

Abstract
Hypoxia promotes stem cell maintenance and tumor progression, but it remains unclear how it regulates long-term adaptation toward these processes. We reveal a striking downregulation of the hypoxia-inducible histone H3 lysine 9 (H3K9) demethylase JMJD1A as a hallmark of clinical human germ cell-derived tumors, such as seminomas, yolk sac tumors, and embryonal carcinomas. Jmjd1a was not essential for stem cell self-renewal but played a crucial role as a tumor suppressor in opposition to the hypoxia-regulated oncogenic H3K9 methyltransferase G9a. Importantly, loss of Jmjd1a resulted in increased tumor growth, whereas loss of G9a produced smaller tumors. Pharmacological inhibition of G9a also resulted in attenuation of tumor growth, offering a novel therapeutic strategy for germ cell-derived tumors. Finally, Jmjd1a and G9a drive mutually opposing expression of the antiangiogenic factor genes Robo4, Igfbp4, Notch4, and Tfpi accompanied by changes in H3K9 methylation status. Thus, we demonstrate a novel mechanistic link whereby hypoxia-regulated epigenetic changes are instrumental for the control of tumor growth through coordinated dysregulation of antiangiogenic gene expression.
INTRODUCTION
Growing solid tumors develop shortages of oxygen (hypoxia) and nutrient supplies. Cancer cells induce the formation of new blood vessels from surrounding host tissues to overcome these adverse conditions. Hypoxia-inducible factors (HIFs) drive the transcriptome of cells to adapt to low-oxygen conditions (1–3). It is now increasingly evident that cancer cells adapted to hypoxia have more-malignant phenotypes (4–7). These findings raise the possibility that hypoxia effectuates long-term adaptation to low-oxygen conditions by altering the epigenetic landscape of cancer cells. However, how hypoxia, HIFs, and possibly other coregulated proteins affect the epigenetic status of oxygen-deprived cancer cells to promote malignancy remains largely unknown.
To examine the impact of hypoxia on cancers, we previously examined global gene expression profiles of seven human cell lines of diverse cancer origins and found that JMJD1A (Jumonji domain-containing 1A, also known as JHDM2A and KDM3A) is one of the epigenetic factors most commonly induced by hypoxia (3). Consistent with this result, other studies have shown that JMJD1A and other Jumonji domain-containing proteins are upregulated in response to hypoxia and that HIF-1α is directly involved in this activation (8, 9). Jumonji domain-containing proteins have been identified as histone lysine demethylases and require Fe(II) and α-ketoglutarate for their catalytic activity (10). Among them, JMJD1A demethylates mono- and dimethylated histone H3 lysine 9 (H3K9) residues. JMJD1A also acts as a coactivator in cooperation with the androgen receptor (11), and it has been proposed to operate downstream of HIF-1α to activate target genes under hypoxic conditions (9). While Jmjd1a−/− mice are viable, males show defects in spermatogenesis leading to infertility, and both genders display an obese phenotype (12–14). In addition to its roles in hypoxia and obesity, Jmjd1a has also been proposed to play an essential role in the maintenance of pluripotency in murine embryonic stem (ES) cells (15), implicating an important link with the regulation of stemness (16).
In contrast to JMJD1A, the histone H3K9 methyltransferase G9A (also known as Ehmt2 and KMT1C) is the major H3K9 mono- and dimethyltransferase in mammals and cooperatively functions by forming a heterodimer with the closely related G9a-like protein (GLP; also known as Ehmt1 and KMT1D) protein (17–19). Loss of either G9a or GLP leads to a global reduction of H3K9 dimethylation, and disruption of either gene in mice results in embryonic lethality (17, 18). G9a is known to repress gene expression by functioning together with histone deacetylases and DNA methyltransferases to downregulate pluripotency-associated genes such as Oct4 (also known as Oct3 and Pou5f1) during development (20, 21). With regard to its role in cancer, G9A has been shown to be posttranscriptionally upregulated in response to hypoxia (22), and recent studies propose that G9A acts positively on tumor growth by silencing tumor suppressor genes (23, 24).
In the present study, we found that JMJD1A was consistently and significantly downregulated at both RNA and protein levels in human germ cell tumors. To determine how this may be important in cancer development, we show that antiangiogenesis-related genes were epigenetically dysregulated in both Jmjd1a- and G9a-deficient ES cells under hypoxic conditions, accompanied by corresponding changes in H3K9 dimethylation and H3K4 trimethylation levels in the proximal promoter regions of these target genes. Furthermore, these genetic alterations led to opposing tumor phenotypes: loss of Jmjd1a resulted in increased tumor growth involving excessive microvessel formation and proliferation of poorly differentiated stem cell populations, whereas loss of G9a produced smaller tumors. Strikingly, tumors derived from Jmjd1a- or G9a-deficient cells show mutually opposing patterns of expression of the transcription factors Nanog and Oct4, which are required for the pluripotency and self-renewal of stem cells. These data suggest that Jmjd1a and G9a may have opposing effects on the maintenance of immature, stem cell-like tumor cell populations, providing a mechanistic link between epigenetic changes in hypoxia and the control of tumor growth, tumor angiogenesis, and stem cell status. Importantly, pharmacological inhibition of G9a resulted in inhibition of tumor growth, offering a novel therapeutic strategy for germ cell-derived tumors.
MATERIALS AND METHODS
Human testicular cancer progression patient samples.
Testicular cancer tissue arrays (TE2081; US Biomax Inc., Rockville, MD) were used for immunohistochemical detection of JMJD1A. Sixty-seven cases of germ cell tumors, including 45 cases of seminomas, 14 cases of embryonal carcinomas, and 8 cases of yolk sac tumors, together with 17 normal specimens consisting of 13 normal tissue sections adjacent to cancer and 4 cases of normal testis were analyzed. The expression of JMJD1A was graded at three different levels: ++ (>30% of tumor cells show strong or diffused immunopositivity in nuclei), + (10 to 30% of tumor cells have moderate or patchy immunopositivity in nuclei), and − (<10% of the tumor cells show weak or focal immunopositivity or no staining in nuclei). All the analyses were assessed by a pathologist. Possible correlations between JMJD1A immunopositivity and pathological grading of the analyzed tumor samples compared to those of the normal tissue were determined using Fisher's exact test with P values of <0.05 required for significance.
Cell culture.
Mouse ES cells were maintained in medium containing 10% fetal calf serum and 500 U/ml leukemia inhibitory factor (LIF). For hypoxia treatments, cells were cultured in 1% O2–5% CO2 at 37°C in an Invivo2 hypoxia workstation 400 (Ruskinn Technology, Leeds, United Kingdom). Genetically manipulated mouse Jmjd1a/G9a knockout (KO) ES cells as well as G9a KO plus transgene (Tg) ES cells have been described previously (14, 17). For detection of alkaline phosphatase activity, cells were plated on 6-well plates at low density (300 cells/well) and stained as previously described (25). For in vitro growth assays, 3 × 105 cells were plated in 6-well plates and counted every 3 days. For microarray analysis, each cell line was exposed to either normoxia (21% O2, 24 h [N]), acute hypoxia (1% O2, 4 h [A]), or chronic hypoxia (1% O2, 24 h [C]). Acute and chronic hypoxia were defined as previously described (26). The cells were plated at three different densities in 6-cm culture dishes, i.e., high (6 × 105 cells/dish [H]), medium (4 × 105 cells/dish [M]), and low (2 × 105 cells/dish [L]), to control for the effects of cell confluence on hypoxic response.
ES cell derivation and genotyping.
Jmjd1a+/− male and Jmjd1a−/− female mice were mated to obtain embryos. Blastocysts were flushed from oviducts at 3.5 days postcoitum and cultured in potassium modified simplex optimized medium (KSOM) (MR-020P-5D; Millipore, Watford, United Kingdom). The zona pellucida was removed using acidic Tyrode's solution, and the blastocysts were cultured in ES cell derivation medium. After 8 to 10 days of culture, outgrown colonies were trypsinized and passaged in ES cell maintenance medium (27). Genotypes were verified using the following three primers: TSGA-G1475R, 5′-GAA CTG CAC CAT TAG CTG TCA CTT CC-3′; TSGA-G2150F, 5′-CAT ACT GGT CTC CAG GAG CCA GAG G-3′; and TSGA-G6540F, 5′-TCA GAC AGT CCT GGG ATC AGA CAC AC-3′.
Microarray analysis.
Total RNA was harvested using the RNeasy Mini kit (Qiagen, Düsseldorf, Germany) and resolved on the 2100 Bioanalyzer (Agilent, CA) for determination of RNA quality. High-purity and -integrity samples with 260/280 and 260/230 absorbance ratios of >1.8 and RNA integrity numbers (RIN) of >8.0 were reverse transcribed into cDNA and in vitro transcribed into biotin-labeled cRNA using the Illumina TotalPrep RNA amplification kit (Ambion, TX). This was hybridized on MouseRef-8 v2.0 Expression BeadChips (Illumina, CA) and scanned on the BeadArray Reader (Illumina) at scan factor 1. Raw intensity values were subjected to background subtraction on the BeadStudio data analysis software (Illumina) and normalized using the cross-correlation method (28). Differential gene expression was identified based on a fold change cutoff of >1.5 compared to the average of the wild-type normoxic controls.
Q-PCR analysis.
Total RNA was isolated using the RNeasy Mini kit (Qiagen), and cDNA was synthesized using SuperScript III reverse transcriptase (Invitrogen, CA) as suggested by the manufacturers. Primer sequences for quantitative PCR (Q-PCR) analyses were designed using the Primer Express software (Applied Biosystems, CA) and are shown in Table S2 in the supplemental material. All samples were preamplified using the TaqMan PreAmp master mix (Applied Biosystems), and Q-PCR was performed using the Power SYBR green PCR master mix (Applied Biosystems) supplemented with AmpliTaq Gold DNA polymerase (Applied Biosystems) on the BioMark real-time PCR system (Fluidigm, CA) according to the manufacturer's specifications. All statistical analyses are t tests relative to the wild-type control unless otherwise indicated.
