

Abstract
Scaffold proteins play a critical role in controlling the activity of the extracellular signal-regulated kinase 1/2 (ERK1/2) pathway. Shoc2 is a leucine-rich repeat scaffold protein that acts as a positive modulator of ERK1/2 signaling. However, the precise mechanism by which Shoc2 modulates the activity of the ERK1/2 pathway is unclear. Here we report the identification of the E3 ubiquitin ligase HUWE1 as a binding partner and regulator of Shoc2 function. HUWE1 mediates ubiquitination and, consequently, the levels of Shoc2. Additionally, we show that both Shoc2 and HUWE1 are necessary to control the levels and ubiquitination of the Shoc2 signaling partner, RAF-1. Depletion of HUWE1 abolishes RAF-1 ubiquitination, with corresponding changes in ERK1/2 pathway activity occurring. Our results indicate that the HUWE1-mediated ubiquitination of Shoc2 is the switch that regulates the transition from an active to an inactive state of the RAF-1 kinase. Taken together, our results demonstrate that HUWE1 is a novel player involved in regulating ERK1/2 signal transmission through the Shoc2 scaffold complex.
INTRODUCTION
The multitiered extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathway is a highly conserved signaling cascade that initiates a diverse range of cellular responses (1). Scaffold proteins play an essential role in the regulation of the ERK1/2 signaling network (2, 3). In addition to their main function in the assembly of protein complexes, scaffolds of the ERK1/2 pathway are thought to deliver signal specificity, regulate accessibility to substrates, target signals to a specific cellular location, and determine the biological outputs of the ERK1/2 pathway (4–6). Despite the important role of scaffolds in the biological activities of ERK1/2 signals, the mechanisms by which scaffolds exert their functions and the role for scaffolds in regulating the dynamics of ERK1/2 signaling remain poorly understood (2, 6, 7).
The scaffold protein Shoc2, initially identified in Caenorhabditis elegans as SUR-8/SOC2, is a critical positive regulator of the ERK1/2 signaling pathway that integrates the Ras and RAF-1 components of the ERK1/2 pathway into a multiprotein complex (8, 9). Aberrant targeting of Shoc2 to the plasma membrane (PM) is found in patients with Noonan-like syndrome with loose anagen hair (NS/LAH) (10). Ablation of Shoc2 in mice causes embryonic lethality due to severe heart defects, indicating that this leucine-rich repeat (LRR) protein is critical for embryonic development (11). Depletion of this protein in cells also has a striking effect on ERK1/2 signaling, particularly evident under physiological conditions of epidermal growth factor receptor (EGFR) activation (12, 13). Several recent studies have suggested that Shoc2 modulates Ras-dependent RAF-1 activation through accelerating the association and the dissociation of the Ras–RAF-1 complex, although the mechanism(s) remains unclear (14, 15). We have previously demonstrated that upon the activation of the ERK1/2 pathway Shoc2 translocates from the cytosol to late endosomes (LEs), possibly as part of the spatiotemporal regulation of signaling through the Ras-RAF module (13). Given the essential role of this scaffold in modulating the ERK1/2 signal, it is important to understand the mechanisms by which Shoc2 controls the ERK1/2 pathway activity.
Ubiquitination, along with phosphorylation, is one of the best-studied regulatory posttranslational modifications (16). The biological processes of protein degradation, cargo trafficking, gene transcription, and immune response are controlled through ubiquitination (17–19). A growing body of evidence also suggests that ubiquitination is a mechanism that contributes to the regulation of the cellular signaling cascades and catalytic activities of signaling proteins (20–22). Ubiquitination is a multistep process that ultimately results in the attachment of ubiquitin (Ub) chains to lysine residues within target proteins. This process involves enzymatic activities of a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin ligase (E3). The specificity of ubiquitination is achieved by a large number of distinct E3 ligases that are responsible for highly specific substrate recognition (16).
In the current study, we have identified the E3 ubiquitin ligase HUWE1 to be a new partner in the Shoc2–Ras–RAF-1 scaffold complex. HUWE1 (also called ARF-BP or MULE) is a large E3 ligase and a member of the homologous to E6-AP carboxyl terminus (HECT) domain-containing family of E3 ubiquitin ligases that is implicated in the regulation of cell proliferation, apoptosis, neural differentiation, and the DNA damage response (23–25). It is mainly expressed in heart, placenta, and brain tissues. Elevated levels of HUWE1 have been found in lung, breast, and colorectal carcinomas, and HUWE1-mediated ubiquitination has been linked to cancer by its ability to target substrates such as p53 and c-Myc for degradation (26–31). Missense mutations and gross duplications in HUWE1 have been reported in patients with X-linked Turner-type mental retardation (32, 33). However, relatively little is known about the mechanisms regulating HUWE1 function and, in particular, its role in regulating the ERK1/2 cascade.
We have determined that HUWE1 is the E3 ligase that modulates ubiquitination of Shoc2. We also established that ubiquitination of Shoc2-associated RAF-1 is controlled by HUWE1 and that Shoc2 depletion abrogates RAF-1 ubiquitination. A Shoc2 ubiquitination-deficient mutant was utilized to examine how impairments in Shoc2 ubiquitination affect its function as a positive regulator of the ERK1/2 pathway. Diminished Shoc2 ubiquitination leads to impaired RAF-1 ubiquitination, increased ERK1/2 phosphorylation, and higher rates of cell proliferation. Our studies provide evidence that Shoc2 serves as a scaffold platform for HUWE1. HUWE1 acts as the ERK1/2 signal tuning component in the scaffold complex, thereby controlling the intensity of ERK1/2 signaling flow through Ras and RAF-1. Our studies also present a distinct mechanism by which ubiquitination is utilized as a negative-feedback mechanism to control the ability of the noncatalytic scaffold Shoc2 to control the signaling activity of the ERK1/2 pathway.
MATERIALS AND METHODS
Reagents and antibodies.
Epidermal growth factor (EGF) was obtained from BD Bioscience. Cycloheximide and MG132 were purchased from Enzo; N-ethylmaleimide (NEM) was from Thermo Scientific/Pierce. Specific proteins were detected using primary antibodies to the following: EGFR, the A isoform of RAF (A-RAF), RAF-1 (S338), the B isoform of RAF (B-RAF), ERK1/2, phospho-ERK1/2, MEK1/2, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Cell Signaling); Shoc2 (Proteintech); hemagglutinin (HA; SydLabs Inc.); tag red fluorescent protein (tRFP; Evrogen); glutathione S-transferase (GST), M-Ras, RAF-1, and phospho-MEK1/2 (Santa Cruz); HUWE1 (Bethyl Laboratories Inc.); ubiquitin (Covance); PP1c (Millipore); and cyclin D1 (kindly provided by Mark Evers).
Yeast two-hybrid screening assays.
The full-length human Shoc2 cDNA (see Fig. 1A) was cloned into the lexA vector pB27 as an N-LexA-Shoc2-C fusion and screened against a human embryo ventricle and heart prey cDNA library. Yeast two-hybrid screens were performed by Hybrigenics SA (Paris, France).
FIG 1.

Shoc2 interacts with HUWE1. (A) Bait and prey regions of Shoc2 and HUWE1 in the yeast two-hybrid screening. (B) 293FT cells were cotransfected with the HECT domain of HUWE1 (HA-HECT), HA–M-Ras, HA-laforin, and Shoc2-tRFP. HA-HECT was immunoprecipitated and analyzed by immunoblotting using anti-HA and anti-Shoc2 antibodies. (C) 293FT cells were cotransfected with Shoc2-GST and HA-HECT. Shoc2 was immunoprecipitated using anti-GST antibodies, and the immunoprecipitate was analyzed by immunoblotting using anti-HA and anti-Shoc2 antibodies. (D) Reciprocal coimmunoprecipitation of endogenous Shoc2 and HUWE1. Shoc2 and HUWE1 antibodies were used to detect Shoc2 in HUWE1 immunoprecipitates, HUWE1 in Shoc2 immunoprecipitates, and Shoc2 and HUWE1 in total lysates of 293FT cells. (E) Recombinant Shoc2-MBP was mixed with GST-HECT coupled to glutathione-Sepharose beads. The beads were washed, eluted, and detected by Western blotting using anti-Shoc2 and anti-GST antibodies. The results in panels B to E are representative of those from three independent experiments. IP, immunoprecipitation; IB, immunoblotting.
Expression plasmids.
tRFP-tagged Shoc2 (Shoc2-tRFP) was described previously (34, 35). The pLVTHM-Shoc2 constructs expressing Shoc2 short hairpin RNA (shRNA) were described previously (13). Truncated Shoc2-tRFP-tagged mutants were generated as described previously for the full-length Shoc2 (13). The MRAS construct carrying 3 HA moieties (3× HA-MRAS) was purchased from Missouri S&T cDNA Resource Center (www.cdna.org). The plasmid carrying yellow fluorescent protein (YFP)-tagged RAF-1 (YFP–RAF-1) was kindly provided by Alexander Sorkin (University of Pittsburgh). The plasmid carrying the HECT domain of HUWE1 (plasmid 25211) was obtained from Addgene. The plasmids carrying HA-tagged ubiquitin and ubiquitin mutants were provided by M. S. Gentry (University of Kentucky). A mammalian GST expression vector was provided by Haining Zhu (University of Kentucky). A maltose-binding protein (MBP) expression vector for protein purification was provided by Craig Vander Kooi (University of Kentucky). Full-length HA-HUWE1 was provided by K. Helin (Biotech Research & Innovation Centre, Denmark). To generate a plasmid expressing HA-HECT, a forward primer containing an EcoRI site and a reverse primer containing a BamHI site after the stop codon were used to amplify the human HECT sequence by PCR. To generate a plasmid expressing Shoc2-MBP, a forward primer containing a BamHI site and a reverse primer containing a NotI site after the stop codon were used to amplify the human Shoc2 sequence by PCR. All constructs were verified by dideoxynucleotide sequencing.
Cell culture and DNA transfections.
293FT cells (Invitrogen), Cos1 cells (ATCC), HeLa cells (ATCC), and stable cell lines (derivatives of Cos1 and HeLa cells) were grown in Dulbecco modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) supplemented with sodium pyruvate, minimal essential medium with nonessential amino acids (MEM-NEAA), penicillin, streptomycin, and l-glutamate (Invitrogen). T47D cells were grown in RPMI 1640 medium containing 10% FBS supplemented with sodium pyruvate, MEM-NEAA, penicillin, streptomycin, and l-glutamate (Invitrogen).
