

Abstract
Staphylococcus lugdunensis is an emergent virulent coagulase-negative staphylococcus responsible for severe infections similar to those caused by Staphylococcus aureus. To understand its potentially pathogenic capacity and have further detailed knowledge of the molecular traits of this organism, 93 isolates from various geographic origins were analyzed by multi-virulence-locus sequence typing (MVLST), targeting seven known or putative virulence-associated loci (atlLR2, atlLR3, hlb, isdJ, SLUG_09050, SLUG_16930, and vwbl). The polymorphisms of the putative virulence-associated loci were moderate and comparable to those of the housekeeping genes analyzed by multilocus sequence typing (MLST). However, the MVLST scheme generated 43 virulence types (VTs) compared to 20 sequence types (STs) based on MLST, indicating that MVLST was significantly more discriminating (Simpson's index [D], 0.943). No hypervirulent lineage or cluster specific to carriage strains was defined. The results of multilocus sequence analysis of known and putative virulence-associated loci are consistent with a clonal population structure for S. lugdunensis, suggesting a coevolution of these genes with housekeeping genes. Indeed, the nonsynonymous to synonymous evolutionary substitutions (dN/dS) ratio, the Tajima's D test, and Single-likelihood ancestor counting (SLAC) analysis suggest that all virulence-associated loci were under negative selection, even atlLR2 (AtlL protein) and SLUG_16930 (FbpA homologue), for which the dN/dS ratios were higher. In addition, this analysis of virulence-associated loci allowed us to propose a trilocus sequence typing scheme based on the intragenic regions of atlLR3, isdJ, and SLUG_16930, which is more discriminant than MLST for studying short-term epidemiology and further characterizing the lineages of the rare but highly pathogenic S. lugdunensis.
INTRODUCTION
Staphylococcus lugdunensis is a coagulase-negative staphylococcus (CoNS) belonging to the normal human skin flora (1) that is increasingly recognized as a virulent pathogen in both community-acquired and nosocomial infections (2–4). It colonizes several distinct niches primarily in the lower part of the body (such as perineal and inguinal areas) but also the lower extremities (including feet and nails) (1). Described as a “wolf in sheep's clothing,” S. lugdunensis resembles Staphylococcus aureus in its clinical presentation and course of infection (tissue destruction and aggressive clinical course) (2). Even if it can cause many types of infections ranging from localized to systemic (2, 5, 6), this opportunistic pathogen has been mainly associated with serious infections, such as skin and soft tissue infections (SSTI) (3, 4, 7, 8), infective endocarditis (5, 6, 9, 10), abscesses (11, 12), and bone and joint infections (13, 14). S. lugdunensis SSTI have been reported to be superficial, painful, often prolonged, and recurrent in patients with skin diseases or after trauma or surgery (7, 15). S. lugdunensis infective endocarditis can occur in native or prosthetic valves as well as on cardiac implantable electronic devices (10), and surgery with valve replacement is often indicated (16, 17). Endocarditis is typically characterized by an aggressive and a rapidly progressive clinical course, with mortality rates higher than those for S. aureus (14.5%) or Staphylococcus epidermidis (20%), ranging from 18.2% (10) to 75% (5, 18). Of note, the prevalence of S. lugdunensis infections is low but may be underestimated due to a frequent lack of reliable identification of staphylococcal species (2, 7, 14) before the matrix-assisted laser desorption ionization–time of flight mass spectrometry-based approach was used (19).
Although this CoNS species was first described >2 decades ago (20), remarkably little is currently known about the virulence factors and molecular mechanisms underpinning the virulence of S. lugdunensis. Furthermore, genetic tools for the manipulation of S. lugdunensis were limited until very recently, when efficient protoplast transformation (21) and electroporation (22) were reported. So, in contrast to S. aureus, only very few pathogenicity factors have been characterized. Among them, the fibrinogen binding protein Fbl is similar in structure and organization to S. aureus clumping factor A (ClfA) (23–25) and is probably the only fibrinogen-binding surface protein of S. lugdunensis (21). A von Willebrand factor-binding protein, denoted vWbl, has also been described (26). The ability of S. lugdunensis to bind von Willebrand factor may play a critical role in the bacterial attachment to vascular lesions, and thereby to colonization preceding infections (22). In 1997, Donvito et al. (27) observed synergistic hemolytic activity of S. lugdunensis with the S. aureus β-hemolysin. This synergistic hemolysis is a function of three small peptides, named S. lugdunensis synergistic hemolysins A (SLUSH-A), SLUSH-B, and SLUSH-C, encoded by the slush operon (27), which is controlled by agr activity (28). As phenol-soluble modulin β-like peptides, SLUSH peptides attract and stimulate human leukocytes in a formyl peptide receptor 2-dependent manner (29) and thereby play a proinflammatory role. Besides, S. lugdunensis is unique among the CoNS in that it contains a gene encoding an iron-regulated surface determinant (Isd) system (30), which shares significant structural and functional homology with the Isd receptors of S. aureus. Under iron-restricted conditions, the Isd proteins cooperate to capture heme and transfer it across the wall into the cytoplasm, where it is degraded to iron for use in cellular processes (31, 32).
Interestingly, the genome sequencing of two strains of S. lugdunensis, N920143 (30) and HKU09-01 (33), revealed genes encoding putative virulence factors, such as adhesins, toxins, and hemolysins. Among them is a gene denoted SLUG_16930 in the N920143 genome that encodes a putative fibrinogen/fibronectin binding adhesin named FbpA homologue, which is homologous to FbpA of S. aureus (30, 34, 35). The hlb gene encodes a putative sphingomyelinase β-toxin of 329 amino acids (aa), which shares 85% identity with that of S. aureus (30, 36), and a putative hemolysin III (denoted SLUG_09050 in the N920143 genome [30]) is encoded as well.
Likewise, bifunctional autolysins may contribute to bacterial pathogenesis by releasing virulence factors (37), and they may serve as adhesins, allowing cells to bind to surfaces and/or the host extracellular matrix and plasma proteins, as described for AtlA of S. aureus (38) and AtlE of S. epidermidis (39). Like AtlA and AtlE, AtlL of S. lugdunensis displays two enzymatic domains separated by three repeat domains (AtlLR1 to AtlLR3) and undergoes proteolytic processing to generate two extracellular peptidoglycan hydrolases, an N-acetylmuramoyl-l-alanine amidase (AM) and an N-acetylglucosaminidase (GL) with two (AtlLR1R2) and one (AtlLR3) repeated sequences harboring glycine tryptophan motifs, respectively (40). We recently reported that a ΔatlL mutant showed a significant reduction in biofilm formation and virulence attenuation using the Caenorhabditis elegans model (41). Since the overall organization of the bifunctional precursor protein is highly conserved in all staphylococcal species (42), the virulence findings for AtlA and AtlE may thus also apply for the entire Staphylococcus genus (43).
In an attempt to better understand the genetic background and population structure of S. lugdunensis, we recently developed the first multilocus sequence typing (MLST) scheme for this species (44). This phylogenetic analysis revealed a clonal population structure, a mutational evolution of this pathogen, and a lack of hypervirulent lineages.
