

Abstract
Nontuberculous mycobacterial infections caused by Mycobacterium abscessus are responsible for a range of disease manifestations from pulmonary to skin infections and are notoriously difficult to treat, due to innate resistance to many antibiotics. Previous population studies of clinical M. abscessus isolates utilized multilocus sequence typing or pulsed-field gel electrophoresis, but high-resolution examinations of genetic diversity at the whole-genome level have not been well characterized, particularly among clinical isolates derived in the United States. We performed whole-genome sequencing of 11 clinical M. abscessus isolates derived from eight U.S. patients with pulmonary nontuberculous mycobacterial infections, compared them to 30 globally diverse clinical isolates, and investigated intrapatient genomic diversity and evolution. Phylogenomic analyses revealed a cluster of closely related U.S. and Western European M. abscessus subsp. abscessus isolates that are genetically distinct from other European isolates and all Asian isolates. Large-scale variation analyses suggested genome content differences of 0.3 to 8.3%, relative to the reference strain ATCC 19977T. Longitudinally sampled isolates showed very few single-nucleotide polymorphisms and correlated genomic deletion patterns, suggesting homogeneous infection populations. Our study explores the genomic diversity of clinical M. abscessus strains from multiple continents and provides insight into the genome plasticity of an opportunistic pathogen.
INTRODUCTION
Nontuberculous mycobacteria (NTM) represent a diverse group of environmental and pathogenic bacteria that are increasing in clinical prevalence in the United States (1) and other countries (2, 3). NTM are thought to be acquired primarily through environmental exposure (4), as they reside in water, biofilms, and soil environments (5, 6), although a few recent studies provide evidence of possible person-to person transmission among individuals with cystic fibrosis (CF) (7, 8). Mycobacterium abscessus is the second most clinically prevalent NTM species in pulmonary NTM infections (1, 9, 10), with Mycobacterium avium complex (MAC) being the most prevalent. M. abscessus infections are challenging to treat because they are innately resistant to many antimicrobials, including some that are effective against Mycobacterium tuberculosis and other mycobacteria (11–13). Acquired antibiotic resistance has been observed in some M. abscessus strains, with mechanisms of resistance ranging from mutational resistance to aminoglycosides (14) to mutational (15) and inducible (16) resistance to macrolides.
The current taxonomy of M. abscessus recognizes two subspecies, M. abscessus subsp. abscessus and M. abscessus subsp. bolletii (17), although recent population and comparative genomic studies support the three previously recognized subspecies, i.e., M. abscessus subsp. abscessus, M. abscessus subsp. massiliense, and M. abscessus subsp. bolletii (18–21). This delineation is important because clinical studies suggest differing treatment outcomes for patients infected with M. abscessus subsp. abscessus versus M. abscessus subsp. massiliense (22, 23). The increased recognition of M. abscessus as an emerging pathogen has reinforced the need to better understand the population structure of clinical M. abscessus at the subspecies and genome levels. Previous population studies of clinical M. abscessus used primarily multilocus sequencing typing (MLST) or pulsed-field gel electrophoresis (PFGE), but these methods have limited resolution for within-subspecies strain typing. We hypothesized that a high-resolution analysis of globally diverse clinical M. abscessus isolates could reveal distinct pathogen lineages and might uncover genetic components relevant to mycobacterial disease.
Whole-genome sequencing (WGS) and phylogenomic analysis are important methods for studying population structure and genomic evolution of bacterial pathogens, as they enable high-resolution analysis of genetic variants ranging from single-nucleotide polymorphisms (SNPs) to large-scale deletions. Analogous to MLST, core genome analyses compare genomic positions that are shared among all isolates, to create phylogenies of isolate populations. For example, a recent retrospective study of M. abscessus isolates derived from cystic fibrosis (CF) patients in the United Kingdom utilized WGS and core genome analyses to compare patient isolates and provided the first suggestive evidence of person-to-person transmission of M. abscessus subsp. massiliense (8). Our subsequent studies using WGS and core genome analysis revealed high levels of genetic relatedness between a subgroup of United Kingdom CF clinic isolates and M. abscessus subsp. massiliense strains from an epidemic of skin infections in Brazil (24) and an M. abscessus outbreak in Seattle, Washington (25).
In addition to phylogenomic comparisons of the core genome, WGS enables the identification of large-scale insertions and deletions (often referred to as the accessory genome). These genomic regions can include phage genes and plasmid elements and may represent genomic islands acquired through horizontal gene transfer (26). Their loss or gain can be attributed to environmental adaptations that can influence antibiotic resistance, virulence, or host range restrictions (26, 27). A recent study of M. abscessus subsp. massiliense isolates, for example, found that the absence of a cluster of glycopeptidolipid genes within a 24.8-kb genomic deletion conferred rough versus smooth colony morphology (28). Genome content variations in lung-associated opportunistic pathogens range from 22% in Staphylococcus aureus (29) to 10% in Pseudomonas aeruginosa (30) but are currently uncharacterized for M. abscessus clinical isolates. Knowledge of genome content variations among M. abscessus isolates could provide clues to genetic mechanisms contributing to virulence, antibiotic resistance, and transmission and may suggest biomarkers of diagnostic utility.
Here we report the genome sequences and genomic features of 11 Mycobacterium abscessus isolates derived from eight patients with pulmonary NTM disease in the United States. The sequenced strains were evaluated for in vitro susceptibility to 19 drugs. Using the complete genome of the M. abscessus subsp. abscessus type strain ATCC 19977T as a reference, we identified core genome SNPs in the United States-derived isolates as well as isolates from the United Kingdom, France, Brazil, Malaysia, and China, to evaluate the global genetic population structure of clinical M. abscessus. In our analysis, we detected large-scale genomic polymorphisms, reflecting gene content variations and genome plasticity, among closely and distantly related isolates. Lastly, we performed longitudinal genomic comparisons of isolates derived from individual patients, to examine infection homogeneity and genetic mutations that may arise during M. abscessus lung infections.
MATERIALS AND METHODS
Bacterial DNA isolation and PCR genotyping.
M. abscessus isolates were grown in Middlebrook 7H9 liquid medium for 5 days, and genomic DNA was extracted using standard protocols (31). Gene-specific primers were used to amplify a partial segment of the RNA polymerase beta subunit (rpoB) gene (32) and the hsp65 gene (19) for DNA sequencing using an ABI 3730xL genetic analyzer. The erythromycin ribosomal methylase gene erm(41) target was amplified using specific primers (16), and gel electrophoresis was carried out using the Lonza FlashGel system, to determine the absence or presence of a 273-bp deletion within the erm(41) gene.
In vitro drug susceptibility testing.
National Jewish Health (NJH) isolates were evaluated for drug susceptibility to a panel of 19 drugs using the microdilution method (33), and results are reported as MICs. NJH2 and NJH3 were not included in the testing because they did not grow in the testing medium.
Genome resequencing.
