

Abstract
Congenital dyserythropoietic anemia type II (CDAII) is an autosomal recessive disease of ineffective erythropoiesis characterized by increased bi/multinucleated erythroid precursors in the bone marrow. CDAII results from mutations in SEC23B. The SEC23 protein is a core component of coat protein complex II-coated vesicles, which transport secretory proteins from the endoplasmic reticulum to the Golgi apparatus. Though the genetic defect underlying CDAII has been identified, the pathophysiology of this disease remains unknown. We previously reported that SEC23B-deficient mice die perinatally, exhibiting massive pancreatic degeneration, with this early mortality limiting evaluation of the adult hematopoietic compartment. We now report that mice with SEC23B deficiency restricted to the hematopoietic compartment survive normally and do not exhibit anemia or other CDAII characteristics. We also demonstrate that SEC23B-deficient hematopoietic stem cells (HSC) do not exhibit a disadvantage at reconstituting hematopoiesis when compared directly to wild-type HSC in a competitive repopulation assay. Secondary bone marrow transplants demonstrated continued equivalence of SEC23B-deficient and WT HSC in their hematopoietic reconstitution potential. The surprising discordance in phenotypes between SEC23B-deficient mice and humans may reflect an evolutionary shift in SEC23 paralog function and/or expression, or a change in a specific COPII cargo critical for erythropoiesis.
INTRODUCTION
Congenital dyserythropoietic anemia type II (CDAII), also known as hereditary erythroblastic multinuclearity with a positive acidified serum lysis test (HEMPAS), is an autosomal recessive disease characterized clinically by mild to moderate anemia resulting from ineffective erythropoiesis (median hemoglobin, 9.1 to 9.8 g/dl), the presence of bi/multinucleated erythroblasts in the bone marrow (BM), jaundice from indirect hyperbilirubinemia, and splenomegaly (1, 2). CDAII is distinguished from other congenital anemias by a characteristic double-membrane appearance in red blood cell (RBC) electron microscopy resulting from the residual endoplasmic reticulum (ER) (3), a faster-migrating and narrower band on SDS-PAGE for the RBC membrane protein band 3, and lysis of RBCs in some, but not all, acidified normal sera (Ham's test) (1, 2).
CDAII results from homozygous or compound heterozygous mutations in SEC23B (4), one of the two mammalian paralogs of SEC23. SEC23 is a core component of the coat protein complex II (COPII)-coated vesicles that transport cargo proteins from the ER to the Golgi apparatus (2, 5). More than 60 different SEC23B mutations have been identified in CDAII patients (4, 6–14), affecting every domain of the protein (7). No patient with two nonsense SEC23B mutations has been reported, suggesting that complete loss of SEC23B may be lethal. Homozygosity for specific missense mutations in SEC23A, the paralog of SEC23B, results in cranio-lenticulo-sutural dysplasia (15), which is thought to be due to defective collagen secretion.
Despite the identification of the genetic defect, the molecular mechanism by which deficiency of SEC23B results in the CDAII phenotype remains unknown (2). Nearly all proteins destined for secretion from the cell or export to the cell membrane or lysosome (∼1/3 of the mammalian proteome) are dependent on COPII vesicles for transport from the ER to the Golgi apparatus. Thus, it is surprising that deficiency of a key, ubiquitously expressed component of the COPII coat, SEC23B, results in a phenotype that is apparently restricted to the red blood cell. Reports of curative allogeneic bone marrow transplantation for CDAII suggest that the mechanism responsible for the erythroid defect is intrinsic to the hematopoietic compartment (9, 16, 17).
We previously reported that mice homozygous for a Sec23b gene trap allele (Sec23bgt/gt) die within 24 h of birth, exhibiting degeneration of the pancreas and other professional secretory tissues (18). The perinatal lethality precluded assessment of adult hematopoietic function in these mice. We now report that chimeric mice with SEC23B deficiency restricted to the hematopoietic compartment can support the normal production of adult RBCs, with no apparent abnormality in hematopoiesis. Competitive hematopoietic stem cell (HSC) transplantation assays also fail to demonstrate a disadvantage of Sec23bgt/gt HSCs at reconstituting hematopoiesis compared to wild-type (WT) HSCs.
MATERIALS AND METHODS
Generation of SEC23B-deficient mice.
Two Sec23b mutant mouse lines, one with a gene trap (gt) cassette insertion into Sec23b intron 19 (Sec23bgt) and the second with a conditional gt insertion in Sec23b intron 4 (Sec23bcgt), were generated as previously described (Fig. 1A and B) (18). All Sec23bgt mice used in this study were generated from heterozygous mice backcrossed to C57BL/6J mice for >10 generations. The Sec23bcgt allele was derived from C57BL/6J embryonic stem cells (18) and maintained on a pure background by backcrosses exclusively to C57BL/6J mice. Sec23bcgt/+ mice were crossed to a mouse ubiquitously expressing FLPe under the control of the human β-actin promoter (β-actin FLP; stock number 005703; Jackson Laboratory) to excise the gt cassette and generate the Sec23b floxed allele (Sec23bfl), with exons 5 and 6 flanked by loxP sites (Fig. 1B). Mice with complete deficiency of SEC23B (Sec23b−) were generated by crossing the Sec23bfl allele to a mouse expressing Cre recombinase driven by an EIIA promoter (EIIA Cre; stock number 003724; Jackson Laboratory). Deletion of Sec23b exons 5 and 6 results in a frameshift and downstream stop codon in exon 7. Mice were housed at the University of Michigan and all procedures were in accordance with the regulations of the Animal Care and Use Committee.
FIG 1.

Sec23b mutant alleles. (A) Schematic of the first Sec23b gene trap allele, demonstrating a gene trap insertion into intron 19 (18). SA, splice acceptor cassette; β-Geo, β-galactosidase-neo fusion; pA, polyadenylation sequence. (B) The Sec23b conditional gene trap allele (Sec23bcgt) contains a gene trap insertion in intron 4 flanked by 2 FRT sites. Mice carrying this allele were crossed to β-actin FLP transgenic mice. Mice heterozygous for the resulting Sec23b floxed allele (Sec23b+/fl) were crossed to EIIACre transgenic mice to excise exons 5 and 6 and generate the Sec23b null allele (Sec23b−). Gray boxes represent exons (with the exon number indicated in the box). F1, F2, R1, cgtB1, and cgtR1 represent Sec23b genotyping primers. En2 SA, splice acceptor of mouse En2 exon 2; IRES, encephalomyocarditis virus internal ribosomal entry site; lacZ, Escherichia coli β-galactosidase gene; pA, simian virus polyadenylation signal; βact:neo, human β-actin promoter-driven neomycin cassette. (C) A three-primer PCR assay (with F1, cgtB1, cgtR1) distinguished the Sec23bcgt allele (F1:cgtB1; 344 bp) and the WT allele (F1:cgtR1; 475 bp). (D) A three-primer PCR assay (with F1, F2, R1) distinguished the alleles Sec23b+ (F2:R1; 235 bp), Sec23bfl (F2:R1; 269 bp), and Sec23b− (F1:R1; 336 bp). Locations of the primers are indicated in panels A and B. The gene structures depicted in panels A and B are not according to scale.
PCR genotyping.
Genotyping for the Sec23bgt allele was performed as previously described (18). The Sec23bcgt allele was genotyped in a three-primer PCR assay using a forward primer (F1) located in Sec23b intron 4 upstream of the insertion site and two reverse primers, one (cgtB1) located in the gene trap insertion cassette between the two FRT sites and the second (cgtR1) located in intron 4 downstream of the FRT sites. This PCR results in a 475-bp product from the WT allele (F1:cgtR1) and a 344-bp product from the Sec23bcgt allele (F1:cgtB1), which are resolved by 2% (wt/vol) agarose gel electrophoresis (Fig. 1C). Genotyping for the Sec23bfl and Sec23b− alleles was performed with a three-primer competitive PCR assay consisting of the forward primer F1, a second forward primer (F2) located in intron 6 between the two loxP sites, and a common reverse primer (R1) located in intron 6 downstream of the insertion site. This reaction produces a 235-bp product from the WT allele (F2:R1), a 269-bp product from the Sec23bfl allele (F2:R1), and a 336-bp product from the Sec23b− allele (F1:R1), which are resolved by 3% agarose gel electrophoresis (Fig. 1D). Locations of the genotyping primers are indicated in Fig. 1A and B. Primer sequences are shown in Table 1.
TABLE 1.