ChIP assays.
Chromatin immunoprecipitation (ChIP) assays were performed using the protein A ChIP kit (Abcam, MA). Cells were fixed in 1% formaldehyde for 10 min. Genomic DNA was sonicated to 250- to 600-bp fragments. Three micrograms of each antibody was incubated overnight with the sheared chromatin for immunoprecipitation followed by reverse cross-linking overnight at 65°C and purification of immunoprecipitated DNA according to the manufacturer's protocol (version from 1 May 2009). A 1.25-μl volume of the final eluate (total volume, approximately 200 μl) corresponding to 1 × 104 cells was subjected to preamplification and subsequent Q-PCR quantification. Primers used for ChIP analysis are described in Table S3 in the supplemental material.
Immunohistochemistry.
Tumor samples were fixed in 4% paraformaldehyde and paraffin embedded by standard methods. Antigen retrieval was performed with citrate buffer (10 mM citric acid–0.05% Tween 20, pH 6.0), and sections were stained by standard immunohistochemistry techniques, using antibodies as indicated and 1 g/liter hematoxylin solution for counterstaining.
Antibodies.
For Western blot experiments, anti-HIF-1α (NB100-479; Novus, CO), anti-Jmjd1a (NB100-77282; Novus), anti-G9a (8620A; Invitrogen), anti-Oct4 (sc-5279; Santa Cruz Biotechnology, TX), anti-Sox2 (AB5603; Millipore), anti-Nanog (RCAB002P; ReproCell, Japan), anti-histone H3 (06-755; Millipore), and antiactin (sc-1616; Santa Cruz Biotechnology) antibodies were used. For ChIP experiments, anti-H3K9me2 (MABI0307; MBL, Japan) and anti-H3K4me3 (MABI0304; MBL) antibodies were used. For immunohistochemistry, anti-CA-9 (clone AM35-1; kind gift of Silvia Pastorekova), anti-CD34 (RAM34; BD Biosciences Pharmingen, CA), anti-cleaved caspase-3 (Asp175) (catalog number 9661; Cell Signaling Technology, MA), anti-JMJD1A (12835-1-AP; Proteintech Group, Inc., IL) (29), anti-Ki-67 (M7249; DakoCytomation, Glostrup, Denmark), and anti-Oct4 (sc-5279; Santa Cruz Biotechnology) antibodies were used.
Animal studies.
Animal work was performed in accordance with protocols approved by the National University of Singapore Institutional Animal Care and Usage Committee (IACUC). Age-matched nonobese diabetic and severe combined immunodeficiency (NOD-SCID) mice (6 to 8 weeks old) were used. For allograft experiments, 1 × 106 Jmjd1a knockout or 5 × 106 G9a knockout ES cells were resuspended in 0.2 ml of Dulbecco's modified Eagle medium (DMEM; Invitrogen) and injected subcutaneously into the dorsal area of mice. Tumors were measured every 3 or 4 days with calipers to calculate tumor growth over a 21-day period. For BIX-01294-treated tumor formation analyses, cells were cultured in medium containing 10% fetal calf serum, 500 U/ml LIF, and either 0.1% dimethyl sulfoxide (DMSO) or 0.1% DMSO containing 1 μM BIX-01294 (Stemgent, MA) for 2 days prior to injection. For histological analysis, tumors were harvested 20 days postinjection. After being photographed and weighed, tumors were fixed in 4% paraformaldehyde and processed for immunohistochemistry. For time course experiments (see Fig. 8C and D), tumors were harvested at days 12, 17, and 21 postinjection. Tumors were snap-frozen in liquid nitrogen and homogenized prior to total RNA extraction using the RNeasy Mini kit (Qiagen). The numbers of tumors used for Q-PCR analysis at each time point are as follows: day 12, 11 WT tumors and 11 Jmjd1a KO tumors; day 17, 12 WT tumors and 12 Jmjd1a KO tumors; day 21, 11 WT tumors and 17 Jmjd1a KO tumors.
FIG 8.

Loss of Jmjd1a increases microvessels and immature cell populations in tumors. (A) Jmjd1a KO tumors contain more proliferative cells than do WT tumors, as indicated by anti-Ki-67 antibody staining (brown); scale bars, 200 μm. (B) Jmjd1a KO tumors show increased vascular density as indicated by immunohistological staining for the endothelial cell-specific marker CD34 (brown). Arrows indicate branch points within the microvessels; scale bars, 200 μm. The average number of branch points observed for each tumor section is given below the panels. (C) Q-PCR analysis of gene expression in time course experiments of tumor growth. Gene expression levels were normalized against Ywhaz and Ubc2 housekeeping genes, and fold change values were calculated as ratios against the WT day 12 tumors. Vegf-A, Hmox1, Nanog, and Oct4 genes were significantly upregulated at day 21 in Jmjd1a KO tumors, but no significant change was observed for Adm, Igfbp4, Notch4, and Robo4 (*, P < 0.05; N.S., not significant). (D) Nanog and Oct4 are maintained at high protein levels of expression in Jmjd1a KO tumors. Western blots show WT and Jmjd1a KO tumors; #1 and #2 on each lane represent independent tumor samples. (E) Enhanced expression of Oct4 (brown) in Jmjd1a KO tumors, indicating increased abundance of immature stem cell-like populations. Scale bars, 200 μm.
shRNA experiments.
Specific knockdown of mRNA expression was performed using retrovirally expressed short hairpin RNAs (shRNAs; OriGene Technologies, MD). Recombinant viruses were prepared by cotransfection of shRNA vectors with the pCL-10A1 packaging plasmid into HEK 293T cells. Viral supernatants were filtered through a 0.45-μm Millex HA filter (Millipore), and infections were performed in the presence of 4 μg/ml Polybrene (Sigma, MO). Drug selections in ES cells were carried out with 2 μg/ml puromycin. The sequences used for knockdown of G9a are 5′-CTG AAC TCT GGT AGC CTG TCC GAG GAC TT-3′ (G9a number 1 shRNA) and 5′-TCG TGT AGC TCA CCG CTT CCA TAA GGC CT-3′ (G9a number 2 shRNA). The sequence 5′-GCA CTA CCA GAG CTA ACT CAG ATA GTA CT-3′ was used as a knockdown control for comparison.
Statistical analyses.
Data are presented as means ± standard errors of the means (SEM). Statistical significance was calculated using either Student's t test or one-way analysis of variance (ANOVA). P values of less than 0.05 were considered to be statistically significant.
Microarray data accession number.
The microarray data were deposited in NCBI GEO under accession number GSE35061.
RESULTS
JMJD1A is significantly downregulated in human germ cell tumors.
G9A is consistently upregulated across a broad spectrum of different cancer types compared to their normal counterparts (30). As G9A and JMJD1A have opposing catalytic activities (11, 17), we hypothesized that downregulation of JMJD1A may be important for tumorigenesis. To determine the expression of JMJD1A in relevant cancers, we first performed meta-analyses of publicly available human cancer expression data sets in Oncomine (http://www.oncomine.org/) and NextBio. To address where JMJD1A may play important roles as a tumor suppressor, we examined cancer types that demonstrated consistent downregulation of JMJD1A expression levels as a hallmark feature.
Intriguingly, we noted that human germ cell tumors show downregulation of JMJD1A mRNA expression up to 8-fold with high statistical significance compared to normal testicular tissues in all data sets examined (Fig. 1A; see also Table S1 in the supplemental material) (31–33). To confirm that JMJD1A protein levels were similarly and consistently downregulated in germ cell tumors, we performed immunohistochemical staining for JMJD1A protein expression on tissue microarrays consisting of 67 germ cell tumor cores and 17 normal tissue controls in duplicates. In agreement with the mRNA expression data, JMJD1A protein levels were significantly downregulated in multiple subtypes of human primary germ cell tumors (seminomas, yolk sac tumors, and embryonal carcinomas) compared to the adjacent normal testicular tissues (Fig. 1B and C; Table 1). Therefore, these data implicate the downregulation of JMJD1A as a conserved and prominent feature characteristic of germ cell tumor malignancies.
FIG 1.

JMJD1A is downregulated in human germ cell tumors. (A) Box-and-whisker plot of JMJD1A gene expression in human germ cell tumors (teratomas) compared to normal testes (31) analyzed using the Oncomine compendium of cancer transcriptome microarray profiles (http://www.oncomine.org/). (B) Box-and-whisker plot of JMJD1A expression scores in different subtypes of germ cell tumors graded as described in Materials and Methods (P < 0.01). (C) Human germ cell tumors contain fewer JMJD1A-positive cells. Representative tissue sections from normal testes adjacent to cancer, seminomas, yolk sac tumors, and embryonal carcinomas were immunohistologically stained with anti-JMJD1A antibodies (brown) and counterstained with hematoxylin (blue). Scale bars, 200 μm.
TABLE 1.
Analysis of results for human germ cell tumor tissue microarray stained with anti-JMJD1A antibodya
| Tissue or tumor subtype | No. of cases | No. of cases with tumor core classification of: |
P value by Fisher's exact test | ||
|---|---|---|---|---|---|
| − | + | ++ | |||
| Overall cancer tissue | 67 | 38 | 16 | 13 | <0.0000001 |
| Seminoma | 45 | 30 | 9 | 6 | <0.0000001 |
| Yolk sac tumor | 8 | 3 | 3 | 2 | <0.01 |
| Embryonal carcinoma | 14 | 5 | 4 | 5 | <0.01 |
| Normal tissue | 17 | 0 | 2 | 15 | |
Each tumor core was classified according to the percentage and expression levels of JMJD1A-positive cells: ++, >30% of tumor cells show strong or diffused immunopositivity in nuclei; +, 10 to 30% of tumor cells have moderate or patchy immunopositivity in nuclei; −, <10% of the tumor cells show weak or focal immunopositivity or no staining in nuclei. Significant association of JMJD1A positivity in each tumor subtype compared to normal testicular tissue was determined using extended Fisher's exact test with significance at P values of <0.01.