Due to their exposure to frequent environmental changes, e.g., immune system responses, virulence and virulence-associated genes may evolve more rapidly than housekeeping genes (45, 46). Consequently, these genes may provide a higher degree of nucleotide sequence polymorphisms, a higher discriminatory power for epidemiology studies (45, 46), and some data on the specific evolution of virulence- or putative virulence-associated loci in comparison with that of housekeeping genes. Chen et al. (47) showed that MVLST provided subtyping data to define the genetic lineages in epidemic clones of Listeria monocytogenes. In this study, we analyzed the polymorphisms of virulence-associated genes (vwbl and isdJ) and putative virulence-associated loci (atlLR2, atlLR3, hlb, SLUG_16930, and SLUG_09050), determined the agr alleles, and analyzed the presence of virulence genes (fbl and slush) by PCR for 93 S. lugdunensis clinical and carriage isolates. The goals of this work were to: (i) develop a multi-virulence-locus sequence typing (MVLST) approach, (ii) compare the discriminatory power of MVLST to that of MLST, and (iii) generate informative sequence data for studying the virulence of S. lugdunensis.
MATERIALS AND METHODS
Bacterial strains.
Ninety-three S. lugdunensis isolates were studied. The set included 3 S. lugdunensis reference strains (ATCC 49576, ATCC 43809, and ATCC 700328), 84 human clinical isolates collected between 1991 and 2011 from 8 different geographic sources and 3 countries (France, Belgium, and Slovenia) (44), and 6 S. lugdunensis carriage strains isolated from the groin, perineum, nail, toe, and axilla of healthy subjects from Kronoberg County in Sweden (1) or from Tours in France (48). The genome sequence data of the S. lugdunensis N920143 strain (30) were included.
MLST.
MLST was performed for the 6 carriage strains, as previously described (44). Allelic profiles and sequence types (ST) were assigned by using the international MLST database (www.pasteur.fr/mlst). The STs were clustered into different clonal complexes (CCs) using the BURST program (http://pubmlst.org/analysis/burst/burst.shtml).
Multilocus sequence analysis of virulence-associated loci.
A first set of 10 isolates representative of the main clusters previously defined by MLST (44) were screened for 10 known or putative S. lugdunensis virulence loci: agr, atlLR2 (AM cell wall-anchoring [CWA] domain), atlLR3 (GL CWA domain), fbl, hlb, isdJ, locus SLUG_09050, locus SLUG_16930, slush, and vwbl.
A locus was validated as a good candidate for multilocus sequence typing analysis if it (i) was present in all strains, (ii) required a single primer pair for PCR amplification and sequencing of the internal fragment, and (iii) contributed to a high discriminatory power.
Finally, seven putative virulence-associated loci were selected (atlLR2, atlLR3, hlb, isdJ, SLUG_09050, SLUG_16930, and vwbl) for MVLST (Table 1). The genomic locations of all loci studied are shown in Fig. 1.
TABLE 1.
Primers used for the amplification and sequencing of the 10 loci investigated
| Gene | PCR and sequencing primers (5′→3′) | Study |
|---|---|---|
| atlLR2 | Forward: GCTATAAACAATGATTCATCATC | This study |
| Reverse: GTGCGTATGCCTGAATTATT | ||
| atlLR3 | Forward: GCGTACATTAGCTGTTAAAACAA | This study |
| Reverse: CGTCATAGAAATAGCTATCC | ||
| hlb | Forward: AAAGCGGATTATATCCAAGGTCAAG | This study |
| Reverse: GCGTTGCCCTCGTATTGTTTAG | ||
| isdJ | Forward: TGTTAAACCAGAAACATTGGCACAA | This study |
| Reverse: GCTGGTTTGGTTTTTGGTG | ||
| SLUG_09050 | Forward: TAATGCTGTTTCGCACGGAGTTGC | Szabados et al. (35) |
| Reverse: GACGCCTACCCATCCCATTACAA | ||
| SLUG_16930 | Forward: GAGATTACTGGACAACAAACG | Szabados et al. (35) |
| Reverse: GTATTGTGACGTCGTTTCCTG | ||
| vwbl | Forward: TGGCGGGATGATTTGGACG | This study |
| Reverse: CCTCATTAAGATGGAGATGAATGC | ||
| slush | Forward: TCGTAGATGCAATTTCAAAAGC | This study |
| Forward: TTTCGTCTTTGCACACACATTTCCAa | Szabados et al. (35) | |
| Reverse: ACAGCACAAAGCCTTAACTATCTCA | ||
| fbl | Forward: TGTAACAGCTACTGCTAAAGCACC | This study |
| Reverse: ATCCCATGCCATAGAAACTCTAGTG | ||
| agrBDC | Forward: GGTCATCATTGTTGTGCCACATTC | This study |
| Reverse 1: GCGCAGAACCAAAGATTGCTAAAACb | ||
| Reverse 2: CTGCCAAAAACAACAAATAATACGc |
FIG 1.
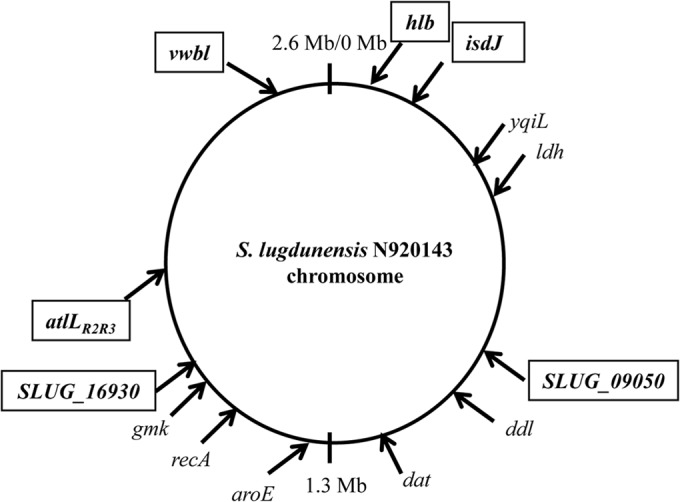
Genomic locations of the loci analyzed in the S. lugdunensis N920143 genome. The seven selected virulence loci are boxed and shown in bold type. The housekeeping genes of the MLST scheme are shown in italics (44).
The genomic DNA of S. lugdunensis was extracted from a single bacterial colony using the InstaGene matrix kit (Bio-Rad), as recommended by the manufacturer. The PCRs were performed in a final volume of 25 μl containing 0.50 μM each primer (Table 1), 12.5 μl of GoTaq (green master mix [Promega]), and 3 μl of extracted DNA. The PCR mixtures were heated for 3 min at 95°C, followed by 35 cycles of a denaturation step at 95°C for 30 s, an annealing step for 30 s at a temperature of 55°C, an elongation step at 72°C for 30 s, and ending with an extension step at 72°C during 10 min. The PCR products were then purified with a NucleoSpin gel and PCR clean-up kit (Macherey-Nagel) and sequenced with the same primers using the GenomeLab DTCS quick start kit on a GenomeLab GeXP analyzer (both Beckman Coulter). Different sequences of a given locus were given allele numbers, and each unique combination of alleles (multilocus allelic profile) was assigned to a virulence type (VT). Single point polymorphisms were assessed by resequencing DNA from two separate PCR experiments.