Transfections of DNA constructs were performed using the polyethyleneimine (Neo Transduction Laboratories, Lexington, KY) or TransIT (Mirus Bio LLC) reagent. Expression of tag RFP-fused proteins was confirmed by Western blotting, as described below. Stably expressing cell lines were selected by growing the transfected cells in the presence of puromycin (0.4 μg/ml).
siRNA transfections.
To silence protein expression by RNA interference, cells were seeded in 12-well plates (at 50 to 60% confluence with 1 ml of DMEM-FBS per well) at least 20 h before transfection. Small interfering (siRNA) transfections were performed at 24- to 36-h intervals according to the manufacturer's recommendations, using Dharmafect reagent 2 (Thermo Fisher Scientific/Dharmacon). The siRNA sequence used to target the HUWE1 transcripts was as follows: 5′-GAGUUUGGAGUUUGUGAAGTT-3′ (36). The efficiency of the siRNA knockdown was validated by Western blotting.
Real-time quantitative PCR (qPCR).
Seventy-two hours after the transfection of HUWE siRNA, total RNA was isolated from HeLa cells using a PureZOL/Aurum total RNA isolation kit (Bio-Rad) according to the manufacturer's instructions. Aliquots containing equal amounts of RNA were subjected to reverse transcription-PCR (RT-PCR) analysis. Quantitative RT-PCR was performed using SoAdvanced SYBR green Supermix and a Bio-Rad CFX detection system (Bio-Rad). Sequence-specific primer sets for the Shoc2 gene were 5′-TGCAGTCCCTCCCAGCAGAGG-3′ and 5′-GCCGTAAATCAAGCATCCGCAGC-3′, and sequence-specific primer sets for the hypoxanthine phosphoribosyltransferase 1 (HPRT1) gene were 5′-GGCGGCTGCGACGAGCCCTCA-3′ and 5′-CGCGCGGGCTGACTGCTCAGG-3′. HPRT1 mRNA was used as a reference gene. The relative amounts of RNAs were calculated using the comparative threshold cycle method (37). The values for the samples were normalized against those for the reference gene, and the results are presented as the fold change in the amount of Shoc2 mRNA recovered from cells transfected with nontargeting siRNA. The data represent the means ± standard deviations (SDs) from two independent experiments.
Immunoprecipitation and Western blot analysis.
The cells grown in 35-mm dishes were placed on ice and washed with calcium- and magnesium-free phosphate-buffered saline (PBS), and the proteins were solubilized in 20 mM HEPES (Sigma), pH 7.6, containing 10 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA (Sigma), 1 mM EGTA (Sigma), 0.5 mM phenylmethylsulfonyl fluoride (Sigma), 10 μg/ml of leupeptin (Roche), 10 μg/ml of aprotinin (Roche), 5 μg/ml of pepstatin A (Sigma), and 50 mM β-glycerophosphate (Sigma) (lysis buffer) for 15 min at 4°C (38). Lysates were then centrifuged at 2,500 × g for 15 min to remove insoluble material. Lysates were incubated with appropriate antibodies for 2 h, and the immunocomplexes were precipitated using protein A- or G-Sepharose. Immunoprecipitates and aliquots of cell lysates were denatured in the sample buffer at 95°C, resolved by electrophoresis, and probed by Western blotting with various antibodies, followed by chemiluminescence detection.
Western blotting was done as described previously (39). Proteins transferred from SDS-polyacrylamide gels to nitrocellulose membranes were visualized using a ChemiDoc analysis system (Bio-Rad). Several exposures were analyzed to determine the linear range of the chemiluminescence signals. Quantification was performed using the densitometry analysis mode of Image Lab software (Bio-Rad, Inc.).
Denaturing immunoprecipitation for in vivo ubiquitination assay.
Denaturing immunoprecipitation was performed as described previously (40). Cells were lysed in denaturing buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton, 1% SDS, 1 mM Na3VO4, 10 mM NaF, 5 mM NEM, 10 μM MG132) and boiled for 10 min. Lysates were diluted 1:10 with the same buffer without SDS and incubated with the appropriate antibody overnight with rotation at 4°C. Protein G-agarose (GE Healthcare Life Sciences) was added, and the beads-agarose were washed four times in lysis buffer (without SDS). Proteins were eluted at 95°C in SDS loading buffer, separated by SDS-PAGE, and transferred to nitrocellulose.
Interaction of recombinant proteins.
GST- and MBP-tagged proteins were affinity purified and stored in PBS containing 10% glycerol. Equal aliquots of human HECT coupled to glutathione-Sepharose beads were incubated with recombinant Shoc2-MBP at 4°C for 16 h. The beads were washed 4 times with cell lysis buffer and eluted with 2× Laemmli sample buffer.
RESULTS
Shoc2 interacts with HUWE1.
Our earlier studies suggested that, in addition to tethering and enforcing physical proximity between Ras and RAF-1, the Shoc2 function extends to the coordination and integration of positive and negative ERK1/2 feedback loops (41). Thus, we set out to identify the proteins of the Shoc2 feedback loops and the mechanism that regulates Shoc2 function. We performed a yeast two-hybrid screen using full-length Shoc2 as bait. Noonan-like syndrome with loose anagen hair (NS/LAH) patients that carry the Shoc2 S2G substitution are predisposed to congenital heart defects; thus, we chose the human adult/fetal heart library for screening (10). Twenty-one of the 128 clones identified to be positive were the isolates of the previously known Shoc2-interacting partner M-Ras (41, 42). Additionally, we identified 41 isolates of the E3 ligase HUWE1. HUWE1 is a large protein with a molecular mass of 482 kDa, and all of the isolates were mapped and contained the C-terminal catalytic domain of HUWE1, the HECT domain (amino acid residues 4161 to 4374) (Fig. 1A). To confirm the Shoc2-HUWE1 interaction under more physiological conditions, we ectopically expressed tRFP-tagged Shoc2 (Shoc2-tRFP) (41) and the HA-tagged HECT domain of HUWE1 (HA-HECT) in 293FT cells. HA-tagged M-Ras was used as a positive control and HA-tagged laforin (HA-laforin), which is not a HUWE1 substrate (41), was used as a negative control in these experiments (Fig. 1B) (41). The HA-HECT domain but not HA-laforin coprecipitated with Shoc2-tRFP (Fig. 1B). To ensure that the interaction was not due to the tag, we also tested glutathione S-transferase (GST)-fused Shoc2 (Shoc2-GST) with HA-HECT. We found that Shoc2-GST coprecipitated with the HA-HECT domain (Fig. 1C).
To further evaluate the Shoc2 and HUWE1 association, we analyzed endogenous Shoc2 and HUWE1 proteins from 293FT cells by immunoprecipitations. The immunoprecipitates were then evaluated by immunoblotting with antibodies specific for endogenous Shoc2 and HUWE1. Shoc2 readily precipitated with endogenous HUWE1, and HUWE1 precipitated with endogenous Shoc2 (Fig. 1D). Moreover, we found that Shoc2 binds to the catalytically inactive version of HA-HECT (HA-HECT [C4341A]) as efficiently as its binds to wild-type (WT) HA-HECT (see Fig. S1A in the supplemental material). To determine that Shoc2 and HUWE1 are directly binding partners, we employed purified recombinant MBP-tagged full-length Shoc2 and the GST-tagged HECT domain. We found that Shoc2 and HUWE1 interact in vitro, demonstrating the direct binding of Shoc2 and HUWE1 (Fig. 1E; see also Fig. S1B and C in the supplemental material).
HUWE1 is a part of the Shoc2–Ras–RAF-1 signaling complex.
To determine whether HUWE1 is a part of the Shoc2–Ras–RAF-1 signaling complex, we expressed and precipitated HA–M-Ras from cells in which endogenous Shoc2 was reconstituted with shRNA-insensitive Shoc2-tRFP (43). HA–M-Ras readily precipitated endogenous HUWE1 and RAF-1 as well as Shoc2-tRFP (Fig. 2A). Similarly, we examined whether HUWE1 coprecipitates with RAF-1. We pulled down GST-tagged RAF-1 from cells expressing the full-length HA-tagged HUWE1 and detected a substantial amount of HA-HUWE1 and endogenous Shoc2 precipitate with GST–RAF-1 (Fig. 2B). Cumulatively, these results indicate that HUWE1 is a novel interacting partner in the Shoc2–Ras–RAF-1 scaffold complex. Furthermore, HUWE1 catalytic activity is not required for its interaction with Shoc2.
FIG 2.
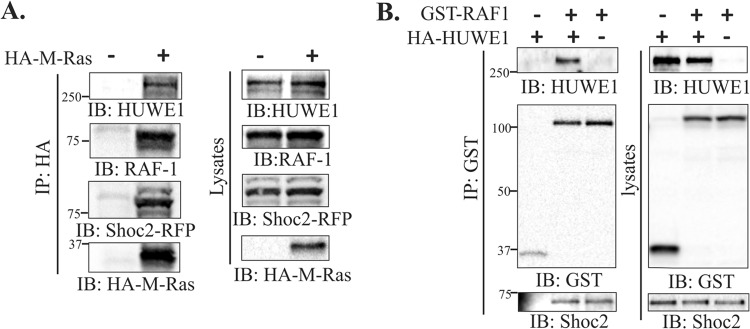
Shoc2 and HUWE1 form a complex with M-Ras and RAF-1. (A) Cos1-SR cells were transfected with HA–M-Ras. HA–M-Ras was immunoprecipitated, and precipitates were then analyzed by immunoblotting using anti-HUWE1, anti-RAF-1, and anti-tRFP antibodies. (B) 293FT cells were cotransfected with GST–RAF-1 and HA-HUWE1. GST–RAF-1 was immunoprecipitated, and precipitates were then analyzed by immunoblotting. HUWE1 and Shoc2 were detected in GST–RAF-1 immunoprecipitates and lysates using anti-HUWE1 and anti-Shoc2 antibodies.
HUWE1 binds to the LRR12 to LRR14 region of Shoc2.