JMJD1A is not required for the maintenance of pluripotency.
As ES cells share the same embryonal origin as germ cell tumors (34, 35), we utilized murine ES cells as a model system for Jmjd1a-dependent tumor development. Jmjd1a knockdown experiments have previously suggested that this epigenetic regulator plays a critical function in maintaining the pluripotency of murine ES cells (15). To confirm the roles of Jmjd1a in stem cell function, we first examined if there were any defects in the maintenance of pluripotency in Jmjd1a homozygous knockout ES cells (14). Q-PCR analysis (Fig. 2A) showed that Jmjd1a knockout ES cells continue to express the key pluripotency factor genes Oct4, Sox2, Klf4, and Nanog at mRNA levels comparable to those of wild-type ES cells. The same result was obtained when examining the protein levels of these factors by Western blotting (Fig. 2B). Furthermore, microarray expression profiling showed no significant changes in additional pluripotency-associated genes, such as Tcl1 (Fig. 2C). However, in G9a knockout ES cells, a significant difference in the expression of pluripotency-associated genes (including a 1.9-fold increase in Nanog and a 2.4-fold increase in Klf4 mRNA levels) was observed (Fig. 2A and C). These results suggest that G9a may be involved in epigenetic silencing of these genes. Interestingly, global H3K9 dimethylation levels did not show any significant overall changes in Jmjd1a knockout ES cells compared to wild-type cells (Fig. 2B). This result was in sharp contrast with that of G9a knockout ES cells, which showed a global reduction of H3K9 dimethylation levels (17). Taken together, our data suggest that Jmjd1a acts in a localized or gene-specific manner instead of globally across the genome as in the case of G9a and that both Jmjd1a and G9a may have common as well as unique epigenetically regulated downstream target genes.
FIG 2.

Loss of Jmjd1a does not affect the expression of pluripotency factors in murine ES cells. (A) Homozygous Jmjd1a-deficient ES cells express key pluripotency genes at a level comparable to that of wild-type (WT) ES cells. Bar charts show Q-PCR analysis of mRNA expression in Jmjd1a-deficient (Jmjd1a KO), G9a-deficient (G9a KO), and reconstituted G9a knockout (G9a KO + Tg) cells compared to WT ES cells. Error bars show standard errors of the means for 6 technical replicates. ***, P < 0.001. (B) Western blot analysis of pluripotency markers Oct4, Sox2, and Nanog shows that their protein levels are comparable in Jmjd1a KO and in WT ES cells. KO1 and KO2 denote independent Jmjd1a KO ES cell lines. (C) Loss of Jmjd1a does not affect expression of pluripotency genes, while loss of G9a dysregulates their expression. The microarray heat map depicts expression changes of selected pluripotency-associated genes in wild-type (WT1 and WT2), Jmjd1a knockout (Jmjd1a KO1 and KO2), G9a knockout (G9a KO1 and KO2), and G9a knockout reconstituted with G9a cDNA (G9a KO + Tg) ES cells. Cell plating density is denoted as high (H; 6 × 105 cells), medium (M; 4 × 105 cells), or low (L; 2 × 105 cells). The scale bar shows the fold change, with red representing upregulation and green downregulation of gene expression compared to the average of the wild-type controls.
We next plated Jmjd1a knockout ES cells at low cell density to assess their ability to self-renew in both normoxic and hypoxic conditions. In contrast to an earlier report (15), there was no obvious defect in the ability of Jmjd1a knockout cells to self-renew during clonal propagation (Fig. 3A and B). Jmjd1a knockout cells stained positive for alkaline phosphatase, forming tight, compact, and dome-shaped colonies typical of ES cells. However, colony numbers in both wild-type and Jmjd1a−/− ES cells increased significantly under hypoxic conditions in the absence of LIF (P < 0.05), suggesting that hypoxia can compensate partially for the effect of LIF on clonogenicity (Fig. 3C). Finally, we confirmed these results by establishing Jmjd1a knockout ES cells from Jmjd1a-deficient blastocysts produced by mating male Jmjd1a heterozygous and female homozygous knockout mice that were viable and fertile. As shown in Fig. 3D to F, multiple Jmjd1a knockout ES cell lines were successfully derived from Jmjd1a null embryos. Thus, we conclude that the loss of Jmjd1a alone has no major role in the maintenance of pluripotency in self-renewing murine ES cells.
FIG 3.

Loss of Jmjd1a does not affect the self-renewing capacity of murine ES cells. (A) Jmjd1a-deficient murine ES cells show no obvious defect in self-renewal. Plates showing alkaline phosphatase staining of colonies from two independent Jmjd1a KO ES cell lines compared to WT ES cell colonies. Bottom panels show closeup morphology of ES cell colonies for each ES cell line. Scale bars, 1 mm. (B, C) Hypoxia substitutes for LIF and rescues colony formation of ES cells in the absence of LIF. (B) Plates showing alkaline phosphatase and Giemsa staining of ES cell colonies formed under the indicated conditions after plating at an initial density of 300 cells/well and culturing either in normoxia (21% O2) or hypoxia (1% O2) for 11 days with or without LIF. (C) Bar chart showing colony numbers for each condition after staining with Giemsa and counting of colonies. The means of colony numbers for wild-type (WT) and Jmjd1a knockout (KO1 and KO2) ES cells are indicated (Average). Statistical significance was calculated using Student's t test. Similar results were obtained from two independent experiments. (D to F) Jmjd1a KO ES cells can be derived from preimplantation embryos. (D) Diagram showing crossing of heterozygous Jmjd1a+/− male and homozygous Jmjd1a−/− female mice to obtain blastocyst stage embryos. (E) Genotypes of wild-type (+/+) and Jmjd1a heterozygous (+/−) and knockout (−/−) ES cell lines were verified by PCR (upper panel), and the table shows the number of Jmjd1a heterozygous and homozygous mutant ES cell lines that were derived from 18 blastocysts obtained from the matings (lower panel). (F) Typical morphology of embryo-derived Jmjd1a+/− and Jmjd1a−/− ES cells in feeder-free cultures; scale bars, 100 μm.
Hypoxia target genes are dysregulated in Jmjd1a and G9a knockout ES cells.
To address how hypoxia mediates epigenetic changes, we next determined if both Jmjd1a and G9a are downstream targets of hypoxia in murine ES cells. Both proteins were upregulated under hypoxic conditions. Protein upregulation was delayed and observed only in chronic hypoxia (24 h) (Fig. 4A), whereas Jmjd1a mRNA levels increased during acute hypoxia (4 h) (Fig. 5A). G9a mRNA expression increased moderately during chronic hypoxia (24 h) (Fig. 5A), which corresponds to significant protein upregulation only in the chronic phase of hypoxia (1.66-fold over normoxia) (Fig. 4A), consistent with a previous report (22). The differences in the timing between upregulation of gene expression and protein production may be due to known hypoxia effects on translation efficiency. Hypoxia is able to downregulate protein synthesis through ER stress and the unfolded protein response (36), which has also been found to vary between the acute and chronic phases of hypoxia (37).
FIG 4.

Identification of Jmjd1a- and G9a-dependent hypoxia transcriptomes in wild-type (WT1 and WT2), Jmjd1a knockout (Jmjd1a KO1 and KO2), G9a knockout (G9a KO1 and KO2), and G9a reconstituted (G9a KO + Tg) ES cells. (A) Hypoxia induces both Jmjd1a and G9a protein levels in mouse ES cells. Western blots show protein extracts of wild-type ES cells cultured in normoxia (21% O2), acute hypoxia (1% O2, 4 h), or chronic hypoxia (1% O2, 24 h) after probing with antibodies as indicated. The upper (150-kDa) and lower (100-kDa) bands for Jmjd1a correspond to long (Ensembl protein ID ENSMUSP00000065716) and short (Ensembl protein ID ENSMUSP00000098862) isoforms, respectively. HIF-1α was used as a positive control for hypoxic induction, and actin was used as a loading control. Band intensities were quantified, and their respective fold changes over the normoxia values of each protein are indicated. (B) Fold changes in gene expression over the average of the WT normoxic controls are as indicated on the scale bar with red for upregulation and green for downregulation. Gene categories 1 and 2 are highlighted in boxes. (C) Number of genes differentially expressed in Jmjd1a- and G9a-deficient cells >1.5-fold over control WT cells. (D and E) Gene ontology classification of genes differentially expressed at least 1.5-fold in Jmjd1a and G9a knockout ES cells compared to the wild-type control. The top ten most significant functional categories in the Jmjd1a knockout (D) and G9a knockout (E) cells were identified using Ingenuity Pathway Analysis. Significant associations with the functional categories were identified using Fisher's exact test at a P value cutoff of 0.05.
FIG 5.

Identification of Jmjd1a- and G9a-dependent hypoxia target genes. Validation of microarray data by Q-PCR. Gene expression levels were normalized against the housekeeping reference gene Ywhaz, and fold change values were calculated against the WT normoxia control. Functional classifications of genes are indicated in boxes. (A) Category 1 genes that show dysregulated response to hypoxia in Jmjd1a KO cells. The Npy gene was upregulated in the absence of Jmjd1a both in normoxia and in hypoxia compared to the wild-type control cells. No significance (N.S.) was observed for any difference in Stc2 and Vegf-a levels of gene expression between Jmjd1a knockout and wild-type ES cells. Jmjd1a expression was verified with primers against the 5′ region of the long isoform of Jmjd1a (Ensembl protein ID ENSMUST00000065509) away from the JmjC domain deletion and shows regulation of expression in both Jmjd1a knockout and wild-type cells. (B) Category 2 genes that become activated in the absence of G9a during hypoxia.*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
To determine how Jmjd1a and G9a regulate the expression of their target genes on a transcriptome-wide scale, we performed microarray analysis of wild-type, Jmjd1a, and G9a knockout ES cells both in normoxia and at different hypoxia time points (4 and 24 h) and analyzed global expression profiles for genes that were differentially expressed >1.5-fold.