Computer analysis of sequence data.
Clustering of the 92 isolates and of S. lugdunensis N920143, whose gene sequences were extracted from the available genome, was determined using the BioEdit sequence alignment editor (http://www.mbio.ncsu.edu/bioedit/bioedit.html) for alignment of the nucleotide sequences, and gene trees were constructed from the sequence data using the neighbor-joining method and bootstrapping algorithms contained in the MEGA 5.1 software (49). Simpson's index of diversity was calculated as proposed by Hunter and Gaston (50). The average numbers of nucleotide differences between the alleles and the ratios of nonsynonymous to synonymous substitutions (dN/dS) were calculated using the S.T.A.R.T.2 program (51). The codon alignments were then submitted to a suite of phylogenetic analysis tools (http://www.datamonkey.org/) for selection analysis (52, 53). In addition, Tajima's test of neutrality was performed using DnaSP version 5 (54) for each alignment.
The index of association (IA) between alleles (55) was used to test for linkage disequilibrium between the alleles of the seven loci analyzed (http://www.mlst.net).
The BURST program (http://pubmlst.org/analysis/burst/burst.shtml) was used to define groups in which every isolate shares at least five identical alleles with at least one other isolate (CCvs) and to characterize ancestral genotypes and their corresponding single-locus variants (isolates that differ at only one of the 7 loci) within these clonal complexes.
Detection of agr alleles and virulence factor genes.
All isolates were screened for fbl and the slush operon, and the agr alleles were determined by PCR and sequencing of an internal fragment. Genomic DNA extraction, PCR amplification, and sequencing were performed as described above, using the primers listed in Table 1. The PCR products were electrophoresed on 1% agarose gels and stained with ethidium bromide to visualize the DNA bands, whose sizes were determined using a 100-bp DNA ladder (New England BioLabs).
Synergistic δ-hemolysis.
All strains were tested for synergistic hemolysis, as previously described (56) by measuring synergistic hemolysis in the presence of β-toxin secreted by S. aureus strain RN4220 on Trypticase soy agar plates supplemented with 5% sheep blood (bioMérieux, France). Briefly, strains of S. lugdunensis were streaked down perpendicular to but not touching the center streak. The test was positive when a zone of complete hemolysis was observed after incubation at 37°C for 24 h and held at room temperature for an additional 24 h (2).
Nucleotide sequence accession numbers.
The nucleotide sequences of the internal fragment loci (atlLR2, atlLR3, hlb, isdJ, SLUG_09050, SLUG_16930, and vwbl) analyzed in this work have been deposited in GenBank under accession no. KJ816790 to KJ816797 for atlLR2, KJ816798 to KJ816806 for atlLR3, KJ816826 to KJ816835 for hlb, KJ816836 to KJ816849 for isdJ, KJ816821 to KJ816825 for SLUG_09050, KJ816807 to KJ816820 for SLUG_16930, and KJ816850 to KJ816858 for vwbl.
RESULTS
Distribution of agr alleles, fbl gene, and slush operon.
All S. lugdunensis isolates tested were shown to be fbl and slush positive. Interestingly, the slush amplicon of S. lugdunensis strain SL9 was shorter than the others. The sequencing of this locus showed a 125-bp deletion between slushB and slushC.
The polymorphism rate of the agr locus was too high to design a unique primer pair for PCR amplification and double-strand sequencing. However, an internal fragment was amplified and sequenced for all strains in the study. Of the 93 strains, 53 (6 carriage strains, 3 reference strains, and 44 clinical strains) had agr-1SL, and 40 clinical strains had agr-2SL (57). The genomic data available for S. lugdunensis N920143 allowed us to determine an agr-1SL allele.
Delta-like synergistic hemolytic activity.
Seventy-two strains (77.4%) gave a clear zone of synergistic complete hemolysis when tested against the β-toxin of S. aureus RN4220. Of note, the SL9 strain was positive for this test in spite of an incomplete slush locus.
Allelic variation in known and putative virulence-associated loci of S. lugdunensis.
Seven known and putative virulence-associated loci were selected (atlLR2, atlLR3, hlb, isdJ, SLUG_09050, SLUG_16930, and vwbl) for MVLST. Primers successfully amplified all seven MVLST loci (2,967 nucleotides in total) from the 93 strains. The data reporting the allelic variations in the seven virulence-associated loci studied are summarized in Table 2. The number of individual alleles for each of the seven genes ranged from 5 (for SLUG_09050) to 14 (for isdJ and SLUG_16930). The number of polymorphic sites on a given locus varied from 4 (1.20%) for SLUG_09050 to 12 (2.72%) for hlb. Polymorphisms resulted for most genes in synonymous substitutions, with a ratio of nonsynonymous to synonymous substitutions ranging from 0 (for SLUG_09050) to 0.133 (for vwbl). The higher dN/dS ratio for atlLR3 (0.258), atlLR2 (0.416), and SLUG_16930 (0.256) than that of hlb and isdJ might suggest a potential role of environmental selective pressure in their evolution, but in Tajima's test, no significant difference from neutral evolution was displayed for these 3 loci compared to that of the others. Single-likelihood ancestor counting (SLAC) analysis of the data (Table 2) and the mixed-effects model episodic (MEME) program (data not shown) also provided no evidence for positive selection of any amino acid of either protein.
TABLE 2.
Genetic polymorphisms of the seven virulence-associated genes analyzed by MVLST
| Gene | Size of PCR product (bp) | Positions of the fragment analyzed within each genea | No. of alleles | No. (%) of polymorphic sites | dN/dSb | dN | dS | Tajima's D testc | SLAC (95% CI)d |
|---|---|---|---|---|---|---|---|---|---|
| atlLR2 | 650 | 1879–2418 | 8 | 8 (1.47) | 0.416 | 0.0042 | 0.01 | −0.13385 | 0.647 (0.277–1.250) |
| atlLR3 | 590 | 2419–2859 | 9 | 9 (2.04) | 0.258 | 0.0046 | 0.0179 | −0.43781 | 0.338 (0.121–0.728) |
| hlb | 554 | 274–714 | 10 | 12 (2.72) | 0.024 | 0.0012 | 0.0471 | 0.38138 | 0.092 (0.015–0.285) |
| isdJ | 498 | 971–1382 | 14 | 10 (2.42) | 0.103 | 0.0034 | 0.033 | 0.90961 | 0.166 (0.066–0.336) |
| SLUG_09050 | 406 | 124–457 | 5 | 4 (1.20) | 0 | 0 | 0.0282 | 0.95707 | 0 (0–0.203) |
| SLUG_16930 | 559 | 586–1037 | 14 | 9 (1.99) | 0.256 | 0.0044 | 0.0171 | 0.42415 | 0.345 (0.158–0.643) |
| vwbl | 461 | 932–1275 | 9 | 7 (2.03) | 0.133 | 0.0025 | 0.0192 | 0.70819 | 0.178 (0.044–0.463) |
Fragment positions are based on the genome of S. lugdunensis N920143.
dN/dS, ratio of nonsynonymous to synonymous substitutions.