To better characterize the interaction between HUWE1 and Shoc2, we utilized truncated forms of Shoc2 and mapped the domain of Shoc2 that is required for binding to the HA-tagged HECT domain of HUWE1. Shoc2 is comprised of two functional domains: an N-terminal domain that mediates Ras and RAF-1 binding and a domain containing a core of 21 leucine-rich repeats (LRRs) (Fig. 3A) (41). We found that deletion of C-terminal LRRs 12 to 21 of Shoc2 (Δ12-C) abolished the Shoc2-HECT interaction (Fig. 3B, lane 3), whereas deletion of the N-terminal region of Shoc2 together with LRRs 1 to 11 (ΔN-11) had no effect on Shoc2-HECT binding (Fig. 3B, lane 4). Deletion of only the N-terminal region of Shoc2 (LRRs) had no effect on binding to the HECT domain of HUWE1 (Fig. 3B, lane 5). To further define the interaction, additional truncation mutants with mutations in the C terminus of Shoc2 were used to coimmunoprecipitate HECT (Fig. 3C and D). A fragment of 71 residues that corresponds to LRRs 12 to 14 was sufficient for HECT domain binding (Fig. 3E, lane 2). Altogether, our data show that HUWE1 binds repeats 12 to 14 of the Shoc2 LRR domain. Repeats 12 to 14 are dispensable for the Shoc2–M-Ras interaction (41). Therefore, the Shoc2 and HUWE1 association is independent of Shoc2–M-Ras or RAF-1 binding.
FIG 3.

Mapping the interacting domains of HUWE1 and Shoc2. (A) Schematic representation of the Shoc2 truncated mutants used in the assays whose results are presented in panel B. (B) 293FT cells were cotransfected with HA-HECT and the Shoc2-tRFP truncated mutants depicted in panel A. HA was immunoprecipitated and analyzed by immunoblotting using anti-HA and anti-tRFP antibodies. (C) Schematic representation of Shoc2 truncated mutants used in the assays whose results are presented in panels D and E. (D and E) 293FT cells were cotransfected with HA-HECT and the Shoc2 truncated mutants depicted in panel C. HA was immunoprecipitated and analyzed by immunoblotting using anti-HA, anti-tRFP, and anti-GST antibodies. The results in panels B, D, and E are representative of those from three independent experiments.
HUWE1 mediates ubiquitination of Shoc2 and RAF-1.
HUWE1 is an E3 ubiquitin ligase that ubiquitinates multiple signaling proteins (44–46). Therefore, we examined whether HUWE1 catalyzes the ubiquitination of Shoc2 or its signaling partner, RAF-1. While modification of RAF-1 by ubiquitin has been reported (47–49), ubiquitination of Shoc2 has not been previously demonstrated. To determine whether Shoc2 undergoes ubiquitination, we precipitated GST-Shoc2 from cells that were lysed under denaturing conditions and examined Shoc2 ubiquitination using anti-Ub antibodies. The analysis revealed that Ub exhibiting a molecular mass greater than >250 kDa is associated with Shoc2, suggesting that multiple ubiquitin moieties are incorporated into Shoc2 (see Fig. S2A in the supplemental material). Similar results were obtained when ubiquitination of GST-Shoc2 was examined in cells coexpressing HA-tagged ubiquitin (HA-Ub). Shoc2-HA-Ub species also accumulated at the high molecular mass (see Fig. S2B in the supplemental material). Interestingly, treatment of cells with the proteasome inhibitor MG132 increased the level of Shoc2 ubiquitination only moderately, and we did not observed a substantial increase in Shoc2. Our findings of a minimal change in Shoc2 ubiquitination in the presence of the proteasome inhibitor imply that most of the ubiquitinated Shoc2 is not an immediate target for the proteasome. To confirm that ubiquitin was conjugated to Shoc2 and not to the Shoc2 binding partner RAF-1, blots were reprobed with antibodies against RAF-1. We did not detect RAF-1 in the Shoc2 immunoprecipitates under the conditions of denaturing lysis (not shown).
To determine whether Shoc2 serves as a substrate for HEWE1 in vitro, we performed in vitro ubiquitination assays using recombinant, purified proteins. We generated 35S-labeled in vitro-transcribed Shoc2 (see Fig. S2C in the supplemental material) along with the HECT domain of HUWE1 purified from Escherichia coli. The addition of recombinant Ub E1 (E1), GST-UbcH5 (E2), ATP, and His6-Ub resulted in no detectable ubiquitination via Western analysis. Similarly, the addition of either Shoc2 or HUWE1 to this mixture yielded little to no ubiquitination. However, the addition of the HUWE1 HECT domain and Shoc2 resulted in dramatic Shoc2 ubiquitination (see Fig. S2D in the supplemental material).
To further probe the role of HUWE1 in the ubiquitination of either Shoc2 or RAF-1, we silenced the expression of HUWE1 using a specific HUWE1 siRNA (36). Knockdown of HUWE1 dramatically reduced the ubiquitination of endogenous Shoc2 (Fig. 4A), GST-Shoc2 (Fig. 4B), and YFP–RAF-1 (Fig. 4C). We also observed that HUWE1 knockdown led to an increase in RAF-1 proteins levels, indicating that HUWE1 may be involved in the regulation of RAF-1 stability (Fig. 4C, lysate). Interestingly, treatment of cells with the proteasome inhibitor MG132 significantly increased the ubiquitination of RAF-1, suggesting that ubiquitinated RAF-1 is a target for the proteasome (Fig. 4C, lanes 1 and 3). This result is addressed in detail below. Expression of full-length HA-HUWE1 or the HA-HECT domain of HUWE1 induced the ubiquitination of Shoc2 (Fig. 4D and E) and RAF-1 (Fig. 4F and G). Conversely, overexpression of the catalytically defective mutant with a mutation in the HA-HECT domain (C4341A) (50) did not increase the ubiquitination of Shoc2 (Fig. 4D, lane 3) or RAF-1 (Fig. 4F, lane 3). Cumulatively, these results indicate that HUWE1 is the E3 ligase that mediates the ubiquitination of both Shoc2 and RAF-1.
FIG 4.

HUWE1 regulates ubiquitination of Shoc2 and RAF-1. (A) Shoc2 was precipitated from HeLa cells using Shoc2 antibodies. Shoc2 ubiquitination was detected with anti-UB antibodies. Immunoprecipitates and lysates were analyzed by immunoblotting using anti-HUWE1 and anti-Shoc2 antibodies. (B) GST-Shoc2 was precipitated from HeLa cells transfected with HA-Ub using glutathione-Sepharose. Shoc2 ubiquitination was detected with anti-HA antibodies. Immunoprecipitates and lysates were analyzed by immunoblotting with anti-HUWE1 and anti-GST antibodies. (C) HeLa cells were transfected with YFP–RAF-1 and HA-Ub. Cells were treated with the proteasome inhibitor MG132 (10 μM). YFP–RAF-1 was precipitated using anti-green fluorescent protein (anti-GFP) antibodies that cross-react with YFP. RAF-1 ubiquitination was detected with anti-HA antibodies. Immunoprecipitates and lysates were analyzed by immunoblotting with anti-HUWE1 and anti-RAF-1 antibodies. (D) 293FT Shoc2-GST-expressing cells were cotransfected with WT HA-HECT or HA-HECT with the C4341A (C/A) substitution. GST-Shoc2 was precipitated using glutathione-Sepharose, and Shoc2 ubiquitination was detected with anti-Ub antibodies. (E) 293FT Shoc2-GST-expressing cells were transfected with the full-length HA-HUWE1. GST-Shoc2 was precipitated using glutathione-Sepharose, and Shoc2 ubiquitination was detected with anti-Ub antibodies. (F) 293FT cells were transfected with WT HA-HECT, HA-HECT with the C4341A substitution, or empty vector. RAF-1 was precipitated using anti-RAF-1 antibody, and RAF-1 ubiquitination was detected with anti-Ub antibodies. (G) 293FT cells were transfected with the full-length HA-HUWE1. RAF-1 was precipitated using anti-RAF-1 antibody, and RAF-1 ubiquitination was detected with anti-Ub antibodies. The results in each panel are representative of those from three independent experiments. Lanes MW, molecular weight markers (numbers to the left of the gels are in thousands).
Shoc2 is essential for HUWE1-mediated ubiquitination of RAF-1.
Given that Shoc2 is a scaffold that tethers signaling proteins in close proximity, we speculated that Shoc2 tethers HUWE1 to ubiquitinate RAF-1. Thus, we investigated whether the loss of Shoc2 in cells affects HUWE1-mediated ubiquitination of RAF-1. In these experiments, we utilized stable cells constitutively depleted of Shoc2 (LV1) or depleted and then reconstituted with the shRNA-insensitive Shoc2-tRFP (SR) (13, 35, 41). Cells expressing nontargeting (NT) siRNA were utilized as an additional control (41). As expected, HUWE1 knockdown induced a clear decrease in RAF-1 ubiquitination in HeLa cells expressing Shoc2 (NT) (Fig. 5, lanes 1 and 2). Conversely, we found that in cells lacking Shoc2 (LV1), depletion of HUWE1 had no effect on already reduced basal levels of RAF-1 ubiquitination (Fig. 5, lanes 3 and 4). Reconstitution of endogenous Shoc2 with Shoc2-tRFP (SR) rescued the effect of HUWE1 depletion on the ubiquitination of RAF-1 (Fig. 5, lanes 5 and 6). These results demonstrate that the presence of Shoc2 is necessary for HUWE1-mediated RAF-1 ubiquitination. We again observed that in cells expressing Shoc2, HUWE1 knockdown leads to an increased abundance of RAF-1, suggesting that HUWE1 depletion may affect RAF-1 stability. Reminiscent of our previous findings (13), depletion of Shoc2 also led to an increase in the level of RAF-1. To demonstrate that the involvement of HUWE1 in the ubiquitination of RAF-1 in the presence of Shoc2 is a part of a general mechanism that controls the function of the Shoc2 signaling module, we recapitulated our findings in different cells (see Fig. S3 in the supplemental material).
FIG 5.