In the absence of Jmjd1a, there was predominantly downregulation of gene expression, consistent with the failure to remove the silencing H3K9 methylation mark. Under normoxic conditions, only a relatively small number of genes were differentially expressed in Jmjd1a knockout cells compared to the wild-type, further confirming that Jmjd1a does not have a prominent role in regulating the ES cell transcriptome or in the maintenance of pluripotency (Fig. 4C, upper panel). However, under hypoxic conditions, there was altered induction of more hypoxia targets in Jmjd1a knockout ES cells than in the wild-type cells (Fig. 4B and C). Comparing wild-type and Jmjd1a knockout ES cells, there was only a marginal increase in the number of dysregulated genes in acute hypoxia compared to normoxia, but this markedly increased (∼250%) upon longer exposure to hypoxia (Fig. 4C, upper panel). The dysregulated genes in Jmjd1a knockout ES cells were classified as category 1 targets (Fig. 4B and 5A), and our gene ontology analysis showed that factors related to cellular growth and proliferation were the most dysregulated in Jmjd1a knockout cells compared to wild-type cells under baseline normoxic conditions (Fig. 4D).
In contrast to the relatively small number of Jmjd1a targets, over 3,000 genes were dysregulated in G9a knockout cells (Fig. 4C, lower panel). Consistent with the role of G9a as a transcriptional repressor, its loss in ES cells led to predominant upregulation of gene expression. We therefore focused on genes that became activated in the absence of G9a under hypoxic conditions. These included genes that were related to tissue development (Fgf10) and cell death regulation (Bcl11b) and were classified as category 2 targets (Fig. 4B and E and 5B).
We next confirmed our microarray data by Q-PCR analysis. Consistent with observations in human renal and colon carcinoma cells following shRNA-mediated knockdown of JMJD1A expression (9), hypoxia-dependent upregulation of Adrenomedullin (Adm) and Hmox1 was affected by Jmjd1a deficiency but Stanniocalcin 2 (Stc2) expression was not (Fig. 5A). Furthermore, only induction of Adm and Hmox1 during chronic hypoxia (24 h) was affected by Jmjd1a deficiency, whereas the acute hypoxia (4 h) induction response was not significantly changed (Fig. 5A). Interestingly, transcriptional upregulation of G9a by chronic hypoxia was abolished in Jmjd1a knockout cells, suggesting a cross talk between these epigenetic modifying enzymes in their modulation of the cellular hypoxic response (Fig. 5A).
Intriguingly, the expression of a cluster of angiogenesis-related factor genes, including Igfbp4, Notch4, Robo4, Tfpi, and Zap-70, was downregulated in Jmjd1a knockout ES cells in normoxia and hypoxia (Fig. 5A). Most of these genes are known to have a function in antiangiogenesis (38–40). Notably, Robo4 (Roundabout 4) has been shown to counteract vascular endothelial growth factor (VEGF) signaling (41). Consistent with this functional relationship, the positive angiogenesis regulator Npy (Neuropeptide Y) (42) was upregulated in Jmjd1a knockout cells (Fig. 5A). Collectively, these results suggest a link between loss of Jmjd1a and a concerted upregulation of angiogenesis.
The epigenetic status of target genes is altered in Jmjd1a- and G9a-deficient ES cells during hypoxia.
Since Jmjd1a and G9a alter gene expression by removing or adding methylation marks to histone H3K9, we next examined if the epigenetic status of Jmjd1a or G9a target genes was affected by hypoxia. We therefore performed chromatin immunoprecipitation (ChIP) experiments on the proximal promoter regions of these target genes. Within all the promoter regions examined, loss of Jmjd1a led to an increase in H3K9 dimethylation and, conversely, loss of G9a led to the reduction of H3K9 dimethylation levels (Fig. 6A to C). Consistent with previous reports (9, 22), hypoxia also induces an overall increase in H3K9 dimethylation of target genes over normoxia in wild-type cells. This suggests that during hypoxic induction of Jmjd1a and G9a (Fig. 4A), it is G9a and possibly other H3K9 methyltransferases that prevail over Jmjd1a and other H3K9 demethylases in the regulation of downstream hypoxia target genes.
FIG 6.

H3K9 and H3K4 methylation status of target genes are regulated by hypoxia, Jmjd1a, and G9a. Representative data from 3 independent rounds of ChIP assays performed with anti-H3K4me3 or anti-H3K9me2 antibodies quantified in Igfbp4 (A), Notch4 (B), and Robo4 (C) gene promoter loci by Q-PCR and expressed as fold change values over the average of control IgG at 1 for all panels (n = 3 technical replicates). (D) The Mlh1 promoter region was used as a negative control for the ChIP assays, as it displays no significant change in H3K4 trimethylation status in the presence (WT Normoxia) or absence (Jmjd1a KO Normoxia) of Jmjd1a. Horizontal bars, Q-PCR amplicons on promoter regions; boxes, exons with UTRs in white and coding regions in black; vertical bars, TATA boxes and putative hypoxia-responsive elements (HRE).
To monitor transcriptional activation, we assessed the H3K4 trimethylation status of the selected promoters, as this mark is known to correlate with gene expression (43). The antiangiogenesis genes Igfbp4, Notch4, and Robo4, which were upregulated in response to hypoxia, also showed a significant increase in H3K4 trimethylation levels in wild-type cells, whereas this activation mark was greatly reduced in Jmjd1a knockout cells (Fig. 6A to C). These epigenetic changes correspond to reduced hypoxic induction levels of these genes, as they gain H3K9 dimethylation and lose H3K4 trimethylation in the absence of Jmjd1a, and this was not observed for the nontarget gene Mlh1 (Fig. 6D). Therefore, transcriptional activation of antiangiogenesis genes during hypoxia was accompanied by epigenetic changes mediated by Jmjd1a demethylating H3K9 and, as in the case of other bona fide target genes that are responsive to hypoxia, correlated with an opposing gain in H3K4 trimethylation.
Interestingly, while H3K9me2 levels are similar in hypoxic wild-type cells and normoxic Jmjd1a knockout cells (Fig. 6A to C, right panels, broken blue lines versus solid green lines), activation of gene expression is greater in the former than the latter (Fig. 5A). This suggests that hypoxia-driven HIF transcription may be able to override H3K9me2 suppression to a certain extent, as there is much more HIF activation in hypoxic cells than in normoxic cells that have no activated HIFs. Therefore, although H3K9 methylation is influential in the hypoxic response of target genes, this effect is not absolute and suggests that suppressive H3K9 methylation versus activating HIF transcription exists in an equilibrium that is shifted by hypoxia and the presence or absence of Jmjd1a/G9a.
Jmjd1a and G9a have opposing effects on tumor formation.
To investigate the functional roles of Jmjd1a and G9a in hypoxia-dependent gene regulation in vivo, we induced tumor formation by subcutaneous injection of Jmjd1a and G9a knockout ES cells into immunodeficient mice. Since germ cell tumors are themselves derived from cells of embryonic origin, we used mouse ES cells as a model for tumor development. Wild-type and knockout ES cells were each injected into one side of each mouse, enabling pairwise comparisons of the final tumor weight within the same animal. The resulting tumors showed induction of a hypoxic phenotype when derived from wild-type and Jmjd1a- and G9a knockout ES cells, as indicated by staining for the hypoxia marker CA-9 (Fig. 7A). Jmjd1a knockout cells formed significantly larger tumors than those generated from wild-type cells (P < 0.05) both in volumetric growth measurements and final tumor weights (Fig. 7B to E). In support of these observations, a significantly increased number of Ki-67-positive cells was detected in Jmjd1a knockout tumors compared to the wild type, indicating that there were more proliferating and possibly self-renewing cells within the knockout tumors (Fig. 8A). The increase in tumor size was not due to an increased growth rate of the Jmjd1a knockout ES cells, as these cells showed comparable proliferation to wild-type cells in vitro, under both normoxic and hypoxic conditions (Fig. 7F and G). Interestingly, macroscopic inspection of Jmjd1a knockout tumors showed that they were more hemorrhagic than the wild-type tumors (Fig. 7C), suggesting differences in tumor vascularization. Indeed, microvessels were more densely networked and abundant in the Jmjd1a knockout tumors (Fig. 8B), as assessed by staining for the endothelial cell-specific marker CD34. Moreover, the number of vascular branch points increased significantly in the Jmjd1a knockout tumors, leading to a more complex vascular structure (Fig. 8B, right panel). Consistent with these phenotypes, time course Q-PCR analysis of the tumor samples revealed that although the positive regulators of angiogenesis Vegf-A and Hmox1 were expressed at lower levels at an earlier time point (day 12), they became significantly upregulated at the later time point (day 21) in Jmjd1a knockout tumors compared to those generated from wild-type cells (Fig. 8C). In contrast, expression of the negative regulators of angiogenesis Robo4, Igfbp4, and Notch4 was lower at the early time point (day 12), consistent with the ES cell expression data, but the differences were gradually abolished in the later stage (day 21) (Fig. 8C). Therefore, downregulation of the antiangiogenesis program seems to precede induction of the proangiogenic gene expression drive. Moreover, we made the striking observation that expressions of Nanog and Oct4, key transcription factor genes required for the maintenance of ES cell pluripotency and self-renewal that are highly expressed in immature, aggressive tumor cells, were also significantly higher in Jmjd1a knockout tumors at both mRNA and protein levels (Fig. 8C and D). This was further supported by immunostaining of tumor samples with Oct4 antibody, showing upregulation of this master regulator of ES cell self-renewal in tumors derived from Jmjd1a-deficient cells (Fig. 8E). Taken together, these data suggest that Jmjd1a-deficient tumors may contain more immature and stem cell-like populations that are supportive of increased tumor growth than wild-type tumors.