Genotyping and qPCR primer sequences
| Primer | Primer sequence (5′ → 3′) |
|---|---|
| Sec23b F1 | ATAGACCAGGCTGGCCTCAGTC |
| Sec23b cgt B1 | CCACAACGGGTTCTTCTGTT |
| Sec23b cgt R1 | CGAGCACAGAGAGACCCAAT |
| Sec23b R | CAAGTGAGTGCCTCCTCACA |
| Sec23b F2 | AACAGCCCAGGTGACTAGGA |
| Sec23b qPCR F | CCCTACGTCTTTCAGATTGTCA |
| Sec23b qPCR R | CGGGCAAAATGGTGTCTATAA |
| Sec24a qPCR F | GAGCAGAGATGGAGCGTTCCT |
| Sec24a qPCR R | TTCTTCCAACCCAAAGCATCA |
| Sec24b qPCR F | GACCCGAGAAGGCGCTTT |
| Sec24b qPCR R | TTTGCCAACCCAAATGTAGAAA |
| Sec24c qPCR F | TTATGCGGGTTCGGACAAG |
| Sec24c qPCR R | CTCATATAGAAAGCGCCAAAGAAAT |
| Sec24d qPCR F | CTCATATAGAAAGCGCCAAAGAAAT |
| Sec24d qPCR R | TCATGTACACAGGCAGCACCTT |
| Sar1a qPCR F | TCGGTGGGCATGAGCAA |
| Sar1a qPCR R | GCCATTAATCGCTGGGAGATAA |
| Sar1b qPCR F | GGGTGGGCACGTGCAA |
| Sar1b qPCR R | TGCCATTGATAGCAGGAAGGT |
| TRAPPC3 qPCR F | GCACGGAGAGCAAGAAAATGA |
| TRAPPC3 qPCR R | GGTGACAAGCGCTCCATAGG |
| Beta-actin qPCR F | GATCTGGCACCACACCTTCT |
| Beta-actin qPCR R | GGGGTGTTGAAGGTCTCAAA |
| GAPDH qPCR F | TGTGTCCGTCGTGGATCTGA |
| GAPDH qPCR R | ACCACCTTCTTGATGTCATCATACTT |
FLC transplants.
Timed matings were performed by intercrossing Sec23b+/gt mice or Sec23b+/− mice. The following morning, designated embryonic day 0.5 (E0.5), mating pairs were separated. Pregnant female mice were euthanized at E17.5 postcoitus. Recovered fetuses were separated and placed individually in petri dishes on ice under sterile conditions. A tail biopsy specimen was obtained from each fetus for genotyping. Fetal livers were individually disrupted, and dispersed cells were suspended in RPMI 1640 (Gibco) supplemented with 2% fetal bovine serum (FBS) at 4°C. Fetal liver cells (FLC) were washed twice in RPMI 1640 with 2% FBS and then suspended in 65% RPMI 1640, 25% FBS, and 10% dimethyl sulfoxide, frozen at −80°C overnight, and stored in vapor-phase liquid nitrogen at −186°C.
Six- to 12-week-old C57BL/6J recipient mice were lethally irradiated with two doses of 550 rads separated by 3 h in a Cs Gammacell 40 Exactor irradiator (MDS Nordion). Three hours after completion of irradiation, frozen WT and SEC23B-deficient FLC (Sec23bgt/gt or Sec23b−/− FLC) were thawed in a 37°C water bath. A total of 106 cells were suspended in 300 μl RPMI 1640 with 2% FBS and injected into the retro-orbital venous sinuses of recipient mice. For each transplant experiment, control mice were injected with medium only. No control mice survived beyond 12 days. Transplanted mice were provided with acidified water (pH 2.35) for 3 weeks posttransplantation.
Competitive FLC transplants.
C57BL/6J mice expressing high levels of green fluorescent protein (GFP) in all tissues, including hematopoietic cells (19) (under the control of the human ubiquitin C promoter [UBC-GFP mice]) were obtained from the Jackson Laboratory (stock number 004353). FLC from crosses between male mice homozygous for the UBC-GFP transgene (UBC-GFPtg/tg) and female C57BL/6J mice were harvested at day 17.5 postcoitus and stored as described above. UBC-GFPtg/+ FLC were mixed in a 1:1 ratio with either Sec23bgt/gt FLC (experimental arm) or WT FLC (control arm) and transplanted into lethally irradiated C57BL/6J recipients as described above.
Bone marrow cells were isolated from the hind limbs of each chimeric mouse. The number of GFP-negative cells per 2 hind limbs was calculated for each hematopoietic lineage by multiplying the ratio of GFP− to GFP+ cells in each lineage by the total number of cells per lineage. The number of GFP− cells per two hind limbs should be proportional to the contribution of GFP− cells to each lineage, corresponding to Sec23bgt/gt cells in the experimental arm and WT cells in the control arm.
GFP− mature myeloid cells (Mac1+ Gr1+) were sorted by fluorescence-activated cell sorting (FACS) from bone marrow samples harvested from chimeric recipient mice. Myeloid cells were genotyped for Sec23b.
For secondary transplants, whole BM cells were harvested from these chimeric recipient mice 20 weeks after the competitive FLC transplant, and 2 × 106 cells were transplanted into lethally irradiated secondary C57BL/6J recipients.
CBC and BM analyses.
Twenty microliters of blood was drawn from the retro-orbital venous sinuses of mice anesthetized with isoflurane. Blood was diluted 1:10 in 5% bovine serum albumin (BSA) in phosphate-buffered saline (PBS; pH 7.4. Complete blood counts (CBC) were performed on the Advia120 whole-blood analyzer (Siemens) according to the manufacturer's instructions.
Following pentobarbital-induced anesthesia, BM was flushed from femurs and tibias of each mouse with either Hanks' balanced salt solution (Gibco) or RPMI 1640 supplemented with 5% FBS. BM cells were used for flow cytometry (below), and a subset (∼160,000 cells) was collected by centrifugation in a Cytospin 4 cytocentrifuge (Thermo Scientific), stained with the HEMA 3 kit (Fisher), and examined under light microscopy. BM cytospin samples were evaluated by an investigator blinded to mouse genotype.
Flow cytometry.
Peripheral blood or BM single-cell suspensions were incubated with various antibodies. The following antibodies were obtained from BioLegend, eBiosciences, or BD Biosciences: anti-Ter119, Gr1 (RB6-8C5), Mac1 (M1/70), CD3 (145-2C11), CD16/CD32 (2.4G2), CD45R/B220 (RA3-6B2), CD150 (TC15-12F12.2), Sca1 (D7), CD117 (2B8), CD48 (BCM1), CD19 (6D5), T cell receptor β (TCRβ; H57-597), CD8 (53-6.7), CD11c (N418), CD4 (RM4-4), NK1.1 (PK136), and TCRγ/δ (GL3). The following antibody cocktail was used to exclude lineage-positive (Lin+) cells: anti-Ter119, CD11b, CD11c, Gr1, C220, CD19, CD3, TCRβ, TCRγ/δ, CD8, and NK1.1. Stained cells were analyzed by flow cytometry using FACSCanto II, FACSAria II, or FACSAria III flow cytometers (Becton Dickinson Biosciences). Dead cells were excluded via 4′,6-diamidino-2-phenylindole staining where appropriate (Sigma). Files were analyzed with the FlowJo software (Tree Star).
Cell sorting.
Ter119+ erythroid precursors were sorted from reconstituted bone marrow samples of recipient mice using a FACSAria II. Ter119+ erythroid precursors were also purified from E17.5 FLC. Mononuclear cells prepared from E17.5 livers were incubated with allophycocyanin (APC)-conjugated anti-Ter119+ antibody (Biolegend) for 30 min on ice, washed twice with PBS containing 4% FBS, and then treated with anti-APC-conjugated magnetic beads (Miltenyi Biotech) for 15 min. Cells were then washed once and suspended in PBS plus 4% FBS. Ter119+ cells were collected using LS MACS separation columns (Miltenyi Biotech) mounted on a magnet stand, according to the manufacturer's instructions.
RBC ghost preparation.
Seventy microliters of peripheral blood was centrifuged at 2,300 × g. The pellet was washed twice with PBS (pH 7.4) and then lysed by suspension in ghost lysis buffer (5 mM Na2PO4, 1.3 mM EDTA; pH 7.6) containing protease inhibitor (one protease inhibitor tablet [stock number 11873580001; Roche] per 50 ml ghost lysis buffer). Lysates were centrifuged at 16,000 × g, and the supernatants containing the RBC membrane fraction were collected and washed 4 to 6 times in ghost lysis buffer. RBC ghosts were stored at −80°C in lysis buffer.
Electron microscopy.
Cells were fixed in 2.5% glutaraldehyde in 0.1 M Sorensen's buffer (pH 7.4) overnight at 4°C. After 2 rinses with 10 to 20 ml of Sorensen's buffer, cells were fixed with 1% osmium tetroxide in 0.1 M Sorensen's buffer, rinsed in double-distilled water, and then en bloc stained with aqueous 3% uranyl acetate for 1 h. Cells were dehydrated in ascending concentrations of ethanol, rinsed twice in 100% ethanol, and embedded in epoxy resin. Samples were ultrathin sectioned at 70 nm in thickness and stained with uranyl acetate and lead citrate. Sections were examined on a Philips CM100 electron microscope at 60 kV. Images were recorded digitally using a Hamamatsu ORCA-HR digital camera system operated with AMT software (Advanced Microscopy Techniques Corp., Danvers, MA).