P > 0.1 for all Tajima's D test results.
SLAC, single-likelihood ancestor counting; CI, confidence interval.
Multilocus sequence data and composite sequence-based analysis of virulence-associated loci.
Multilocus allelic profile analysis based on the compilation of the seven loci, as well as the 2,967-bp composite sequence-based analysis, generated 43 different VTs among the 93 strains and the DNA sequence data of S. lugdunensis N920143. The phylogenetic relationships between the 93 isolates and the in silico data of S. lugdunensis N920143 are shown in Fig. 2.
FIG 2.

Dendrogram showing genetic relationships of 93 S. lugdunensis isolates and the genome sequence data from S. lugdunensis N920143 based on the composite sequence of seven virulence-associated loci (atlLR2, atlLR3, hlb, isdJ, SLUG_09050, SLUG_16930, and vwbl). N920143 represents S. lugdunensis N920143 strain (in silico data). VTs, virulence types; STs, sequence types previously described (44). Abbreviations (for clinical sources): OA, osteoarticular; SST, skin and soft tissue; M, material device; B, blood; Ca, carriage. Abbreviations (for origins): A, Rouen (France); B, Nantes (France); C, Nancy (France); D, Bordeaux (France); E, Montpellier (France); F, Versailles (France); G, Louvain (Belgium); H, Maribor (Slovenia); I, Tours (France); J, Kronoberg County (Sweden); K, Lyon (France). CCv, clonal complexes. The asterisks represent the ancestral VT for each CCv. REF1 (reference sequence 1), S. lugdunensis ATCC 700328; REF2, S. lugdunensis ATCC 43809; REF3, S. lugdunensis ATCC 49576. NC, unknown.
Four major VTs (VT-1, VT-18, VT-31, and VT-35) were shared by five to 14 isolates, and eight VTs (VT-4, VT-5, VT-7, VT-20, VT-26, VT-36, VT-38, and VT-40) were shared by two to four isolates, but the majority (32 of 43 VTs) of these VTs were represented by a single isolate (Fig. 2), reflecting the genetic diversity of the population studied.
No correlation was found between the virulence-associated locus pattern and geographic origin, or even with clinical presentation. Indeed, 4 VTs were shared by isolates from different geographic and clinical sources; for example, for VT-1, including isolates from Belgium, France (Bordeaux, Nantes, and Rouen), and Sweden, and carriage isolates, as well as isolates collected from patients with a wide spectrum of infections (bacteremia, osteoarticular infections, SSTI, and material device-associated infections). Similarly, the carriage strains did not cluster in a separate lineage; 5 of 6 commensal strains were clustered with clinical isolates in VT-1 (Fig. 2). Of note, the 6 isolates exhibiting glycopeptide tolerance (44) belonged to 4 VTs (VT-1, VT-17, VT-25, and VT-31) and did not define a specific lineage.
The analysis using the BURST algorithm (58) defined six CCvs, CCv1 to CCv6, including almost all VTs and two singletons (VT-25 and VT-29). Two majority CCvs (CCv1 and CCv2) contain 27 VTs, including 71 strains. Ancestral genotypes and their single-locus variants were identified for each CCv (Fig. 2). Of note, the strains associated with infections on material devices clustered mainly (11/12) in CCv2.
Contribution of recombination and mutations to genomic evolution of S. lugdunensis.
Monolocus dendrograms were globally congruent, highlighting that recombination events are scarce but can exist for the SLUG_16930 locus of S. lugdunensis strains SL78 and SL91. However, no statistically significant evidence for recombination was found by SplitsTree analysis and phi test (data not shown). A quantitative analysis of the linkage between the alleles from the seven loci studied was performed by calculating the index of association (IA) to test the probability that recombination occurred (55). A significant linkage disequilibrium was detected when all isolates were included (IA, 3.74) and when one isolate per VT was used (IA, 2.56). Likewise, the G+C contents of each locus were closed to each other and similar to the G+C percentage of the whole genome, with a G+CN920143 of 33.8%, and that of the loci as follows: G+CatlLR2, 34.4%; G+CatlLR3, 32.32%; G+Chlb, 34.12%; G+CisdJ, 34.79%; G+CSLUG_09050, 34.59%; G+CSLUG_16930, 32.07%; and G+Cvwbl, 35.48%.
The congruence of monolocus dendrograms associated with the linkage disequilibrium observed and the lack of intragenic recombination from extraspecies sources argue for a clonal population structure for S. lugdunensis.
Comparison of MVLST and MLST.
A total of 87 strains (3 reference strains and 84 clinical isolates) investigated in this study were previously analyzed by MLST (44). In the present study, the STs of S. lugdunensis N920143 and of six carriage strains of S. lugdunensis were determined using the Institut Pasteur MLST databases (www.pasteur.fr/mlst). Five of them (4 from Sweden and one from France) clustered in ST1, whereas the sixth one from Sweden belonged to ST10 (Fig. 2). Therefore, a total of 20 STs were generated for the 93 strains and N920143.
The MVLST scheme allowed us to define 43 VTs, which is much more discriminant than the MLST scheme (44). Indeed, virulence-associated loci generated more allelic variations (5 to 14 alleles) than did the housekeeping genes (4 to 9). A slightly higher polymorphism rate (1.20 to 2.72%) was observed for the virulence-associated loci compared to that of housekeeping genes (0.9% to 1.8%). The comparison of dendrograms based on the concatenated gene sequences from the MLST gene set and the MVLST gene set showed that strains were distributed in the same clusters, leading to a congruence of dendrograms (Fig. 2). The congruence of the MLST and MVLST monolocus and multilocus dendrograms associated with a lack of recombination events (IA was similar for both schemes) further argue for a clonal structure of S. lugdunensis.
However, MVLST further resolved all the clusters in which the isolates were indistinguishable by MLST, as a given ST included 3 (in STs 4, 5, 12, 13, and 15) to 8 VTs (in ST1). Isolates from a given VT belonged to 1 to 4 STs that were very closely related, and all were included in CC1 (44). Of note, the 9 isolates that represented one ST as determined by MLST were also found to be singletons by MVLST. Together, these results show that MVLST provides more discrimination than does MLST, as confirmed by the Simpson's index values (DMVLST, 0.943; DMLST, 0.890) (Table 3). The classification of the isolates into MVLST clonal complexes by the BURST program showed groupings that were highly concordant with those determined by MLST. In addition, CCv2 defined by MVLST gathered isolates with STs included in two MLST-defined CCs (CC2 and CC3) (44). Furthermore, two new CCvs (MVLST-defined CCv3 and CCv6) were defined; CCv3 consists of ST13 isolates, whereas CCv6 contains ST10 isolates.
TABLE 3.
Comparative genetic diversity characteristics of MVLST, MLST, and trilocus analysis
| Sequencing typing method | Size of concatenated nucleotidic sequences (bp) | No. (%) of polymorphic sites | Simpson's index (95% CI)a | P value |
|---|---|---|---|---|
| MLST | 3,037 | 39 (1.28) | 0.890 (0.862–0.917) | |
| MVLST | 2,967 | 59 (1.99) | 0.943 (0.918–0.967) | 0.001b |
| Trilocus | 1,407 | 27 (1.92) | 0.922 (0.893–0.952) | 0.075c/0.016d |
CI, confidence interval.
