

Abstract
Staphylococcus aureus is a versatile bacterial pathogen that produces T cell-activating toxins known as superantigens (SAgs). Although excessive immune activation by SAgs can induce a dysregulated cytokine storm as a component of what is known as toxic shock syndrome (TSS), the contribution of SAgs to the staphylococcal infection process is not well defined. Here, we evaluated the role of the bacterial superantigen staphylococcal enterotoxin A (SEA) in a bacteremia model using humanized transgenic mice expressing SAg-responsive HLA-DR4 molecules. Infection with S. aureus Newman induced SEA-dependent Vβ skewing of T cells and enhanced bacterial survival in the liver compared with infection by sea knockout strain. SEA-induced gamma interferon, interleukin-12, and chemokine responses resulted in increased infiltration of CD11b+ Ly6G+ neutrophils into the liver, promoting the formation of abscesses that contained large numbers of viable staphylococci. Hepatic abscesses occurred significantly more frequently in S. aureus Newman-infected livers than in livers infected with the Newman sea knockout strain, promoting the survival of S. aureus in vivo. This represents a novel mechanism during infection whereby S. aureus utilizes SAgs to form a specialized niche and manipulate the immune system.
INTRODUCTION
Staphylococcus aureus is a common human commensal equipped with numerous virulence factors that allow this organism to successfully colonize and infect host tissues. Staphylococcal diseases most frequently manifest as skin and soft-tissue infections with a high propensity for abscess formation (1, 2); however, S. aureus is also readily capable of disseminating into deeper tissues to cause invasive and life-threatening infections, including endocarditis, osteomyelitis, and sepsis (3). Moreover, S. aureus also can induce toxin-driven diseases, such as food poisoning, staphylococcal scalded skin syndrome, and toxic shock syndrome (TSS) (4). The versatility of this bacterium as a successful commensal and pathogen, coupled with the development of resistance to a wide array of antibiotics, has led to the establishment of S. aureus as a leading cause of both hospital- and community-associated infections (5, 6).
Many of the specialized S. aureus virulence factors have evolved to target innate immune mechanisms, primarily neutrophils and macrophages, which are key cells involved in the clearance of S. aureus (7–9). In contrast, S. aureus also secretes superantigens (SAgs) that directly target and activate cells of the adaptive immune system (10, 11). The family of SAgs in S. aureus now includes over 20 genetically distinct SAg variants that comprise the staphylococcal enterotoxins (SEs), staphylococcal enterotoxin-like (SEls) toxins, and toxic shock syndrome toxin-1 (TSST-1) (12). These functionally unique exotoxins circumvent antigen presentation by engaging lateral surfaces of major histocompatibility complex class II (MHC-II) molecules (13–16) and complementarity determining region 2 (CDR2) of the T cell receptor (TCR) β-chain variable region (Vβ) (17–20). Thus, SAgs alter the conventional TCR-peptide-MHC-II activation complex to prevent antigen recognition by the CDR loops (21), leading to the activation and expansion of numerous T cells in a Vβ-restricted manner (22). In cases of severe SAg intoxication, excessive T cell activation can result in a cytokine storm that leads to the development of TSS (11, 23).
In vivo mouse experiments using the injection of purified SAgs have demonstrated many important features of SAg biology, yet these experiments cannot recapitulate the complex interactions between S. aureus and the host. Although S. aureus has been studied intensively using live in vivo infection models, relatively few reports have examined the role of SAgs using genetically controlled SAg knockout strains. Early work by Tarkowski and colleagues has demonstrated a pathogenic role of TSST-1 for the onset of dermatitis, arthritis, and septic mortality in mice (24, 25). In addition, vaccination with SAg toxoids or neutralization of SAgs with monoclonal antibodies has prevented or reduced mortality from experimental S. aureus sepsis (26–28). Rabbits are particularly sensitive to the effects of SAgs; using this animal species, deletion of the gene encoding SEl-X from S. aureus USA300 demonstrated reduced mortality from necrotizing pneumonia (29), and deletion of the gene encoding staphylococcal enterotoxin C (sec) from S. aureus MW2 prevented mortality in a rabbit model of sepsis/infective endocarditis (30). Furthermore, engineered high-affinity SAg inhibitors or vaccination with SAg toxoids can protect rabbits from S. aureus pneumonia, infective endocarditis, and sepsis (31–33). Collectively, these studies show unequivocally that SAgs enhance the severity and lethality of staphylococcal infection.
The majority of the human population has circulating antibodies against SAgs that are protective against TSS, which rarely develops (34, 35), indicating that SAg exposure does not usually result in overt disease. Furthermore, at least 80% of clinical strains of S. aureus are genetically positive for at least one SAg gene (36), and the recently discovered selx has been found in ∼95% of all S. aureus strains (29). Thus, the high prevalence and widespread distribution of SAgs in S. aureus suggests these toxins provide an evolutionary advantage to S. aureus. Although SAg-induced virulence has been attributed to the cytokine storm that results in immune cell infiltration, pyrexia, hypotension, endothelial damage (29, 30), and ultimately death, enhanced host mortality may not provide an evolutionarily prudent tactic for bacterial survival and propagation. We reasoned that there are other biologically relevant SAg functions that contribute to S. aureus fitness, and given that S. aureus is one of the most common sources of bacteremia (37), we set out to study the role of SAgs in this context. Using an isogenic sea knockout strain of S. aureus, we found that SEA manipulates the immune system and recruits neutrophils to promote formation of hepatic abscesses, forming a protective niche for staphylococcal survival in vivo.
MATERIALS AND METHODS
Mice.
Six-to-twelve-week-old male and female HLA-DR4-IE (DRB1*0401) humanized transgenic mice lacking endogenous mouse MHC-II on a C57BL/6 (B6) background (here referred to as DR4-B6 mice) (38) were used for all in vivo infection experiments. B6 mice were purchased from Charles River. All animal experiments were in accordance with the Canadian Council on Animal Care Guide to the Care and Use of Experimental Animals, and the animal protocol was approved by the Animal Use Subcommittee at Western University.
Bacterial strains, media, and growth conditions.
S. aureus strains listed in Table 1 were grown aerobically at 37°C in tryptic soy broth (TSB) (Difco) with shaking (250 rpm) or on tryptic soy agar (TSA) supplemented with the appropriate antibiotics (Sigma-Aldrich). Escherichia coli DH5α was used as a cloning host and was grown in Luria-Bertani (LB) broth (Difco) or LB agar supplemented with appropriate antibiotics at 37°C with shaking (250 rpm). Growth curves were done using a Bioscreen C MBR system (Thermo Labsystems).