FIG 7.

Loss of Jmjd1a increases tumor growth. (A) Teratomas have hypoxic cores. Wild-type and Jmjd1a and G9a knockout ES cell-derived teratoma tumor sections were stained for the hypoxia marker CA-9 (carbonic anhydrase 9) with an anti-CA-9 antibody (brown) and counterstained with hematoxylin (blue). Scale bars, 200 μm. (B) Jmjd1a KO ES cells form tumors with an increased growth rate (n = 14). cc, cubic centimeters. (C) Gross morphology of tumors resected from NOD-SCID mice 20 days after subcutaneous injection with WT or Jmjd1a KO ES cells. The scale shows length in centimeters. (D) Final tumor masses of WT and Jmjd1a KO tumors formed after 20 days of growth. (E) Jmjd1a knockout ES cells produce larger tumors than those formed by wild-type cells. The chart shows comparisons of the tumor weights in grams produced by matching wild-type and Jmjd1a knockout ES cell injections at 2 dorsal sites for each mouse (n = 18). Black lines show mice in which Jmjd1a knockout ES cells formed larger tumors, while blue lines show exceptions. The red line indicates the average tumor size for both wild-type and Jmjd1a knockout ES cell injections. (F and G) Loss of Jmjd1a has no significant effect on the growth rate of ES cells both in normoxia and in hypoxia. (F) Graph showing the in vitro growth rate of Jmjd1a knockout and wild-type ES cells plated with an initial seeding density of 3 × 105 cells per well of a 6-well plate in normoxia, with cells counted every 3 to 4 days. (G) Cell numbers after plating 3 × 105 cells on each well of a 6-well plate under hypoxic or normoxic conditions and counting after 3 days of growth for 4 replicate cultures. Error bars denote standard errors of the means.
In contrast to the Jmjd1a results, G9a knockout cells formed significantly smaller tumors than wild-type ES cells (P < 0.01) (Fig. 9A to D), and this effect was reversible with the reconstitution of G9a cDNA in knockout cells, indicating that G9a acts positively on tumor formation. The reduction in tumor size was not due to decreased growth rates of G9a knockout cells, as they proliferated in a manner comparable with that of wild-type cells (data not shown). Moreover, G9a knockout tumors had fewer microvessels than the wild-type tumors, as assessed by immunostaining using CD34 antibodies (Fig. 9E). Furthermore, immunohistochemical analysis for cleaved caspase-3 and Ki-67 expression revealed that G9a knockout tumors had more apoptotic and fewer proliferative cells, respectively, than wild-type cells, suggesting that the reduced tumor size observed for G9a knockout cells was consistent with increased apoptosis and a decreased number of self-renewing cells within the tumors (Fig. 9F and G). Q-PCR analysis of tumor samples revealed that G9a knockout tumors showed significantly higher levels of expression of the tumor suppressor gene Bcl11b (Fig. 9H), reproducing the results of the microarray analysis of G9a knockout ES cells compared to wild-type (Fig. 4B). Bcl11b is known to be either mutated or deleted in T-cell acute lymphoblastic leukemia (44). Remarkably, G9a knockout tumors showed downregulation of Oct4 and Nanog mRNA expression (Fig. 9H), in contrast to Jmjd1a knockout cells (Fig. 8C). In agreement with the expression results, there was also a dramatic decrease of Nanog protein levels in G9a knockout tumors, as assessed by Western blotting (Fig. 9I), and a similarly striking decrease in expression of Oct4 protein levels (Fig. 9I and J). These results suggest that G9a knockout tumors may be unable to maintain significant immature, self-renewing stem cell-like populations required for tumorigenesis.
FIG 9.

G9a acts positively to promote tumor formation. (A) Loss of G9a decreases the tumor growth rate. (B) Gross morphology of tumors resected from NOD-SCID mice 20 days after subcutaneous injections with WT, G9a KO, and reconstituted G9a KO ES cells (G9a KO + Tg). 2-3 and 22-10, tumors derived from two independent G9a KO ES cell lines. (C) Final tumor masses of WT, G9a KO, and reconstituted G9a KO ES cells. Tumors were resected after 20 days of growth. **, P < 0.01 for comparisons versus WT; ††, P < 0.001 for the reconstituted G9a KO versus the G9a KO tumors. (D) G9a KO ES cells produce smaller tumors than those derived from wild-type cells. Comparison of tumor weights for wild-type and G9a KO cells injected into different dorsal sites within the same mouse (n = 14). Line graphs indicate the decrease in tumor size for G9a KO ES cell injections; exceptions showing increased tumor sizes are in blue. (E) G9a KO tumors have fewer microvessels compared to WT tumors as indicated by anti-CD34 staining (brown). Scale bars, 200 μm. (F) There is increased induction of apoptosis in G9a KO tumors as indicated by immunohistochemical staining for the apoptosis marker cleaved caspase-3 (brown). Scale bars, 200 μm. (G) G9a KO tumors contain fewer proliferating cells as indicated by anti-Ki-67 staining (brown). Scale bars, 200 μm. (H) Q-PCR analysis of tumor samples harvested at 21 days postinjection for target gene expression. Gene expression levels were normalized against the Ywhaz housekeeping gene, and fold change values were calculated against the WT tumors. (I) Protein levels of Nanog and Oct4 were downregulated in G9a KO tumors. #1 and #2, independent biological replicates of tumor protein extracts. (J) Oct4 expression was greatly reduced in G9a KO tumors as indicated by anti-Oct4 staining (brown). Scale bars, 200 μm.
Inhibition of G9A reduces tumor growth.
To demonstrate that Jmjd1a and G9a are acting antagonistically in tumor formation, we knocked down G9a using virally delivered shRNA in a Jmjd1a knockout background. G9a was successfully downregulated using two independent lentiviral shRNA constructs (Fig. 10A). We then injected these cells together with control shRNA-treated Jmjd1a knockout cells into immunodeficient mice to monitor tumor growth. Consistent with our model that G9a is a positive regulator of tumor growth, knockdown of G9a in a Jmjd1a-deficient background led to a statistically significant reduction in tumor size compared to the control tumors (P < 0.05) (Fig. 10B and C). Furthermore, there was a general upregulation of the antiangiogenic factor genes Igfbp4, Tfpi, Notch4, and Robo4 in the Jmjd1a KO + G9a knockdown (KD) tumors, of which upregulation levels for Igfbp4 and Tfpi were significant (P < 0.05) (Fig. 10D; data not shown for Notch4 and Robo4) compared to control tumors. The residual presence of endogenous G9a in the knockdowns may mitigate more significant changes in antiangiogenic gene expression levels than complete loss-of-function in G9a knockout cells. Indeed, a baseline level of G9a protein (Fig. 10A) persists in the G9a KD cells that may present less severe conditions than the G9a knockout cells. Nevertheless, although only two of four antiangiogenic factors are significantly dysregulated individually, the overall synergistic effect of increasing the expression of multiple antiangiogenic factors may collectively account for the phenotypic reduction of the Jmjd1a KO + G9a KD tumors compared to control tumors.
FIG 10.

Gain in tumor size with loss of Jmjd1a is rescued by loss of G9a. (A) Western blot analysis of Jmjd1a KO ES cells stably transfected with G9a shRNA (independent cell lines #1 and #2) shows decreased G9a protein expression. (B) Morphological appearance of tumors resected from NOD-SCID mice 24 days after subcutaneous injection of Jmjd1a KO ES cells expressing either control or G9a shRNA. (C) Final tumor masses of Jmjd1a KO tumors with either control or G9a shRNA knockdown formed after 24 days of growth. (D) Antiangiogenic genes were upregulated in Jmjd1a KO + G9a KD tumors. Gene expression levels were normalized against the Ywhaz reference gene, and the fold change over the Jmjd1a KO + Control KD was calculated. *, P < 0.05. (E) Morphological appearance of tumors resected from NOD-SCID mice 20 days after subcutaneous injection of wild-type ES cells pretreated either with control (DMSO only) or with 1 μM BIX-01294-containing medium. (F) Final masses of wild-type tumors following treatments as described for panel E. (G) Morphological appearance of tumors resected from NOD-SCID mice 20 days after subcutaneous injection of Jmjd1a-deficient ES cells pretreated either with control (DMSO only) or with 1 μM BIX-01294-containing medium. (H) Final masses of Jmjd1a-deficient tumors under the same treatment conditions as for panel G.
To independently confirm the knockdown results, we inhibited G9a enzymatic activity using the chemical compound BIX-01294 to determine if this could similarly reduce tumor growth. BIX-01294 blocks G9a histone methyltransferase activity by binding to the substrate peptide groove of the catalytic SET domain, thereby reducing H3K9me2 levels (45, 46). We first optimized the BIX-01294 concentration and chose the moderate dose of 1 μM, at which the 50% inhibitory concentration (IC50) was attained for the attenuation of cell numbers. Subsequently, both wild-type and Jmjd1a-deficient cells were treated with medium containing either 1 μM BIX-01294 in DMSO or only DMSO for 2 days prior to injection of equal numbers of cells into immunodeficient mice. Jmjd1a-deficient cells were used in comparisons against wild-type cells, as they were more representative of human germ cell-derived tumors that had downregulated JMJD1A expression (Fig. 1). Importantly, the BIX-01294-pretreated cells generated greatly reduced tumors both in the wild-type and Jmjd1a-deficient genetic backgrounds with statistical significance (Fig. 10E to H). These results clearly indicate that G9a acts positively on tumor growth and that inhibition of G9a either by knockdown or pharmacological inhibition can reduce tumor growth in germ cell-derived tumor models.