Western blotting.
Proteins were separated by SDS gel electrophoresis using 4 to 20% gradient Tris-glycine gels (Invitrogen) and Tris-glycine running buffer or by using 4 to 12% gradient bis-Tris gels (Invitrogen) and morpholinepropanesulfonic acid running buffer (Invitrogen). Proteins were then transferred onto nitrocellulose membranes (Bio-Rad). For X-ray development, membranes were blocked in 5% (wt/vol) milk–Tris-buffered saline with Tween (TBST), probed with primary antibody, washed 3 times in TBST, probed with peroxidase-coupled secondary antibodies (Thermo Scientific), washed 3 times in TBST, and developed using the Western Lightning Plus-ECL system (Perkin-Elmer). Quantitative Western blot assays were performed using the Odyssey system (Licor Biosciences) according to the manufacturer's instructions. Secondary antibodies utilized were IRDye 680RD and IRDye 800 CW. Band intensities were quantified using the Odyssey software. SEC23A band intensity was normalized to that of beta-actin or RalA.
Antibodies.
Anti-SEC23B and anti-SEC23A antibodies were generated in rabbit against peptides LTKSAMPVQQARPAQPQEQP and DNAKYVKKGTKHFEA, respectively. Anti-Band3 and anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) antibodies were obtained from Millipore. Antiactin antibody was obtained from Santa Cruz Biotechnology.
Quantitative RT-PCR.
RNA was isolated with TRIzol. Reverse transcription (RT) was performed using the SuperScript first-strand synthesis system for RT-PCR (Invitrogen) with random primers. Real-time PCR amplification was performed in triplicates with the Power SYBR green PCR master mix (Applied Biosystems) and the Applied Biosystems 7900HT Fast real-time PCR system. Relative gene expression was calculated using the 2−ΔΔCT method. Beta-actin and GAPDH were used as internal controls. Two samples of each genotype were analyzed, each in triplicate. Quantitative PCR (qPCR) primer sequences are listed in Table 1.
RESULTS
Transplantation of SEC23B-deficient HSC does not result in a CDAII phenotype.
SEC23B-deficient mice die perinatally, exhibiting degeneration of their professional secretory tissues but no evidence of anemia at birth (18). To assess the impact of SEC23B deficiency on adult mouse hematopoietic function, equal numbers of FLC collected from either Sec23bgt/gt or WT E17.5 embryos were transplanted into lethally irradiated C57BL/6J recipient mice. Livers harvested from Sec23bgt/gt and WT E17.5 embryos exhibited no significant differences in total cell counts or numbers of long-term HSC (ckit+ Sca1+ CD48− CD150+ Lin−) (20) as measured by flow cytometry (Fig. 2A, B, and C). Hemoglobin (Fig. 3A) and hematocrit (Fig. 3B) levels measured at weeks 6, 8, 12, and 25 posttransplantation were all within the normal range and indistinguishable between mice transplanted with Sec23bgt/gt FLC and recipients of WT FLC, as were spleen weights (Fig. 3C) and bone marrow myeloid-to-erythroid ratios (Fig. 3D). There was also no increase in the number of bi/multinucleated RBC precursors observed in the recipients of Sec23bgt/gt BM (Fig. 3E).
FIG 2.

FACS analysis of E17.5 Sec23bgt/gt and WT FLC. (A) Livers were harvested from 3 Sec23bgt/gt and 3 WT E17.5 embryos. Flow cytometry on FLC single-cell suspensions demonstrated equivalent total numbers of recovered cells from Sec23bgt/gt and WT fetal livers. Each liver is represented by 1 point, and horizontal lines indicate mean values for each group. (B) The number of long-term hematopoietic stem cells (ckit+ Sca1+ CD48− CD150+ Lin−) was equivalent for Sec23bgt/gt and WT fetal livers. (C) FACS distributions used to calculate numbers of long-term hematopoietic stem cells in panel B.
FIG 3.

Transplant recipients of Sec23bgt/gt FLC do not exhibit CDAII. (A and B) Mice transplanted with Sec23bgt/gt FLC demonstrated equivalent hemoglobin (A) and hematocrit (B) levels compared to mice transplanted with control WT FLC over the course of 25 weeks of posttransplantation follow-up (P > 0.05 for all time points). n = 5 to 7 mice per group. Error bars represent standard deviations. (C) Spleens harvested from transplant recipients of Sec23bgt/gt and WT FLC were equivalent in weights. Recipients of Sec23bgt/gt FLC exhibited equivalent myeloid:erythroid ratios (D) and number of bi/multinucleated RBC precursors (E; evaluated independently by two investigators) compared to control mice transplanted with WT FLC. Each dot represents results from one mouse. Horizontal lines indicate means, and error bars indicate standard deviations.
Characteristic RBC abnormalities in humans with CDAII include a “double-membrane” appearance on transmission electron microscopy and narrower band size together with a shift in the mobility of membrane protein band 3 on sodium dodecyl sulfate-polyacrylamide gel electrophoresis. RBC from mice transplanted with Sec23bgt/gt FLC did not exhibit either of these abnormalities (Fig. 4A to C).
FIG 4.

RBC from mice transplanted with Sec23bgt/gt FLC do not exhibit a band 3 glycosylation defect or a double RBC membrane. (A) RBC ghosts were isolated from Sec23bgt/gt and WT RBC and fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Coomassie blue stain revealed no difference in the appearance of the RBC membrane protein band 3 in Sec23bgt/gt RBC ghosts. Each lane represents a sample from a different individual mouse. (B) Similarly, band 3 protein appeared indistinguishable on Western blotting between Sec23bgt/gt and WT RBC ghosts. (C) Sec23bgt/gt RBC lack the “double-membrane” appearance on transmission electron microscopy characteristic of human CDAII. RBC were evaluated at three different magnifications (indicated in the right lower corner of each image). Arrows indicate RBC membranes.
SEC23B-deficient FLC and WT FLC are equivalent in reconstituting erythropoiesis.
To assess whether there was a more subtle hematopoietic defect, SEC23B-deficient FLC were tested for their ability to reconstitute hematopoiesis compared to WT FLC in a competitive repopulation assay. In this experiment, Sec23bgt/gt FLC were mixed with UBC-GFPtg/+ Sec23b+/+ FLC in a 1:1 ratio and cotransplanted into lethally irradiated C57BL/6J recipient mice. Following engraftment, hematopoietic cells from recipient mice were characterized by GFP expression to distinguish cells of Sec23bgt/gt or WT FLC origin. Control mice were cotransplanted with a 1:1 ratio of WT FLC with or without the UBC-GFPtg/+ transgene.
Over the course of 18 weeks of follow-up, WT FLC exhibited no competitive advantage at reconstituting erythropoiesis compared to SEC23B-deficient FLC (Fig. 5A). Similarly, no defects were observed in the ability of Sec23bgt/gt FLC to differentiate into neutrophils (Fig. 6A) or lymphocytes (Fig. 6B and C).
FIG 5.
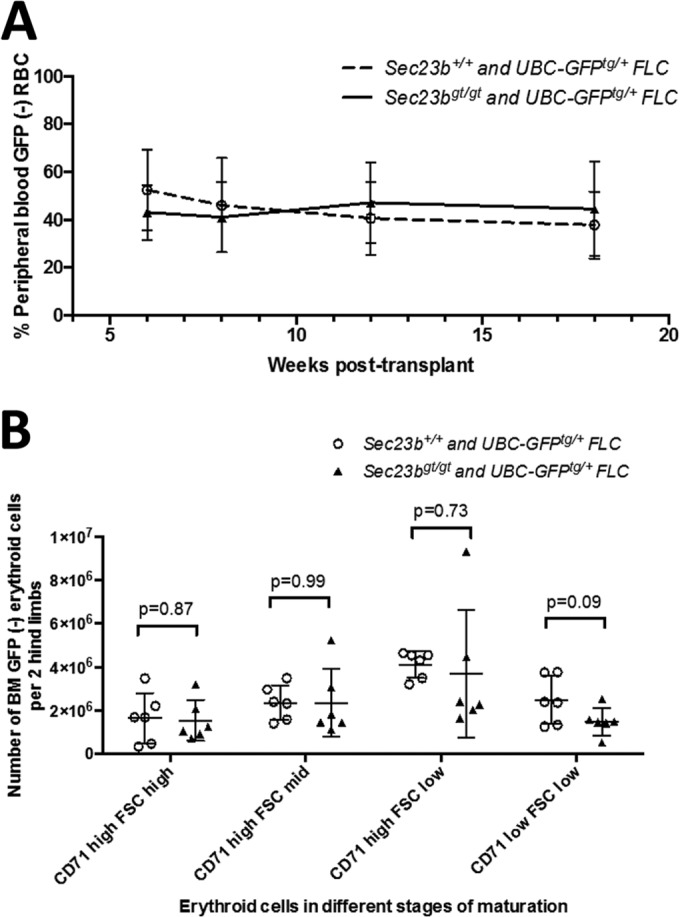
Sec23bgt/gt FLC do not exhibit a competitive disadvantage at reconstituting erythropoiesis compared to WT FLC. C57BL/6J mice were cotransplanted with a 1:1 mix of GFP− Sec23bgt/gt FLC and UBC-GFPTg+ Sec23b+/+ FLC in a competitive transplant assay (experimental arm). Following engraftment, the percentage of GFP− cells in the peripheral blood/BM of recipient mice indicated the percentage of cells derived from Sec23bgt/gt FLC. Control mice were cotransplanted with a 1:1 mix of WT GFP− and GFPTg+ Sec23b+/+ FLC. (A) By peripheral blood FACS, Ter119+ RBC were determined to be derived from both Sec23bgt/gt and WT FLC. Sec23bgt/gt RBC persisted at a stable level throughout the 18-week follow-up period, suggesting no competitive advantage to WT FLC over Sec23bgt/gt FLC at reconstituting erythropoiesis. (B) In the BM, the contribution of GFP− Sec23bgt/gt cells to the populations of Ter119+ erythroid precursors was equivalent to the contribution of the GFP− WT cells in the control arm. Erythroid cells were further stratified by forward scatter and CD71 expression, to identify more primitive progenitors as larger cells expressing higher levels of CD71, more mature smaller cells with lower expression of CD71, and intermediate cells. Sec23bgt/gt cells contributed to all subsets of erythroid cells.