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| S. aureus Newman | Early methicillin-sensitive isolate from secondary infection in a patient with tubercular osteomyelitis | 90 |
| S. aureus RN4220 | Restriction-deficient derivation of NCTC8325-4 | 91 |
| S. aureus Newman Δsea | sea-null S. aureus Newman | This study |
| S. aureus Newman Δsea(pSEA) | sea-null S. aureus Newman complemented with wild-type sea | This study |
| E. coli DH5α | Cloning strain | Invitrogen |
| E. coli BL21(DE3) | Protein expression strain | New England Biolabs |
| Plasmids | ||
| pET28 | Protein expression vector | Novagen |
| pET28::sea | Recombinant SEA expression plasmid | This study |
| pDG1513 | Source of tetR gene | 41 |
| pMAD | Integration plasmid | 40 |
| pALC2073 | Complementation vector | 42 |
Construction of recombinant S. aureus strains.
Restriction enzymes were purchased from New England BioLabs, and primers were designed using Primer3 software (39) and supplied by Sigma-Aldrich. The gene encoding staphylococcal enterotoxin A (SEA) in S. aureus strain Newman was insertionally inactivated with a tetracycline-resistant cassette using the pMAD integration vector by following an established protocol (40). Wild-type sea along with its corresponding upstream (Up) and downstream (Down) fragments were PCR amplified from the genome of Newman using seaFP (5′-AACGGGATCCCATGTGCTTGAACTTAGAGAGGAA-3′) and seaRP (5′-TTCGGTCGACCCCAATAGCTTTTGCGATGT-3′) and directionally cloned into pMAD via BamHI and SalI sites. A 262-bp fragment was excised from the middle of sea using ClaI and EcoRI and replaced with a tetracycline resistance marker (tetR) excised from pDG1513 (41). This construct then was transformed into S. aureus Newman after undergoing methylation in S. aureus RN4220. Allelic replacement of the wild-type sea with tetR via homologous recombination was conducted as described previously (40). The sea-null Newman Δsea strain was complemented by amplifying the native sea promoter and complete sea gene from Newman using the primers seaFP and seaRP and cloned into the BamHI and SalI sites of the plasmid pALC2073 (42). This construct (pALC2073::sea) was electroporated into the Newman Δsea mutant, generating the S. aureus Newman Δsea(pSEA) complementation strain.
Construction and purification of recombinant SEA.
Wild-type sea lacking the signal peptide was PCR amplified from the genome of S. aureus Newman using the primers 5′-GGGCCATGGGCAGCCATCATCATCATCATCACAGCAGCGGCGAAAACTTGTATTTCCAAAGCGAGAAAAGCGAAGAAAT-3′ and 5′-GGGGGATCCTTAACTTGTATATAAATATATATC-3′, introducing nucleotide sequences encoding a His6 tag and a tobacco etch virus (TEV) protease cleavage site (ENLYFQ↓G) onto the N terminus of sea. This PCR product was inserted into pET28a (Novagen) via BamHI and NcoI sites to create pET28a::sea and transformed into E. coli BL21(DE3) for protein purification. Cells were induced with isopropyl β-d-1-thiogalactopyranoside (Sigma-Aldrich) to express His6-tagged SEA and purified using nickel column chromatography as previously described (43). The His6 tag was removed with TEV protease and dialyzed in HEPES or PBS before use.
Cellular proliferation and cytokine quantification.
The ability of B6 and DR4-B6 mice to respond to SEA was assessed using the incorporation of [3H]thymidine as described previously (44). Mouse spleens were collected and broken into a single-cell suspension, followed by erythrocyte lysis in ammonium-chloride-potassium (ACK) buffer. The remaining cells were suspended in RPMI (Invitrogen Life Technologies) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich), 100 μg/ml streptomycin, 100 U/ml penicillin (Gibco), 2 mM l-glutamine (Gibco), 1 mM MEM sodium pyruvate (Gibco), 100 μM nonessential amino acid (Gibco), and 25 mM HEPES (pH 7.2) (Gibco) and then seeded into 96-well plates at a density of 1.1 × 106 cells/ml. Various concentrations of recombinant SEA were added to cells and incubated for 72 h at 37°C. Cells then were pulsed with 1 μCi/well of [3H]thymidine for an additional 18 h prior to harvesting on fiberglass filters. Counts were measured using a 1450 Microbeta liquid scintillation counter (Wallac).
Supernatants from the S. aureus strains were tested for SAg activity using DR4-B6 splenocytes seeded into 96-well plates as described above. Titrations of recombinant SEA and supernatants from overnight cultures of S. aureus Newman and the Newman Δsea mutant diluted 1:10 were added to splenocytes for 18 h at 37°C, and supernatants were assayed for interleukin-2 (IL-2) by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (eBioscience).
Staphylococcal bacteremia model.
Single bacterial colonies were picked from a TSA plate and grown in 5 ml TSB overnight (16 to 18 h), and the optical density at 600 nm (OD600) was adjusted to 1.0. Cells subsequently were subcultured into (2%) TSB and grown to exponential phase (OD600 of ∼3.0 to 3.5). The bacterial pellet was washed 3× with Hanks balanced salt solution (HBSS) (HyClone) and resuspended in HBSS to an OD600 of 0.15, corresponding to ∼5 × 107 CFU/ml. Mice were injected via the tail vein with 5 × 106 CFU of S. aureus in a total volume of 100 μl. Mice were weighed and monitored daily. At 8 or 96 h postinfection, mice were sacrificed and the heart, lungs, kidneys, and liver were aseptically harvested. All organs were homogenized, plated on mannitol salt agar (Difco), and incubated at 37°C overnight. S. aureus colonies were enumerated the following day with a limit of detection determined to be 3 CFU per 10 μl.
Determination of Vβ populations targeted by SAgs using flow cytometry.
Lymph nodes (cervical, axillary, brachial, inguinal, and popliteal) were isolated in toto from mice and pushed through a cell strainer to create a single-cell suspension. Cells were stained with allophycocyanin (APC)-conjugated anti-CD3 (clone 145-2C11) (eBioscience) and fluorescein isothiocyanate (FITC)-conjugated anti-Vβ3 (clone KJ25) (BD Pharmingen) or FITC-conjugated anti-Vβ8 (clone KJ16) (eBioscience). Events were acquired using a FACSCanto II (BD Biosciences), and data were analyzed using FlowJo v.8.7 (TreeStar).
Detection of cytokines and chemokines in vivo.
Eight hours postinfection, serum supernatants and livers were collected. Supernatants were obtained from whole livers by homogenization in HBSS supplemented with the complete protease inhibitor cocktail (Roche). Samples were analyzed using a 32-multiplex array against mouse cytokines and chemokines (Eve Technologies).
Liver leukocyte isolation, staining, and cytofluorometric analysis.
Livers were extracted from mice and pushed through a fine mesh. Leukocytes were isolated from livers as previously described using a 33.75% Percoll gradient (GE Healthcare) (45). Cells were stained with FITC-conjugated anti-F4/80 (clone BM8), FITC-conjugated anti-Ly6G (clone RB6-8C5), phycoerythrin (PE)-conjugated anti-CD11b (clone M1/70), or APC-conjugated anti-CD3 (eBioscience). Events were acquired and data analyzed as outlined above.
Histological analysis.