Approximately 1 μg of total genomic DNA was used for library preparation for SOLiD fragment sequencing, according to the manufacturer's protocol. Fragments were size selected between 100 and 250 bp, and genome sequencing was performed using the SOLiD 5500 platform (Life Technologies, Carlsbad, CA); 75-bp single-end reads were produced, and sequence reads were filtered using the default purity filter threshold in the Lifescope genome analysis software (Life Technologies). Only purity-filtered reads were used for downstream analyses.
Single-nucleotide polymorphism detection and annotation.
For strains with next-generation resequencing data, including all NJH isolates and United Kingdom isolates reported by Bryant et al. (8) (see Table 2), sequence reads were mapped to the M. abscessus reference genome sequence (34), which includes a 5,067,172-bp chromosome and a 23,319-bp plasmid, using the Lifescope genome analysis software (Life Technologies, Carlsbad, CA) whole-genome resequencing pipeline for SOLiD data or the Bowtie mapping algorithm for Illumina data (35). SNP and reference base calls were identified with the pileup program in SAMtools version 0.1.7 (36). SNPs were filtered using a custom Perl script and the following parameters: SNP quality score of >20, minimum of 10× read depth, >50% of base calls supporting the variant base, and <25% of variant calls occurring at the beginning or end of fragment reads.
TABLE 2.
Globally diverse clinical M. abscessus isolates used for phylogenomic analyses
| Subspeciesa | Isolate | Isolation date | Country of origin | Sequence accession no.b | Reference |
|---|---|---|---|---|---|
| MAB | ATCC 19977T | 1957 | France | NC_010397.1 | 34 |
| MAB | 6G0728 | 2003 | USA | AKUS00000000 | Unpublished |
| MAB | 6G0125 | 2010 | USA | AKUF00000000 | Unpublished |
| MAB | 3A0119R | 2010 | USA | AKUX01000000 | Unpublished |
| MAB | 3A0122R | 2002 | USA | AKUY00000000 | Unpublished |
| MAB | V06705 | 2005 | France | AUMY00000000 | 48 |
| MAB | 6a | 2007 | UK | ERR114968 | 8 |
| MAB | 7a | October 2007 | UK | ERR115041 | 8 |
| MAB | 7b | October 2007 | UK | ERR114965 | 8 |
| MAB | 7c | October 2007 | UK | ERR114966 | 8 |
| MAB | 9a | November 2007 | UK | ERR114986 | 8 |
| MAB | 9b | November 2007 | UK | ERR115102 | 8 |
| MAB | 9c | December 2007 | UK | ERR114974 | 8 |
| MAB | 9d | December 2007 | UK | ERR115104 | 8 |
| MAB | 1b | 2008 | UK | ERR119107 | 8 |
| MAB | 23a | 2010 | UK | ERR115039 | 8 |
| MAB | M152 | NRc | Malaysia | AKVT00000000 | 49 |
| MAB | M94 | NR | Malaysia | AJGG00000000 | 50 |
| MAB | M93 | 2010 | Malaysia | AJGF00000000 | 51 |
| MAB | 9808 | 1998 | China | ANAR00000000 | Unpublished |
| MBOL | BD | 2004 | France | AHAS00000000 | 52 |
| MBOL | M24 | NR | Malaysia | AJLY00000000 | 53 |
| MMAS | 5S0304 | 1998 | USA | AKTX00000000 | Unpublished |
| MMAS | 4S0116S | 2008 | USA | AKVE00000000 | Unpublished |
| MMAS | 4S0116R | 2008 | USA | AKVD00000000 | Unpublished |
| MMAS | 47J26 | 2009 | UK | AGQU00000000 | 54 |
| MMAS | CRM0020 | 2006 | Brazil | ATFQ00000000 | 55 |
| MMAS | 19m | 2010 | UK | ERR115051 | 8 |
| MMAS | M154 | 2010 | Malaysia | AJMA00000000 | 20 |
| MMAS | CCUG49998T | 2004 | France | AKVF00000000 | 56 |
MAB, M. abscessus subsp. abscessus; MBOL, Mycobacterium abscessus subsp. bolletii; MMAS, M. abscessus subsp. massiliense.
Genome sequences were obtained from the National Center for Biotechnology Information (NCBI) or the European Nucleotide Archive (ENA).
NR, not reported.
For M. abscessus strains with publically available draft genomes (including strains 6G0728, 6G0125, 3A0119R, 3A0122R, V06705, M152, M94, M93, 9808, BD, M24, 5S0304, 4S0116S, 4S0116R, 47J26, CRM0020, M154, and CCUG49998T) (see Table 2), we performed multigenome alignments of each draft genome to the M. abscessus reference genome sequence (34). Whole-genome alignments and single-nucleotide polymorphism (SNP) identification were performed with the progressiveMauve algorithm in Mauve 2.3.1 (37).
Genotype matrices for core genome comparisons were created for chromosomal positions for which high-quality variant and/or reference calls were available for all isolates. Genomic positions with ambiguous bases and/or missing data were excluded from the analyses. High-confidence SNP sites were annotated as genic or intergenic with a SQLite database and custom DBI-Perl scripts, using the gene annotation provided for the reference genome (34). Genic SNPs were further annotated for amino acid-level changes using ANNOVAR software (38).
Phylogenomic analysis.
To elucidate the phylogeny of all 41 isolates, high-confidence genotype data at 2,479 core genomic positions were concatenated into a FASTA sequence for each strain using a custom Perl script. A neighbor-joining (NJ) phylogeny was estimated using the observed differences between the concatenated sequences, with 1,000 bootstrap replicates, in SeaView 4.4.2 (39). To further resolve the clade of 18 U.S./European isolates, a high-resolution genotype data set of 128,074 core genomic positions was generated and a NJ phylogeny was estimated as described above.
Large-scale polymorphism analyses.
For 11 NJH isolates and 9 United Kingdom isolates with next-generation sequence data, sequence reads were mapped to the complete genome of the type strain ATCC 19977T. Read counts were normalized for GC content bias using a modified version of a previous method (40). Sequence coverage values were estimated with a custom Perl script that counts all uniquely mapped reads in sliding, nonoverlapping, 1-kb windows across the entire ATCC 19977T chromosome (5,068 total windows). Read counts for all 18 strains were converted to z-scores, and the genome-wide normalized read counts were clustered by hierarchical clustering using a Pearson correlation distance metric and average linkage with the Multiple Experiment Viewer (MeV) Java package (41). Pearson correlations of genome-wide deletion patterns of same-patient isolates were performed with the R statistical package (42).
Putative large-scale deletions were identified as contiguous regions greater than 30 kb with z-scores of less than −2.0. To determine the sequence homology of six identified deletion regions, genomic sequences were extracted from the reference genome with a custom Perl script and were queried against the NCBI nonredundant nucleotide database using the Blastn algorithm. BLAST results were filtered and sequence homology was defined with the following criteria: E value of 0.0, ≥70% sequence identity, and ≥30% query coverage.