DISCUSSION
Jmjd1a has been proposed to play an essential role in the maintenance of pluripotency in murine ES cells (15). Interestingly, a recent study has demonstrated that Jmjd1a-deficient mice display defects in sex determination and abnormal spermiogenesis (47), indicating that Jmjd1a is critical for germ cell development. Given that germ cell tumors are themselves derived from cells of embryonic origin, we therefore decided to study the molecular basis of hypoxia-regulated epigenetics using mouse ES cells as the common denominator for both tumor and stem cell development. We found that Jmjd1a-deficient ES cells continue to express the key pluripotency genes Oct4, Sox2, Klf4, and Nanog at mRNA and protein levels comparable to those of wild-type ES cells. Furthermore, our genome-wide gene expression microarray analysis showed no significant perturbation of pluripotency-associated genes, and there was no obvious self-renewal defect in Jmjd1a-deficient ES cells. Finally, we successfully derived Jmjd1a knockout ES cells from Jmjd1a null embryos produced by viable and fertile Jmjd1a null and heterozygous adult mice. The discrepancies between our results and those of previous studies may have arisen from the different strategies that have been employed in the study of Jmjd1a function, i.e., the use of knockout ES cells versus shRNA-based knockdown procedures. The transient knockdown of Jmjd1a may lead to an acute loss of pluripotency in ES cells, but the permanent knockout of Jmjd1a results in more-long-term adaptive compensation for the loss of pluripotency. We conclude that Jmjd1a alone does not play an essential role in the maintenance of pluripotency in murine ES cells under self-renewal conditions.
In the case of G9a, there are severe gastrulation defects in the epiblast of postimplantation knockout embryos, resulting in early lethality (17). Consistent with the in vivo role of G9a in epiblast development, G9a knockout ES cells also display significant disruption in the expression of stemness genes such as Nanog and Klf4. Despite this, G9a null ES cells can still be maintained in culture, suggesting that G9a may instead play greater roles at later stages of epiblast development during gastrulation. When we injected Jmjd1a- and G9a-deficient ES cells into immunodeficient mice, there was robust reciprocal regulation of Oct4 and Nanog expression whereby loss of Jmjd1a resulted in activation and loss of G9a led to downregulation of these genes. This regulation is unexpected of a direct Jmjd1a/G9a epigenetic function and may instead be due to changes in the H3K9 methylation status or expression of antiangiogenic genes that produce microenvironments conducive or detrimental to the maintenance of stem cell-like populations, respectively. The significant effects of Jmjd1a/G9a with the onset of hypoxia during tumor formation, in contrast to the lack of pluripotency defects in ES cells, highlight the effect of hypoxia on the expression of pluripotency genes and further suggest that Jmjd1a/G9a may become important only upon exit from self-renewal and the subsequent onset of differentiation, rather than at ES cell self-renewal stages (48), affecting the status of stem cell-like populations needed for tumor growth.
Previously, JMJD1A was thought to act downstream of HIF-1α to upregulate hypoxia target genes for the long-term adaptation of cells to hypoxic conditions. Xenograft studies using shRNA-mediated knockdown of JMJD1A in human colon carcinoma cells led to decreased tumor size (9, 29), suggesting that JMJD1A may function as an oncogenic driver. In the present study, we observed increased tumor formation by Jmjd1a-deficient cells coupled with increased expression of Nanog and Oct4, consistent with the maintenance of immature, stem cell-like tumor cells. Thus, JMJD1A may regulate target genes differently depending on the cell context, thereby having disparate roles on tumor growth in cancers of different origins. Indeed, germ cell tumors show consistent and specific downregulation of JMJD1A expression compared to their normal counterparts. Therefore, our results may reflect properties of immature, poorly differentiated tumors with functional Nanog and Oct4 expression such as those of germ cell provenance (49, 50) but not of differentiated somatic tumors, which rarely express these stem cell factors (51, 52). Our finding that tumors derived from Jmjd1a- or G9a-deficient cells show mutually opposing regulation of Nanog and Oct4 suggests that Jmjd1a and G9a may play antagonistic roles in the maintenance of immature, stem cell-like populations in Nanog- and Oct4-positive tumors.
To investigate how hypoxia alters the epigenetic histone methylation status of ES cells, we chose to study hypoxia-dependent regulation of not only Jmjd1a but also G9a. These two hypoxia-regulated enzymes have opposing catalytic functions and hence titrate the equilibrium between mono- and dimethylation and demethylation of H3K9. Previously, it was shown that JMJD1A forms a protein complex with HIF-1α to activate target genes in vascular endothelial cells (53). This offers a mechanistic explanation regarding target gene specificity of JMJD1A in hypoxia, which is facilitated by direct HIF recognition and hypoxia-dependent recruitment to target genes. In contrast, G9a is known to have a broader, more diverse range of target genes than Jmjd1a (54). Although the mechanism of G9a targeting many of these genes is not fully understood, G9a has been reported to interact with many zinc finger proteins (19, 55, 56). This suggests that Jmjd1a/G9a interactions may influence the genome-wide distribution and degree of H3K9 dimethylation in hypoxia, where Jmjd1a and G9a share some common targets but also regulate specific subsets of target genes. These differing mechanisms behind Jmjd1a/G9a specificity may explain the overlapping but also distinct target genes obtained in the transcriptomic analysis of Jmjd1a and G9a knockout cells.
The small number of Jmjd1a-regulated target genes compared to that of G9a identified in our microarray studies is indicative of the differences in transcriptional output mediated by the two epigenetic factors. This is further supported by our ChIP studies showing that during hypoxia, loss of Jmjd1a has a clear effect on the H3K9 methylation status of only a specific subset of antiangiogenesis-related gene promoters. In the case of Igfbp4 and Robo4, there were higher levels of the H3K9 dimethylation mark in Jmjd1a-deficient cells, probably due to a failure in removing H3K9 methylation marks from the promoters, and this in turn correlates with low levels of H3K4 trimethylation and reduced expression of these genes.
Although Jmjd1a is a 2-oxoglutarate-dependent dioxygenase and requires dioxygen for its catalytic activity, it has previously been shown that JMJD1A is able to retain its demethylase activity even when subjected to severe hypoxia of as little as 0.2% oxygen, and its expression is directly upregulated by HIF-1α binding to hypoxia-responsive elements (HREs) present in the promoter region (57). Thus, the increase in demethylase activity of Jmjd1a may be facilitated by increased protein and RNA production during hypoxia (Fig. 4A and 5A), and this is not compromised by the depletion of oxygen.
In addition to Jmjd1a and G9a, it is also possible that other hypoxia-regulated methyltransferases and demethylases provide additional levels of control over target gene expression (8, 58). The repertoire of hypoxia-regulated epigenetic factors may extend beyond direct transcriptional control by the HIFs and may also be regulated by posttranslational mechanisms (59) that may not be evident in gene expression studies. Hypoxia-regulated microRNAs have been shown to bind to the 3′ untranslated region (UTR) of Ezh2, an H3K27 histone methyltransferase, thereby inhibiting its translation into protein (60). It is possible that other epigenetic factors are also regulated at the posttranslational stage by RNA binding proteins, thus providing an additional layer of control over the expression of target genes. The H3K9 methylation status may therefore not be the sole determinant of gene expression, and this may account for the differing transcriptional output upon hypoxic induction despite similar H3K9 methylation levels (Fig. 6A to C, right panels, broken blue lines versus solid green lines, and Fig. 5A).
Our data suggest that loss of Jmjd1a unleashes an angiogenic drive leading to increased tumor growth. The angiogenic drive is in part governed by inactivating antagonists against angiogenesis, such as the inhibition of the Slit-Robo4 signaling pathway through the downregulation of Robo4, coupled with increased expression of bona fide proangiogenic factor genes such as Vegf-A and Hmox1 (9, 61). In human germ cell-derived tumor development, Jmjd1a-deficient cells may thus be able to generate a microenvironmental niche that is more suitable for both immature and differentiated cells to proliferate than wild-type cells, resulting in larger tumors (Fig. 11).
FIG 11.
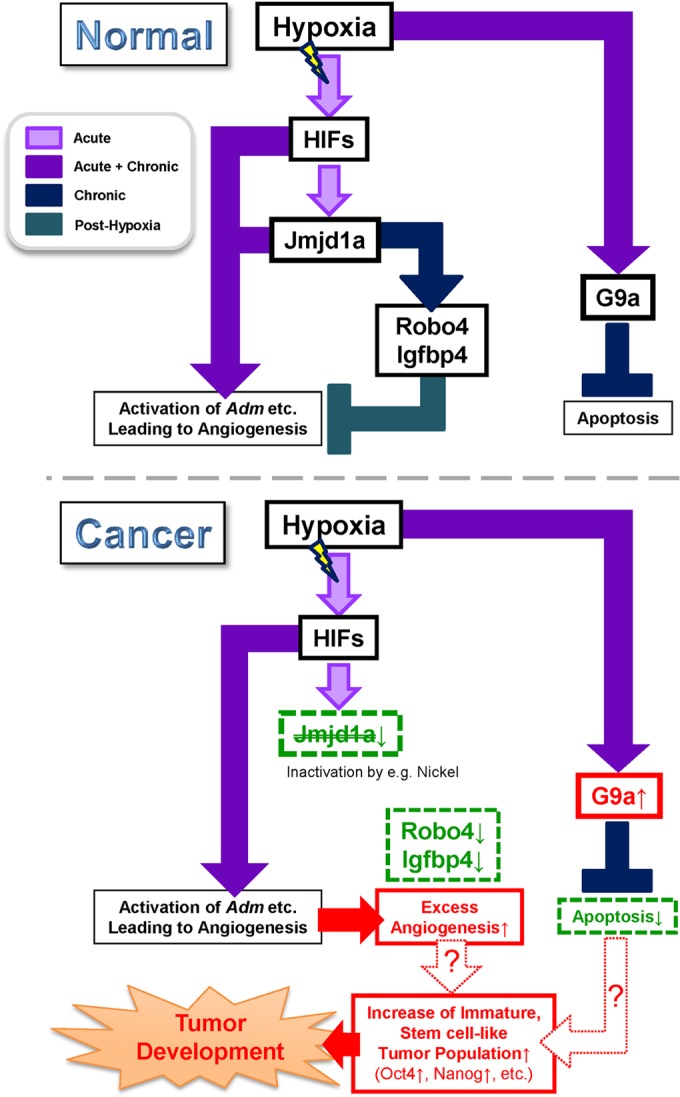
Schematic diagram showing how Jmjd1a and G9a modulate hypoxia signaling pathways in normal and germ cell-derived tumor development. HIFs directly activate Jmjd1a transcription, and some of the HIFs also target genes responsible for the induction of angiogenesis, such as Adm in the acute phase. However, in the later stages (chronic hypoxia), Jmjd1a derepresses expression of antiangiogenic factors as a negative-feedback mechanism to prevent excess vascular formation. In contrast, G9a is upregulated during acute hypoxia and suppresses apoptosis in the chronic hypoxia phase (upper panel). In the absence of Jmjd1a, the induction of antiangiogenic factors is lost or severely attenuated, leading to excess vascular formation. This may facilitate the maintenance of immature, stem cell-like tumor populations that eventually lead to tumor development (lower panel).