Standard histology techniques were used. Briefly, tissues were fixed in 10% formalin, embedded in paraffin, and thin sectioned. Sections were stained with a combination hematoxylin and eosin (H&E)/Gram stain, and images were captured using a BX-61 upright microscope (Olympus).
Statistical analyses.
Data were analyzed using unpaired Student's t test or one-way analysis of variance (ANOVA) with Tukey's posttest analysis. All statistical analyses were performed using Prism v5.0 (GraphPad), with P < 0.05 being considered significant.
RESULTS
Construction of isogenic sea-null S. aureus Newman mutant.
A general feature of most bacterial SAgs is that these toxins do not efficiently bind mouse MHC-II molecules (46, 47). S. aureus Newman encodes the SEA SAg (48), so we first tested the ability of recombinant SEA protein to activate splenocytes isolated from both B6 and DR4-B6 transgenic mice. SEA resulted in a dose-dependent proliferative response as low as 1 pg for splenocytes from DR4-B6 mice, while proliferation of B6 splenocytes was not detected (Fig. 1A). Thus, the remaining experiments were conducted in DR4-B6 mice.
FIG 1.

Construction of S. aureus Newman Δsea mutant strain that does not display superantigenic activity in SAg-sensitive DR4-B6 mice. (A) Splenocytes isolated from DR4-B6 (black circles) and B6 (black squares) mice were treated with various concentrations of recombinant SEA for 72 h, followed by the addition of [3H]thymidine. Proliferation was recorded as radioactive counts per minute. (B) Schematic diagram of the double homologous recombination method used to generate the S. aureus Newman Δsea strain from wild-type S. aureus Newman. Up and Down designate the region upstream and downstream, respectively, of sea from the genome of S. aureus Newman. (C) Growth curve analysis of S. aureus Newman (open circles) and the Newman Δsea mutant (black circle) in TSB and TSB only (black triangle). (D) IL-2 production from DR4-B6 splenocytes activated with various concentrations of recombinant SEA (white bars) and bacterial supernatants diluted 1:10 from S. aureus Newman and the Newman Δsea mutant (black bars).
We next aimed to evaluate the role of SEA in a model of S. aureus bacteremia. To accomplish this, we generated an isogenic SEA-negative S. aureus strain. Using an allelic replacement strategy, we integrated a tetracycline resistance marker into a deleted portion of the sea gene in S. aureus Newman (Fig. 1B). The resulting sea-null S. aureus strain was confirmed to be tetracycline resistant and erythromycin sensitive, with the tetR insertion verified by PCR and DNA sequencing. Growth curve analysis demonstrated no apparent defect for the Newman Δsea mutant compared to wild-type Newman (Fig. 1C). We next tested the supernatants from both S. aureus Newman and Newman Δsea strains for SAg activity on DR4-B6 splenocytes using IL-2 production as a measure of T cell activation. Ten-fold-diluted supernatants from wild-type S. aureus Newman induced ∼50 pg/ml IL-2 from DR4-B6 splenocytes, which extrapolates to secreted SEA concentrations of ∼100 ng/ml. In contrast, we did not detect any IL-2 production from Newman Δsea mutant supernatants, confirming both the genetic deletion and that other functional DR4-B6-reactive SAgs, such as the genome-encoded SEl-X, which is the only other known SAg encoded by Newman (29), do not display superantigenic activity under these growth conditions (Fig. 1D).
SEA is produced in vivo during staphylococcal bacteremia.
S. aureus Newman is known to produce SEA during the exponential phase of growth in vitro (49); however, the exact environmental triggers in vivo are not well defined. We aimed to determine if SEA was produced during S. aureus Newman infection by examining the Vβ profiles of infected mice, as SEA is known to target Vβ3+ T cells but not Vβ8+ T cells (50, 51). Vβ-specific T cell subpopulations from lymph nodes were measured using flow cytometry from mice inoculated with S. aureus strain Newman, the Newman Δsea mutant, or the Newman Δsea(pSEA) mutant or from vehicle-treated mice. Ninety-six hours postinoculation, mice infected with the S. aureus Newman Δsea mutant did not show a difference in Vβ3+ CD3+ lymphocyte populations compared to vehicle-treated mice. Conversely, wild-type S. aureus Newman and Newman Δsea(pSEA) mutant infection demonstrated a significant decrease in Vβ3+ CD3+ cells compared to levels for vehicle-treated mice, indicating Vβ-specific targeting by SEA (Fig. 2A). Concurrent analysis of Vβ8+ CD3+ cells was used as an internal control, since it is an irrelevant T cell subpopulation that is not targeted by SEA. Thus, the significant decrease in the ratio of Vβ3+ CD3+ to Vβ8+ CD3+ cells from 0.33 (vehicle) to 0.16 (Newman) during infection with S. aureus Newman showed that SEA was specifically targeting the Vβ3+ CD3+ population (Fig. 2B), thereby confirming the production of SEA in vivo during infection in our model.
FIG 2.
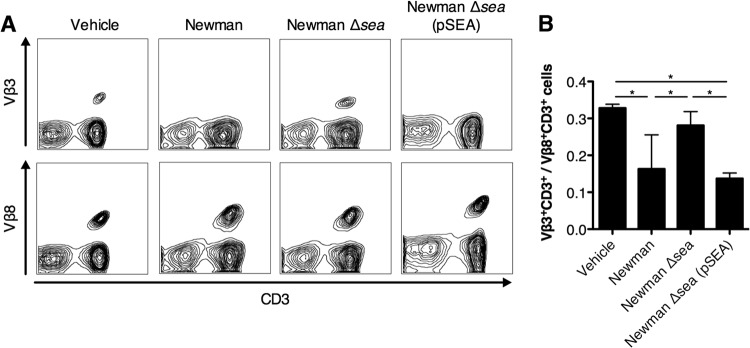
SEA is produced during S. aureus bacteremia and interacts specifically with the Vβ3+ subset of T cells. Flow cytometry analysis of lymph node populations 96 h postinfection [vehicle, n = 3; Newman strain, n = 4; Newman Δsea mutant, n = 4; and Newman Δsea(pSEA) mutant, n = 5]. (A) Representative fluorescence-activated cell sorter plots from each infection group stained with antibodies against either CD3 and Vβ3 or CD3 and Vβ8. Vβ3 and Vβ8 staining were from the same mouse, with Vβ8 acting as the internal control for each mouse. Each sample was gated for the CD3+ Vβ+ population. (B) Ratio of CD3+ Vβ3+ to CD3+ Vβ8+ cells per mouse for each infection group. Data are shown as means ± standard errors of the means (SEM). Significant differences (P < 0.05) as determined by one-way ANOVA with Tukey's posttest are denoted with an asterisk.
Bacterial survival is enhanced in the livers of mice infected with SEA-producing S. aureus.