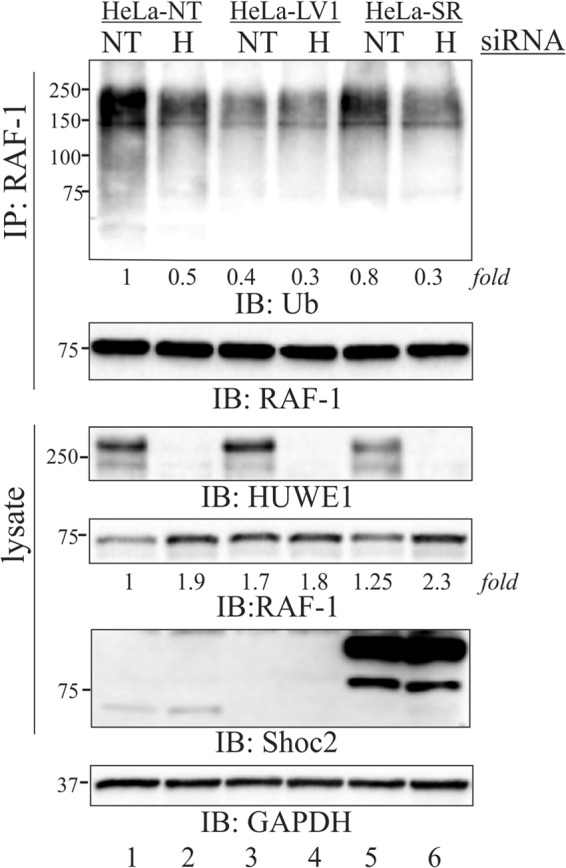
HUWE1 regulates the ubiquitination of RAF-1 in a Shoc2-dependent manner. HeLa-NT, HeLa-LV1, and HeLa-SR cells were transiently transfected with HUWE1 siRNA. At 48 h posttransfection, the cells were treated with MG132. RAF-1 was precipitated using anti-RAF-1 antibodies. RAF-1 ubiquitination was detected with anti-Ub antibodies. The expression of HUWE1, RAF-1, and Shoc2 was analyzed using specific antibodies. The results in each panel are representative of those from three independent experiments. Numbers to the left of the gels are molecular weights (in thousands). NT, nontargeting siRNA; H, HUWE1 siRNA.
HUWE1 regulates Shoc2-dependent ERK1/2 phosphorylation.
We next sought to determine whether HUWE1-mediated Shoc2 and RAF-1 ubiquitination affects ERK1/2 activity. To determine this, we examined ERK1/2 phosphorylation in response to EGF stimulation in cells depleted of HUWE1. We found that silencing of HUWE1 induced a clear increase in ERK1/2 phosphorylation in cells expressing Shoc2, with the maximum increase being observed 7 min after the stimulation of cells with EGF (Fig. 6A, lanes 4 to 6; see also Fig. S4 in the supplemental material). Silencing of HUWE1 in the Shoc2-depleted cells did not lead to a significant change in already dramatically reduced ERK1/2 phosphorylation (Fig. 6A, lanes 10 to 12). These results indicate that HUWE1 mediates ERK1/2 phosphorylation in the presence of Shoc2. To confirm that HUWE1 regulates ERK1/2 phosphorylation in the context of the Shoc2 scaffold complex, we utilized stable cells differentially expressing Shoc2 (Fig. 6B). As expected, HUWE1 knockdown led to a significant increase in RAF-1 and ERK1/2 phosphorylation after stimulation of cells with EGF for 7 min (Fig. 6B, lanes 3 and 4). Conversely, in cells depleted of Shoc2 (LV1), HUWE1 silencing had essentially no effect on the levels of phospho-ERK1/2 (Fig. 6B, lanes 7 and 8). Constitutive expression of Shoc2-tRFP rescued the effect of HUWE1 depletion on the amplitude of ERK1/2 phosphorylation (Fig. 6B, lanes 11 and 12). These data support the conclusion that HUWE1 is a part of the mechanism that modulates ERK1/2 activity transmitted through the Shoc2 scaffold complex.
FIG 6.

HUWE1-mediated ubiquitination is required to modulate the ERK1/2 pathway. (A) HeLa cells were depleted of HUWE1, Shoc2, or both by siRNA. At 48 h posttransfection, the cells were serum starved for 16 h and stimulated with EGF (10 ng/ml) for 7 or 15 min. The expression of the indicated proteins was analyzed using specific antibodies. Multiple blots from the experiments whose results are exemplified in panel A were analyzed. Bars represent the mean values ± SDs (n = 3) for phosphorylated ERK1/2 (pERK1/2; 7 min of EGF treatment) normalized to the value for GAPDH in arbitrary units (phosphorylated ERK1/2/GAPDH). (B) Cos1 cells stably depleted of Shoc2 (Cos-LV1) were transiently transfected with HUWE1 siRNA. In Cos-SR cells, Shoc2 was rescued with Shoc2-tRFP. At 48 h posttransfection, the cells were serum starved for 16 h and stimulated with EGF (0.2 ng/ml) for 7 min. The expression of the indicated proteins was analyzed using specific antibodies. Multiple blots from the experiments for which the results are exemplified in panel B were analyzed. Bars represent the mean values ± SDs (n = 3) for phosphorylated ERK1/2 (7 min of EGF treatment) normalized to the value for GAPDH in arbitrary units (phosphorylated ERK1/2/GAPDH). The results in each panel are representative of those from three independent experiments. *, a proteolytic fragment of Shoc2-tRFP that is often detected by immunoblotting in cells expressing full-length Shoc2-tRFP. NT, nontargeting siRNA; tERK1/2, total ERK1/2; H, HUWE1 siRNA.
HUWE1 regulates the stability of RAF-1.
As mentioned earlier, HUWE1 depletion led to an increase in Shoc2 and RAF-1 protein levels (Fig. 4 and 6). Therefore, we investigated whether HUWE1 targets Shoc2 and RAF-1 for degradation. First, we tested whether silencing of HUWE1 affected the levels of the B and A isoforms of RAF (B-RAF and A-RAF, respectively), as well as those of EGFR, MEK1/2, and the earlier reported Shoc2-interacting partner PP1c in different cell types (Fig. 7A) (42). We found that only Shoc2 and RAF-1 levels were dramatically increased when HUWE1 was depleted.
FIG 7.

HUWE1 regulates the stability of RAF-1. (A) 293FT, Cos1, or HeLa cells were transiently transfected with HUWE1 siRNA. At 48 h after transfection, cells were harvested for immunoblotting. The expression of Shoc2, HUWE1, B-RAF, A-RAF, RAF-1, EGFR, MEK1/2, PP1c, and GAPDH was analyzed. (B) T47D cells stably depleted of Shoc2 (T47D-LV1) were transiently transfected with HUWE1 siRNA. In T47D-SR cells, Shoc2 was rescued with Shoc2-tRFP. At 48 h after transfection, cells were harvested for immunoblotting. The expression of Shoc2, HUWE1, RAF-1, and GAPDH was analyzed. (C) 293FT cells were transfected with HUWE1 siRNA and subsequently treated with cycloheximide (CHX). Cell lysates were analyzed for HUWE1, Shoc2, RAF-1, cyclin D1, and GAPDH expression using specific antibodies. (D) Graph showing the relative amount of Shoc2 that remained in cells following cycloheximide treatment. The proteins in the immunoblots shown in panel C were quantified by normalizing the amounts of total Shoc2 to the amount of GAPDH. (E) Graph showing the relative amount of RAF-1 that remained in cells following cycloheximide treatment. The proteins in the immunoblots shown in panel C were quantified by normalizing the amounts of total RAF-1 to the amount of GAPDH. (F) Total RNA was extracted from HeLa cells transfected with either NT siRNA (siNT) or an siRNA duplex targeting HUWE1 (siHUWE1), and quantitative RT-PCR was performed using Shoc2-specific primers. The data are presented as the fold change of the Shoc2 mRNA levels in cells transfected with NT siRNA versus the Shoc2 mRNA levels in cells transfected with HUWE1 siRNA (mean ± SD, n = 2). HPRT1 is a control mRNA.
We then determined that the dramatic increase in the protein levels of RAF-1 upon depletion of HUWE1 was observed only in cells expressing Shoc2 (NT and SR) (Fig. 7B, lanes 1, 2, 5, and 6). Depletion of HUWE1 in cells in which Shoc2 was silenced (LV1) had no significant effect on the already elevated levels of RAF-1 due to the loss of Shoc2 (Fig. 7B, lanes 3 and 4). This result indicates that HUWE1 controls RAF-1 stability only when HUWE1 is associated with Shoc2.
We next inhibited protein translation by incubating the cells with cycloheximide and examined the stability of endogenous Shoc2 and RAF-1 by immunoblotting. Although the Shoc2 levels were increased after HUWE1 was knocked down, we could not detect any changes in the stability of Shoc2 in HUWE1-depleted cells (Fig. 7C and D). In contrast, HUWE1 silencing led to an increased abundance and an increased half-life of RAF-1 (Fig. 7C and E), indicating that HUWE1 modulates the stability of RAF-1. To test whether HUWE1 regulates the level of Shoc2 indirectly by controlling its transcription, Shoc2 mRNA levels were tested using real-time PCR. We found that Shoc2 mRNA expression was not affected by HUWE1 depletion (Fig. 7F).
We then hypothesized that HUWE1-catalyzed Shoc2 ubiquitination mediates a function other than controlling the stability of Shoc2. To gain further insights into the role of Shoc2 ubiquitination, we determined the type of Shoc2-conjugated ubiquitin linkages. Specific types of ubiquitination can regulate protein function, trafficking, or stability (21, 51). While K11- and K48-linked polyubiquitination targets substrates to the proteasome, K63-ubiquitin linkages are utilized to modify protein function in a proteasome-independent manner (52–54). To determine the type of ubiquitination, we took advantage of a series of Ub mutants with lysines (K) mutated to arginines (R). We transfected Shoc2-GST and HA-tagged WT Ub or Ub with a mutation at K11 only, K48 only, or K63 only into cells. Robust ubiquitination of Shoc2 by WT, K48-mutated-only, and K63-mutated-only HA-ubiquitin was observed (Fig. 8A). In contrast, Shoc2-GST was only weakly ubiquitinated by the K11-mutated-only ubiquitin. Thus, we concluded that K48- and K63-ubiquitin linkages are the predominant linkages that covalently attached to Shoc2. These data suggest that controlling Shoc2 stability is not the only role for Shoc2 ubiquitination.
FIG 8.