In conclusion, we have provided novel insights into how two hypoxia-regulated epigenetic factors, Jmjd1a and G9a, have opposing effects not only on the epigenetic status of target promoters in normoxia and hypoxia but also on tumor formation and stem cell regulation. This provides a mechanistic link between epigenetic changes driven by hypoxia and the control of stem cell status and tumor angiogenesis and growth. This regulatory scenario seems to be of particular relevance in human germ cell-derived tumors, and pharmacological inhibition provides a promising novel strategy in the treatment of these tumor types.
Supplementary Material
ACKNOWLEDGMENTS
We thank E. Teng and L. M. T. Chia for their help with mouse allograft experiments and S. Pastorekova for the kind gift of the CA-9 antibody. We express our gratitude to H. Okawa, L. Y. Yit, and H. Shimura for their assistance in tumor RNA and protein extractions.
The meta-analysis of human germ cell tumor data was supported by the DNA-chip Development Center for Infectious Diseases, Research Institute for Microbial Diseases, Osaka University. This work was supported by the Singapore National Research Foundation and the Singapore Ministry of Education under the Research Center of Excellence Programme, the Swedish Research Council, the Swedish Cancer Society, the Swedish Childhood Cancer Foundation, and the European Union.
Author contributions were as follows: J.U., J.C.H., K.L.L., S.K., H.K., and L.P. designed research; J.U., J.C.H., K.L.L., S.K., W.S., N.F., N.Z., S.L.C., and H.K. performed research; H.Y., S.L.C., M.T., Y.S., H.K., and L.P. contributed new reagents/analytic tools; J.U., J.C.H., K.L.L., S.K., H.Y., W.S., N.F., N.Z., S.L.C., M.T., Y.S., H.K., and L.P. analyzed data; and J.U., J.C.H., K.L.L., H.K., and L.P. wrote the paper.
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 28 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00099-14.
REFERENCES
- 1.Semenza GL. 2014. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 9:47–71. 10.1146/annurev-pathol-012513-104720 [DOI] [PubMed] [Google Scholar]
- 2.Pugh CW, Ratcliffe PJ. 2003. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med. 9:677–684. 10.1038/nm0603-677 [DOI] [PubMed] [Google Scholar]
- 3.Lendahl U, Lee K, Yang H, Poellinger L. 2009. Generating specificity and diversity in the transcriptional response to hypoxia. Nat. Rev. Genet. 10:821–832. 10.1038/nrg2665 [DOI] [PubMed] [Google Scholar]
- 4.Höckel M, Vaupel P. 2001. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 93:266–276. 10.1093/jnci/93.4.266 [DOI] [PubMed] [Google Scholar]
- 5.Harris AL. 2002. Hypoxia—a key regulatory factor in tumour growth. Nat. Rev. Cancer 2:38–47. 10.1038/nrc704 [DOI] [PubMed] [Google Scholar]
- 6.Chan DA, Giaccia AJ. 2007. Hypoxia, gene expression, and metastasis. Cancer Metastasis Rev. 26:333–339. 10.1007/s10555-007-9063-1 [DOI] [PubMed] [Google Scholar]
- 7.Keith B, Johnson RS, Simon MC. 2011. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 12:9–22. 10.1038/nrc3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia X, Lemieux ME, Li W, Carroll JS, Brown M, Liu XS, Kung AL. 2009. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc. Natl. Acad. Sci. U. S. A. 106:4260–4265. 10.1073/pnas.0810067106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krieg A, Rankin E, Chan D, Razorenova O, Fernandez S, Giaccia A. 2010. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol. Cell. Biol. 30:344–353. 10.1128/MCB.00444-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. 2006. Histone demethylation by a family of JmjC domain-containing proteins. Nature 439:811–816. 10.1038/nature04433 [DOI] [PubMed] [Google Scholar]
- 11.Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. 2006. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 125:483–495. 10.1016/j.cell.2006.03.027 [DOI] [PubMed] [Google Scholar]
- 12.Okada Y, Scott G, Ray M, Mishina Y, Zhang Y. 2007. Histone demethylase JHDM2A is critical for Tnp1 and Prm1 transcription and spermatogenesis. Nature 450:119–123. 10.1038/nature06236 [DOI] [PubMed] [Google Scholar]
- 13.Tateishi K, Okada Y, Kallin E, Zhang Y. 2009. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 458:757–761. 10.1038/nature07777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inagaki T, Tachibana M, Magoori K, Kudo H, Tanaka T, Okamura M, Naito M, Kodama T, Shinkai Y, Sakai J. 2009. Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes Cells 14:991–1001. 10.1111/j.1365-2443.2009.01326.x [DOI] [PubMed] [Google Scholar]
- 15.Loh Y, Zhang W, Chen X, George J, Ng H. 2007. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 21:2545–2557. 10.1101/gad.1588207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niwa H. 2007. How is pluripotency determined and maintained? Development 134:635–646. 10.1242/dev.02787 [DOI] [PubMed] [Google Scholar]
- 17.Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, Shinkai Y. 2002. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 16:1779–1791. 10.1101/gad.989402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, Sakihama T, Kodama T, Hamakubo T, Shinkai Y. 2005. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 19:815–826. 10.1101/gad.1284005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ueda J, Tachibana M, Ikura T, Shinkai Y. 2006. Zinc finger protein Wiz links G9a/GLP histone methyltransferases to the co-repressor molecule CtBP. J. Biol. Chem. 281:20120–20128. 10.1074/jbc.M603087200 [DOI] [PubMed] [Google Scholar]
- 20.Tachibana M, Matsumura Y, Fukuda M, Kimura H, Shinkai Y. 2008. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J. 27:2681–2690. 10.1038/emboj.2008.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Epsztejn-Litman S, Feldman N, Abu-Remaileh M, Shufaro Y, Gerson A, Ueda J, Deplus R, Fuks F, Shinkai Y, Cedar H, Bergman Y. 2008. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat. Struct. Mol. Biol. 15:1176–1183. 10.1038/nsmb.1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H, Yan Y, Davidson T, Shinkai Y, Costa M. 2006. Hypoxic stress induces dimethylated histone H3 lysine 9 through histone methyltransferase G9a in mammalian cells. Cancer Res. 66:9009–9016. 10.1158/0008-5472.CAN-06-0101 [DOI] [PubMed] [Google Scholar]
- 23.Lee S, Kim J, Kim W, Lee Y. 2009. Hypoxic silencing of tumor suppressor RUNX3 by histone modification in gastric cancer cells. Oncogene 28:184–194. 10.1038/onc.2008.377 [DOI] [PubMed] [Google Scholar]
- 24.Chen M, Hua K, Kao H, Chi C, Wei L, Johansson G, Shiah S, Chen P, Jeng Y, Cheng T, Lai T, Chang J, Jan Y, Chien M, Yang C, Huang M, Hsiao M, Kuo M. 2010. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 70:7830–7840. 10.1158/0008-5472.CAN-10-0833 [DOI] [PubMed] [Google Scholar]
- 25.Cox WG, Singer VL. 1999. A high-resolution, fluorescence-based method for localization of endogenous alkaline phosphatase activity. J. Histochem. Cytochem. 47:1443–1456. 10.1177/002215549904701110 [DOI] [PubMed] [Google Scholar]
- 26.Holmquist-Mengelbier L, Fredlund E, Lofstedt T, Noguera R, Navarro S, Nilsson H, Pietras A, Vallon-Christersson J, Borg A, Gradin K, Poellinger L, Pahlman S. 2006. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell 10:413–423. 10.1016/j.ccr.2006.08.026 [DOI] [PubMed] [Google Scholar]
- 27.Yamagata K, Ueda J, Mizutani E, Saitou M, Wakayama T. 2010. Survival and death of epiblast cells during embryonic stem cell derivation revealed by long-term live-cell imaging with an Oct4 reporter system. Dev. Biol. 346:90–101. 10.1016/j.ydbio.2010.07.021 [DOI] [PubMed] [Google Scholar]
- 28.Chua SW, Vijayakumar P, Nissom PM, Yam CY, Wong VV, Yang H. 2006. A novel normalization method for effective removal of systematic variation in microarray data. Nucleic Acids Res. 34:e38. 10.1093/nar/gkl024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uemura M, Yamamoto H, Takemasa I, Mimori K, Hemmi H, Mizushima T, Ikeda M, Sekimoto M, Matsuura N, Doki Y, Mori M. 2010. Jumonji domain containing 1A is a novel prognostic marker for colorectal cancer: in vivo identification from hypoxic tumor cells. Clin. Cancer Res. 16:4636–4646. 10.1158/1078-0432.CCR-10-0407 [DOI] [PubMed] [Google Scholar]