To evaluate a role for SEA in S. aureus bacteremia, we injected 5 × 106 CFU of S. aureus Newman or the S. aureus Newman Δsea mutant into the tail vein of DR4-B6 mice and assessed the bacterial burden in multiple organs at 96 h postinfection. Bacterial loads were highest in the kidneys and livers but also were found in the heart and lungs (Fig. 3A to D). Although the bacterial load was not statistically different in the kidneys or lungs, we observed an ∼100-fold decrease in bacterial burden in the livers of mice infected with the S. aureus Newman Δsea mutant compared with wild-type S. aureus Newman-infected mice. There was also a significant difference between the bacterial loads in the heart (Fig. 3D) between S. aureus Newman- and Newman Δsea mutant-infected mice. In order to confirm this pronounced phenotype was SEA dependent on and not due to an inadvertent secondary site mutation in the S. aureus Newman Δsea mutant, we restored SEA expression in trans using the pSEA plasmid. The Newman Δsea(pSEA) complemented strain restored the virulence phenotype in both the liver and heart, as seen with wild-type S. aureus Newman (Fig. 3). These data indicate that expression of SEA by S. aureus Newman promotes infection within the liver and heart but apparently does not alter bacterial burden in other organs tested.
FIG 3.

Septic infection with SEA-producing S. aureus results in higher bacterial loads in the liver than SEA-deficient S. aureus. Bacterial counts of mice infected with S. aureus Newman (n = 17), the Newman Δsea mutant (n = 17), or the Newman Δsea(pSEA) mutant (n = 12) from liver (A), kidneys (B), lungs (C), and heart (D) 96 h postinfection. Each point represents data from one mouse. Results reflect 3 independent experiments. The line in each treatment group represents the mean, and counts below the limit of detection are interpreted as being negligible. Significant differences (P < 0.05) as determined by unpaired Student's t test are denoted with an asterisk. NS, no significance.
SEA induces production of IFN-γ and other inflammatory cytokines and chemokines both locally and systemically during S. aureus infection.
Since it is well known that SAgs function to induce cytokine production, we reasoned that the survival advantage seen during infection with S. aureus Newman was a downstream result of SAg-mediated immune activation. We investigated early cytokine production to assess both local and systemic inflammation of infected mice 8 h postinfection. Liver homogenate supernatants and sera from Newman- and Newman Δsea mutant-infected mice were analyzed for 32 cytokines and chemokines (see Table S1 in the supplemental material). Systemically, gamma interferon (IFN-γ) and IL-12p70 were upregulated in wild-type-infected mice sera compared to Newman Δsea mutant infection, as was the chemokine interferon-induced protein 10 (IP-10) (Fig. 4A). Elevated levels of IFN-γ, tumor necrosis factor alpha (TNF-α), IL-6, and IL-12p40 were detected from mouse livers infected with S. aureus Newman compared to Newman Δsea mutant-infected mice (Fig. 4B), which are known to be induced by SAgs (23, 52). Additionally, the chemokines MIP-2 and MCP-1 were upregulated in Newman-infected livers (Fig. 4B). Bacterial burdens in the liver at 8 h postinfection were not significantly different between wild-type and Newman Δsea mutant-infected mice (Fig. 4C), and no bacteria were detected in blood from any mice (data not shown), indicating that the differences in chemokine and cytokine production likely are not due to differences in bacterial load. Overall, these data demonstrate that SEA is an important driver of SAg-induced inflammation during our model of S. aureus bacteremia in DR4-B6 mice.
FIG 4.
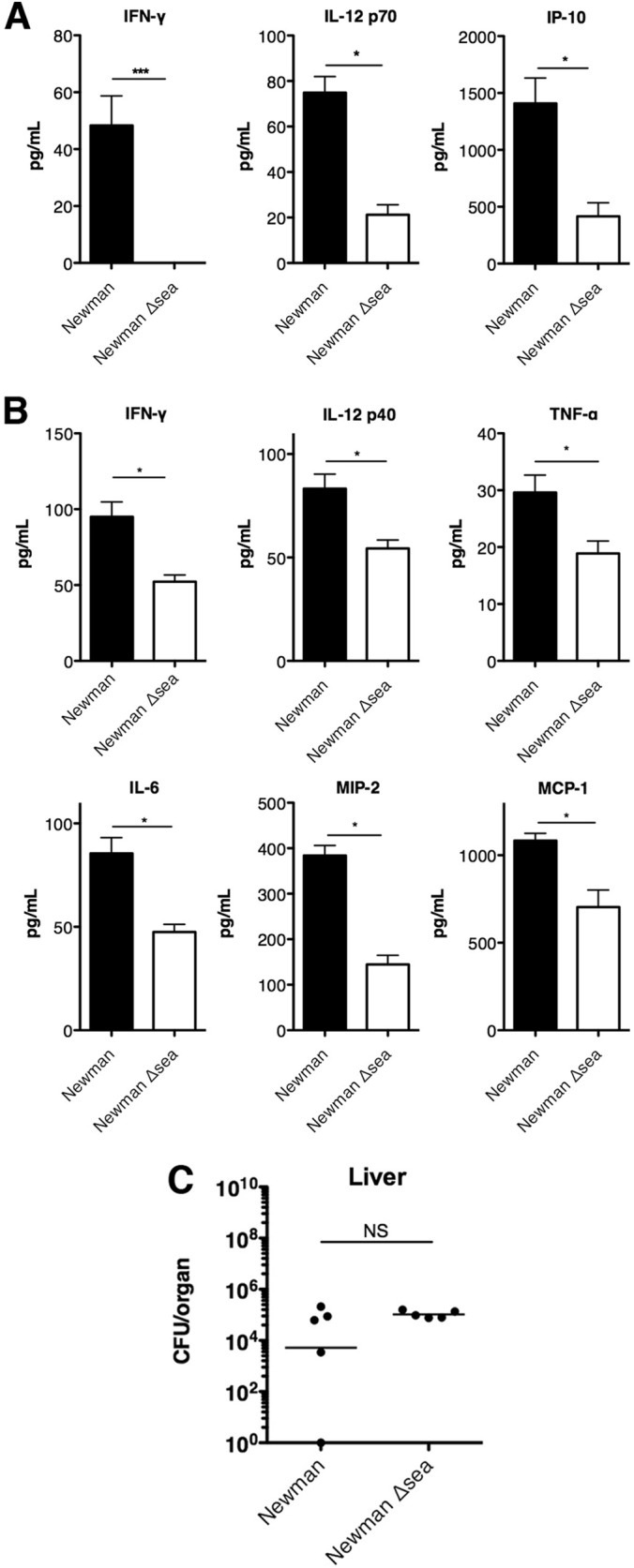
Cytokines and chemokines induced by S. aureus Newman and Newman Δsea mutant infection. Serum and liver supernatants were collected from mice 8 h postinfection from S. aureus Newman- and Newman Δsea mutant-infected mice. Blinded samples were sent for multiplex cytokine array analysis (n = 3 to 4 per experimental group). (A) Serum levels of cytokines and chemokines were significantly different in Newman and Newman Δsea mutant infection. (B) Local production of liver chemokines and cytokines were significantly different during infection with Newman compared to that during infection with the Newman Δsea mutant. (C) Bacterial burdens in the liver at 8 h postinfection (n = 5 per group). Data are shown as means ± SEM. Significant differences (P < 0.05) as determined by unpaired Student's t test are denoted with an asterisk. ***, P < 0.001.