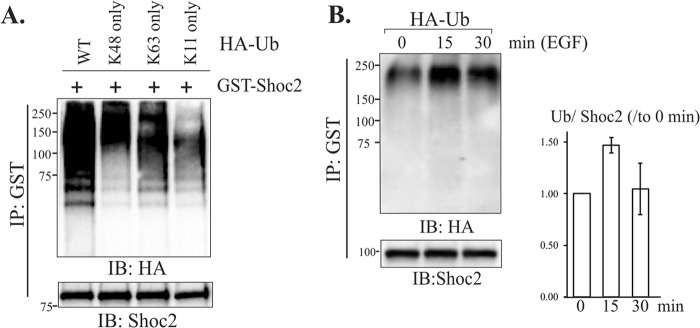
Ubiquitination of Shoc2 in cells stimulated with EGF. (A) 293FT cells were transiently cotransfected with GST-Shoc2 and HA-tagged WT Ub or mutants with ubiquitin mutations at K11 only, K48 only, or K63 only. Shoc2 was precipitated using anti-GST antibodies, followed by immunoblotting with the indicated antibodies. (B) HeLa cells were transfected with GST-Shoc2 and HA-Ub. At 48 h posttransfection, the cells were serum starved for 16 h and stimulated with EGF (10 ng/ml) for 15 and 30 min. GST-Shoc2 was precipitated using glutathione-Sepharose. Shoc2 ubiquitination was detected with HA antibodies. The expression of Shoc2 was analyzed using Shoc2-specific antibodies. The mean amount of Ub normalized to the total amount of Shoc2 ubiquitination at 0 min ± SD from three experiments is presented on the graph. The results in each panel are representative of those from three independent experiments. Numbers to the left of the gels are molecular weights (in thousands).
We also tested whether ubiquitination of Shoc2 is stimulated by activation of the EGFR-ERK1/2 pathway. We found that Shoc2 is basally ubiquitinated, but its ubiquitination is increased when cells are stimulated with EGF for 15 min (Fig. 8B, second lane). The increase in ubiquitination was then followed by a reduction in Shoc2 ubiquitination, which was observed within 30 min of cell stimulation with EGF (Fig. 8B, third lane). Interestingly, the maximum in the ubiquitination of Shoc2 corresponds to a decline in phosphorylation of ERK1/2 (Fig. 6A). Thus, EGF-induced activation of the ERK1/2 pathway appears to stimulate the capacity of HUWE1 to catalyze the reversible ubiquitination of Shoc2. Our findings that HUWE1 knockdown has very little effect on Shoc2 stability, as well as our findings that the polyubiquitin chains that formed on Shoc2 include K63-linked chains, a modification that generally does not target a substrate protein for proteasomal degradation, agree with our hypothesis that HUWE1-mediated Shoc2 ubiquitination has a role in addition to controlling Shoc2 stability.
Ubiquitin-conjugated sites of Shoc2.
As an approach to assess the functional consequences of HUWE1-mediated ubiquitination of Shoc2, we mapped the lysine residues in Shoc2 that are modified by ubiquitin conjugation. Shoc2-FLAG was precipitated from cells using anti-FLAG antibodies and stringently washed to minimize coprecipitation with other proteins. Immunoprecipitated Shoc2-FLAG was separated by SDS-PAGE and visualized by SYPRO Ruby protein gel stain (not shown). In order to analyze ubiquitinated Shoc2 by mass spectrometry (MS), the gel regions containing ubiquitin-modified Shoc2 protein were excised, digested with trypsin, and analyzed by liquid chromatography-tandem MS (LC-MS/MS) (55, 56). Shoc2 was the predominant protein identified in these samples. Moreover, no other ubiquitinated proteins were identified in these samples (not shown). A database search using parameters allowing an additional mass of 114.1 Da, corresponding to the ubiquitin remnant on lysine residues, identified eight distinct ubiquitination sites on Shoc2 at lysines 169, 170, 216, 228, 369, 439, 475, and 542 (Fig. 9A). Representative spectra that demonstrate ubiquitination at Lys 369 are shown in Fig. S5A in the supplemental material. In each case, manual validation independently confirmed the ubiquitination site assignment.
FIG 9.

Identification of multiple ubiquitination sites in the LRR domain of Shoc2 by LC-MS/MS. (A) FLAG-Shoc2 was precipitated from transiently transfected 293FT cells. Eight ubiquitination sites were identified within the LRR domain. Each peptide was characterized by a GG-modified lysine, which caused them to bear an additional mass of 114.1 Da. Peptides matching the MS/MS spectra were accepted only if their masses varied by less than 10 ppm from the expected monoisotopic mass of the parent ion. (B) Ub-modified lysines were mapped on a previously reported modeled structure of the Shoc2 LRR domain. (C) Lysines depicted in panel A were mutated to arginines (5KR, 6KR, 7KR, and 8KR). The WT and mutants of Shoc2 were transiently coexpressed with HA-Ub in 293FT cells. Shoc2 immunoprecipitates were probed with HA and Shoc2 antibodies. The mean amount of Ub (%) normalized to the total amount of WT Shoc2 ± SD from three experiments is presented on the graph. (D) 293FT cells were cotransfected with Shoc2 WT-YFP, Shoc2 7KR mutant-YFP, and HA-HECT. The HECT domain was detected in WT Shoc2-YFP and Shoc2 7KR mutant-YFP immunoprecipitates and lysates from transfected 293FT cells. Immunoprecipitates were analyzed by immunoblotting using HA and Shoc2 antibodies. Numbers to the left of the gels are molecular weights (in thousands).
Sequence analysis revealed that all identified ubiquitination sites map to the LRR region of Shoc2. Using a previously reported model of Shoc2, we found that the eight lysines are clustered into two groups at the N and C termini of the Shoc2 solenoid (Fig. 9B). To examine the effects of Shoc2 ubiquitination on its function, each of the eight lysines (K) of Shoc2 was replaced with arginines (R), to minimally perturb the structure but prevent ubiquitination of the mutant proteins. Single and multiple (nKR) point mutations were generated and expressed in 293FT cells. Western blot analysis revealed that ubiquitination of the 7KR mutant was reduced by ∼70% to 80% compared to that of WT Shoc2 (Fig. 9C). Despite multiple Lys → Arg mutations, the extent of Shoc2 binding to the HECT domain of HUWE1, as well as to M-Ras and RAF-1, was essentially similar to that of WT Shoc2 (Fig. 9D; see also Fig. S5B in the supplemental material). The cellular distribution as well as the half-life of the Shoc2(7KR)-YFP mutant was largely similar to that of WT Shoc2-YFP (see Fig. S5C and D in the supplemental material). These data suggested that multiple lysine-to-arginine substitutions had a direct effect on ubiquitination but did not result in an unfolded protein, nor did it affect the Shoc2 capacity to assemble the scaffold complex or the cellular distribution.
HUWE1 regulates EGFR/RAF-1-induced cell proliferation.
To determine how HUWE1-mediated Shoc2 ubiquitination regulates the activity of the ERK1/2 pathway, we generated HeLa and Cos1 cells in which endogenous Shoc2 was depleted and reconstituted with either shRNA-resistant WT Shoc2-YFP (HeLa-SY-WT) or the Shoc2 7KR mutant-YFP (HeLa-SY-7KR) (13). In order to prevent clonal variations due to the different sites of viral genome incorporation in the following experiments, we utilized pooled populations of cells. We examined the ubiquitination of RAF-1 in cells expressing either WT Shoc2 or the Shoc2 7KR mutant. Immunoprecipitation experiments showed that ubiquitination of RAF-1 was significantly lower in cells expressing the Shoc2 7KR mutant than in cells expressing WT Shoc2 (Fig. 10A). We then tested whether decreased RAF-1 ubiquitination affects RAF-1 phosphorylation and, subsequently, that of the kinases MEK1/2 and ERK1/2. We found that the amplitude of RAF-1 phosphorylation at Ser338 as well as the phosphorylation of MEK1/2 and ERK1/2 was higher in cells expressing the Shoc2 7KR mutant than in cells expressing WT Shoc2 when stimulated with EGF (Fig. 10B). We also observed that depletion of HUWE1 in cells expressing the Shoc2 7KR mutant did not affect the already increased ERK1/2 activity (see Fig. S6A in the supplemental material). Similarly, when Cos1-SY-WT or Cos1-SY-7KR cells were used, we observed increased ERK1/2 and RAF-1 phosphorylation (see Fig. S6B in the supplemental material). These findings indicate that reduced Shoc2 ubiquitination leads to abridged RAF-1 ubiquitination, followed by an increased amplitude of ERK1/2 phosphorylation.
FIG 10.

Diminished Shoc2 ubiquitination alters the ubiquitination of RAF-1 and ERK1/2 signaling. (A) RAF-1 was immunoprecipitated from HeLa cells stably expressing either WT Shoc2-YFP or Shoc2 7KR mutant-YFP. The ubiquitination of RAF-1 was detected with anti-Ub antibodies. Numbers to the left of the gels are molecular weights (in thousands). (B) HeLa-SY cells stably expressing either WT Shoc2-YFP or Shoc2 7KR mutant-YFP were serum starved and treated with 10 ng/ml of EGF for the indicated times. The cell lysates were probed for phosphorylated RAF-1 (S338), phosphorylated MEK1/2, phosphorylated ERK1/2, RAF-1, and GAPDH. (C) Equal numbers of HeLa-SY cells constitutively expressing either WT Shoc2-YFP or the Shoc2 7KR mutant-YFP were plated onto 24 wells, and the numbers were counted 24, 48, and 72 h after seeding. The graph depicts the mean number from triplicate experiments ± SD. (D) The viability of HeLa-SY cells constitutively expressing either WT Shoc2-YFP or Shoc2 7KR mutant-YFP was measured using a CellTiter 96 AQueous One solution cell proliferation assay. The graph depicts the mean number from triplicate experiments ± SD. The results in each panel are representative of those from three independent experiments. OD, optical density.