P value between Simpson's index values of MLST and MVLST.
P value between Simpson's index values of MLST and trilocus analysis.
P value between Simpson's index values of MVLST and trilocus analysis.
Multilocus sequence data based on two up to six loci.
In order to provide a more suitable tool for studying the virulence characteristics of S. lugdunensis, a multilocus allelic profile analysis based on the compilation of two up to six loci was performed for the 93 isolates and the DNA sequence data of S. lugdunensis N920143. The allelic variation of isdJ and the SLUG_16930 locus generated a maximum of 27 different VTs, which is seven more than that of the seven housekeeping gene analysis. The number of VTs identified for the 93 isolates and N920143 was 33 (of which 22 singletons) when analyzing the polymorphisms of atlLR2 or atlLR3 and isdJ and SLUG_16930, and up to 37 (of which 23 singletons) when analyzing atlLR3, hlb, isdJ, and SLUG_16930. The combination of five and six loci generated 41 (with atlLR2, atlLR3, hlb, isdJ, and SLUG_16930) and 43 VTs (with atlLR2, atlLR3, hlb, isdJ, SLUG_16930, and vwbl), respectively. In conclusion, a trilocus sequence typing scheme based on the intragenic regions of atl (encoding a bifunctional autolysin), isdJ (encoding an iron-regulated surface determinant protein), and SLUG_16930 locus (encoding an FbpA homologue) were successful for strain differentiation within S. lugdunensis species and may provide an alternative to MLST and MVLST for discriminating isolates (Dtrilocus, 0.922) (Table 3).
DISCUSSION
Because S. lugdunensis is more virulent than other CoNS, it is worthwhile to examine the differences in the repertoire and polymorphisms of genes encoding virulence factors. The aim of the present study was thus to design a MVLST scheme based on the allelic polymorphisms of seven virulent-associated loci, which may evolve more rapidly than housekeeping genes (45, 46) and should therefore be more discriminative than MLST. We wanted to further delineate the lineages and clonal groups previously defined by MLST (44) and characterize the links between within-species genetic variations and characteristics, such as pathogenic potential, virulence, and epidemiology.
Ninety-three S. lugdunensis strains from diverse clinical and geographic sources were studied; 6 carriage strains and the genome sequence of N920143 (30) were added to the collection of 87 strains previously characterized by MLST (44). As systems to genetically manipulate S. lugdunensis succeeded only very recently (21, 22), a few virulent factors are known to date. We selected genes encoding proteins that can partly explain the particular virulence of S. lugdunensis, such as adhesins (Fbl, vWbl, a putative fibrinogen binding protein [SLUG_16930 and FbpA homologue]) (25, 26, 35), hemolysins (SLUSH, a putative β-hemolysin [SLUG_16930], and a putative hemolysin III [SLUG_09050]) (27, 30) and IsdJ, a protein of an iron acquisition system, which of the CoNS, is only used by S. lugdunensis (30, 32). Besides theses virulence-associated loci, we analyzed the polymorphisms of the repeated sequences (atlLR2 and atlLR3) of the putative CWA domains of the AM and GL autolysins, respectively, generated by the proteolytic cleavage of the bifunctional murein hydrolase, AtlL (40). Indeed, AtlL might play a role in virulence, as it has been shown in vitro through biofilm formation and in vivo in the C. elegans model (41). As AtlL presents high identity percentage with all staphylococcal bifunctional autolysins (40, 42, 43), it might be involved in the internalization of the bacteria in the eukaryote cell (59). Furthermore, the agr quorum-sensing and signal transduction system, which has been described as a global regulator of virulence gene expression in S. aureus, was also studied (60).
The fbl gene, which was detected in all 93 isolates, was not selected for the MVLST scheme because of its very low polymorphism rate (<0.65%, data not shown). This result confirms that fbl is a good PCR target for the S. lugdunensis identification (61). The slush operon was also detected in all the strains of our collection, unlike in Svabados et al. (35), who detected it only in 28 strains of 58 with the same primers. Shown to mediate the synergistic δ-like hemolytic activity of S. lugdunensis (27) and to stimulate human leukocytes via interaction with the formyl-peptide receptor 2 (29) as phenol-soluble modulin peptides, the SLUSH peptides display an important role in the virulence of this species. Interestingly, 21 of 93 (22.6%) of the strains did not exhibit hemolytic activity on an agar plate despite the presence of the slush operon; of note, hemolysis was observed for the strain with an incomplete operon (SL9), suggesting that (i) the deleted portion of the slush gene is not directly involved in the hemolytic activity or (ii) hemolysis is regulated, implying the presence of compensatory hemolysins (62). This does not seem to involve agr allelic variation, as hemolysis was observed irrespective of the agr-1SL or agr-2SL variants. The detection of the only two agr types (agr-1SL and agr-2SL) originally described by Dufour et al. (57) in this large set of strains suggests a limited allelic variation of this locus for S. lugdunensis compared to that of S. aureus (57, 60).
Thus, a total of seven loci were chosen (atlLR2, atlLR3, hlb, isdJ, SLUG_09050, SLUG_16930, and vwbl) to design the first MVLST scheme of this rare but aggressive pathogen. The analysis of an internal fragment of each virulence-associated locus showed a moderate polymorphism rate (1.20% to 2.72%), but this was higher than that of housekeeping genes (44). The low apparent rates of recombination associated with a high index of association between the alleles of the different loci and the congruence of the dendrograms of individual virulence-associated genes and of that of both schemes confirm a clonal population structure for S. lugdunensis. Thus, if homologous recombination does exist, it rarely contributes to the evolution of S. lugdunensis. The dN/dS ratio obtained for atlLR2, atlLR3, and SLUG_16930 (FbpA homologue) was higher than that observed for the other loci, but the different tests of selection performed provided no evidence for positive selection of any amino acid in either protein. Of note, the nonsynonymous mutation rates were higher in the AtlLR2 domain (dN/dS, 0.416) than in the AtlLR3 domain (dN/dS, 0.258), unlike the data of AtlE of S. epidermidis, for which the AtlER3 was the most polymorphic domain (63). The allelic variation in these repeated sequences might reflect differences in the target sites of both hydrolytic enzymes, AM-R1-R2 and GL-R3, at the murein structure (42), and it might affect their adhesion ability. Interestingly, Bose et al. (64) recently showed that S. aureus AM and GL have nonredundant functions in cell division, autolysis, and biofilm formation. By homology with S. aureus AtlA-AM, S. lugdunensis AtlL-AM might have different contributions from those of GL, but the different statistical tests used did not confirm any environmental selective pressure on the atlLR2 locus. In this context, it would be interesting to further explore whether AtlLR2 and AtlLR3 are pathogenicity markers.