CD11b+ Ly6G+ neutrophils are recruited to the liver during S. aureus infection in an SEA-dependent manner.
Given the production of the MIP-2 and MCP-1 chemokines in the liver induced by SEA from S. aureus Newman-infected mice, we predicted that there also would be a difference in the number of immune cells trafficking to the liver. Since macrophages and neutrophils are the primary cells responsible for the clearance of S. aureus, we examined these populations to evaluate if there was a defect in phagocyte recruitment during staphylococcal infection with SAgs. Additionally, the liver is known to contain large numbers of resident macrophages (Kupffer cells), so we hypothesized that SEA would have an effect on the macrophage population. Leukocytes were isolated from mouse livers 96 h postinfection and stained for various surface markers. Analysis of F4/80+ macrophages showed no significant difference between mice infected with S. aureus Newman and the Newman Δsea mutant (Fig. 5A). Similarly, levels of CD3+ T cells were not significantly different (Fig. 5B) despite the decreased number of Vβ3+ T cells detected in lymph nodes (Fig. 3). However, mice infected with S. aureus Newman showed an increased frequency of CD11b+ Ly6G+ neutrophils (Fig. 5C), suggesting that SEA-induced chemokines (Fig. 4) resulted in the recruitment of neutrophils to the liver.
FIG 5.
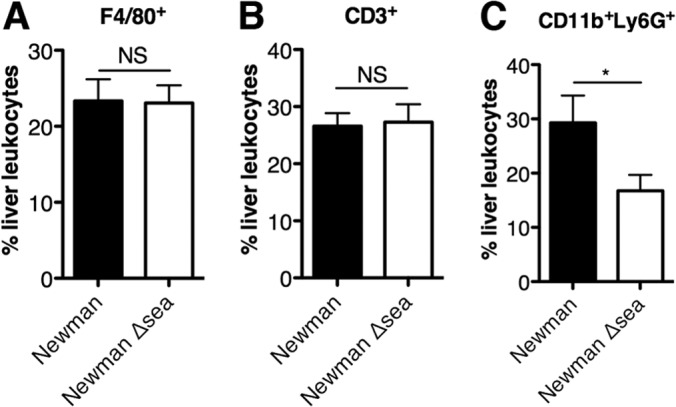
Livers of mice infected with S. aureus Newman show an increase in CD11b+ Ly6G+ neutrophils but not F4/80+ macrophages or CD3+ T cells. Livers of infected mice 96 h postinfection were broken into single-cell suspensions, and leukocytes were isolated by Percoll gradient. Samples were stained with antibodies against F4/80, CD3, or CD11b and Ly6G and analyzed by flow cytometry. Samples underwent doublet discrimination with debris gated out. Subsequently, cells were gated on F4/80+ macrophages (A), CD3+ T cells (B), or CD11b+ Ly6G+ neutrophils (C). Data are shown as the means ± SEM from three independent experiments, with n = 12 for each group. Significant differences (P < 0.05) as determined by unpaired Student's t test are denoted with an asterisk. NS, no significance.
SEA promotes the formation of hepatic abscesses that contain viable bacteria at high densities.
During organ retrieval following the bacteremia model, we observed numerous white hepatic lesions that commonly formed on the surface of livers of S. aureus Newman-infected mice (Fig. 6A). An abscess score was established whereby livers were examined on a lobe-by-lobe basis for visible surface lesions and enumerated. We observed a significant increase in the number of abscesses formed in the livers of S. aureus Newman-infected mice compared with mice infected with the S. aureus Newman Δsea mutant. The number of abscesses from the S. aureus Newman Δsea strain complemented with pSEA was similar to that of wild-type S. aureus Newman-infected mice, demonstrating that this phenotype was SEA dependent (Fig. 6B). H&E/Gram staining of thin sections from both groups showed high neutrophilic infiltration into the abscess with polymorphonuclear cells and associated tissue damage. Abscesses contained large numbers of Gram-positive cocci in the center (Fig. 6C, insets i and ii). However, the overall abscess structure appeared to be very similar between wild-type S. aureus Newman and Newman Δsea mutant infections (Fig. 6C). Abscesses also were excised from the liver, homogenized, and compared to hepatic immune cells isolated from the surrounding liver tissue. Compared to nonabscessed liver tissue, abscesses contained few live host cells as assessed by trypan blue staining and had lost forward and side scatter when analyzed with flow cytometry (data not shown). Additionally, individually excised abscesses yielded high counts of viable S. aureus (106 to 107 CFU/abscess) (data not shown). We did not detect staphylococci distant from the abscesses within the surrounding liver parenchyma or in sham-infected mice (Fig. 6D). These data indicated that the enhanced fitness phenotype of S. aureus Newman we observed (Fig. 3A) is attributed to an increase in abscess formation that confers greater bacterial survival and growth in the liver.
FIG 6.

Infection with S. aureus Newman results in greater abscess formation than infection with the S. aureus Newman Δsea mutant. (A) Visible white lesions (abscesses) on a representative liver of a Newman-infected mouse, indicated by white arrows. (B) Liver abscess score from mice infected with Newman (n = 14), the Newman Δsea mutant (n = 13), and the Newman Δsea(pSEA) mutant (n = 5). Data are shown as means ± SEM from at least three independent experiments. Significant differences (P < 0.05) as determined by unpaired Student's t test are denoted with an asterisk. (C) Representative H&E/Gram-stained histological sections of abscesses from Newman- and Newman Δsea mutant-infected mice. The black bar indicates 100 μm and 10 μm on insets i and ii, respectively. (D) Representative sections of Gram-stained liver parenchyma surrounding abscesses from sham-, Newman-, and Newman Δsea mutant-infected mice. The black bar indicates 50 μm.
DISCUSSION
In this work, we combined SAg-sensitive humanized transgenic DR4-B6 mice with an isogenic sea knockout strain of S. aureus to study the role of SAgs during staphylococcal bacteremia. By using a live infection model, we were able to study not only the detrimental effects of SAg intoxication on the host but also the advantageous effects of SAg expression for S. aureus. We demonstrated an SEA-specific downstream effect that enhanced the number of abscesses formed in the liver, although individual abscesses appeared similar in both morphology and bacterial counts from both strains. This in turn increased bacterial persistence in the liver overall, since staphylococcal abscess communities are sustained within a fibrin pseudocapsule that is protective against immune cells and permits bacterial survival in vivo (53, 54). Abscess formation is an important host immune response during infection for limiting the spread of infection to other tissues. Host immunity against S. aureus infection is dependent on abscess formation by neutrophils (55), and suppurative abscesses have long been recognized as a hallmark of S. aureus infection (54, 56). However, successful eradication of S. aureus by neutrophils exists in a balance, with staphylococci actively subverting neutrophil responses in order to persist in vivo (9, 53). The presence of abscesses during staphylococcal bacteremia is clinically significant, since hematogenous spread from the abscess is well documented (54, 57, 58).