Nucleotide sequence accession numbers.
Sequences were submitted to GenBank under accession numbers SRX641283 (NJH2), SRX641284 (NJH3), SRX641291 (NJH4), SRX641292 (NJH5), SRX641293 (NJH6), SRX641294 (NJH7), SRX641295 (NJH9), SRX339602 (NJH8), SRX641281 (NJH1), SRX641302 (NJH10), and SRX339603 (NJH11).
RESULTS AND DISCUSSION
Clinical and microbiological attributes of patients and isolates.
M. abscessus isolates examined in this study were obtained from sputum or bronchoalveolar lavage (BAL) fluid samples from eight patients referred to National Jewish Health (NJH), in Denver, Colorado, for management of chronic M. abscessus-related pulmonary infections between 2009 and 2011 (Table 1). All except one patient had a ≥2-year history of M. abscessus pulmonary infection, and all patients had received prolonged treatment with multiple antimycobacterial drugs, including macrolides and aminoglycosides. Isolates NJH1 to NJH4 were sampled longitudinally from the same patient at three time points during a 6-month period; isolates NJH2 and NJH3 were individual colonies collected at the second time point. The primary residences of all patients were in the northern or eastern United States, with the exception of one individual from Puerto Rico (isolate NJH7). Patients were primarily female (6/8 patients) and greater than 60 years of age (7/8 patients), and the majority of patients (6/7 patients) were smear negative at the time of sputum collection. Four of the patients had adult cystic fibrosis (isolates NJH1, NJH5, NJH8, and NJH9), and two were carriers of a CFTR polymorphic allele (isolates NJH7 and NJH11).
TABLE 1.
Clinical Mycobacterium abscessus isolates sequenced in this study and patient information
| Patient | Isolate | Sample collection date | Species identificationa | Sourceb | Sexc | AFB smear resultd | Age (yr) | Cystic fibrosis |
|---|---|---|---|---|---|---|---|---|
| 1 | NJH1 | September 2010 | MAB | Sputum | F | − | 66 | Yes |
| 1 | NJH2 | October 2010 | MAB | Sputum | F | − | 66 | Yes |
| 1 | NJH3 | October 2010 | MAB | Sputum | F | − | 66 | Yes |
| 1 | NJH4 | March 2011 | MAB | Sputum | F | − | 66 | Yes |
| 2 | NJH5 | 2009 | MAB | Sputum | F | − | 62 | Yes |
| 3 | NJH6 | 2011 | MAB | Sputum | F | − | 75 | NR |
| 4 | NJH7 | 2010 | MAB | BAL fluid | F | + | 71 | Carrier |
| 5 | NJH8 | 2011 | MAB | Sputum | M | NR | 41 | Yes |
| 6 | NJH9 | 2011 | MAB | BAL fluid | F | − | 66 | Yes |
| 7 | NJH10 | 2010 | MAB | Sputum | F | − | 72 | No |
| 8 | NJH11 | 2009 | MMAS | Sputum | M | − | 79 | Carrier |
Isolates were identified as either M. abscessus subsp. abscessus (MAB) or M. abscessus subsp. massiliense (MMAS) by sequencing portions of the rpoB and hsp65 genes and by amplification of the erm(41) gene.
Isolates were derived from sputum or bronchoalveolar lavage (BAL) fluid samples.
F, female; M, male.
AFB, acid-fast bacillus; NR, not reported.
All isolates were initially identified to the subspecies level by Sanger sequencing of the rpoB and hsp65 genes. Based on sequence homology to type strains, isolates NJH1 to NJH10 were identified as M. abscessus subsp. abscessus, and isolate NJH11 was identified as M. abscessus subsp. massiliense. All isolates were also evaluated for the erm(41) deletion (16). In agreement with the rpoB and hsp65 results, isolates NJH1 to NJH10 exhibited a full-length erm(41) amplicon consistent with M. abscessus subsp. abscessus, while NJH11 showed a smaller, deletion-containing amplicon consistent with M. abscessus subsp. massiliense.
Genomic sequencing and core genome relationships among diverse clinical isolates.
Genomic sequence reads from the pulmonary-derived NJH clinical isolates were mapped to the reference genome of the M. abscessus type strain ATCC 19977T (34), which was initially isolated from a knee infection with abscess-like subcutaneous lesions (43). The NJH isolates were deep-sequenced at levels of 97- to 214-fold coverage (see Table S1 in the supplemental material). Sequence reads from only one isolate, NJH9, showed substantial read mapping to a mercury resistance plasmid present in the reference strain, indicating that this plasmid is not widespread in the isolates studied.
To investigate the phylogenomic relationships of the NJH M. abscessus isolates, compared to globally diverse clinical isolates, we assembled 30 publically available M. abscessus genomes from the United States, the United Kingdom, France, China, Malaysia, and Brazil for our analysis (Table 2), including 11 representative M. abscessus isolates from a recent retrospective study of CF patients in the United Kingdom (8). First, we performed a neighbor-joining (NJ) phylogenetic reconstruction for 2,479 core genome positions (2,320 genic and 159 intergenic) that were shared across all 41 isolates (Fig. 1A). The resulting phylogeny supports the three previously recognized M. abscessus subspecies as monophyletic, consistent with population studies utilizing MLST genotyping (19, 21), and the NJH isolates grouped into expected clades consistent with subspecies identifications based on rpoB, hsp65, and erm(41) findings. Our phylogeny is also consistent with previously reported, independent, phylogenomic comparisons (8, 20, 24, 25, 44).
FIG 1.

Phylogenomic comparison of the National Jewish Health (NJH) M. abscessus clinical isolates from the United States, compared with global strains. (A) Genotype data at 2,479 core genome positions, relative to the M. abscessus subsp. abscessus ATCC 19977T reference genome, were used to estimate the phylogeny among 41 M. abscessus isolates using the neighbor-joining algorithm. The phylogenomic tree supports previously recognized subspecies as monophyletic groups. Gray bars and asterisks, isolates acquired from the same patients. (B) Higher-resolution phylogeny of the 18-isolate U.S./European M. abscessus subsp. abscessus cluster was created using genotype information at 128,074 core genome positions. Isolation dates are included. Only 320 (0.25%) of 128,074 core genome sites vary among these isolates.
Within the subspecies of M. abscessus subsp. abscessus included in our analysis, we observed two major subclades (Fig. 1A). The basal subclade included all Asian isolates, one NJH isolate (NJH8), and six isolates from three CF patients in the United Kingdom. This clade was genetically distinct from a cluster of 18 isolates exclusively from the United States or Europe, including the ATCC 19977T type strain (43), nearly all of the NJH isolates, four additional U.S. isolates, one French isolate, and four isolates from the United Kingdom. In this analysis, the 18 clustered isolates were distinguished by only 9 of 2,479 core genome SNP sites (0.36%). An additional 198 SNP sites discriminated the adjacent Puerto Rican isolate (NJH7). These results suggest that, among the M. abscessus subsp. abscessus strains analyzed, the Asian isolates are divergent from most U.S. isolates, while the United Kingdom isolates are divided between the two genetic subgroups. Further sampling and future studies are needed to validate these regional subgroups.