Forty-three VTs were defined among the 93 isolates and the genome sequence data of N920143 by MVLST, demonstrating additional resolution compared to that found in a previous MLST study (44); six divergent lineages were characterized, among which 2 (CCv3 and CCv6) were newly generated compared to those characterized with MLST. The distribution of the clinical strains in the MVLST dendrogram did not allow us to identify hypervirulent lineages, but the majority of strains collected from infections associated with material devices were distributed in CCv2. The 6 carriage strains that belonged to ST1 (CC1) and ST10 were clustered with clinical strains in CCv1 and CCv6 by MVLST. To further characterize the virulence traits of S. lugdunensis, it might be interesting now to explore the allelic variation of other virulence determinants (other surface-anchored proteins, for example) or regulatory genes whose polymorphisms can affect the expression of virulence genes (65). Of note, the monolocus MVLST analysis showed that the polymorphisms of three loci, such as atlR2, isdJ, and SLUG_16930, generated 33 different allelic profiles and therefore is well suited to short-term epidemiology. atlR3 was preferred to atlR2 because of the number of polymorphic sites.
Finally, this work provides the first MVLST scheme described for S. lugdunensis. This MVLST analysis (i) confirms the clonal population structure and the mutational evolution of this species and (ii) reveals a coevolution of the virulence-associated loci studied with housekeeping genes. However, MVLST offers higher resolution than MLST for S. lugdunensis typing for macroepidemiological purposes, and a trilocus sequence typing scheme might be proposed as a tool for microepidemiological ones. Such methods provide unambiguous and portable data, unlike pulsed-field gel electrophoresis (PFGE), which until recently was the only other typing method described (48) for this species. It would be informative then to enrich these results with sequence data from worldwide strains (in particular carriage ones) and to perform a comparative analysis of the whole-genome sequence of carriage and invasive strains of S. lugdunensis to further characterize the virulence determinants of this unusually aggressive CoNS.
ACKNOWLEDGMENTS
We thank L. Bieber, M. Delmée, F. Doucet-Populaire, V. Dubois, G. Grise, H. Marchandin, A. Morel, F. Mory, A. Reynaud, M. Rupnik, and N. van der Mee-Marquet for kindly supplying strains.
Footnotes
Published ahead of print 30 July 2014
REFERENCES
- 1.Bieber L, Kahlmeter G. 2010. Staphylococcus lugdunensis in several niches of the normal skin flora. Clin. Microbiol. Infect. 16:385–388. 10.1111/j.1469-0691.2009.02813.x [DOI] [PubMed] [Google Scholar]
- 2.Frank KL, Del Pozo JL, Patel R. 2008. From clinical microbiology to infection pathogenesis: how daring to be different works for Staphylococcus lugdunensis. Clin. Microbiol. Rev. 21:111–133. 10.1128/CMR.00036-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kleiner E, Monk AB, Archer GL, Forbes BA. 2010. Clinical significance of Staphylococcus lugdunensis isolated from routine cultures. Clin. Infect. Dis. 51:801–803. 10.1086/656280 [DOI] [PubMed] [Google Scholar]
- 4.Tan TY, Ng SY, Ng WX. 2006. Clinical significance of coagulase-negative staphylococci recovered from nonsterile sites. J. Clin. Microbiol. 44:3413–3414. 10.1128/JCM.00757-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel R, Piper KE, Rouse MS, Uhl JR, Cockerill FR, III, Steckelberg JM. 2000. Frequency of isolation of Staphylococcus lugdunensis among staphylococcal isolates causing endocarditis: a 20-year experience. J. Clin. Microbiol. 38:4262–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vandenesch F, Etienne J, Reverdy ME, Eykyn SJ. 1993. Endocarditis due to Staphylococcus lugdunensis: report of 11 cases and review. Clin. Infect. Dis. 17:871–876. 10.1093/clinids/17.5.871 [DOI] [PubMed] [Google Scholar]
- 7.Böcher S, Tønning B, Skov RL, Prag J. 2009. Staphylococcus lugdunensis, a common cause of skin and soft tissue infections in the community. J. Clin. Microbiol. 47:946–950. 10.1128/JCM.01024-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delaunay F, Pegot A, Coquerel-Beghin D, Aktouf A, Auquit-Auckbur I. 2014. Fasciites nécrosantes à Staphylococcus lugdunensis après dermolipectommie abdominale: à propos de deux cas et revue de la littérature. Ann. Chir. Plast. Esthét. 59:136–139. 10.1016/j.anplas.2013.12.002 [DOI] [PubMed] [Google Scholar]
- 9.Liu PY, Huang YF, Tang CW, Chen YY, Hsieh KS, Ger LP, Chen YS, Liu YC. 2010. Staphylococcus lugdunensis infective endocarditis: a literature review and analysis of risk factors. J. Microbiol. Immunol. Infect. 43:478–484. 10.1016/S1684-1182(10)60074-6 [DOI] [PubMed] [Google Scholar]
- 10.Tsao YT, Wang WJ, Lee SW, Hsu JC, Ho FM, Chen WL. 2012. Characterization of Staphylococcus lugdunensis endocarditis in patients with cardiac implantable electronic devices. Int. J. Infect. Dis. 16:e464–e467. 10.1016/j.ijid.2012.02.010 [DOI] [PubMed] [Google Scholar]
- 11.Bellamy R, Barkham T. 2002. Staphylococcus lugdunensis infection sites: predominance of abscesses in the pelvic girdle region. Clin. Infect. Dis. 35:E32–E34. 10.1086/341304 [DOI] [PubMed] [Google Scholar]
- 12.Papapetropoulos N, Papapetropoulou M, Vantarakis A. 2013. Abscesses and wound infections due to Staphylococcus lugdunensis: report of 16 cases. Infection 41:525–528. 10.1007/s15010-012-0381-z [DOI] [PubMed] [Google Scholar]
- 13.Shah NB, Osmon DR, Fadel H, Patel R, Kohner PC, Steckelberg JM, Mabry T, Berbari EF. 2010. Laboratory and clinical characteristics of Staphylococcus lugdunensis prosthetic joint infections. J. Clin. Microbiol. 48:1600–1603. 10.1128/JCM.01769-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szabados F, Anders A, Kaase M, Marlinghaus L, Gatermann SG, Teske W, Lichtinger T. 2011. Late periprosthetic joint infection due to Staphylococcus lugdunensis identified by matrix-assisted laser desorption/ionisation time of flight mass spectrometry: a case report and review of the literature. Case Rep. Med. 2011:608919. 10.1155/2011/608919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arias M, Tena D, Apellániz M, Asensio MP, Caballero P, Hernández C, Tejedor F, Bisquert J. 2010. Skin and soft tissue infections caused by Staphylococcus lugdunensis: report of 20 cases. Scand. J. Infect. Dis. 42:879–884. 10.3109/00365548.2010.509332 [DOI] [PubMed] [Google Scholar]