A basal level of abscess formation still could be observed during S. aureus Newman Δsea mutant infection, albeit at a lower frequency than that of wild-type infection, since the former still retains essential cell surface proteins required for abscess formation (53). The lower bacterial counts seen in the Newman Δsea mutant likely are not due to an inherent growth defect (Fig. 1C) or an inability to survive within neutrophils, since viable bacteria were observed within both Newman and Newman Δsea mutant abscesses. To our knowledge, this model is the first to describe a liver tropism for S. aureus related to SAg expression. Although we also observed renal abscesses in the infected mice, no differences were detected in bacterial counts between SEA-expressing and sea-null mutant infections. We speculate that given the paucity of resident T cells in the kidney (59), the initial infection within the kidney remained independent of SEA function. Additionally, high densities of staphylococcal cell walls (such as the loads observed in the kidneys) have been shown to downregulate SAg-mediated T cell activation (60), which may nullify SAg activity locally.
Consistent with our findings, SEC has been shown to increase renal damage during experimental infective endocarditis/sepsis in rabbits, including the formation of kidney abscesses, although this was attributed to the embolization of valve vegetations (30). Similarly, blocking SEC function using a high-affinity SEC binding inhibitor resulted in a drastic reduction of vegetation size (32). Although the Newman Δsea mutant demonstrated decreased counts within the hearts, we did not observe any obvious aortic valve vegetations from wild-type Newman, although our protocol is not an endocarditis model as valve damage is not actively induced. Neutralization of SEB also decreased abscess size using a murine thigh infection model (28). Although it is difficult to aggregate these collective findings, an overall picture is now emerging that SAg-induced inflammation contributes to the formation and severity of S. aureus abscesses in multiple experimental settings.
Compared to Newman Δsea mutant infection, wild-type Newman infection produced significantly higher quantities of cytokines and chemokines that correspond to those induced by SAgs reported in the literature (23, 52, 61, 62). Although IL-2 is a cytokine typically used to measure T cell-dependent superantigenic activity in vitro, we did not detect differences in IL-2 production from the in vivo liver samples. This finding may be explained by IL-2 levels peaking at 2 to 4 h in vivo in response to SAg (63) and by the very short half-life of IL-2 in vivo (64, 65). The SEA-driven inflammatory milieu likely mediates the promotion of abscesses and seems to be driven by the early production of both IL-12 and IFN-γ, which are detected in both serum and liver supernatants 8 h postinfection. IL-12 enhances production of IFN-γ after SAg challenge (63), productively boosting the cytokine and chemokine response. McLoughlin et al. have shown that IFN-γ is a master regulator during S. aureus infections, mediating chemokine responses that allow for neutrophil recruitment and trafficking (66). This is consistent with our observations in Newman-infected mice where we observed an increase in IFN-γ and chemokines that are chemotactic for neutrophils. Purified SAgs have been shown to recruit neutrophils (but not T cells) mediated by TNF-α and chemokines (67). Notably, our study is the first report showing SEA increases trafficking of CD11b+ Ly6G+ neutrophils during a live infection. Given that abscess formation is largely driven by neutrophils, the infiltration into the liver correlates well with the increased incidence of hepatic abscesses.
It appears paradoxical that an increased influx of CD11b+ Ly6G+ neutrophils had an inverse correlation to bacterial survival, considering the important role of neutrophils in staphylococcal clearance. As a successful human pathogen, S. aureus has evolved many mechanisms to counteract neutrophils (9). While neutrophils are absolutely necessary for the eradication of staphylococcal infections (68, 69), their presence during infection has also been described as pathogenic (66, 70). IP-10, which we showed to be upregulated systemically by SEA, can promote phagocytosis (71); however, MIP-2, which is also upregulated, is capable of enhancing intracellular bacterial survival within neutrophils (66, 70). The avoidance of neutrophil bactericidal activity likely contributed to S. aureus survival during early abscess formation and, subsequently, the staphylococcal community in the mature abscess. This supports the paradigm that neutrophils can be pathogenic during systemic infection due to SAgs usurping the immune system to form abscesses, thereby conferring staphylococcal fitness and survival in vivo. The sea gene is located on the same immune evasion cluster (IEC) of β-hemolysin converting phage, which includes staphylococcal complement inhibitor (SCIN), chemotaxis inhibitory protein of S. aureus (CHIPS), and staphylokinase (SAK) (72, 73). It has been proposed that CHIPS and SCIN stall early neutrophil recruitment to successfully establish an infection. This is due to their inhibitory action against complement proteins, an early innate response (73, 74). While this seems counterintuitive to the neutrophil recruiting activities of SEA, it also has been proposed that this early blockade of neutrophils allows later modulation of the immune system by SEA and SAK. This theory fits with our model where we see an accumulation of neutrophils later on at 96 h in response to SEA. SCIN may work in tandem with SEA by inhibiting phagocytosis and bactericidal activity of recruited neutrophils (75), which may help form neutrophilic abscesses that S. aureus can survive in. SAK may be involved in dissemination from abscesses due to its ability to cleave fibrin (76), which is characteristic of abscesses. However, it should be noted that the IEC factors, SEA included, are highly human specific (72); thus, they may not be active in our murine model, which is sensitized to SEA only.
SAg function typically has been attributed to crippling the adaptive arm of the immune system by inducing T cell anergy and deletion of T cell-dependent B cell responses (77, 78). Indeed, an inability to form neutralizing antibodies has been linked to many cases of TSS (11, 79); however, SAgs also are highly immunogenic, and the majority of the population is able to form both anti-SAg and anti-staphylococcal antibodies (80). Although purified SAgs have long been shown to induce T cell anergy (81–84), to our knowledge, the role of SAg-mediated T cell anergy has not been demonstrated during a live infection. In our model, SEA-expressing S. aureus caused a decrease in the detectable Vβ3+ CD3+ cells, although SAg-activated T cells usually undergo early expansion (85, 86). This decrease may be a result of Vβ-specific TCR internalization (87, 88), T cell deletion (85), or a combination thereof. Injection of mice with purified SEA similarly resulted in Vβ3-specific CD4+ T cell suppression mediated by IFN-γ and myeloid-derived suppressor cells (89), and this may represent an additional role for SAgs to subvert the immune response. The effect of T cell anergy during staphylococcal disease may inhibit numerous T cells in the context of chronic infection; however, SAgs do not target T cells in an antigen-specific manner, so it is unclear how Vβ-specific anergy would contribute to staphylococcal infections. Thus, it will be important to dissect the role of SAg-mediated T cell suppression during live infections in future studies. Given that SAgs have an inherent ability to impact numerous immune cells, it is highly likely that these toxins are multifunctional virulence factors and are able to influence both the adaptive and the innate immune systems. Overall, this work shows that SAgs are used by S. aureus during infection to target not only T cells directly but also neutrophils as a result of the SAg-elicited cytokines. While the recruitment of neutrophils appears to be counterintuitive to survival, our work demonstrates that SAg expression by S. aureus enables a sophisticated method of in vivo survival by subverting the neutrophil response into a protective niche, demonstrating a biologically relevant and highly novel role for SAgs during infection.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by operating grants from the Canadian Institutes of Health Research (CIHR) to J.K.M. (MOP-64176) and S.M.M.H. (MOP-130465). S.X.X. was supported by an Ontario Graduate Scholarship, S.M.M.H. holds a Canada Research Chair in Viral Immunity and Pathogenesis, and J.K.M. was the recipient of a New Investigator Award from the CIHR.