The lack of SNP variations in the U.S./Europe M. abscessus subsp. abscessus cluster is intriguing, given the temporal and geographic disparities among the isolates. To further resolve the 18 clustered isolates and to test for temporal or geographic groupings, a high-resolution phylogeny was constructed using genotype information at 128,074 core genome nucleotide positions (Fig. 1B). A total of 320 SNPs (320/128,074 positions = 0.25%) were observed in this data set, similar to the proportion of variations observed in the initial phylogeny. The majority of SNP variations occurred in the French isolate, V06705, which was the most divergent in the cluster. Overall, the branching patterns did not distinguish temporal or geographic groups, suggesting independent acquisitions of the isolates. Therefore, the high genetic relatedness may represent a dominant clinical M. abscessus subsp. abscessus subgroup or may reflect similarities in the isolates' geographic or environmental niches.
In vitro drug susceptibility results and variants associated with resistance.
Nine of 11 NJH clinical isolates were evaluated for in vitro drug susceptibility to 19 antimicrobial drugs. These drugs span several drug classes, and the results are reported as MICs (Table 3). Most isolates exhibited resistance to imipenem, amoxicillin-clavulanic acid (Augmentin), two of four aminoglycosides (tobramycin and gentamicin), four generations of cephalosporins, two of three tetracyclines (doxycycline and minocycline), and two fluoroquinolones (ciprofloxacin and moxifloxacin), confirming the innate resistance of M. abscessus to several drug classes (11, 13). Drugs that showed in vitro activity against the M. abscessus strains included the aminoglycosides amikacin and kanamycin and the macrolides clarithromycin and azithromycin, which are commonly used to treat pulmonary M. abscessus infections (12), as well as the less commonly prescribed tigecycline.
TABLE 3.
Drug susceptibility of NJH M. abscessus clinical isolates to a 20-drug panel, reported as MICs
| Drug | MIC (μg/ml) |
No. of resistant isolates/total no. of isolatesa | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NJH1 | NJH4 | NJH5 | NJH6 | NJH7 | NJH8 | NJH9 | NJH10 | NJH11 | ||
| Amikacin | 16 | ≤8 | ≤8 | ≤8 | >64 | 32 | 16 | ≤8 | 32 | 1/9 |
| Kanamycin | 16 | ≤8 | ≤8 | ≤8 | >64 | 32 | 16 | ≤8 | 16 | 1/9 |
| Tobramycin | 16 | 16 | 8 | 8 | >16 | 16 | 8 | 16 | >16 | 6/9 |
| Gentamicin | >16 | 8 | 4 | 8 | >16 | >16 | 16 | 8 | >16 | 5/9 |
| Imipenem | 16 | >16 | >16 | 8 | 8 | 8 | 16 | >16 | >16 | 6/9 |
| Cefoxitin | 64 | 64 | 64 | 32 | 64 | 64 | 32 | 64 | 64 | 9/9 |
| Ceftriaxone | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | 9/9 |
| Cefotaxime | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | 9/9 |
| Cefepime | 32 | >32 | >32 | >32 | >32 | 32 | >32 | >32 | >32 | 9/9 |
| Clarithromycin | 0.5 | 0.5 | 1 | 2 | 1 | 1 | 1 | >32 | 0.5 | 1/9 |
| Azithromycin | ≤16 | ≤16 | 32 | 32 | 32 | 32 | ≤16 | >256 | ≤16 | 1/9 |
| Amoxicillin/clavulanic acid | >32/16 | >32/16 | >32/16 | >32/16 | >32/16 | >32/16 | >32/16 | >32/16 | >32/16 | 9/9 |
| Ciprofloxacin | 8 | >8 | 8 | 4 | >8 | >8 | 8 | >8 | 8 | 9/9 |
| Moxifloxacin | >4 | >4 | >4 | >4 | >4 | >4 | >4 | >4 | >4 | 9/9 |
| Trimethoprim/sulfa | >4/76 | >4/76 | >4/76 | >4/76 | >4/76 | >4/76 | >4/76 | >4/76 | >4/76 | 9/9 |
| Doxycycline | 16 | >16 | >16 | >16 | >16 | >16 | >16 | >16 | >16 | 9/9 |
| Minocycline | 8 | >8 | >8 | >8 | >8 | >8 | >8 | >8 | >8 | 9/9 |
| Tigecycline | 1 | 0.5 | ≤0.25 | 2 | 1 | 1 | 0.5 | 2 | 1 | 0/9 |
| Linezolid | >16 | 16 | >16 | 16 | >16 | >16 | 16 | >16 | >16 | 9/9 |
Determination of isolates with resistant phenotypes was based on previously defined MIC thresholds specific to each drug.
Among the NJH isolates tested, only two exhibited different susceptibility phenotypes. NJH7 was the only isolate with observed in vitro resistance to amikacin and kanamycin. Based on our analysis, this resistance can be explained by the presence of a 16S rRNA mutation that was previously associated with aminoglycoside resistance in M. abscessus and Mycobacterium chelonae isolates and corresponds to the A1408G mutation in Escherichia coli (14). NJH10 was the only isolate with observed resistance to clarithromycin and azithromycin. This isolate does not have a resistance-conferring mutation in the 23S rRNA (15) and therefore may contain a novel mechanism of resistance.
Large-scale polymorphic genomic regions in M. abscessus clinical isolates.
Given the high levels of genetic relatedness in the core genomes of the clustered U.S./European isolates, we also investigated large-scale genomic variations among a subset of clustered and nonclustered isolates. We analyzed sequence coverage in 1-kb windows relative to the ATCC 19977T reference genome and identified 1-kb windows with substantially low coverage (<2-fold), indicative of deletions or highly divergent regions of the genome. Among the 19 isolates analyzed, we observed a range of 0.3 to 8.3% of the reference genome that was not represented in the clinical isolates (Table 4). The least divergence was observed among clustered isolates (0.3 to 2.8%), and greater divergence was observed among nonclustered isolates (5.9 to 7.1%). As expected, the most divergence was observed in the M. abscessus subsp. massiliense isolate (NJH11; 8.3%). This diversity is similar to genome content differences observed among clinical M. tuberculosis isolates (45) and illustrates the genome plasticity and diversity among U.S. and European M. abscessus isolates.
TABLE 4.