FIG 6.

Analysis of peripheral blood and BM hematopoietic compartments of mice cotransplanted with a 1:1 mix of GFP− Sec23bgt/gt FLC and UBC-GFPTg+ Sec23b+/+ FLC (experimental arm) and control mice cotransplanted with a 1:1 mix of GFP− and GFPTg+ Sec23b+/+ FLC. (A to C) By peripheral blood FACS, Mac1+ Gr1+ neutrophils (A), B220+ B lymphocytes (B), and CD3+ T lymphocytes (C) were found to be derived from both Sec23bgt/gt and WT FLC. The Sec23bgt/gt peripheral blood cells persisted at a stable level throughout the 18-week follow-up period, suggesting no competitive advantage to WT FLC over Sec23bgt/gt FLC at reconstituting hematopoiesis. (D and E) In the BM, the contributions of GFP− Sec23bgt/gt FLC to the long-term hematopoietic stem cells (D; ckit+ Sca1+ CD48− CD150− Lin−) and to myeloid cells (E; Mac1+ GR1+) in the experimental arm were equivalent to the contributions of the GFP− WT cells in the control arm. (F) Similarly, the contribution of GFP(-) Sec23bgt/gt T lymphocytes in the thymus was equivalent to that of GFP− WT cells in the control arm. *, there was a trend for some subsets of GFP− Sec23bgt/gt T lymphocytes to be underrepresented; however, this did not reach statistical significance after correction for multiple observations, based on the Holm-Sidak method or the Bonferroni method. ISP, immature single-positive cells; DP, CD4+ CD8+ double-positive T lymphocytes; NS, not significant. (G) In contrast, GFP− Sec23bgt/gt CD19+ CD220+ BM B lymphocytes were underrepresented. Each point represents one mouse. Lines represent mean values for each group, and error bars indicate standard deviations.
Eighteen weeks after transplantation, reconstituted bone marrows and thymi were harvested from transplant recipients, and the relative contributions of Sec23bgt/gt and WT cells to each hematopoietic compartment were evaluated. Erythroid cells were stratified by forward scatter and CD71 expression to identify primitive progenitors (larger cells expressing higher levels of CD71), mature cells (smaller cells expressing low levels of CD71), and erythroid cells in intermediate stages of development (average or small-sized cells expressing high levels of CD71) (21). Sec23bgt/gt and WT cells contributed similarly to all populations of erythroid cells examined (Fig. 5B).
The contributions of GFP− Sec23bgt/gt cells to the populations of long-term HSC (Fig. 6D) and myeloid cells (Fig. 6E) in the BM and to all subgroups of T lymphocytes (Fig. 6F) in the thymus were equivalent to those of GFP+ WT cells. There was a trend for some subsets of T lymphocytes to be underrepresented (Sec23bgt/gt CD8+ TCRβ immature single-positive cells, CD4+ CD8+ double-positive cells, and CD4+ T lymphocytes); however, this did not reach statistical significance after correction for multiple observations. In contrast, BM Sec23bgt/gt B lymphocytes (Fig. 6G) were underrepresented relative to their WT counterparts (P = 0.005).
To exclude the possibility that the reconstituted GFP− hematopoietic cells in recipient mice were derived from host reconstitution rather than Sec23bgt/gt FLC, GFP− mature myeloid cells (Mac1+ Gr1+) were FACS sorted from bone marrow samples of mice cotransplanted with GFP− Sec23bgt/gt FLC and GFP+ WT FLC. The genotype of the isolated myeloid cells was confirmed to be Sec23bgt/gt (Fig. 7).
FIG 7.
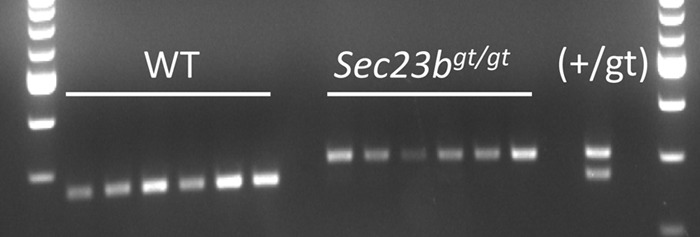
Reconstituted GFP− hematopoietic cells in mice cotransplanted with a 1:1 mix of GFP− Sec23bgt/gt FLC and UBC-GFPTg+ Sec23b+/+ FLC were derived from Sec23bgt/gt FLC and not from host reconstitution. GFP− myeloid cells isolated from mice transplanted with a 1:1 ratio of UBC-GFPtg/+ FLC and Sec23bgt/gt FLC and from control mice transplanted with 1:1 ratio of UBC-GFPtg/+ FLC and WT FLC were genotyped for Sec23bgt/gt. Six mice from each group were examined. Each lane represents the genotype of myeloid cells isolated from a single mouse. The genotype of a Sec23b+/gt control DNA is shown. The lower and upper bands correspond to the expected PCR products for the Sec23b WT and gt alleles, respectively.
SEC23B-deficient HSC and WT HSC are equivalent in their hematopoietic reconstitution potentials.
To further test for the hematopoietic reconstitution potential of Sec23bgt/gt HSC, bone marrows chimeric for Sec23bgt/gt and WT HSC were harvested from the competitive FLC transplant recipients (described above) and transplanted into secondary WT recipients. Over the course of 18 weeks of follow-up, the contributions of Sec23bgt/gt and WT HSC to the reconstituted erythroid, myeloid, B cell, and T cell compartments were equivalent (Fig. 8A and Fig. 9A to C). Bone marrows from a subset of secondary transplant recipients were analyzed at 26 weeks posttransplantation. Persistence of GFP− Sec23bgt/gt erythroid cells in all stages of erythroid differentiation (Fig. 8B) and Sec23bgt/gt long-term HSC (Fig. 8C) was observed.
FIG 8.