Footnotes
Published ahead of print 9 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02110-14.
REFERENCES
- 1.Pallin DJ, Egan DJ, Pelletier AJ, Espinola JA, Hooper DC, Camargo CA., Jr 2008. Increased US emergency department visits for skin and soft tissue infections, and changes in antibiotic choices, during the emergence of community-associated methicillin-resistant Staphylococcus aureus. Ann. Emerg. Med. 51:291–298. 10.1016/j.annemergmed.2007.12.004 [DOI] [PubMed] [Google Scholar]
- 2.Johnson JK, Khoie T, Shurland S, Kreisel K, Stine OC, Roghmann MC. 2007. Skin and soft tissue infections caused by methicillin-resistant Staphylococcus aureus USA300 clone. Emerg. Infect. Dis. 13:1195–1200. 10.3201/eid1308.061575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lowy FD. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339:520–532. 10.1056/NEJM199808203390806 [DOI] [PubMed] [Google Scholar]
- 4.Dinges MM, Orwin PM, Schlievert PM. 2000. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 13:16–34. 10.1128/CMR.13.1.16-34.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chambers HF. 2001. The changing epidemiology of Staphylococcus aureus? Emerg. Infect. Dis. 7:178–182. 10.3201/eid0702.010204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.David MZ, Daum RS. 2010. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 23:616–687. 10.1128/CMR.00081-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nizet V. 2007. Understanding how leading bacterial pathogens subvert innate immunity to reveal novel therapeutic targets. J. Allergy Clin. Immunol. 120:13–22. 10.1016/j.jaci.2007.06.005 [DOI] [PubMed] [Google Scholar]
- 8.Foster TJ. 2005. Immune evasion by staphylococci. Nat. Rev. Microbiol. 3:948–958. 10.1038/nrmicro1289 [DOI] [PubMed] [Google Scholar]
- 9.Spaan AN, Surewaard BG, Nijland R, van Strijp JA. 2013. Neutrophils versus Staphylococcus aureus: a biological tug of war. Annu. Rev. Microbiol. 67:629–650. 10.1146/annurev-micro-092412-155746 [DOI] [PubMed] [Google Scholar]
- 10.Proft T, Fraser J. 1998. Superantigens: just like peptides only different. J. Exp. Med. 187:819–821. 10.1084/jem.187.6.819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCormick JK, Yarwood JM, Schlievert PM. 2001. Toxic shock syndrome and bacterial superantigens: an update. Annu. Rev. Microbiol. 55:77–104. 10.1146/annurev.micro.55.1.77 [DOI] [PubMed] [Google Scholar]
- 12.Xu SX, McCormick JK. 2012. Staphylococcal superantigens in colonization and disease. Front. Cell. Infect. Microbiol. 2:52. 10.3389/fcimb.2012.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jardetzky TS, Brown JH, Gorga JC, Stern LJ, Urban RG, Chi YI, Stauffacher C, Strominger JL, Wiley DC. 1994. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature 368:711–718. 10.1038/368711a0 [DOI] [PubMed] [Google Scholar]
- 14.Kim J, Urban RG, Strominger JL, Wiley DC. 1994. Toxic shock syndrome toxin-1 complexed with a class II major histocompatibility molecule HLA-DR1. Science 266:1870–1874. 10.1126/science.7997880 [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Li H, Dimasi N, McCormick JK, Martin R, Schuck P, Schlievert PM, Mariuzza RA. 2001. Crystal structure of a superantigen bound to the high-affinity, zinc-dependent site on MHC class II. Immunity 14:93–104. 10.1016/S1074-7613(01)00092-9 [DOI] [PubMed] [Google Scholar]
- 16.Petersson K, Hakansson M, Nilsson H, Forsberg G, Svensson LA, Liljas A, Walse B. 2001. Crystal structure of a superantigen bound to MHC class II displays zinc and peptide dependence. EMBO J. 20:3306–3312. 10.1093/emboj/20.13.3306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fields BA, Malchiodi EL, Li H, Ysern X, Stauffacher CV, Schlievert PM, Karjalainen K, Mariuzza RA. 1996. Crystal structure of a T-cell receptor beta-chain complexed with a superantigen. Nature 384:188–192. 10.1038/384188a0 [DOI] [PubMed] [Google Scholar]
- 18.Li H, Llera A, Tsuchiya D, Leder L, Ysern X, Schlievert PM, Karjalainen K, Mariuzza RA. 1998. Three-dimensional structure of the complex between a T cell receptor beta chain and the superantigen staphylococcal enterotoxin B. Immunity 9:807–816. 10.1016/S1074-7613(00)80646-9 [DOI] [PubMed] [Google Scholar]
- 19.Andersen PS, Schuck P, Sundberg EJ, Geisler C, Karjalainen K, Mariuzza RA. 2002. Quantifying the energetics of cooperativity in a ternary protein complex. Biochemistry 41:5177–5184. 10.1021/bi0200209 [DOI] [PubMed] [Google Scholar]
- 20.Nur-ur Rahman AK, Bonsor DA, Herfst CA, Pollard F, Peirce M, Wyatt AW, Kasper KJ, Madrenas J, Sundberg EJ, McCormick JK. 2011. The T cell receptor beta-chain second complementarity determining region loop (CDR2beta) governs T cell activation and Vbeta specificity by bacterial superantigens. J. Biol. Chem. 286:4871–4881. 10.1074/jbc.M110.189068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sundberg EJ, Deng L, Mariuzza RA. 2007. TCR recognition of peptide/MHC class II complexes and superantigens. Semin. Immunol. 19:262–271. 10.1016/j.smim.2007.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marrack P, Kappler J. 1990. The staphylococcal enterotoxins and their relatives. Science 248:1066. [PubMed] [Google Scholar]
- 23.Krakauer T. 2000. Coordinate suppression of superantigen-induced cytokine production and T-cell proliferation by a small nonpeptidic inhibitor of class II major histocompatibility complex and CD4 interaction. Antimicrob. Agents Chemother. 44:1067–1069. 10.1128/AAC.44.4.1067-1069.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdelnour A, Bremell T, Tarkowski A. 1994. Toxic shock syndrome toxin 1 contributes to the arthritogenicity of Staphylococcus aureus. J. Infect. Dis. 170:94–99. 10.1093/infdis/170.1.94 [DOI] [PubMed] [Google Scholar]