Large sequence polymorphisms between 19 clinical M. abscessus isolates and the reference type strain, ATCC 19977T
| Isolate | Country of origin | No. of genomic windows with <2-fold coveragea (% of genome) | Genetic groupb |
|---|---|---|---|
| NJH1 | USA | 141 (2.8) | MAB clustered |
| NJH2 | USA | 142 (2.8) | MAB clustered |
| NJH3 | USA | 142 (2.8) | MAB clustered |
| NJH4 | USA | 142 (2.8) | MAB clustered |
| NJH5 | USA | 82 (1.6) | MAB clustered |
| NJH6 | USA | 15 (0.3) | MAB clustered |
| NJH9 | USA | 25 (0.5) | MAB clustered |
| NJH10 | USA | 16 (0.3) | MAB clustered |
| 7a | UK | 85 (1.7) | MAB clustered |
| 7b | UK | 85 (1.7) | MAB clustered |
| 7c | UK | 85 (1.7) | MAB clustered |
| NJH7 | USA | 71 (1.4) | MAB clustered |
| NJH8 | USA | 297 (5.9) | MAB nonclustered |
| 1b | UK | 317 (6.3) | MAB nonclustered |
| 9b | UK | 360 (7.1) | MAB nonclustered |
| 9c | UK | 359 (7.1) | MAB nonclustered |
| 9d | UK | 359 (7.1) | MAB nonclustered |
| 9a | UK | 360 (7.1) | MAB nonclustered |
| NJH11 | USA | 419 (8.3) | MMAS |
Genomic windows with significantly low sequence read coverage (less than 2-fold), compared to the reference strain.
The three genetic groups identified in our phylogenomic analysis were clustered M. abscessus subsp. abscessus (MAB) isolates, nonclustered M. abscessus subsp. abscessus isolates, and M. abscessus subsp. massiliense (MMAS) isolates.
To test whether the clustered isolates shared the same deletions, we performed a cluster analysis of genome-wide coverage patterns (Fig. 2). The clustering results were similar to the initial core genome phylogeny (Fig. 1A), as the U.S./European clustered isolates grouped into a major clade distinct from nonclustered isolates. One exception was the Puerto Rican isolate (NJH7) which clustered with the U.S. isolates based on deletion patterns but was somewhat divergent from the U.S. and United Kingdom isolates in the core genome phylogeny (Fig. 1A). In contrast to the high-resolution phylogeny (Fig. 1B), the U.S./European clustered isolates grouped together largely by geographic region. This finding, along with the grouping of NJH7 with other North American isolates, suggests that evolution of the accessory genome may be driven by events within local microenvironments.
FIG 2.

Large-scale genomic variations among M. abscessus genomes. Large-scale genomic variations, including deletions and duplications, were identified by sequence read mapping and coverage assessment along a reference genome. Chromosome coordinates of three regions of the reference genome are indicated on the x axis. Next-generation sequence read counts were assessed along nonoverlapping 1-kb sliding windows of the reference genome (5,068 total windows). Contiguous windows with z-scores (y axes) less than −2.0 indicate putative genomic deletions or highly divergent regions, while contiguous stretches of high z-scores, greater than 2.0, indicate putative genomic duplications. The dendrogram represents the result of average linkage hierarchical clustering of genome-wide coverage values, using a Pearson correlation metric. Arrows A to F, large-scale genomic differences (>30 kb) among the clinical M. abscessus isolates.
Further analyses of six large-scale deletions (>30 kb) (Fig. 2, arrows A to F), relative to the reference M. abscessus genome, revealed potential mechanisms of accessory genome evolution among clinical M. abscessus strains (see Table S2 in the supplemental material). Regions A and F, which were deleted in all nonclustered isolates, include operons for copper ion and phosphate ion transport, respectively. Both genomic regions contain sequences that are homologous to plasmids and transposons, providing evidence for recombination or horizontal gene transfer (26). Region C, which was absent in 14 of 19 isolates, shows sequence homology only to mycobacteriophages, consistent with previous annotation as a prophage (34). Two U.S. isolates (NJH7 and NJH10) had excessive sequence coverage in this region, suggesting multiple genomic copies of prophage sequences. Regions B, D, and E show no evidence of recombination or homology to phage DNA. Region E, which was deleted in nonclustered isolates and M. abscessus subsp. massiliense (NJH11), includes a cassette of biphenyl and aromatic hydrocarbon metabolic enzymes that functionally enable degradation of environmental chemicals (46) in some cases. Overall, the results suggest that gene content variations in clinical M. abscessus are due, in part, to recombination and/or metabolic adaptations.
Genomic comparisons of longitudinally sampled isolates from individual patients.
To evaluate same-patient strain variations, we compared genomic information derived from three sets of isolates from individual patients. From the United Kingdom CF M. abscessus study (8), we studied isolates from patients 7 and 9. Isolates 7a, 7b, and 7c were collected at a single time point. Isolates 9a and 9b were collected at an initial time point during treatment, and isolates 9c and 9d were collected 1 month later. From NJH, four isolates (NJH1 to NJH4) were longitudinally isolated from a single patient at three time points during a 6-month period, including two colonies (NJH2 and NJH3) that were collected at the second time point.
In the previous core genome phylogenies (Fig. 1) with limited numbers of genomic positions (2,479 bp and 128,074 bp), all three sets of isolates showed identical genotypes among themselves within each set. Therefore, we performed in-depth core genome analyses for each patient isolate group, to explore infection homogeneity and the potential for genomic mutations during infection and treatment. Using all high-quality base calls within each group of patient isolates, we found 4 SNPs among the three patient 7 isolates across 4,971,231 genomic positions (0.00008%), 12 SNPs among the four patient 9 isolates across 4,503,047 positions (0.0002%), and 10 SNPs among the four NJH patient 1 isolates across 4,587,198 positions (0.0002%). In contrast to SNPs in the overall phylogeny (Fig. 1A), in which the minority of coding SNPs were nonsynonymous (20.9%) versus synonymous (79.5%), the majority of SNPs in the longitudinal groups were nonsynonymous changes (55.6% to 87.5%), suggesting positive selective pressures as possible driving forces of genetic mutation. Examples of genes with nonsynonymous SNPs observed among NJH1 patient isolates include an amino acid permease family protein (NCBI gene locus tag MAB_0950c), a probable DNase TatD (NCBI gene locus tag MAB_1129), a putative membrane protein MmpL (NCBI gene locus tag MAB_2303), sulfate adenylate transferase CysD (NCBI gene locus tag MAB_4181), and arsenic-transport integral membrane protein ArsC (NCBI gene locus tag MAB_4863).
To test for large-scale variations among same-patient isolates, correlations of genome-wide sequence coverage patterns were performed for all pairwise combinations of isolates within and between patient groups (Fig. 3). Results demonstrated that all same-patient isolate pairs were highly correlated, compared to different-patient isolate pairs, and we saw no evidence of isolate-specific deletions within patient groups. These data, along with minimal SNP variations, suggest homogeneous populations in the patients studied, consistent with previous longitudinal studies of M. abscessus (8, 47). The high-resolution comparisons enabled by WGS provide evidence for genetic mutations and possible adaptation within the host.
FIG 3.