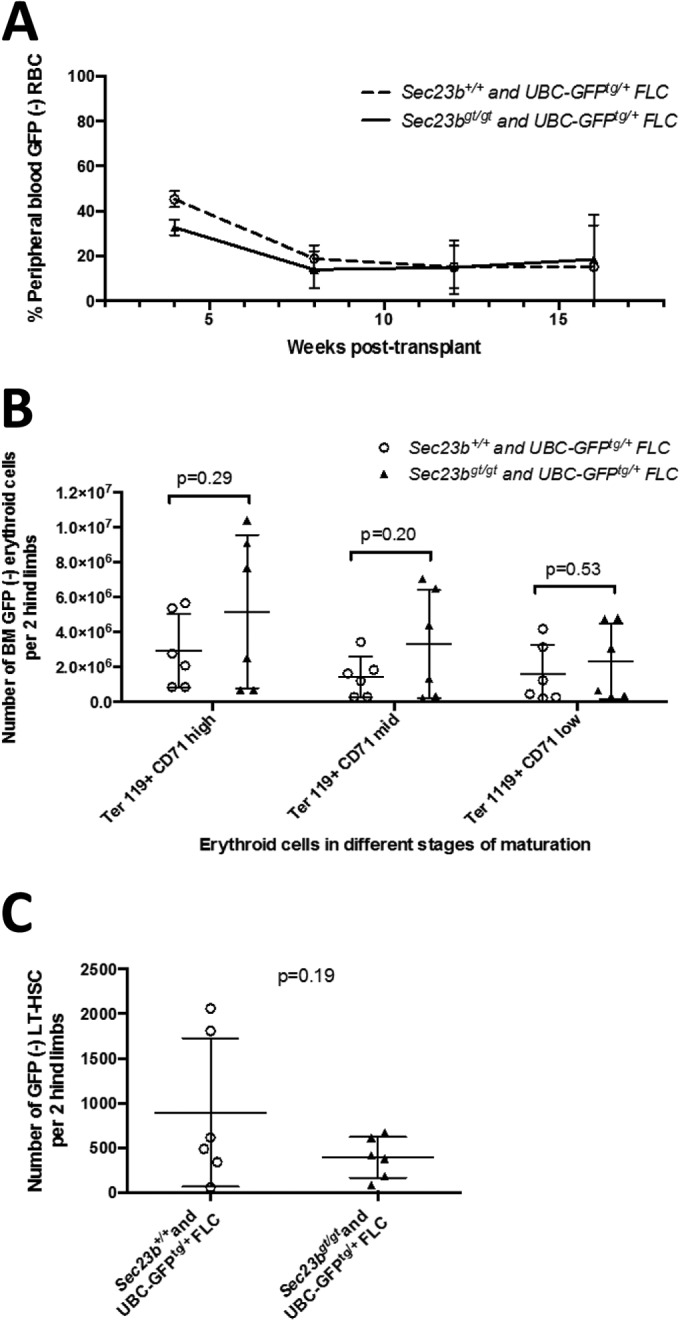
Secondary bone marrow transplantation experiments demonstrate continued equivalence of Sec23bgt/gt and WT FLC at reconstituting erythropoiesis. BM harvested from mice chimeric for GFP− Sec23bgt/gt and GFP+ WT hematopoietic cells were transplanted into lethally irradiated secondary recipients (experimental arm). Control mice were transplanted with BM harvested from mice chimeric for GFP− and GFP+ WT hematopoietic cells. (A) By peripheral blood FACS, the contribution of Sec23bgt/gt GFP− cells to the population of Ter119+ RBC in the experimental arm was equivalent to the contribution of WT GFP− cells in control mice over the course of 18 weeks of follow-up. Peripheral blood analysis was performed on 13 to 15 mice per group. Error bars represent standard deviations. (B and C) Bone marrow cells were isolated from both hind limbs of each secondary transplant recipient mouse. The contributions of Sec23bgt/gt GFP− Ter119+ erythroid cells at various stages of differentiation (B) and hematopoietic stem cells (C; ckit+ Sca1+ CD48− CD150− Lin−) in the experimental mice were equivalent to the contributions of WT GFP− cells in the control arm. Each point represents one mouse. Lines represent mean values for each group. P values were calculated with Student's t test.
FIG 9.
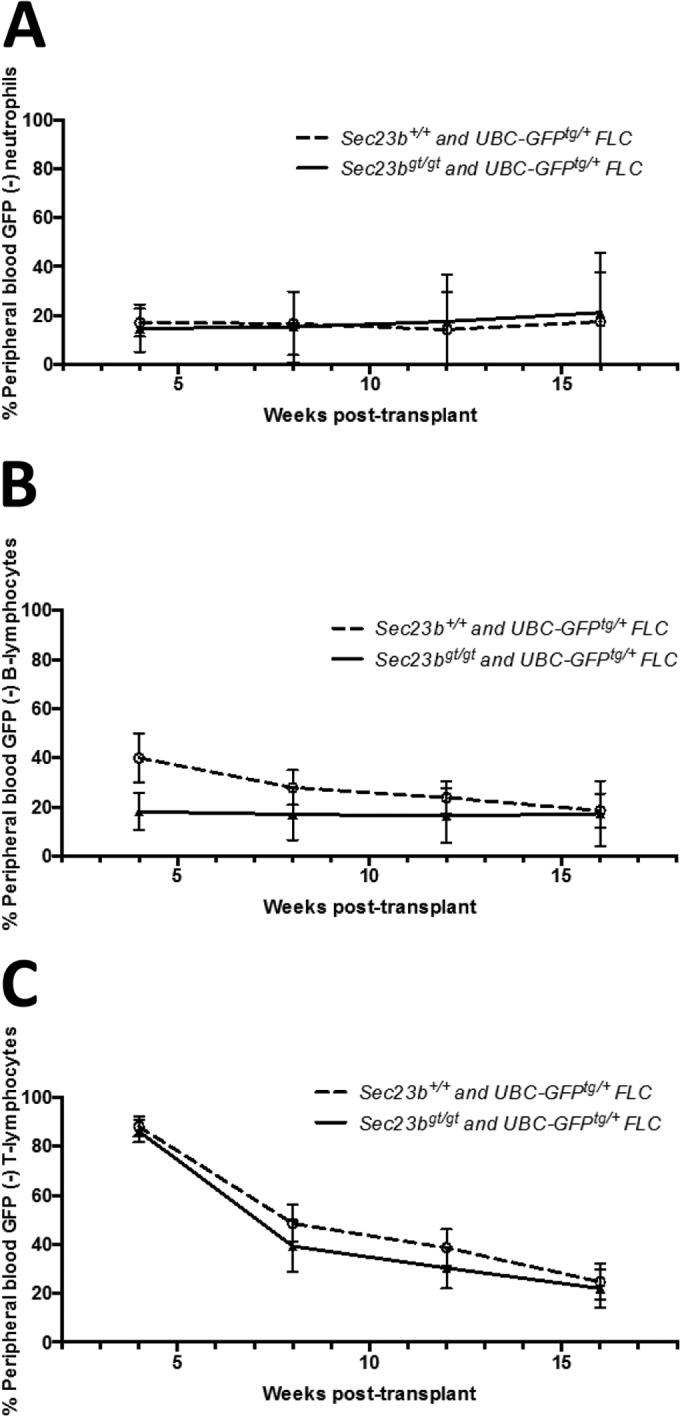
Secondary bone marrow transplantation experiments demonstrated continued equivalence of Sec23bgt/gt and WT FLC at reconstituting hematopoiesis. BM harvested from mice chimeric for GFP− Sec23bgt/gt and GFP+ WT hematopoietic cells were transplanted into lethally irradiated secondary recipients (experimental arm). Control mice were transplanted with BM harvested from mice chimeric for GFP− and GFP+ WT hematopoietic cells. Peripheral blood was obtained from recipient mice. By FACS, the contributions of Sec23bgt/gt GFP− cells to the population of Mac1+ Gr1+ neutrophils (A), B220+ B lymphocytes (B), and CD3+ T lymphocytes (C) in the experimental arm were equivalent to the contributions of WT GFP− cells in control mice over the course of 18 weeks of follow-up. n = 13 to 15 mice per group. Error bars represent standard deviations.
A second Sec23b null allele confirmed the absence of an RBC phenotype in SEC23B-deficient mice.
Sec23bgt/gt murine embryonal fibroblasts express a SEC23B/βGEO fusion protein resulting from the gene trap insertion into intron 19 of Sec23b. Though SEC23B/βGEO coimmunoprecipitates with SEC24 (binding partner for SEC23) (18), a similar pancreatic phenotype was observed for Sec23bgt/gt mice and a second targeted allele. However, to rule out any residual function of the Sec23bgt allele masking a hematopoietic phenotype, another set of transplant experiments was performed with FLC derived from a second Sec23b mutant allele, Sec23b− (excision of exons 5 and 6 [Fig. 1]), which should result in a protein truncated after amino acid 130 (the full-length SEC23B protein contains 767 amino acids).
FLC were harvested from E16.5 Sec23b−/− embryos and transplanted into lethally irradiated C57BL/6J recipients ubiquitously expressing GFP (UBC-GFPtg/+). Reconstituted hematopoietic cells in recipient mice were GFP−, confirming donor stem cell engraftment. At 2 months and 5 months posttransplantation, transplant recipients of Sec23b−/− FLC exhibited normal RBC counts (Fig. 10A) and hemoglobin (Fig. 10B) and hematocrit (Fig. 10C) levels that were indistinguishable from those of recipients of control FLC. RBC ghosts prepared from reconstituted Sec23b−/− peripheral blood demonstrated no evidence of an alteration in band 3 glycosylation compared to control ghosts, by Western blotting (Fig. 10D). Additionally, Ter119+ erythroid precursors isolated from Sec23b−/− FLC did not exhibit the “double-membrane” appearance on transmission electron microscopy (Fig. 10 E) that is characteristic of human CDAII.
FIG 10.

Mice transplanted with Sec23b−/− FLC do not exhibit an erythroid phenotype. Lethally irradiated UBC-GFPtg/+ mice were transplanted with GFP− FLC harvested from either Sec23b−/− or WT E.16.5 embryos. Reconstituted hematopoietic cells in recipient mice were GFP−, confirming donor engraftment. Recipients of Sec23b−/− FLC had indistinguishable RBC counts (A), hemoglobin (B), and hematocrit (C) levels compared to control recipients of WT FLC. (D and E) Sec23b−/− RBC ghosts did not exhibit a band 3 glycosylation defect by Western blotting (D), nor did Ter119+ erythroid precursors demonstrate a “double membrane” by transmission electron microscopy (E). (F) By FACS analysis, Ter119+ erythroid cells comprised 36.67% (±16.69% [SD]) and 41.56% (±17.68%) of total live BM cells harvested from mice transplanted with Sec23b−/− and WT FLC, respectively. (G) Mice transplanted with Sec23b−/− FLC did not exhibit an increase in the percentage of bi/multinucleated RBC precursors. (H) BM erythroid compartments were stratified by forward scatter and CD71 expression into 5 stages of erythroid development (stages I through V, in chronological order) (22). The distributions of erythroid cells among stages I through V were comparable in mice transplanted with Sec23b−/− FLC and mice transplanted with WT FLC. Error bars indicate standard deviations. Means and standard deviation are indicated by horizontal lines.