It has been reported that overexpression of wild-type RAF-1 does not confer a proliferative advantage to cells (57). Similarly, we also observed that overexpression of RAF-1 does not lead to an increased ERK1/2 phosphorylation (see Fig. S6C in the supplemental material), yet hyperactivation of RAF-1 up to a certain threshold was reported to induce the cell cycle and cell proliferation (58–60). Thus, we examined whether increased ERK1/2 phosphorylation in cells expressing the ubiquitin-deficient Shoc2 7KR mutant induces cell proliferation. We compared the growth rates of cells expressing the Shoc2 WT and Shoc2 7KR mutant-YFP and found that the rates of growth of Shoc2 7KR-YFP-expressing cells were markedly increased compared with those of control cells (cells with the Shoc2 WT) (Fig. 10C and D; see also Fig. S6D and E in the supplemental material). As the obtained results were not due to the changes in MEK1/2 and ERK1/2 levels, it suggested that increased proliferation is due to increased RAF-1 activation and the reduced ubiquitination of Shoc2.
DISCUSSION
The scaffold protein Shoc2 was shown to accelerate ERK1/2 activity by bringing several signaling proteins in close proximity: H-, N-, K- and M-Ras, RAF-1, PP1c, and the recently identified member of the LAP protein family SCRIB (8, 12, 42, 61). We previously showed that, in addition to its function as an ERK1/2 pathway signaling accelerator, Shoc2 integrates a negative ERK1/2 feedback loop that inhibits ERK1/2 signals. However, the mechanisms of these events have not been determined (41). In this study, we identified the E3 ubiquitin ligase HUWE1 to be an integral component of the Shoc2–Ras–RAF-1 scaffold complex. Our results demonstrate that binding of HUWE1 to Shoc2 provides a molecular mechanism for inhibition of the ERK1/2 signaling activity.
HUWE1 is a part of the Shoc2–Ras–RAF-1 signaling complex.
Several lines of evidence indicate that HUWE1 is a binding partner in the Shoc2 scaffold complex: (i) a pool of endogenous Shoc2 is found in the same molecular complex with HUWE1, (ii) HUWE1 is found in the same molecular complex with M-Ras and RAF-1, (iii) the HECT domain of HUWE1 interacts with Shoc2 in vitro, in the absence of other components of the complex, and (iv) repeats 12 to 14 of Shoc2's LRR domain are sufficient for HUWE1 binding to Shoc2 (Fig. 1 to 3). Although it is unusual for HECT domain E3 ligases to interact with their partners through the catalytic domain, it is not unprecedented, particularly for HUWE1. An essential component of the DNA polymerase CDC6 is modulated by HUWE1 and is also recognized through the HECT domain of HUWE1 (36).
HUWE1 is the E3 ligase for both Shoc2 and RAF-1.
Our results show that the Shoc2 interaction with HUWE1 results in ubiquitination of Shoc2. We found that HUWE1-polyubiquitinated lysine residues are within Shoc2's LRR domain. Consistent with our findings that HUWE1 promoted K63 polyubiquitination of Shoc2, HUWE1 inhibited the ability of Shoc2 to accelerate ERK1/2 phosphorylation. Though HUWE1 has been shown to synthesize K48 chains on several of its substrates, including p53, ARF, Mcl-1, and N-Myc (46, 62, 63), it also preferentially assembles K63 chains on c-Myc (52) and on its recently identified substrate Dvl (64). In both cases, HUWE1-induced ubiquitination was shown to be a part of a negative-feedback loop. We do not have an explanation for the K48-linked ubiquitination or for the increased abundance of Shoc2 in HUWE1-depleted cells. An attractive hypothesis is that HUWE1 controls the turnover of a specific pool of Shoc2, conceivably, the spatially restricted pool of Shoc2 that is found in endosomes (13). Hence, changes in the degradation rates of the Shoc2, when analyzed using a total cell lysate, may be masked by the total Shoc2, thereby preventing detection of the differences in the rates of degradation of specific Shoc2 pools.
In this study, we also show that HUWE1 is a E3 ubiquitin ligase for RAF-1. Importantly, Shoc2 is required to observe the effects of HUWE1 silencing on the stability and phosphorylation of RAF-1, thus placing Shoc2 in the center of a HUWE1-dependent feedback loop (Fig. 5 and 6). The silencing of Shoc2 itself led to an increase in RAF-1, thus supporting the notion that HUWE1 controls RAF-1 stability when it is in complex with Shoc2. The increase in RAF-1 and ERK1/2 phosphorylation that was detected in our experiments cannot be simply attributed to the stabilization of either Shoc2 or RAF-1 proteins (41, 61) (see Fig. S6 in the supplemental material). Therefore, our findings strongly suggest that, in the context of the Shoc2–RAF-1 scaffolding complex, HUWE1 regulates two events: (i) the amplitude of signal transmission in the Shoc2 module and (ii) the stability of both partners. Moreover, our data further support the notion that K63 polyubiquitination by HUWE1 is an important mechanism by which HUWE1 controls the signaling activities of proteins in mammalian cells.
Functional implications of HUWE1-mediated regulation of Shoc2.
We found that ablation of Shoc2 ubiquitination alters the amplitude of RAF-1 and ERK1/2 phosphorylation (Fig. 10; see also Fig. S6 in the supplemental material). Cells expressing a Shoc2 ubiquitination-defective mutant (7KR) showed higher rate of proliferation, likely due to changes in the activity of the ERK1/2 pathway. Thus, our findings place HUWE1 at the negative-feedback loop regulating the ERK1/2 pathway activity.
The results of this study indicate that, in addition to tethering of Ras and RAF-1 to accelerate ERK1/2 activity, Shoc2 also allows HUWE1 to fine-tune its capacity to accelerate signals transmitted through its complex. By coupling Ras–RAF-1 phosphorylation signals with HUWE1-induced Shoc2 ubiquitination, the dynamic range of RAF-1 activity can be fine-tuned within normal parameters to fit a given physiological situation. Thus, ubiquitin modification of a noncatalytic Shoc2 scaffold may play an active role in controlling ERK1/2 activity. How the ubiquitin system may function to control Shoc2 complex assembly, stability, and, ultimately, the degradation of molecules in the complex is yet to be defined. Also, whether RAF-1 ubiquitination is a cause or a consequence of RAF-1 dissociation from the complex and ultimate degradation is not currently known. It is tempting to speculate that ubiquitination is the posttranslational modification that impacts Shoc2 function allosterically by eliciting its conformational change. Changes in Shoc2 conformation will be followed by alterations in the accessibility of RAF-1 to HUWE1. Future studies of ubiquitinated/deubiquitinated Shoc2 complexes will resolve the basis for this hypothesis. It is also possible, if not likely, that Shoc2 provides a binding surface for additional partners and allows signaling selectivity. It may form distinct scaffolding complexes in different tissues or under different physiological conditions. Such complexity predicts that alterations in Shoc2 structure/function may have important effects on cell signaling and behavior. Not surprisingly, the S2G substitution in Shoc2 caused congenital NS/LAH syndrome with pathological conditions ranging from distinctive craniofacial dysmorphisms, a wide spectrum of congenital heart defects, short stature, and variable neurocognitive impairments to brain anomalies and myelofibrosis (65, 66).
Based on our results, we propose the following model with regard to what is currently understood of the regulation of the ERK1/2 pathway through the Shoc2 module (Fig. 11A). In this model, in the context of a Shoc2 scaffolding platform, HUWE1 has a dual role and modifies the ubiquitination of both Shoc2 and RAF-1. Activation of the ERK1/2 pathway induces HUWE1-mediated ubiquitination of Shoc2, which is followed by the subsequent ubiquitination of RAF-1. Ubiquitination of Shoc2 and RAF-1 leads to changes in the dynamics of ERK1/2 signaling and stabilization of both proteins (Fig. 5 to 10). It will be important to determine whether the Shoc2 scaffold is able to recruit a deubiquitinating enzyme (DUB) and what E2 ubiquitin ligase is in the complex. Conceivably, impairments in this mechanism will allow an unrestricted number of functional scaffold complexes and subsequent uncontrolled ERK1/2 signaling, leading to changes in cellular behavior (i.e., increased cell proliferation) (Fig. 10). It would be interesting to examine our model in the presence and absence of constitutively active Ras/RAF-1 and determine how RAF-1 activity changes with expression of ligase-defective HUWE1. Further deciphering of the composition, dynamics, and function of the HUWE1–Shoc2–RAF-1 complex will strengthen our understanding of the mechanisms that are extensively targeted by therapeutics.
FIG 11.
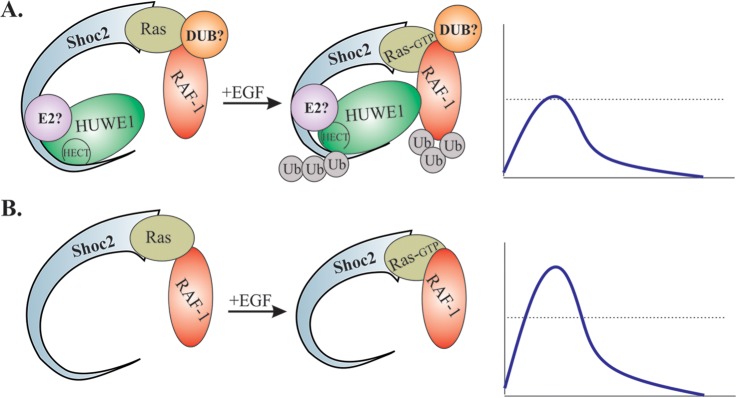
Model recapitulating the role played by HUWE1 in regulating Shoc2-supported ERK1/2 activity. (A) Shoc2 incorporates the E3 ligase HUWE1 into the Ras and RAF-1 signaling complex. HUWE1-mediated ubiquitination of Shoc2 is necessary for the ubiquitination of RAF-1 and fine-tuning of RAF-1 activity and/or its levels in the complex. The identities of the deubiquitinating enzyme (DUB) and the E2 ubiquitin ligase (E2) allowing transient ubiquitination of Shoc2 remain to be determined. (B) The absence of HUWE1 impedes mechanisms modulating the activity of the ERK1/2 pathway.
The implications of the HUWE1-mediated ubiquitination for RAF-1/B-RAF dimerization and propagation of kinase activity may be directly relevant to the clinical application of B-RAF inhibitors. An ability to switch between the activated/deactivated statuses of RAF-1 in the Shoc2 complex would allow additional fine-tuned control of the ERK1/2 pathway and may present an opportunity to enhance the effectiveness of clinically relevant B-RAF inhibitors. Further studies of this complex will also provide important insights into understanding of normal ERK1/2 signaling.