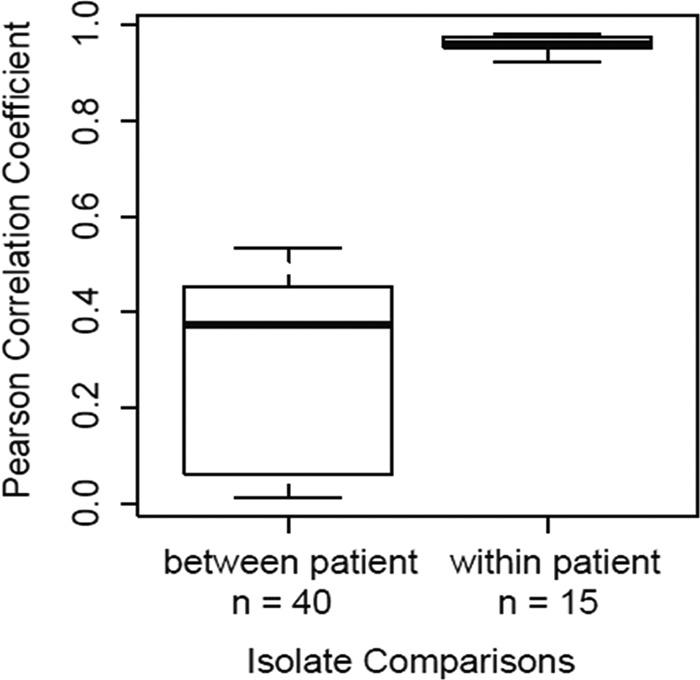
Correlations of genome-wide coverage patterns among 11 M. abscessus isolates collected from three patients. Correlations of genome-wide sequence coverage patterns were performed for all pairwise combinations of 11 isolates (n = 55). Each pair was binned as between-patient isolates (n = 40) or within-patient isolates (n = 15). Isolates obtained from the same patient had much higher correlations than isolates from different patients, suggesting genetically homogeneous infecting populations in individual patients.
Conclusions.
Our study examined the genome sequences of 11 clinically relevant M. abscessus isolates from eight U.S. patients with persistent pulmonary infections that exhibited in vitro resistance to several classes of antimicrobials. Moreover, it provides a comprehensive phylogenomic assessment of U.S. M. abscessus isolates in comparison with globally diverse clinical isolates. Our core genome comparisons, across 41 M. abscessus isolates, revealed three genetic subgroups, consistent with previous subspecies classifications. We observed that most of the NJH isolates examined (9/11 isolates) belonged to a cluster of M. abscessus subsp. abscessus isolates, from primarily U.S. or Western European origins, with low core genomic diversity. This cluster was genetically distinct from most United Kingdom isolates and all Asian isolates examined in the study. Future studies with expanded populations of both clinical and environmental isolates could reveal whether this cluster represents a clinical subtype of M. abscessus or an ecological subgroup related through a shared environmental niche.
Despite the core genome similarities observed among clustered U.S. and European isolates, we found substantial accessory genome variations in the form of multiple large-scale deletions. This illustrates the resolving power of WGS in detecting not only SNPs but also the presence or absence of genomic islands, which can confer clinically important traits associated with virulence and antibiotic resistance. Overall, we observed genome content variations of 0.3 to 7.1% within subspecies (M. abscessus subsp. abscessus) and 8.5% between subspecies (M. abscessus subsp. abscessus versus M. abscessus subsp. massiliense), including one large deletion that was shared across subspecies (Fig. 2, arrow F). Functional annotation analyses of the deletion regions suggested that genome content variations in M. abscessus could be attributed to recombination and/or metabolic adaptations.
Analyses of longitudinally collected samples from individual patients showed low levels of SNP variations and no evidence of large-scale variations, suggesting homogeneous populations in the hosts studied. SNPs in the longitudinal studies were enriched for nonsynonymous mutations, compared to the overall background population, suggesting that pathogen genome evolution in the host may be under positive selective pressure from prolonged exposure to the host immune system, antibiotic treatment, or other selective pressures. Further WGS studies will be needed to explore long-term in vivo mutation rates in the host and potential implications for antibiotic targets.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Jewish Health NTM Center of Excellence, the Colorado Bioscience Discovery Program, the Eppley Foundation, the Potts Memorial Foundation, the Boettcher Foundation Webb-Waring Biomedical Research Program (to M.S.), and NIH Biomedical Informatics training grant 2T15LM009451-06 (to N.A.H. and B.G.).
Footnotes
Published ahead of print 23 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.01144-14.
REFERENCES
- 1.Prevots DR, Shaw PA, Strickland D, Jackson LA, Raebel MA, Blosky M, Oca RM, Shea YR, Seitz AE, Holland SM, Olivier KN. 2010. Nontuberculous mycobacterial lung disease prevalence at four integrated health care delivery systems. Am. J. Respir. Crit. Care Med. 182:970–976. 10.1164/rccm.201002-0310OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore JE, Kruijshaar ME, Ormerod LP, Drobniewski F, Abubakar I. 2010. Increasing reports of non-tuberculous mycobacteria in England, Wales and Northern Ireland, 1995–2006. BMC Public Health 10:612. 10.1186/1471-2458-10-612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marras TK, Chedore P, Ying AM, Jamieson F. 2007. Isolation prevalence of pulmonary non-tuberculous mycobacteria in Ontario, 1997–2003. Thorax 62:661–666. 10.1136/thx.2006.070797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Primm TP, Lucero CA, Falkinham JO., III 2004. Health impacts of environmental mycobacteria. Clin. Microbiol. Rev. 17:98–106. 10.1128/CMR.17.1.98-106.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falkinham JO., III 2011. Nontuberculous mycobacteria from household plumbing of patients with nontuberculous mycobacteria disease. Emerg. Infect. Dis. 17:419–424. 10.3201/eid1703.101510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feazel LM, Baumgartner LK, Peterson KL, Frank DN, Harris JK, Pace NR. 2009. Opportunistic pathogens enriched in showerhead biofilms. Proc. Natl. Acad. Sci. U. S. A. 106:16393–16399. 10.1073/pnas.0908446106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aitken ML, Limaye A, Pottinger P, Whimbey E, Goss CH, Tonelli MR, Cangelosi GA, Dirac MA, Olivier KN, Brown-Elliott BA, McNulty S, Wallace RJ., Jr 2012. Respiratory outbreak of Mycobacterium abscessus subspecies massiliense in a lung transplant and cystic fibrosis center. Am. J. Respir. Crit. Care Med. 185:231–232. 10.1164/ajrccm.185.2.231 [DOI] [PubMed] [Google Scholar]
- 8.Bryant JM, Grogono DM, Greaves D, Foweraker J, Roddick I, Inns T, Reacher M, Haworth CS, Curran MD, Harris SR, Peacock SJ, Parkhill J, Floto RA. 2013. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet 381:1551–1560. 10.1016/S0140-6736(13)60632-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winthrop KL, McNelley E, Kendall B, Marshall-Olson A, Morris C, Cassidy M, Saulson A, Hedberg K. 2010. Pulmonary nontuberculous mycobacterial disease prevalence and clinical features: an emerging public health disease. Am. J. Respir. Crit. Care Med. 182:977–982. 10.1164/rccm.201003-0503OC [DOI] [PubMed] [Google Scholar]