BM SEC23B-deficient erythroid cells are normally distributed among stages of erythroid development.
Ter119+ erythroid cells were determined by flow cytometry to comprise 37% (standard deviation [SD], 17%) and 42% (SD, 18%) of the total number of live BM cells in mice transplanted with Sec23b−/− and WT FLC, respectively (Fig. 10D). Mice transplanted with Sec23b−/− FLC did not exhibit an increase in the percentage of BM bi/multinucleated RBC precursors (Fig. 10E).
To test for stage-specific defects in erythroid maturation resulting from murine SEC23B deficiency, BM Ter119+ erythroid cells from mice transplanted with either Sec23b−/− or WT FLC were stratified by Ter119 expression, CD71 expression, and forward scatter into 5 distinct populations of erythroid development, designated stages I to V (22). Stage I is the earliest stage and consists predominantly of proerythroblasts. Erythroid cells progress through stages I, II, III, and IV in chronological order, and ultimately reach the final stage, stage V, which encompasses primarily mature RBCs.
The distribution of BM erythroid cells among the 5 stages of erythroid development was comparable in mice transplanted with Sec23b−/− FLC and control mice transplanted with WT FLC (Fig. 10F).
The SEC23A protein level is increased in SEC23B-deficient erythroid precursors.
Anti-SEC23A and anti-SEC23B antipeptide antibodies were generated and tested for specificity to their respective paralogs (Fig. 11A and B). The SEC23B protein was undetectable by Western blotting in Sec23b−/− FLC and in sorted Ter119+ Sec23b−/− erythroid precursors (Fig. 11C and D). Sec23b mRNA isolated from Sec23b−/− erythroid precursors was markedly reduced, likely due to nonsense-mediated decay resulting from the exons 5 and 6 deletion and the resulting frameshift (23) (Fig. 11E). To assess any potential change in SEC23A protein levels in response to loss of SEC23B, quantitative Western blot analysis was performed and revealed an ∼50% increase in the steady-state levels of SEC23A protein in SEC23B-deficient compared to WT erythroid cells (Fig. 11F). However, no change in the mRNA expression of the four Sec24 paralogs, the two Sar1 paralogs, or TRAPPC3 were observed between SEC23B-deficient and WT erythroid cells as measured by qPCR (Fig. 11G).
FIG 11.

The SEC23A protein level is increased in Sec23b−/− erythroid precursors. (A and B) Lysates from COS cells transfected with GFP-tagged SEC23A or SEC23B were examined by immunoblotting with antipeptide antibodies raised against SEC23A (A) or SEC23B (B) and demonstrated a high degree of specificity of these antibodies for their respective paralogs. (C and D) Western blotting of whole-cell lysates from WT control and Sec23b−/− FLC (C) and sorted control and Sec23b−/− Ter119+ erythroid precursors (D) demonstrated no detectable SEC23B protein in Sec23b−/− cells. (E) qPCR analysis showed a marked reduction of Sec23b mRNA in Sec23b−/− compared to WT Ter119+ cells. (F) The SEC23A protein level, normalized to β-actin or to RalA, was increased in Ter119+ erythroid precursors compared to WT controls, as determined by quantitative Western blot analysis (P = 0.029 and 0.030 for normalization to β-actin and RalA, respectively). (G) mRNA levels measured by qPCR for the four Sec24 paralogs, the two Sar1 paralogs, and TRAPPC3 were all indistinguishable between Sec23b−/− and WT Ter119+ erythroid cells.
DISCUSSION
Homozygous or compound heterozygous SEC23B mutations in humans result in CDAII, with the clinical phenotype restricted to a characteristic set of RBC abnormalities and no reported nonhematologic clinical manifestations. In contrast, SEC23B-deficient mice die perinatally, exhibiting degeneration of multiple professional secretory tissues, with apparently normal RBCs. However, the failure of these mice to survive beyond the immediate perinatal period precluded detailed RBC analysis and characterization of adult hematopoiesis. To address this issue, we performed FLC transplantation experiments to generate chimeric mice with SEC23B deficiency restricted to the hematopoietic compartment. Surprisingly, no RBC abnormalities characteristic of human CDAII could be detected in these animals. In addition to the absence of anemia, SEC23B-deficient RBCs lacked the duplicated membrane and band 3 glycosylation defects that are characteristic of CDAII in humans. Erythroid hyperplasia and multinucleated RBC precursors were also absent from the BM. Competitive transplants and secondary transplant experiments also failed to uncover even a subtle defect in erythropoiesis or reconstitution of myeloid cells and T lymphocytes. SEC23B-deficient B lineage cells appeared underrepresented in the BM of mice transplanted with equal numbers of Sec23bgt/gt and WT FLC. However, this finding was not associated with a reduction in the number of SEC23B null B lymphocytes in the peripheral blood of these chimeric animals, and thus its significance remains unclear. Furthermore, secondary recipients of BM harvested from these mice did not exhibit a decreased contribution of the peripheral blood Sec23bgt/gt B lymphocytes compared to their WT counterparts. We confirmed the absence of SEC23B protein in Sec23b−/− erythroid precursors, with qPCR analysis of mRNA isolated from Sec23b−/− erythroid precursors demonstrating a marked reduction in Sec23b mRNA, likely due to nonsense-mediated decay (23).
SEC23B is ubiquitously expressed in various tissues (18, 24, 25) and is an integral component of COPII vesicles, which facilitate the transport of ∼8,000 proteins from the ER to the Golgi apparatus (26). Despite this broad and fundamental function, SEC23B deficiency in humans results in a phenotype restricted to the RBC compartment. Though deficiencies of the inner COPII coat components, SEC23, SEC24, and SAR1, are all lethal in yeast, the corresponding COPII proteins for which deficiencies have been reported in mice or humans show a wide range of phenotypes. SEC23A deficiency in humans (discussed below) results in cranio-lenticulo-sutural dysplasia (15), whereas SEC24A, SEC24B, and SEC24D deficiencies in mice result in low plasma cholesterol (27), chraniorachischisis (28), and early embryonic lethality (29), respectively. SAR1B mutations in humans result in a disease of lipid malabsorption and chylomicron accumulation in the enterocytes (30). It is interesting that other genetic deficiencies that affect a large portion of the proteome also selectively disrupt the erythroid compartment, including mutations in genes encoding several ribosomal proteins resulting in Diamond-Blackfan syndrome. These observations suggest that the demanding process of RBC production may be particularly sensitive to perturbations of the basic cell machinery.
The mouse is a well-established model for the study of human hematopoiesis (31), with numerous gene-targeted mice closely recapitulating the eythropoietic phenotypes of the corresponding human diseases (32–36). The lack of conservation in SEC23B-deficient phenotypes between humans and mice is particularly surprising, given the previous report of a CDAII-like phenotype in SEC23B-deficient zebrafish embryos (4).
The mammalian genome encodes 2 SEC23 paralogs, SEC23A and SEC23B. In humans, SEC23A mutations result in cranio-lenticulo-sutural dysplasia (15), an autosomal recessive disease thought to result from abnormal collagen secretion. This disease is characterized by skeletal abnormalities, late closure of fontanelles, dysmorphic features, and sutural cataracts. Though SEC23A-deficient mice have not been reported, Sec23a-deficient zebrafish exhibit abnormal cartilage development reminiscent of the human phenotype (15, 37). The SEC23A and SEC23B proteins exhibit a high degree of sequence similarity (∼85% identical at the amino acid level), suggesting that the 2 SEC23 paralogs may overlap extensively in function and that the disparate phenotypes of SEC23B deficiency in humans and mice could be due to a shift in tissue-specific expression patterns during mammalian evolution. Consistent with this hypothesis, recently reported analyses of SEC23A/B in cultured erythroid progenitors (38) and transcriptomes for human and murine erythroid cells at several stages of terminal maturation (39, 40) demonstrated different patterns of SEC23A/SEC23B expression in humans and mice. This is evident particularly in the latest stage of erythroid maturation, with SEC23B the predominant paralog in humans and SEC23A in mice. However, an additional unique function for SEC23B in the human erythroid compartment, that is not required in mice, cannot be excluded.
Of note, Western blot analysis demonstrates an increase of steady-state SEC23A protein levels in SEC23B-deficient erythroid progenitors, suggesting a balance between the SEC23A and -B cytoplasmic pools, potentially mediated via SEC23/24 heterodimer formation. This is similar to the increase in SEC24B observed in hepatocytes of SEC24A-deficient mice (27). However, there was no apparent change in the mRNA expression of other core components of the COPII vesicles.
