, Band M, Chen J. 2010. Mammalian target of rapamycin regulates miRNA-1 and follistatin in skeletal myogenesis. J. Cell Biol. 189:1157–1169. 10.1083/jcb.200912093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FI, Wang D-Z. 2006. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 38:228–233. 10.1038/ng1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dey BK, Gagan J, Yan Z, Dutta A. 2012. miR-26a is required for skeletal muscle differentiation and regeneration in mice. Genes Dev. 26:2180–2191. 10.1101/gad.198085.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goljanek-Whysall K, Pais H, Rathjen T, Sweetman D, Dalmay T, Munsterberg A. 2012. Regulation of multiple target genes by miR-1 and miR-206 is pivotal for C2C12 myoblast differentiation. J. Cell Sci. 125:3590–3600. 10.1242/jcs.101758 [DOI] [PubMed] [Google Scholar]
- 26.Mukherjee S, Mahata B, Mahato B, Adhya S. 2008. Targeted mRNA degradation by complex-mediated delivery of antisense RNAs to intracellular human mitochondria. Hum. Mol. Genet. 17:1292–1298. 10.1093/hmg/ddn017 [DOI] [PubMed] [Google Scholar]
- 27.Ritchie W, Rasko JEJ. 2014. Refining microRNA target predictions: sorting the wheat from the chaff. Biochem. Biophys. Res. Commun. 445:780–784. 10.1016/j.bbrc.2014.01.181 [DOI] [PubMed] [Google Scholar]
- 28.Kitazawa S, Kitazawa R, Maeda S. 1999. Transcriptional regulation of rat cyclin D1 gene by CpG methylation status in promoter region. J. Biol. Chem. 274:28787–28793. 10.1074/jbc.274.40.28787 [DOI] [PubMed] [Google Scholar]
- 29.Hardie DG. 2011. AMP-activated protein kinase—an energy sensor that regulates all aspects of cell function. Genes Dev. 25:1895–1908. 10.1101/gad.17420111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koo S-H, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussour F, Brindle P, Takemori H, Montminy M. 2005. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437:1109–1114. 10.1038/nature03967 [DOI] [PubMed] [Google Scholar]
- 31.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. 2008. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30:214–226. 10.1016/j.molcel.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma XM, Blenis J. 2009. Molecular mechanisms of mTOR mediated translational control. Nat. Rev. Mol. Cell Biol. 10:307–318. 10.1038/nrm2672 [DOI] [PubMed] [Google Scholar]
- 33.Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, Sonenberg N. 1999. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 13:1422–1437. 10.1101/gad.13.11.1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abmayr SM, Pavlath GK. 2012. Myoblast fusion: lessons from flies and mice. Development 139:641–656. 10.1242/dev.068353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chargé SB, Rudnicki MA. 2004. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 84:209–238. 10.1152/physrev.00019.2003 [DOI] [PubMed] [Google Scholar]
- 36.Harrington LS, Findlay GM, Lamb RF. 2005. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem. Sci. 30:35–42. 10.1016/j.tibs.2004.11.003 [DOI] [PubMed] [Google Scholar]
- 37.Magnuson B, Ekim B, Fingar DC. 2012. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 441:1–21. 10.1042/BJ20110892 [DOI] [PubMed] [Google Scholar]
- 38.Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. 2012. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485:109–113. 10.1038/nature11083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dowling RJO, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj A, Liu Y, Kozma SC, Thomas G, Sonenberg N. 2010. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 328:1172–1176. 10.1126/science.1187532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Averous J, Fonseca BD, Proud CG. 2008. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene 27:1106–1113. 10.1038/sj.onc.1210715 [DOI] [PubMed] [Google Scholar]
- 41.Huntzinger E, Izaurralde E. 2011. Gene silencing by microRNAs: contributions of translational depression and mRNA decay. Nat. Rev. Genet. 12:99–110. 10.1038/nrg2936 [DOI] [PubMed] [Google Scholar]