- 10.Jonsson BE, Gilljam M, Lindblad A, Ridell M, Wold AE, Welinder-Olsson C. 2007. Molecular epidemiology of Mycobacterium abscessus, with focus on cystic fibrosis. J. Clin. Microbiol. 45:1497–1504. 10.1128/JCM.02592-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffith DE. 2011. The talking Mycobacterium abscessus blues. Clin. Infect. Dis. 52:572–574. 10.1093/cid/ciq252 [DOI] [PubMed] [Google Scholar]
- 12.Jarand J, Levin A, Zhang L, Huitt G, Mitchell JD, Daley CL. 2011. Clinical and microbiologic outcomes in patients receiving treatment for Mycobacterium abscessus pulmonary disease. Clin. Infect. Dis. 52:565–571. 10.1093/cid/ciq237 [DOI] [PubMed] [Google Scholar]
- 13.Nessar R, Cambau E, Reyrat JM, Murray A, Gicquel B. 2012. Mycobacterium abscessus: a new antibiotic nightmare. J. Antimicrob. Chemother. 67:810–818. 10.1093/jac/dkr578 [DOI] [PubMed] [Google Scholar]
- 14.Prammananan T, Sander P, Brown BA, Frischkorn K, Onyi GO, Zhang Y, Bottger EC, Wallace RJ., Jr 1998. A single 16S ribosomal RNA substitution is responsible for resistance to amikacin and other 2-deoxystreptamine aminoglycosides in Mycobacterium abscessus and Mycobacterium chelonae. J. Infect. Dis. 177:1573–1581. 10.1086/515328 [DOI] [PubMed] [Google Scholar]
- 15.Wallace RJ, Jr., Meier A, Brown BA, Zhang Y, Sander P, Onyi GO, Bottger EC. 1996. Genetic basis for clarithromycin resistance among isolates of Mycobacterium chelonae and Mycobacterium abscessus. Antimicrob. Agents Chemother. 40:1676–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nash KA, Brown-Elliott BA, Wallace RJ., Jr 2009. A novel gene, erm(41), confers inducible macrolide resistance to clinical isolates of Mycobacterium abscessus but is absent from Mycobacterium chelonae. Antimicrob. Agents Chemother. 53:1367–1376. 10.1128/AAC.01275-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leao SC, Tortoli E, Euzeby JP, Garcia MJ. 2011. Proposal that Mycobacterium massiliense and Mycobacterium bolletii be united and reclassified as Mycobacterium abscessus subsp. bolletii comb. nov., designation of Mycobacterium abscessus subsp. abscessus subsp. nov. and emended description of Mycobacterium abscessus. Int. J. Syst. Evol. Microbiol. 61:2311–2313. 10.1099/ijs.0.023770-0 [DOI] [PubMed] [Google Scholar]
- 18.Macheras E, Roux AL, Bastian S, Leao SC, Palaci M, Sivadon-Tardy V, Gutierrez C, Richter E, Rusch-Gerdes S, Pfyffer G, Bodmer T, Cambau E, Gaillard JL, Heym B. 2011. Multilocus sequence analysis and rpoB sequencing of Mycobacterium abscessus (sensu lato) strains. J. Clin. Microbiol. 49:491–499. 10.1128/JCM.01274-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zelazny AM, Root JM, Shea YR, Colombo RE, Shamputa IC, Stock F, Conlan S, McNulty S, Brown-Elliott BA, Wallace RJ, Jr., Olivier KN, Holland SM, Sampaio EP. 2009. Cohort study of molecular identification and typing of Mycobacterium abscessus, Mycobacterium massiliense, and Mycobacterium bolletii. J. Clin. Microbiol. 47:1985–1995. 10.1128/JCM.01688-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heydari H, Wee WY, Lokanathan N, Hari R, Mohamed Yusoff A, Beh CY, Yazdi AH, Wong GJ, Ngeow YF, Choo SW. 2013. MabsBase: a Mycobacterium abscessus genome and annotation database. PLoS One 8:e62443. 10.1371/journal.pone.0062443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho YJ, Yi H, Chun J, Cho SN, Daley CL, Koh WJ, Jae Shin S. 2013. The genome sequence of ‘Mycobacterium massiliense’ strain CIP 108297 suggests the independent taxonomic status of the Mycobacterium abscessus complex at the subspecies level. PLoS One 8:e81560. 10.1371/journal.pone.0081560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koh WJ, Jeon K, Lee NY, Kim BJ, Kook YH, Lee SH, Park YK, Kim CK, Shin SJ, Huitt GA, Daley CL, Kwon OJ. 2011. Clinical significance of differentiation of Mycobacterium massiliense from Mycobacterium abscessus. Am. J. Respir. Crit. Care Med. 183:405–410. 10.1164/rccm.201003-0395OC [DOI] [PubMed] [Google Scholar]
- 23.Harada T, Akiyama Y, Kurashima A, Nagai H, Tsuyuguchi K, Fujii T, Yano S, Shigeto E, Kuraoka T, Kajiki A, Kobashi Y, Kokubu F, Sato A, Yoshida S, Iwamoto T, Saito H. 2012. Clinical and microbiological differences between Mycobacterium abscessus and Mycobacterium massiliense lung diseases. J. Clin. Microbiol. 50:3556–3561. 10.1128/JCM.01175-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davidson RM, Hasan NA, de Moura VC, Duarte RS, Jackson M, Strong M. 2013. Phylogenomics of Brazilian epidemic isolates of Mycobacterium abscessus subsp. bolletii reveals relationships of global outbreak strains. Infect. Genet. Evol. 20:292–297. 10.1016/j.meegid.2013.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tettelin H, Davidson RM, Agrawal S, Aitken ML, Shallom S, Hasan NA, Strong M, de Moura VC, De Groote MA, Duarte RS, Hine E, Parankush S, Su Q, Daugherty SC, Fraser CM, Brown-Elliott BA, Wallace RJ, Jr., Holland SM, Sampaio EP, Olivier KN, Jackson M, Zelazny AM. 2014. High-level relatedness among Mycobacterium abscessus subsp. massiliense strains from widely separated outbreaks. Emerg. Infect. Dis. 20:364–371. 10.3201/eid2003.131106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Juhas M, van der Meer JR, Gaillard M, Harding RM, Hood DW, Crook DW. 2009. Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 33:376–393. 10.1111/j.1574-6976.2008.00136.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jackson RW, Vinatzer B, Arnold DL, Dorus S, Murillo J. 2011. The influence of the accessory genome on bacterial pathogen evolution. Mob. Genet. Elements 1:55–65. 10.4161/mge.1.1.16432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim BJ, Kim BR, Lee SY, Kook YH. 2013. Rough colony morphology of Mycobacterium massiliense type II genotype is due to the deletion of glycopeptidolipid locus within its genome. BMC Genomics 14:890. 10.1186/1471-2164-14-890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng Y, Chen CJ, Su LH, Hu S, Yu J, Chiu CH. 2008. Evolution and pathogenesis of Staphylococcus aureus: lessons learned from genotyping and comparative genomics. FEMS Microbiol. Rev. 32:23–37. 10.1111/j.1574-6976.2007.00086.x [DOI] [PubMed] [Google Scholar]