- 16.Sabe MA, Shrestha NK, Gordon S, Menon V. 2014. Staphylococcus lugdunensis: a rare but destructive cause of coagulase-negative staphylococcus infective endocarditis. Eur. Heart J. Acute Cardiovasc. Care, in press. 10.1177/2048872614523350 [DOI] [PubMed] [Google Scholar]
- 17.Zinkernagel AS, Zinkernagel MS, Elzi MV, Genoni M, Gubler J, Zbinden R, Mueller NJ. 2008. Significance of Staphylococcus lugdunensis bacteremia: report of 28 cases and review of the literature. Infection 36:314–321. 10.1007/s15010-008-7287-9 [DOI] [PubMed] [Google Scholar]
- 18.Anguera I, Del Río A, Miró JM, Matínez-Lacasa X, Marco F, Gumá JR, Quaglio G, Claramonte X, Moreno A, Mestres CA, Mauri E, Azqueta M, Benito N, García-de la María C, Almela M, Jiménez-Expósito MJ, Sued O, De Lazzari E, Gatell JM, Hospital Clinic Endocarditis Study Group 2005. Staphylococcus lugdunensis infective endocarditis: description of 10 cases and analysis of native valve, prosthetic valve, and pacemaker lead endocarditis clinical profiles. Heart. 91:e10. 10.1136/hrt.2004.040659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szabados F, Woloszyn J, Richter C, Kaase M, Gatermann S. 2010. Identification of molecularly defined Staphylococcus aureus strains using matrix-assisted laser desorption/ionization time of flight mass spectrometry and the Biotyper 2.0 database. J. Med. Microbiol. 59:787–790. 10.1099/jmm.0.016733-0 [DOI] [PubMed] [Google Scholar]
- 20.Freney J, Brun Y, Bes M, Meugnier F, Grimont F, Grimont PAD, Nervi C, Fleurette J. 1988. Staphylococcus lugdunensis sp. nov. and Staphylococcus schleiferi sp. nov., two species from human clinical specimens. Int. J. Syst. Bacteriol. 38:168–172. 10.1099/00207713-38-2-168 [DOI] [Google Scholar]
- 21.Marlinghaus L, Becker K, Korte M, Neumann S, Gatermann SG, Szabados F. 2012. Construction and characterization of three knockout mutants of the fbl gene in Staphylococcus lugdunensis. APMIS 120:108–116. 10.1111/j.1600-0463.2011.02819.x [DOI] [PubMed] [Google Scholar]
- 22.Heilbronner S, Hanses F, Monk IR, Speziale P, Foster TJ. 2013. Sortase A promotes virulence in experimental Staphylococcus lugdunensis endocarditis. Microbiology 159:2141–2152. 10.1099/mic.0.070292-0 [DOI] [PubMed] [Google Scholar]
- 23.Mitchell J, Tristan A, Foster TJ. 2004. Characterization of the fibrinogen-binding surface protein Fbl of Staphylococcus lugdunensis. Microbiology 150:3831–3841. 10.1099/mic.0.27337-0 [DOI] [PubMed] [Google Scholar]
- 24.Geoghegan JA, Ganesh VK, Smeds E, Liang X, Höök M, Foster TJ. 2010. Molecular characterization of the interaction of staphylococcal microbial surface components recognizing adhesive matrix molecules (MSCRAMM) ClfA and Fbl with fibrinogen. J. Biol. Chem. 285:6208–6216. 10.1074/jbc.M109.062208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nilsson M, Bjerketorp J, Guss B, Frykberg L. 2004. A fibrinogen-binding protein of Staphylococcus lugdunensis. FEMS Microbiol. Lett. 241:87–93. 10.1016/j.femsle.2004.10.008 [DOI] [PubMed] [Google Scholar]
- 26.Nilsson M, Bjerketorp J, Wiebensjö A, Ljungh A, Frykberg L, Guss B. 2004. A von Willebrand factor-binding protein from Staphylococcus lugdunensis. FEMS Microbiol. Lett. 234:155–161. 10.1111/j.1574-6968.2004.tb09527.x [DOI] [PubMed] [Google Scholar]
- 27.Donvito B, Etienne J, Denoroy L, Greenland T, Benito Y, Vandenesch F. 1997. Synergistic hemolytic activity of Staphylococcus lugdunensis is mediated by three peptides encoded by a non-agr genetic locus. Infect. Immun. 65:95–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donvito B, Etienne J, Greenland T, Mouren C, Delorme V, Vandenesch F. 1997. Distribution of the synergistic haemolysin genes hld and slush with respect to agr in human staphylococci. FEMS Microbiol. Lett. 151:139–144. 10.1111/j.1574-6968.1997.tb12562.x [DOI] [PubMed] [Google Scholar]
- 29.Rautenberg M, Joo HS, Otto M, Peschel A. 2011. Neutrophil responses to staphylococcal pathogens and commensals via the formyl peptide receptor 2 relates to phenol-soluble modulin release and virulence. FASEB J. 25:1254–1263. 10.1096/fj.10-175208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heilbronner S, Holden MT, van Tonder A, Geoghegan JA, Foster TJ, Parkhill J, Bentley SD. 2011. Genome sequence of Staphylococcus lugdunensis N920143 allows identification of putative colonization and virulence factors. FEMS Microbiol. Lett. 322:60–67. 10.1111/j.1574-6968.2011.02339.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haley KP, Janson EM, Heilbronner S, Foster TJ, Skaar EP. 2011. Staphylococcus lugdunensis IsdG liberates iron from host heme. J. Bacteriol. 193:4749–4757. 10.1128/JB.00436-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zapotoczna M, Heilbronner S, Speziale P, Foster TJ. 2012. Iron-regulated surface determinant (Isd) proteins of Staphylococcus lugdunensis. J. Bacteriol. 194:6453–6467. 10.1128/JB.01195-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tse H, Tsoi HW, Leung SP, Lau SK, Woo PC, Yuen KY. 2010. Complete genome sequence of Staphylococcus lugdunensis strain HKU09-01. J. Bacteriol. 192:1471–1472. 10.1128/JB.01627-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheung AI, Projan SJ, Edelstein RE, Fischetti VA. 1995. Cloning, expression, and nucleotide sequence of a Staphylococcus aureus gene (fbpA) encoding a fibrinogen-binding protein. Infect. Immun. 63:1914–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szabados F, Nowotny Y, Marlinghaus L, Korte M, Neumann S, Kaase M, Gatermann SG. 2011. Occurrence of genes of putative fibrinogen binding proteins and hemolysins, as well as of their phenotypic correlates in isolates of S. lugdunensis of different origins. BMC Res. Notes. 4:113. 10.1186/1756-0500-4-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayashida A, Bartlett AH, Foster TJ, Park PW. 2009. Staphylococcus aureus beta-toxin induces lung injury through syndecan-1. Am. J. Pathol. 174:509–518. 10.2353/ajpath.2009.080394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berry AM, Lock RA, Hansman D, Paton JC. 1989. Contribution of autolysin to virulence of Streptococcus pneumoniae. Infect. Immun. 57:2324–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heilmann C, Hartleib J, Hussain MS, Peters G. 2005. The multifunctional Staphylococcus aureus autolysin aaa mediates adherence to immobilized fibrinogen and fibronectin. Infect. Immun. 73:4793–4802. 10.1128/IAI.73.8.4793-4802.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heilmann C, Hussain M, Peters G, Götz F. 1997. Evidence for autolysin-mediated primary attachment of Staphylococcus epidermidis to a polystyrene surface. Mol. Microbiol. 24:1013–1024. 10.1046/j.1365-2958.1997.4101774.x [DOI] [PubMed] [Google Scholar]