Reports of curative bone marrow transplantation for CDAII (9, 16) indicate that the pathological defect in this disease is confined to a transplanted cell. However, the mechanism by which human SEC23B deficiency results in the unique erythropoietic phenotype of CDAII remains unknown. The role of SEC23 in ER-to-Golgi apparatus transport suggests that CDAII results from the impaired secretion of one or more key cargo proteins that depend on SEC23B for export from the ER. Mutation of scl4a1 (the gene encoding band 3) in zebrafish results in increased binucleated erythroblasts, suggesting that band 3 could be the critical cargo, with CDAII resulting from a selective block in its transport to the membrane (41). However, humans with band 3 mutations exhibit hereditary spherocytosis and other RBC shape disorders but not CDAII (42, 43). The observation that RBC from CDAII patients are lysed in some but not all acidified normal sera (Ham's test) (1, 2) may provide a clue as to the identity of the critical SEC23B-dependent secretory cargo(s).
SEC23B interacts directly with SEC31, a component of the outer layer of the COPII coat. SEC23B also interacts with Bet3 (44), a component of the tethering complex TRAPPI, and with p150Glued (45), a component of the dynactin complex. Whether these direct SEC23B interactions contribute to the pathophysiology of CDAII is unknown.
In conclusion, we have shown that mice with hematopoietic deficiency of SEC23B support a normal erythroid compartment. Future studies aimed at understanding the functional overlap between SEC23A and SEC23B, as well as the specific protein cargos dependent on SEC23A/B for exit from the ER, should provide further insight into the pathophysiology of CDAII.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants R01 HL039693 and P01-HL057346 (D.G.), R01 AI091627 (I.M.), and R01 HL094505 (B.Z.). David Ginsburg is a Howard Hughes Medical Institute investigator.
We thank Sasha Meshinchi and Jeff Harrison from the Microscopy and Image-Analysis Laboratory at the University of Michigan for help with electron microscopy. We acknowledge Elizabeth Hughes, Keith Childs, and Thomas Saunders for preparation of gene-targeted mice and the Transgenic Animal Model Core of the University of Michigan's Biomedical Research Core Facilities. Core support was provided by the University of Michigan Cancer Center (P30 CA046592).
R.K., M.P.V., and D.G. conceived of the study and designed experiments. R.K., M.P.V., and M.J. performed most of the experiments. B.Z., L.E., J.T., and D.S. contributed to the execution of the experiments. R.K. and D.G. wrote the paper. All authors contributed to the integration and discussion of the results.
We declare no conflicts of interest.
Footnotes
Published ahead of print 28 July 2014
REFERENCES
- 1.Heimpel H, Anselstetter V, Chrobak L, Denecke J, Einsiedler B, Gallmeier K, Griesshammer A, Marquardt T, Janka-Schaub G, Kron M, Kohne E. 2003. Congenital dyserythropoietic anemia type II: epidemiology, clinical appearance, and prognosis based on long-term observation. Blood 102:4576–4581. 10.1182/blood-2003-02-0613 [DOI] [PubMed] [Google Scholar]
- 2.Khoriaty R, Vasievich MP, Ginsburg D. 2012. The COPII pathway and hematologic disease. Blood 120:31–38. 10.1182/blood-2012-01-292086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alloisio N, Texier P, Denoroy L, Berger C, Miraglia del Giudice E, Perrotta S, Iolascon A, Gilsanz F, Berger G, Guichard J. 1996. The cisternae decorating the red blood cell membrane in congenital dyserythropoietic anemia (type II) originate from the endoplasmic reticulum. Blood 87:4433–4439 [PubMed] [Google Scholar]
- 4.Schwarz K, Iolascon A, Verissimo F, Trede NS, Horsley W, Chen W, Paw BH, Hopfner KP, Holzmann K, Russo R, Esposito MR, Spano D, De Falco L, Heinrich K, Joggerst B, Rojewski MT, Perrotta S, Denecke J, Pannicke U, Delaunay J, Pepperkok R, Heimpel H. 2009. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat. Genet. 41:936–940. 10.1038/ng.405 [DOI] [PubMed] [Google Scholar]
- 5.Zanetti G, Pahuja KB, Studer S, Shim S, Schekman R. 2012. COPII and the regulation of protein sorting in mammals. Nat. Cell Biol. 14:20–28. 10.1038/ncb2390 [DOI] [PubMed] [Google Scholar]
- 6.Bianchi P, Fermo E, Vercellati C, Boschetti C, Barcellini W, Iurlo A, Marcello AP, Righetti PG, Zanella A. 2009. Congenital dyserythropoietic anemia type II (CDAII) is caused by mutations in the SEC23B gene. Hum. Mutat. 30:1292–1298. 10.1002/humu.21077 [DOI] [PubMed] [Google Scholar]
- 7.Russo R, Esposito MR, Asci R, Gambale A, Perrotta S, Ramenghi U, Forni GL, Uygun V, Delaunay J, Iolascon A. 2010. Mutational spectrum in congenital dyserythropoietic anemia type II: identification of 19 novel variants in SEC23B gene. Am. J. Hematol. 85:915–920. 10.1002/ajh.21866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amir A, Dgany O, Krasnov T, Resnitzky P, Mor-Cohen R, Bennett M, Berrebi A, Tamary H. 2011. E109K is a SEC23B founder mutation among Israeli Moroccan Jewish patients with congenital dyserythropoietic anemia type II. Acta Haematol. 125:202–207. 10.1159/000322948 [DOI] [PubMed] [Google Scholar]
- 9.Fermo E, Bianchi P, Notarangelo LD, Binda S, Vercellati C, Marcello AP, Boschetti C, Barcellini W, Zanella A. 2010. CDAII presenting as hydrops foetalis: molecular characterization of two cases. Blood Cells Mol. Dis. 45:20–22. 10.1016/j.bcmd.2010.03.005 [DOI] [PubMed] [Google Scholar]
- 10.Iolascon A, Russo R, Esposito MR, Asci R, Piscopo C, Perrotta S, Feneant-Thibault M, Garcon L, Delaunay J. 2010. Molecular analysis of 42 patients with congenital dyserythropoietic anemia type II: new mutations in the SEC23B gene and a search for a genotype-phenotype relationship. Haematologicaae 95:708–715. 10.3324/haematol.2009.014985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Punzo F, Bertoli-Avella AM, Scianguetta S, Della Ragione F, Casale M, Ronzoni L, Cappellini MD, Forni G, Oostra BA, Perrotta S. 2011. Congenital dyserythropoietic anemia type II: molecular analysis and expression of the SEC23B gene. Orphanet J. Rare Dis. 6:89. 10.1186/1750-1172-6-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russo R, Gambale A, Esposito MR, Serra ML, Troiano A, De Maggio I, Capasso M, Luzzatto L, Delaunay J, Tamary H, Iolascon A. 2011. Two founder mutations in the SEC23B gene account for the relatively high frequency of CDA II in the Italian population. Am. J. Hematol. 86:727–732. 10.1002/ajh.22096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu G, Niu S, Dong A, Cai H, Anderson GJ, Han B, Nie G. 2012. A Chinese family carrying novel mutations in SEC23B and HFE2, the genes responsible for congenital dyserythropoietic anaemia II (CDA II) and primary iron overload, respectively. Br. J. Haematol. 158:143–145. 10.1111/j.1365-2141.2012.09102.x [DOI] [PubMed] [Google Scholar]
- 14.Russo R, Langella C, Esposito MR, Gambale A, Vitiello F, Vallefuoco F, Ek T, Yang E, Iolascon A. 2013. Hypomorphic mutations of SEC23B gene account for mild phenotypes of congenital dyserythropoietic anemia type II. Blood Cells Mol. Dis. 51:17–21. 10.1016/j.bcmd.2013.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyadjiev SA, Fromme JC, Ben J, Chong SS, Nauta C, Hur DJ, Zhang G, Hamamoto S, Schekman R, Ravazzola M, Orci L, Eyaid W. 2006. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat. Genet. 38:1192–1197. 10.1038/ng1876 [DOI] [PubMed] [Google Scholar]
- 16.Iolascon A, Sabato V, de Mattia D, Locatelli F. 2001. Bone marrow transplantation in a case of severe, type II congenital dyserythropoietic anaemia (CDA II). Bone Marrow Transplant. 27:213–215. 10.1038/sj.bmt.1702764 [DOI] [PubMed] [Google Scholar]
- 17.Remacha AF, Badell I, Pujol-Moix N, Parra J, Muniz-Diaz E, Ginovart G, Sarda MP, Hernandez A, Moliner E, Torrent M. 2002. Hydrops fetalis-associated congenital dyserythropoietic anemia treated with intrauterine transfusions and bone marrow transplantation. Blood 100:356–358. 10.1182/blood-2001-12-0351 [DOI] [PubMed] [Google Scholar]