- 30.Mathee K, Narasimhan G, Valdes C, Qiu X, Matewish JM, Koehrsen M, Rokas A, Yandava CN, Engels R, Zeng E, Olavarietta R, Doud M, Smith RS, Montgomery P, White JR, Godfrey PA, Kodira C, Birren B, Galagan JE, Lory S. 2008. Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. U. S. A. 105:3100–3105. 10.1073/pnas.0711982105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Soolingen D, de Haas PE, Kremer K. 2001. Restriction fragment length polymorphism typing of mycobacteria. Methods Mol. Med. 54:165–203. 10.1385/1-59259-147-7:165 [DOI] [PubMed] [Google Scholar]
- 32.Adekambi T, Colson P, Drancourt M. 2003. rpoB-based identification of nonpigmented and late-pigmenting rapidly growing mycobacteria. J. Clin. Microbiol. 41:5699–5708. 10.1128/JCM.41.12.5699-5708.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clinical and Laboratory Standards Institute. 2011. Susceptibility testing of mycobacteria, nocardiae, and other aerobic actinomycetes; approved standard—2nd ed. CLSI document M42-A2 Clinical and Laboratory Standards Institute, Wayne, PA: [PubMed] [Google Scholar]
- 34.Ripoll F, Pasek S, Schenowitz C, Dossat C, Barbe V, Rottman M, Macheras E, Heym B, Herrmann JL, Daffe M, Brosch R, Risler JL, Gaillard JL. 2009. Non mycobacterial virulence genes in the genome of the emerging pathogen Mycobacterium abscessus. PLoS One 4:e5660. 10.1371/journal.pone.0005660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gouy M, Guindon S, Gascuel O. 2010. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27:221–224. 10.1093/molbev/msp259 [DOI] [PubMed] [Google Scholar]
- 40.Cheung MS, Down TA, Latorre I, Ahringer J. 2011. Systematic bias in high-throughput sequencing data and its correction by BEADS. Nucleic Acids Res. 39:e103. 10.1093/nar/gkr425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. 2003. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34:374–378 http://www.biotechniques.com/multimedia/archive/00010/03342mt01_10634a.pdf [DOI] [PubMed] [Google Scholar]
- 42.R Development Core Team. 2011. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 43.Moore M, Frerichs JB. 1953. An unusual acid-fast infection of the knee with subcutaneous, abscess-like lesions of the gluteal region: report of a case with a study of the organism, Mycobacterium abscessus, n. sp. J. Invest. Dermatol. 20:133–169 [DOI] [PubMed] [Google Scholar]
- 44.Choo SW, Wee WY, Ngeow YF, Mitchell W, Tan JL, Wong GJ, Zhao Y, Xiao J. 2014. Genomic reconnaissance of clinical isolates of emerging human pathogen Mycobacterium abscessus reveals high evolutionary potential. Sci. Rep. 4:4061. 10.1038/srep04061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsolaki AG, Hirsh AE, DeRiemer K, Enciso JA, Wong MZ, Hannan M, Goguet de la Salmoniere YO, Aman K, Kato-Maeda M, Small PM. 2004. Functional and evolutionary genomics of Mycobacterium tuberculosis: insights from genomic deletions in 100 strains. Proc. Natl. Acad. Sci. U. S. A. 101:4865–4870. 10.1073/pnas.0305634101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moody JD, Doerge DR, Freeman JP, Cerniglia CE. 2002. Degradation of biphenyl by Mycobacterium sp. strain PYR-1. Appl. Microbiol. Biotechnol. 58:364–369. 10.1007/s00253-001-0878-3 [DOI] [PubMed] [Google Scholar]
- 47.Wallace RJ, Jr., Zhang Y, Brown BA, Fraser V, Mazurek GH, Maloney S. 1993. DNA large restriction fragment patterns of sporadic and epidemic nosocomial strains of Mycobacterium chelonae and Mycobacterium abscessus. J. Clin. Microbiol. 31:2697–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pang S, Renvoise A, Perret C, Guinier M, Chelghoum N, Brossier F, Capton E, Jarlier V, Sougakoff W. 2013. Whole-genome sequence of Mycobacterium abscessus clinical strain V06705. Genome Announc. 1:e00690–13. 10.1128/genomeA.00690-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ngeow YF, Wong YL, Tan JL, Ong CS, Ng KP, Choo SW. 2012. Genome sequence of Mycobacterium abscessus strain M152. J. Bacteriol. 194:6662. 10.1128/JB.01846-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choo SW, Wong YL, Leong ML, Heydari H, Ong CS, Ng KP, Ngeow YF. 2012. Analysis of the genome of Mycobacterium abscessus strain M94 reveals an uncommon cluster of tRNAs. J. Bacteriol. 194:5724. 10.1128/JB.01407-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choo SW, Wong YL, Yusoff AM, Leong ML, Wong GJ, Ong CS, Ng KP, Ngeow YF. 2012. Genome sequence of the Mycobacterium abscessus strain M93. J. Bacteriol. 194:3278. 10.1128/JB.00492-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi GE, Cho YJ, Koh WJ, Chun J, Cho SN, Shin SJ. 2012. Draft genome sequence of Mycobacterium abscessus subsp. bolletii BDT. J. Bacteriol. 194:2756–2757. 10.1128/JB.00354-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wong YL, Choo SW, Tan JL, Ong CS, Ng KP, Ngeow YF. 2012. Draft genome sequence of Mycobacterium bolletii strain M24, a rapidly growing mycobacterium of contentious taxonomic status. J. Bacteriol. 194:4475. 10.1128/JB.00916-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chan J, Halachev M, Yates E, Smith G, Pallen M. 2012. Whole-genome sequence of the emerging pathogen Mycobacterium abscessus strain 47J26. J. Bacteriol. 194:549. 10.1128/JB.06440-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davidson RM, Reynolds PR, Farias-Hesson E, Duarte RS, Jackson M, Strong M. 2013. Genome sequence of an epidemic isolate of Mycobacterium abscessus subsp. bolletii from Rio de Janeiro, Brazil. Genome Announc. 1:e00617-13. 10.1128/genomeA.00617-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tettelin H, Sampaio EP, Daugherty SC, Hine E, Riley DR, Sadzewicz L, Sengamalay N, Shefchek K, Su Q, Tallon LJ, Conville P, Olivier KN, Holland SM, Fraser CM, Zelazny AM. 2012. Genomic insights into the emerging human pathogen Mycobacterium massiliense. J. Bacteriol. 194:5450. 10.1128/JB.01200-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.