To summarize, our studies are the first to characterize the ubiquitination of Shoc2, identify HUWE1 as a part of the ERK1/2 signaling, and provide insights into the novel mechanism modulating ERK1/2 activity through controlling the signaling scaffold.
Supplementary Material
ACKNOWLEDGMENTS
We thank N. Charles Waechter, Louis Hersh, Alexander Sorkin, Rebecca E. Dutch, Craig Vander Kooi, Mark Evers, M. Kurokawa, and Stacy Smith for providing reagents, critical reading of the manuscript, and insightful discussions; Lina Abdelmoti for technical assistance; the Viral Production Core at the Department of Molecular and Cellular Biochemistry (University of Kentucky) for assistance with the production of lentiviruses; the Protein Structural Core at the Department of Molecular and Cellular Biochemistry (University of Kentucky) for assistance with protein purification; and the Proteomics Core at the Department of Molecular and Cellular Biochemistry (University of Kentucky) for assistance with identification of posttranslational modifications.
The cores mentioned above are supported in part by a grant from the National Institute of General Medical Sciences (P20GM103486). This project was supported in part by grants from the National Cancer Institute (R00CA126161 to E.G.), the National Institute of General Medical Sciences (P20GM103486), and the National Institute of Neurological Disorders and Stroke (R01NS070899 to M.S.G.) and by an American Heart Association postdoctoral award (12POST12030381 to V.D.).
Footnotes
Published ahead of print 14 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00811-14.
REFERENCES
- 1.Katz M, Amit I, Yarden Y. 2007. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim. Biophys. Acta 1773:1161–1176. 10.1016/j.bbamcr.2007.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhattacharyya RP, Remenyi A, Yeh BJ, Lim WA. 2006. Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu. Rev. Biochem. 75:655–680. 10.1146/annurev.biochem.75.103004.142710 [DOI] [PubMed] [Google Scholar]
- 3.Witzel F, Maddison L, Bluthgen N. 2012. How scaffolds shape MAPK signaling: what we know and opportunities for systems approaches. Front. Physiol. 3:475. 10.3389/fphys.2012.00475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pullikuth AK, Catling AD. 2007. Scaffold mediated regulation of MAPK signaling and cytoskeletal dynamics: a perspective. Cell. Signal. 19:1621–1632. 10.1016/j.cellsig.2007.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown MD, Sacks DB. 2008. Compartmentalised MAPK pathways. Handb. Exp. Pharmacol. 186:205–235. 10.1007/978-3-540-72843-6_9 [DOI] [PubMed] [Google Scholar]
- 6.Brown MD, Sacks DB. 2009. Protein scaffolds in MAP kinase signalling. Cell. Signal. 21:462–469. 10.1016/j.cellsig.2008.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Good MC, Zalatan JG, Lim WA. 2011. Scaffold proteins: hubs for controlling the flow of cellular information. Science 332:680–686. 10.1126/science.1198701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sieburth DS, Sun Q, Han M. 1998. SUR-8, a conserved Ras-binding protein with leucine-rich repeats, positively regulates Ras-mediated signaling in C. elegans. Cell 94:119–130. 10.1016/S0092-8674(00)81227-1 [DOI] [PubMed] [Google Scholar]
- 9.Selfors LM, Schutzman JL, Borland CZ, Stern MJ. 1998. soc-2 encodes a leucine-rich repeat protein implicated in fibroblast growth factor receptor signaling. Proc. Natl. Acad. Sci. U. S. A. 95:6903–6908. 10.1073/pnas.95.12.6903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cordeddu V, Di Schiavi E, Pennacchio LA, Ma'ayan A, Sarkozy A, Fodale V, Cecchetti S, Cardinale A, Martin J, Schackwitz W, Lipzen A, Zampino G, Mazzanti L, Digilio MC, Martinelli S, Flex E, Lepri F, Bartholdi D, Kutsche K, Ferrero GB, Anichini C, Selicorni A, Rossi C, Tenconi R, Zenker M, Merlo D, Dallapiccola B, Iyengar R, Bazzicalupo P, Gelb BD, Tartaglia M. 2009. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat. Genet. 41:1022–1026. 10.1038/ng.425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yi J, Chen M, Wu X, Yang X, Xu T, Zhuang Y, Han M, Xu R. 2010. Endothelial SUR-8 acts in an ERK-independent pathway during atrioventricular cushion development. Dev. Dyn. 239:2005–2013. 10.1002/dvdy.22343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li W, Han M, Guan KL. 2000. The leucine-rich repeat protein SUR-8 enhances MAP kinase activation and forms a complex with Ras and Raf. Genes Dev. 14:895–900 [PMC free article] [PubMed] [Google Scholar]
- 13.Galperin E, Abdelmoti L, Sorkin A. 2012. Shoc2 is targeted to late endosomes and required for Erk1/2 activation in EGF-stimulated cells. PLoS One 7:e36469. 10.1371/journal.pone.0036469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsunaga-Udagawa R, Fujita Y, Yoshiki S, Terai K, Kamioka Y, Kiyokawa E, Yugi K, Aoki K, Matsuda M. 2010. The scaffold protein Shoc2/SUR-8 accelerates the interaction of Ras and Raf. J. Biol. Chem. 285:7818–7826. 10.1074/jbc.M109.053975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshiki S, Matsunaga-Udagawa R, Aoki K, Kamioka Y, Kiyokawa E, Matsuda M. 2010. Ras and calcium signaling pathways converge at Raf1 via the Shoc2 scaffold protein. Mol. Biol. Cell 21:1088–1096. 10.1091/mbc.E09-06-0455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Bie P, Ciechanover A. 2011. Ubiquitination of E3 ligases: self-regulation of the ubiquitin system via proteolytic and non-proteolytic mechanisms. Cell Death Differ. 18:1393–1402. 10.1038/cdd.2011.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoeller D, Hecker CM, Dikic I. 2006. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat. Rev. Cancer 6:776–788. 10.1038/nrc1994 [DOI] [PubMed] [Google Scholar]
- 18.Kirkin V, Dikic I. 2007. Role of ubiquitin- and Ubl-binding proteins in cell signaling. Curr. Opin. Cell Biol. 19:199–205. 10.1016/j.ceb.2007.02.002 [DOI] [PubMed] [Google Scholar]
- 19.MacGurn JA, Hsu PC, Emr SD. 2012. Ubiquitin and membrane protein turnover: from cradle to grave. Annu. Rev. Biochem. 81:231–259. 10.1146/annurev-biochem-060210-093619 [DOI] [PubMed] [Google Scholar]
- 20.Marchese A, Trejo J. 2013. Ubiquitin-dependent regulation of G protein-coupled receptor trafficking and signaling. Cell. Signal. 25:707–716. 10.1016/j.cellsig.2012.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grabbe C, Husnjak K, Dikic I. 2011. The spatial and temporal organization of ubiquitin networks. Nat. Rev. Mol. Cell Biol. 12:295–307. 10.1038/nrm3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clark K, Nanda S, Cohen P. 2013. Molecular control of the NEMO family of ubiquitin-binding proteins. Nat. Rev. Mol. Cell Biol. 14:673–685. 10.1038/nrm3644 [DOI] [PubMed] [Google Scholar]
- 23.Pervin S, Tran A, Tran L, Urman R, Braga M, Chaudhuri G, Singh R. 2011. Reduced association of anti-apoptotic protein Mcl-1 with E3 ligase Mule increases the stability of Mcl-1 in breast cancer cells. Br. J. Cancer 105:428–437. 10.1038/bjc.2011.242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shmueli A, Oren M. 2005. Life, death, and ubiquitin: taming the mule. Cell 121:963–965. 10.1016/j.cell.2005.06.018 [DOI] [PubMed] [Google Scholar]
- 25.Zhong Q, Gao W, Du F, Wang X. 2005. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 121:1085–1095. 10.1016/j.cell.2005.06.009 [DOI] [PubMed] [Google Scholar]
- 26.Bernassola F, Karin M, Ciechanover A, Melino G. 2008. The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell 14:10–21. 10.1016/j.ccr.2008.06.001 [DOI] [PubMed] [Google Scholar]
- 27.Chen D, Brooks CL, Gu W. 2006. ARF-BP1 as a potential therapeutic target. Br. J. Cancer 94:1555–1558. 10.1038/sj.bjc.6603119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Confalonieri S, Quarto M, Goisis G, Nuciforo P, Donzelli M, Jodice G, Pelosi G, Viale G, Pece S, Di Fiore PP. 2009. Alterations of ubiquitin ligases in human cancer and their association with the natural history of the tumor. Oncogene 28:2959–2968. 10.1038/onc.2009.156 [DOI] [PubMed] [Google Scholar]
- 29.Adhikary S, Marinoni F, Hock A, Hulleman E, Popov N, Beier R, Bernard S, Quarto M, Capra M, Goettig S, Kogel U, Scheffner M, Helin K, Eilers M. 2005. The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell 123:409–421. 10.1016/j.cell.2005.08.016 [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, Berger FG, Yang J, Lu X. 2011. USP4 inhibits p53 through deubiquitinating and stabilizing ARF-BP1. EMBO J. 30:2177–2189. 10.1038/emboj.2011.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao X, Heng JI, Guardavaccaro D, Jiang R, Pagano M, Guillemot F, Iavarone A, Lasorella A. 2008. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat. Cell Biol. 10:643–653. 10.1038/ncb1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nava C, Lamari F, Heron D, Mignot C, Rastetter A, Keren B, Cohen D, Faudet A, Bouteiller D, Gilleron M, Jacquette A, Whalen S, Afenjar A, Perisse D, Laurent C, Dupuits C, Gautier C, Gerard M, Huguet G, Caillet S, Leheup B, Leboyer M, Gillberg C, Delorme R, Bourgeron T, Brice A, Depienne C. 2012. Analysis of the chromosome X exome in patients with autism spectrum disorders identified novel candidate genes, including TMLHE. Transl. Psychiatry 2:e179. 10.1038/tp.2012.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Froyen G, Corbett M, Vandewalle J, Jarvela I, Lawrence O, Meldrum C, Bauters M, Govaerts K, Vandeleur L, Van Esch H, Chelly J, Sanlaville D, van Bokhoven H, Ropers HH, Laumonnier F, Ranieri E, Schwartz CE, Abidi F, Tarpey PS, Futreal PA, Whibley A, Raymond FL, Stratton MR, Fryns JP, Scott R, Peippo M, Sipponen M, Partington M, Mowat D, Field M, Hackett A, Marynen P, Turner G, Gecz J. 2008. Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. Am. J. Hum. Genet. 82:432–443. 10.1016/j.ajhg.2007.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galperin E, Sorkin A. 2005. Visualization of Rab5 activity in living cells using FRET microscopy. Methods Enzymol. 403:119–134. 10.1016/S0076-6879(05)03011-9 [DOI] [PubMed] [Google Scholar]