- 18.Tao J, Zhu M, Wang H, Afelik S, Vasievich MP, Chen XW, Zhu G, Jensen J, Ginsburg D, Zhang B. 2012. SEC23B is required for the maintenance of murine professional secretory tissues. Proc. Natl. Acad. Sci. U. S. A. 109:E2001–E2009. 10.1073/pnas.1209207109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaefer BC, Schaefer ML, Kappler JW, Marrack P, Kedl RM. 2001. Observation of antigen-dependent CD8+ T-cell/ dendritic cell interactions in vivo. Cell. Immunol. 214:110–122. 10.1006/cimm.2001.1895 [DOI] [PubMed] [Google Scholar]
- 20.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. 2005. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121:1109–1121. 10.1016/j.cell.2005.05.026 [DOI] [PubMed] [Google Scholar]
- 21.Chen ML, Logan TD, Hochberg ML, Shelat SG, Yu X, Wilding GE, Tan W, Kujoth GC, Prolla TA, Selak MA, Kundu M, Carroll M, Thompson JE. 2009. Erythroid dysplasia, megaloblastic anemia, and impaired lymphopoiesis arising from mitochondrial dysfunction. Blood 114:4045–4053. 10.1182/blood-2008-08-169474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen K, Liu J, Heck S, Chasis JA, An X, Mohandas N. 2009. Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc. Natl. Acad. Sci. U. S. A. 106:17413–17418. 10.1073/pnas.0909296106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Losson R, Lacroute F. 1979. Interference of nonsense mutations with eukaryotic messenger RNA stability. Proc. Natl. Acad. Sci. U. S. A. 76:5134–5137. 10.1073/pnas.76.10.5134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fromme JC, Ravazzola M, Hamamoto S, Al-Balwi M, Eyaid W, Boyadjiev SA, Cosson P, Schekman R, Orci L. 2007. The genetic basis of a craniofacial disease provides insight into COPII coat assembly. Dev. Cell 13:623–634. 10.1016/j.devcel.2007.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paccaud JP, Reith W, Carpentier JL, Ravazzola M, Amherdt M, Schekman R, Orci L. 1996. Cloning and functional characterization of mammalian homologues of the COPII component Sec23. Mol. Biol. Cell 7:1535–1546. 10.1091/mbc.7.10.1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanapin A, Batalov S, Davis MJ, Gough J, Grimmond S, Kawaji H, Magrane M, Matsuda H, Schonbach C, Teasdale RD, Yuan Z. 2003. Mouse proteome analysis. Genome Res. 13:1335–1344. 10.1101/gr.978703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen XW, Wang H, Bajaj K, Zhang P, Meng ZX, Ma D, Bai Y, Liu HH, Adams E, Baines A, Yu G, Sartor MA, Zhang B, Yi Z, Lin J, Young SG, Schekman R, Ginsburg D. 2013. SEC24A deficiency lowers plasma cholesterol through reduced PCSK9 secretion. eLife 2:e00444. 10.7554/eLife.00444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merte J, Jensen D, Wright K, Sarsfield S, Wang Y, Schekman R, Ginty DD. 2010. Sec24b selectively sorts Vangl2 to regulate planar cell polarity during neural tube closure. Nat. Cell Biol. 12:41–46. 10.1038/ncb2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baines AC, Adams EJ, Zhang B, Ginsburg D. 2013. Disruption of the Sec24d gene results in early embryonic lethality in the mouse. PLoS One 8:e61114. 10.1371/journal.pone.0061114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones B, Jones EL, Bonney SA, Patel HN, Mensenkamp AR, Eichenbaum-Voline S, Rudling M, Myrdal U, Annesi G, Naik S, Meadows N, Quattrone A, Islam SA, Naoumova RP, Angelin B, Infante R, Levy E, Roy CC, Freemont PS, Scott J, Shoulders CC. 2003. Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat. Genet. 34:29–31. 10.1038/ng1145 [DOI] [PubMed] [Google Scholar]
- 31.Orkin SH, Zon LI. 2008. Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132:631–644. 10.1016/j.cell.2008.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.White RA, Birkenmeier CS, Lux SE, Barker JE. 1990. Ankyrin and the hemolytic anemia mutation, nb, map to mouse chromosome 8: presence of the nb allele is associated with a truncated erythrocyte ankyrin. Proc. Natl. Acad. Sci. U. S. A. 87:3117–3121. 10.1073/pnas.87.8.3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bodine DMt, Birkenmeier CS, Barker JE. 1984. Spectrin deficient inherited hemolytic anemias in the mouse: characterization by spectrin synthesis and mRNA activity in reticulocytes. Cell 37:721–729. 10.1016/0092-8674(84)90408-2 [DOI] [PubMed] [Google Scholar]
- 34.Hughes MR, Anderson N, Maltby S, Wong J, Berberovic Z, Birkenmeier CS, Haddon DJ, Garcha K, Flenniken A, Osborne LR, Adamson SL, Rossant J, Peters LL, Minden MD, Paulson RF, Wang C, Barber DL, McNagny KM, Stanford WL. 2011. A novel ENU-generated truncation mutation lacking the spectrin-binding and C-terminal regulatory domains of Ank1 models severe hemolytic hereditary spherocytosis. Exp. Hematol. 39:305–320. 10.1016/j.exphem.2010.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siatecka M, Sahr KE, Andersen SG, Mezei M, Bieker JJ, Peters LL. 2010. Severe anemia in the Nan mutant mouse caused by sequence-selective disruption of erythroid Kruppel-like factor. Proc. Natl. Acad. Sci. U. S. A. 107:15151–15156. 10.1073/pnas.1004996107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finberg KE, Heeney MM, Campagna DR, Aydinok Y, Pearson HA, Hartman KR, Mayo MM, Samuel SM, Strouse JJ, Markianos K, Andrews NC, Fleming MD. 2008. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat. Genet. 40:569–571. 10.1038/ng.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lang MR, Lapierre LA, Frotscher M, Goldenring JR, Knapik EW. 2006. Secretory COPII coat component Sec23a is essential for craniofacial chondrocyte maturation. Nat. Genet. 38:1198–1203. 10.1038/ng1880 [DOI] [PubMed] [Google Scholar]
- 38.Satchwell TJ, Pellegrin S, Bianchi P, Hawley BR, Gampel A, Mordue KE, Budnik A, Fermo E, Barcellini W, Stephens DJ, van den Akker E, Toye AM. 2013. Characteristic phenotypes associated with congenital dyserythropoietic anemia (type II) manifest at different stages of erythropoiesis. Haematologicaae 98:1788–1796. 10.3324/haematol.2013.085522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.An X, Schulz VP, Li J, Wu K, Liu J, Xue F, Hu J, Mohandas N, Gallagher PG. 2014. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood 123:3466–3477. 10.1182/blood-2014-01-548305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pishesha N, Thiru P, Shi J, Eng Sankaran JCVG, Lodish HF. 2014. Transcriptional divergence and conservation of human and mouse erythropoiesis. Proc. Natl. Acad. Sci. U. S. A. 111:4103–4108. 10.1073/pnas.1401598111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paw BH, Davidson AJ, Zhou Y, Li R, Pratt SJ, Lee C, Trede NS, Brownlie A, Donovan A, Liao EC, Ziai JM, Drejer AH, Guo W, Kim CH, Gwynn B, Peters LL, Chernova MN, Alper SL, Zapata A, Wickramasinghe SN, Lee MJ, Lux SE, Fritz A, Postlethwait JH, Zon LI. 2003. Cell-specific mitotic defect and dyserythropoiesis associated with erythroid band 3 deficiency. Nat. Genet. 34:59–64. 10.1038/ng1137 [DOI] [PubMed] [Google Scholar]
- 42.Grace RF, Lux SE. 2009. Disorders of the red cell membrane, p 659–837 In Orkin SH, Nathan DG, Ginsburg D, Look AT, Fisher DE, Lux E. (ed), Hematology of infancy and childhood. Saunders, Philadelphia, PA [Google Scholar]
- 43.Iolascon A, De Falco L, Borgese F, Esposito MR, Avvisati RA, Izzo P, Piscopo C, Guizouarn H, Biondani A, Pantaleo A, De Franceschi L. 2009. A novel erythroid anion exchange variant (Gly796Arg) of hereditary stomatocytosis associated with dyserythropoiesis. Haematologicaae 94:1049–1059. 10.3324/haematol.2008.002873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cai H, Yu S, Menon S, Cai Y, Lazarova D, Fu C, Reinisch K, Hay JC, Ferro-Novick S. 2007. TRAPPI tethers COPII vesicles by binding the coat subunit Sec23. Nature 445:941–944. 10.1038/nature05527 [DOI] [PubMed] [Google Scholar]
- 45.Watson P, Forster R, Palmer KJ, Pepperkok R, Stephens DJ. 2005. Coupling of ER exit to microtubules through direct interaction of COPII with dynactin. Nat. Cell Biol. 7:48–55. 10.1038/ncb1206 [DOI] [PMC free article] [PubMed] [Google Scholar]