- 30.Huang J, Dorsey J, Chuikov S, Perez-Burgos L, Zhang X, Jenuwein T, Reinberg D, Berger SL. 2010. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J. Biol. Chem. 285:9636–9641. 10.1074/jbc.M109.062588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korkola JE, Houldsworth J, Chadalavada RS, Olshen AB, Dobrzynski D, Reuter VE, Bosl GJ, Chaganti RS. 2006. Down-regulation of stem cell genes, including those in a 200-kb gene cluster at 12p13.31, is associated with in vivo differentiation of human male germ cell tumors. Cancer Res. 66:820–827. 10.1158/0008-5472.CAN-05-2445 [DOI] [PubMed] [Google Scholar]
- 32.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. 2004. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia 6:1–6. 10.1016/S1476-5586(04)80047-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kupershmidt I, Su QJ, Grewal A, Sundaresh S, Halperin I, Flynn J, Shekar M, Wang H, Park J, Cui W, Wall GD, Wisotzkey R, Alag S, Akhtari S, Ronaghi M. 2010. Ontology-based meta-analysis of global collections of high-throughput public data. PLoS One 5(9):pii:e13066. 10.1371/journal.pone.0013066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sperger JM, Chen X, Draper JS, Antosiewicz JE, Chon CH, Jones SB, Brooks JD, Andrews PW, Brown PO, Thomson JA. 2003. Gene expression patterns in human embryonic stem cells and human pluripotent germ cell tumors. Proc. Natl. Acad. Sci. U. S. A. 100:13350–13355. 10.1073/pnas.2235735100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ezeh UI, Turek PJ, Reijo RA, Clark AT. 2005. Human embryonic stem cell genes OCT4, NANOG, STELLAR, and GDF3 are expressed in both seminoma and breast carcinoma. Cancer 104:2255–2265. 10.1002/cncr.21432 [DOI] [PubMed] [Google Scholar]
- 36.Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG. 2002. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol. Cell. Biol. 22:7405–7416. 10.1128/MCB.22.21.7405-7416.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koritzinsky M, Magagnin MG, van den Beucken T, Seigneuric R, Savelkouls K, Dostie J, Pyronnet S, Kaufman RJ, Weppler SA, Voncken JW, Lambin P, Koumenis C, Sonenberg N, Wouters BG. 2006. Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J. 25:1114–1125. 10.1038/sj.emboj.7600998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leong KG, Hu X, Li L, Noseda M, Larrivee B, Hull C, Hood L, Wong F, Karsan A. 2002. Activated Notch4 inhibits angiogenesis: role of beta 1-integrin activation. Mol. Cell. Biol. 22:2830–2841. 10.1128/MCB.22.8.2830-2841.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mousa SA, Mohamed S. 2004. Inhibition of endothelial cell tube formation by the low molecular weight heparin, tinzaparin, is mediated by tissue factor pathway inhibitor. Thromb. Haemost. 92:627–633. 10.1160/Th04-02-0069 [DOI] [PubMed] [Google Scholar]
- 40.Contois LW, Nugent DP, Caron JM, Cretu A, Tweedie E, Akalu A, Liebes L, Friesel R, Rosen C, Vary C, Brooks PC. 2012. Insulin-like growth factor binding protein-4 (IGFBP-4) differentially inhibits growth factor-induced angiogenesis. J. Biol. Chem. 287:1779–1789. 10.1074/jbc.M111.267732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones C, London N, Chen H, Park K, Sauvaget D, Stockton R, Wythe J, Suh W, Larrieu-Lahargue F, Mukouyama Y, Lindblom P, Seth P, Frias A, Nishiya N, Ginsberg M, Gerhardt H, Zhang K, Li D. 2008. Robo4 stabilizes the vascular network by inhibiting pathologic angiogenesis and endothelial hyperpermeability. Nat. Med. 14:448–453. 10.1038/nm1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zukowska-Grojec Z, Karwatowska-Prokopczuk E, Rose W, Rone J, Movafagh S, Ji H, Yeh Y, Chen WT, Kleinman HK, Grouzmann E, Grant DS. 1998. Neuropeptide Y: a novel angiogenic factor from the sympathetic nerves and endothelium. Circ. Res. 83:187–195. 10.1161/01.RES.83.2.187 [DOI] [PubMed] [Google Scholar]
- 43.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39:311–318. 10.1038/ng1966 [DOI] [PubMed] [Google Scholar]
- 44.Gutierrez A, Kentsis A, Sanda T, Holmfeldt L, Chen SC, Zhang J, Protopopov A, Chin L, Dahlberg SE, Neuberg DS, Silverman LB, Winter SS, Hunger SP, Sallan SE, Zha S, Alt FW, Downing JR, Mullighan CG, Look AT. 2011. The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood 118:4169–4173. 10.1182/blood-2010-11-318873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kubicek S, O'Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, Rea S, Mechtler K, Kowalski JA, Homon CA, Kelly TA, Jenuwein T. 2007. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 25:473–481. 10.1016/j.molcel.2007.01.017 [DOI] [PubMed] [Google Scholar]
- 46.Chang Y, Zhang X, Horton JR, Upadhyay AK, Spannhoff A, Liu J, Snyder JP, Bedford MT, Cheng X. 2009. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat. Struct. Mol. Biol. 16:312–317. 10.1038/nsmb.1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuroki S, Matoba S, Akiyoshi M, Matsumura Y, Miyachi H, Mise N, Abe K, Ogura A, Wilhelm D, Koopman P, Nozaki M, Kanai Y, Shinkai Y, Tachibana M. 2013. Epigenetic regulation of mouse sex determination by the histone demethylase Jmjd1a. Science 341:1106–1109. 10.1126/science.1239864 [DOI] [PubMed] [Google Scholar]
- 48.Herzog M, Josseaux E, Dedeurwaerder S, Calonne E, Volkmar M, Fuks F. 2012. The histone demethylase Kdm3a is essential to progression through differentiation. Nucleic Acids Res. 40:7219–7232. 10.1093/nar/gks399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Looijenga LH, Stoop H, de Leeuw HP, de Gouveia Brazao CA, Gillis AJ, van Roozendaal KE, van Zoelen EJ, Weber RF, Wolffenbuttel KP, van Dekken H, Honecker F, Bokemeyer C, Perlman EJ, Schneider DT, Kononen J, Sauter G, Oosterhuis JW. 2003. POU5F1 (OCT3/4) identifies cells with pluripotent potential in human germ cell tumors. Cancer Res. 63:2244–2250 [PubMed] [Google Scholar]
- 50.Hart AH, Hartley L, Parker K, Ibrahim M, Looijenga LH, Pauchnik M, Chow CW, Robb L. 2005. The pluripotency homeobox gene NANOG is expressed in human germ cell tumors. Cancer 104:2092–2098. 10.1002/cncr.21435 [DOI] [PubMed] [Google Scholar]
- 51.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. 2008. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 40:499–507. 10.1038/ng.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cantz T, Key G, Bleidissel M, Gentile L, Han DW, Brenne A, Scholer HR. 2008. Absence of OCT4 expression in somatic tumor cell lines. Stem Cells 26:692–697. 10.1634/stemcells.2007-0657 [DOI] [PubMed] [Google Scholar]
- 53.Mimura I, Nangaku M, Kanki Y, Tsutsumi S, Inoue T, Kohro T, Yamamoto S, Fujita T, Shimamura T, Suehiro J, Taguchi A, Kobayashi M, Tanimura K, Inagaki T, Tanaka T, Hamakubo T, Sakai J, Aburatani H, Kodama T, Wada Y. 2012. Dynamic change of chromatin conformation in response to hypoxia enhances the expression of GLUT3 (SLC2A3) by cooperative interaction of hypoxia-inducible factor 1 and KDM3A. Mol. Cell. Biol. 32:3018–3032. 10.1128/MCB.06643-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. 2009. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat. Genet. 41:246–250. 10.1038/ng.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gyory I, Wu J, Fejer G, Seto E, Wright KL. 2004. PRDI-BF1 recruits the histone H3 methyltransferase G9a in transcriptional silencing. Nat. Immunol. 5:299–308. 10.1038/ni1046 [DOI] [PubMed] [Google Scholar]
- 56.Banck MS, Li S, Nishio H, Wang C, Beutler AS, Walsh MJ. 2009. The ZNF217 oncogene is a candidate organizer of repressive histone modifiers. Epigenetics 4:100–106. 10.4161/epi.4.2.7953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beyer S, Kristensen M, Jensen K, Johansen J, Staller P. 2008. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J. Biol. Chem. 283:36542–36552. 10.1074/jbc.M804578200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, Moon EJ, Kim HS, Lee SK, Chung HY, Kim CW, Kim KW. 2001. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 7:437–443. 10.1038/86507 [DOI] [PubMed] [Google Scholar]
- 59.Gorospe M, Tominaga K, Wu X, Fahling M, Ivan M. 2011. Post-transcriptional control of the hypoxic response by RNA-binding proteins and microRNAs. Front. Mol. Neurosci. 4:7. 10.3389/fnmol.2011.00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cao P, Deng Z, Wan M, Huang W, Cramer SD, Xu J, Lei M, Sui G. 2010. MicroRNA-101 negatively regulates Ezh2 and its expression is modulated by androgen receptor and HIF-1alpha/HIF-1beta. Mol. Cancer 9:108. 10.1186/1476-4598-9-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sunamura M, Duda DG, Ghattas MH, Lozonschi L, Motoi F, Yamauchi J, Matsuno S, Shibahara S, Abraham NG. 2003. Heme oxygenase-1 accelerates tumor angiogenesis of human pancreatic cancer. Angiogenesis 6:15–24. 10.1023/A:1025803600840 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.