- 35.Galperin E, Sorkin A. 2008. Endosomal targeting of MEK2 requires RAF, MEK kinase activity and clathrin-dependent endocytosis. Traffic 9:1776–1790. 10.1111/j.1600-0854.2008.00788.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hall JR, Kow E, Nevis KR, Lu CK, Luce KS, Zhong Q, Cook JG. 2007. Cdc6 stability is regulated by the Huwe1 ubiquitin ligase after DNA damage. Mol. Biol. Cell 18:3340–3350. 10.1091/mbc.E07-02-0173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3:1101–1108. 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- 38.Sorkin A, Duex JE. 2010. Quantitative analysis of endocytosis and turnover of epidermal growth factor (EGF) and EGF receptor. Curr. Protoc. Cell Biol. Chapter 15:Unit 15.14. 10.1002/0471143030.cb1514s46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang X, Sorkin A. 2003. Epidermal growth factor receptor internalization through clathrin-coated pits requires Cbl RING finger and proline-rich domains but not receptor polyubiquitylation. Traffic 4:529–543. 10.1034/j.1600-0854.2003.t01-1-00109.x [DOI] [PubMed] [Google Scholar]
- 40.Cheng A, Zhang M, Gentry MS, Worby CA, Dixon JE, Saltiel AR. 2007. A role for AGL ubiquitination in the glycogen storage disorders of Lafora and Cori's disease. Genes Dev. 21:2399–2409. 10.1101/gad.1553207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jeoung M, Abdelmoti L, Jang ER, Vander Kooi CW, Galperin E. 2013. Functional integration of the conserved domains of Shoc2 scaffold. PLoS One 8:e66067. 10.1371/journal.pone.0066067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodriguez-Viciana P, Oses-Prieto J, Burlingame A, Fried M, McCormick F. 2006. A phosphatase holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M-Ras effector to modulate Raf activity. Mol. Cell 22:217–230. 10.1016/j.molcel.2006.03.027 [DOI] [PubMed] [Google Scholar]
- 43.Jeoung M, Galperin E. 2014. Visualizing of signaling proteins on endosomes utilizing knockdown and reconstitution approach. Methods Enzymol. 534:47–63. 10.1016/B978-0-12-397926-1.00003-2 [DOI] [PubMed] [Google Scholar]
- 44.Kurokawa M, Kim J, Geradts J, Matsuura K, Liu L, Ran X, Xia W, Ribar TJ, Henao R, Dewhirst MW, Kim WJ, Lucas JE, Wang S, Spector NL, Kornbluth S. 2013. A network of substrates of the E3 ubiquitin ligases MDM2 and HUWE1 control apoptosis independently of p53. Sci. Signal. 6:ra32. 10.1126/scisignal.2003741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qi CF, Kim YS, Xiang S, Abdullaev Z, Torrey TA, Janz S, Kovalchuk AL, Sun J, Chen D, Cho WC, Gu W, Morse HC., III 2012. Characterization of ARF-BP1/HUWE1 interactions with CTCF, MYC, ARF and p53 in MYC-driven B cell neoplasms. Int. J. Mol. Sci. 13:6204–6219. 10.3390/ijms13056204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao X, Heng JI, Guardavaccaro D, Jiang R, Pagano M, Guillemot F, Iavarone A, Lasorella A. 2008. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat. Cell Biol. 10:643–653. 10.1038/ncb1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dogan T, Harms GS, Hekman M, Karreman C, Oberoi TK, Alnemri ES, Rapp UR, Rajalingam K. 2008. X-linked and cellular IAPs modulate the stability of C-RAF kinase and cell motility. Nat. Cell Biol. 10:1447–1455. 10.1038/ncb1804 [DOI] [PubMed] [Google Scholar]
- 48.Du J, Zeng J, Ou X, Ren X, Cai S. 2006. Methylglyoxal downregulates Raf-1 protein through a ubiquitination-mediated mechanism. Int. J. Biochem. Cell Biol. 38:1084–1091. 10.1016/j.biocel.2005.10.019 [DOI] [PubMed] [Google Scholar]
- 49.Noble C, Mercer K, Hussain J, Carragher L, Giblett S, Hayward R, Patterson C, Marais R, Pritchard CA. 2008. CRAF autophosphorylation of serine 621 is required to prevent its proteasome-mediated degradation. Mol. Cell 31:862–872. 10.1016/j.molcel.2008.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pandya RK, Partridge JR, Love KR, Schwartz TU, Ploegh HL. 2010. A structural element within the HUWE1 HECT domain modulates self-ubiquitination and substrate ubiquitination activities. J. Biol. Chem. 285:5664–5673. 10.1074/jbc.M109.051805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hurley JH, Lee S, Prag G. 2006. Ubiquitin-binding domains. Biochem. J. 399:361–372. 10.1042/BJ20061138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Inoue S, Hao Z, Elia AJ, Cescon D, Zhou L, Silvester J, Snow B, Harris IS, Sasaki M, Li WY, Itsumi M, Yamamoto K, Ueda T, Dominguez-Brauer C, Gorrini C, Chio II, Haight J, You-Ten A, McCracken S, Wakeham A, Ghazarian D, Penn LJ, Melino G, Mak TW. 2013. Mule/Huwe1/Arf-BP1 suppresses Ras-driven tumorigenesis by preventing c-Myc/Miz1-mediated down-regulation of p21 and p15. Genes Dev. 27:1101–1114. 10.1101/gad.214577.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hurley JH, Stenmark H. 2011. Molecular mechanisms of ubiquitin-dependent membrane traffic. Annu. Rev. Biophys. 40:119–142. 10.1146/annurev-biophys-042910-155404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nathan JA, Kim HT, Ting L, Gygi SP, Goldberg AL. 2013. Why do cellular proteins linked to K63-polyubiquitin chains not associate with proteasomes? EMBO J. 32:552–565. 10.1038/emboj.2012.354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kirkpatrick DS, Denison C, Gygi SP. 2005. Weighing in on ubiquitin: the expanding role of mass-spectrometry-based proteomics. Nat. Cell Biol. 7:750–757. 10.1038/ncb0805-750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Denis NJ, Vasilescu J, Lambert JP, Smith JC, Figeys D. 2007. Tryptic digestion of ubiquitin standards reveals an improved strategy for identifying ubiquitinated proteins by mass spectrometry. Proteomics 7:868–874. 10.1002/pmic.200600410 [DOI] [PubMed] [Google Scholar]
- 57.Papin C, Denouel-Galy A, Laugier D, Calothy G, Eychene A. 1998. Modulation of kinase activity and oncogenic properties by alternative splicing reveals a novel regulatory mechanism for B-Raf. J. Biol. Chem. 273:24939–24947. 10.1074/jbc.273.38.24939 [DOI] [PubMed] [Google Scholar]
- 58.Pumiglia KM, Decker SJ. 1997. Cell cycle arrest mediated by the MEK/mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. U. S. A. 94:448–452. 10.1073/pnas.94.2.448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sewing A, Wiseman B, Lloyd AC, Land H. 1997. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol. Cell. Biol. 17:5588–5597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M. 1997. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol. Cell. Biol. 17:5598–5611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Young LC, Hartig N, Munoz-Alegre M, Oses-Prieto JA, Durdu S, Bender S, Vijayakumar V, Vietri Rudan M, Gewinner C, Henderson S, Jathoul AP, Ghatrora R, Lythgoe MF, Burlingame AL, Rodriguez-Viciana P. 2013. An MRAS, SHOC2, and SCRIB complex coordinates ERK pathway activation with polarity and tumorigenic growth. Mol. Cell 52:679–692. 10.1016/j.molcel.2013.10.004 [DOI] [PubMed] [Google Scholar]
- 62.Yoon SY, Lee Y, Kim JH, Chung AS, Joo JH, Kim CN, Kim NS, Choe IS, Kim JW. 2005. Over-expression of human UREB1 in colorectal cancer: HECT domain of human UREB1 inhibits the activity of tumor suppressor p53 protein. Biochem. Biophys. Res. Commun. 326:7–17. 10.1016/j.bbrc.2004.11.004 [DOI] [PubMed] [Google Scholar]
- 63.Zhao X, D'Arca D, Lim WK, Brahmachary M, Carro MS, Ludwig T, Cardo CC, Guillemot F, Aldape K, Califano A, Iavarone A, Lasorella A. 2009. The N-Myc-DLL3 cascade is suppressed by the ubiquitin ligase Huwe1 to inhibit proliferation and promote neurogenesis in the developing brain. Dev. Cell 17:210–221. 10.1016/j.devcel.2009.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Groot RE, Ganji RS, Bernatik O, Lloyd-Lewis B, Seipel K, Sedova K, Zdrahal Z, Dhople VM, Dale TC, Korswagen HC, Bryja V. 2014. Huwe1-mediated ubiquitylation of dishevelled defines a negative feedback loop in the Wnt signaling pathway. Sci. Signal. 7:ra26. 10.1126/scisignal.2004985 [DOI] [PubMed] [Google Scholar]
- 65.Gripp KW, Zand DJ, Demmer L, Anderson CE, Dobyns WB, Zackai EH, Denenberg E, Jenny K, Stabley DL, Sol-Church K. 2013. Expanding the SHOC2 mutation associated phenotype of Noonan syndrome with loose anagen hair: structural brain anomalies and myelofibrosis. Am. J. Med. Genet. A 161:2420–2430. 10.1002/ajmg.a.36098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoban R, Roberts AE, Demmer L, Jethva R, Shephard B. 2012. Noonan syndrome due to a SHOC2 mutation presenting with fetal distress and fatal hypertrophic cardiomyopathy in a premature infant. Am. J. Med. Genet. A 158A:1411–1413. 10.1002/ajmg.a.35318 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.