- 40.Bourgeois I, Camiade E, Biswas R, Courtin P, Gibert L, Götz F, Chapot-Chartier MP, Pons JL, Pestel-Caron M. 2009. Characterization of AtlL, a bifunctional autolysin of Staphylococcus lugdunensis with N-acetylglucosaminidase and N-acetylmuramoyl-l-alanine amidase activities. FEMS Microbiol. Lett. 290:105–113. 10.1111/j.1574-6968.2008.01414.x [DOI] [PubMed] [Google Scholar]
- 41.Gibert L, Didi J, Marlinghaus L, Lesouhaitier O, Legris S, Szabados F, Pons JL, Pestel-Caron M. 2014. The major autolysin of Staphylococcus lugdunensis, AtlL, is involved in cell separation, stress-induced autolysis and contributes to bacterial pathogenesis. FEMS Microbiol. Lett. 352:78–86. 10.1111/1574-6968.12374 [DOI] [PubMed] [Google Scholar]
- 42.Albrecht T, Raue S, Rosenstein R, Nieselt K, Götz F. 2012. Phylogeny of the staphylococcal major autolysin and its use in genus and species typing. J. Bacteriol. 194:2630–2636. 10.1128/JB.06609-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Götz F, Heilmann C, Stehle T. 2014. Functional and structural analysis of the major amidase (Atl) in Staphylococcus. Int. J. Med. Microbiol. 304:156–163. 10.1016/j.ijmm.2013.11.006 [DOI] [PubMed] [Google Scholar]
- 44.Chassain B, Lemée L, Didi J, Thiberge JM, Brisse S, Pons JL, Pestel-Caron M. 2012. Multilocus sequence typing analysis of Staphylococcus lugdunensis implies a clonal population structure. J. Clin. Microbiol. 50:3003–3009. 10.1128/JCM.00988-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cai S, Kabuki DY, Kuaye AY, Cargioli TG, Chung MS, Nielsen R, Wiedmann M. 2002. Rational design of DNA sequence-based strategies for subtyping Listeria monocytogenes. J. Clin. Microbiol. 40:3319–3325. 10.1128/JCM.40.9.3319-3325.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chowdhury SA, Arias CA, Nallapareddy SR, Reyes J, Willems RJ, Murray BE. 2009. A trilocus sequence typing scheme for hospital epidemiology and subspecies differentiation of an important nosocomial pathogen, Enterococcus faecalis. J. Clin. Microbiol. 47:2713–2719. 10.1128/JCM.00667-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Y, Zhang W, Knabel SJ. 2007. Multi-virulence-locus sequence typing identifies single nucleotide polymorphisms which differentiate epidemic clones and outbreak strains of Listeria monocytogenes. J. Clin. Microbiol. 45:835–846. 10.1128/JCM.01575-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van der Mee-Marquet N, Achard A, Mereghetti L, Danton A, Minier M, Quentin R. 2003. Staphylococcus lugdunensis infections: high frequency of inguinal area carriage. J. Clin. Microbiol. 41:1404–1409. 10.1128/JCM.41.4.1404-1409.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hunter PR, Gaston MA. 1988. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. J. Clin. Microbiol. 26:2465–2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jolley KA, Feil EJ, Chan MS, Maiden MC. 2001. Sequence type analysis and recombinational tests (START). Bioinformatics 17:1230–1231. 10.1093/bioinformatics/17.12.1230 [DOI] [PubMed] [Google Scholar]
- 52.Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL. 2012. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8:e1002764. 10.1371/journal.pgen.1002764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pond SL, Frost SD. 2005. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533. 10.1093/bioinformatics/bti320 [DOI] [PubMed] [Google Scholar]
- 54.Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. 10.1093/bioinformatics/btp187 [DOI] [PubMed] [Google Scholar]
- 55.Smith J, Smith NH, O'Rourke M, Spratt BG. 1993. How clonal are bacteria? Proc. Natl. Acad. Sci. U. S. A. 90:4384–4388. 10.1073/pnas.90.10.4384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hébert GA, Hancock GA. 1985. Synergistic hemolysis exhibited by species of staphylococci. J. Clin. Microbiol. 22:409–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dufour P, Jarraud S, Vandenesch F, Greenland T, Novick RP, Bes M, Etienne J, Lina G. 2002. High genetic variability of the agr locus in Staphylococcus species. J. Bacteriol. 184:1180–1186. 10.1128/jb.184.4.1180-1186.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feil EJ, Maiden MC, Achtman M, Spratt BG. 1999. The relative contributions of recombination and mutation to the divergence of clones of Neisseria meningitidis. Mol. Biol. Evol. 16:1496–1502. 10.1093/oxfordjournals.molbev.a026061 [DOI] [PubMed] [Google Scholar]
- 59.Hirschhausen N, Schlesier T, Schmidt MA, Götz F, Peters G, Heilmann C. 2010. A novel staphylococcal internalization mechanism involves the major autolysin Atl and heat shock cognate protein Hsc70 as host cell receptor. Cell Microbiol. 12:1746–1764. 10.1111/j.1462-5822.2010.01506.x [DOI] [PubMed] [Google Scholar]
- 60.Thoendel M, Kavanaugh JS, Flack CE, Horswill AR. 2011. Peptide signaling in the staphylococci. Chem. Rev. 111:117–151. 10.1021/cr100370n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pereira EM, Oliveira FL, Schuenck RP, Zoletti GO, Dos Santos KR. 2009. Detection of Staphylococcus lugdunensis by a new species-specific PCR based on the fbl gene. FEMS Immunol. Med. Microbiol. 58:295–298. 10.1111/j.1574-695X.2009.00626.x [DOI] [PubMed] [Google Scholar]
- 62.Ji G, Beavis R, Novick RP. 1997. Bacterial interference caused by autoinducing peptide variants. Science 276:2027–2030. 10.1126/science.276.5321.2027 [DOI] [PubMed] [Google Scholar]
- 63.Sivadon V, Rottman M, Quincampoix JC, Prunier E, de Mazancourt P, Bernard L, Lortat-Jacob A, Piriou P, Judet T, Gaillard JL. 2006. Polymorphism of the cell wall-anchoring domain of the autolysin-adhesin AtlE and its relationship to sequence type, as revealed by multilocus sequence typing of invasive and commensal Staphylococcus epidermidis strains. J. Clin. Microbiol. 44:1839–1843. 10.1128/JCM.44.5.1839-1843.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bose JL, Lehman MK, Fey PD, Bayles KW. 2012. Contribution of the Staphylococcus aureus Atl AM and GL murein hydrolase activities in cell division, autolysis, and biofilm formation. PLoS One 7:e42244. 10.1371/journal.pone.0042244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Geisinger E, Chen J, Novick RP. 2012. Allele-dependent differences in quorum-sensing dynamics result in variant expression of virulence genes in Staphylococcus aureus. J. Bacteriol. 194:2854–2864. 10.1128/JB.06685-11 [DOI] [PMC free article] [PubMed] [Google Scholar]