- 25.Molne L, Tarkowski A. 2000. An experimental model of cutaneous infection induced by superantigen-producing Staphylococcus aureus. J. Investig. Dermatol. 114:1120–1125. 10.1046/j.1523-1747.2000.00973.x [DOI] [PubMed] [Google Scholar]
- 26.Nilsson IM, Verdrengh M, Ulrich RG, Bavari S, Tarkowski A. 1999. Protection against Staphylococcus aureus sepsis by vaccination with recombinant staphylococcal enterotoxin A devoid of superantigenicity. J. Infect. Dis. 180:1370–1373. 10.1086/315023 [DOI] [PubMed] [Google Scholar]
- 27.Narita K, Hu DL, Tsuji T, Nakane A. 2008. Intranasal immunization of mutant toxic shock syndrome toxin 1 elicits systemic and mucosal immune response against Staphylococcus aureus infection. FEMS Immunol. Med. Microbiol. 52:389–396. 10.1111/j.1574-695X.2008.00384.x [DOI] [PubMed] [Google Scholar]
- 28.Varshney AK, Wang X, Scharff MD, MacIntyre J, Zollner RS, Kovalenko OV, Martinez LR, Byrne FR, Fries BC. 2013. Staphylococcal enterotoxin B-specific monoclonal antibody 20B1 successfully treats diverse Staphylococcus aureus infections. J. Infect. Dis. 208:2058–2066. 10.1093/infdis/jit421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson GJ, Seo KS, Cartwright RA, Connelley T, Chuang-Smith ON, Merriman JA, Guinane CM, Park JY, Bohach GA, Schlievert PM, Morrison WI, Fitzgerald JR. 2011. A novel core genome-encoded superantigen contributes to lethality of community-associated MRSA necrotizing pneumonia. PLoS Pathog. 7:e1002271. 10.1371/journal.ppat.1002271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salgado-Pabon W, Breshears L, Spaulding AR, Merriman JA, Stach CS, Horswill AR, Peterson ML, Schlievert PM. 2013. Superantigens are critical for Staphylococcus aureus Infective endocarditis, sepsis, and acute kidney injury. mBio 4:e00494–13. 10.1128/mBio.00494-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strandberg KL, Rotschafer JH, Vetter SM, Buonpane RA, Kranz DM, Schlievert PM. 2010. Staphylococcal superantigens cause lethal pulmonary disease in rabbits. J. Infect. Dis. 202:1690–1697. 10.1086/657156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattis DM, Spaulding AR, Chuang-Smith ON, Sundberg EJ, Schlievert PM, Kranz DM. 2013. Engineering a soluble high-affinity receptor domain that neutralizes staphylococcal enterotoxin C in rabbit models of disease. Protein Eng. Des. Sel. 26:133–142. 10.1093/protein/gzs094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spaulding AR, Salgado-Pabon W, Kohler PL, Horswill AR, Leung DY, Schlievert PM. 2013. Staphylococcal and streptococcal superantigen exotoxins. Clin. Microbiol. Rev. 26:422–447. 10.1128/CMR.00104-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonventre PF, Linnemann C, Weckbach LS, Staneck JL, Buncher CR, Vigdorth E, Ritz H, Archer D, Smith B. 1984. Antibody responses to toxic-shock-syndrome (TSS) toxin by patients with TSS and by healthy staphylococcal carriers. J. Infect. Dis. 150:662–666. 10.1093/infdis/150.5.662 [DOI] [PubMed] [Google Scholar]
- 35.Kansal R, Davis C, Hansmann M, Seymour J, Parsonnet J, Modern P, Gilbert S, Kotb M. 2007. Structural and functional properties of antibodies to the superantigen TSST-1 and their relationship to menstrual toxic shock syndrome. J. Clin. Immunol. 27:327–338. 10.1007/s10875-007-9072-4 [DOI] [PubMed] [Google Scholar]
- 36.Holtfreter S, Grumann D, Schmudde M, Nguyen HT, Eichler P, Strommenger B, Kopron K, Kolata J, Giedrys-Kalemba S, Steinmetz I, Witte W, Broker BM. 2007. Clonal distribution of superantigen genes in clinical Staphylococcus aureus isolates. J. Clin. Microbiol. 45:2669–2680. 10.1128/JCM.00204-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naber CK. 2009. Staphylococcus aureus bacteremia: epidemiology, pathophysiology, and management strategies. Clin. Infect. Dis. 48(Suppl 4):S231–S237. 10.1086/598189 [DOI] [PubMed] [Google Scholar]
- 38.Ito K, Bian HJ, Molina M, Han J, Magram J, Saar E, Belunis C, Bolin DR, Arceo R, Campbell R, Falcioni F, Vidovic D, Hammer J, Nagy ZA. 1996. HLA-DR4-IE chimeric class II transgenic, murine class II-deficient mice are susceptible to experimental allergic encephalomyelitis. J. Exp. Med. 183:2635–2644. 10.1084/jem.183.6.2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koressaar T, Remm M. 2007. Enhancements and modifications of primer design program Primer3. Bioinformatics 23:1289–1291. 10.1093/bioinformatics/btm091 [DOI] [PubMed] [Google Scholar]
- 40.Arnaud M, Chastanet A, Debarbouille M. 2004. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 70:6887–6891. 10.1128/AEM.70.11.6887-6891.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guerout-Fleury AM, Shazand K, Frandsen N, Stragier P. 1995. Antibiotic-resistance cassettes for Bacillus subtilis. Gene 167:335–336. 10.1016/0378-1119(95)00652-4 [DOI] [PubMed] [Google Scholar]
- 42.Bateman BT, Donegan NP, Jarry TM, Palma M, Cheung AL. 2001. Evaluation of a tetracycline-inducible promoter in Staphylococcus aureus in vitro and in vivo and its application in demonstrating the role of sigB in microcolony formation. Infect. Immun. 69:7851–7857. 10.1128/IAI.69.12.7851-7857.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brouillard JN, Gunther S, Varma AK, Gryski I, Herfst CA, Rahman AK, Leung DY, Schlievert PM, Madrenas J, Sundberg EJ, McCormick JK. 2007. Crystal structure of the streptococcal superantigen SpeI and functional role of a novel loop domain in T cell activation by group V superantigens. J. Mol. Biol. 367:925–934. 10.1016/j.jmb.2007.01.024 [DOI] [PubMed] [Google Scholar]
- 44.Rahman AK, Herfst CA, Moza B, Shames SR, Chau LA, Bueno C, Madrenas J, Sundberg EJ, McCormick JK. 2006. Molecular basis of TCR selectivity, cross-reactivity, and allelic discrimination by a bacterial superantigen: integrative functional and energetic mapping of the SpeC-Vbeta2.1 molecular interface. J. Immunol. 177:8595–8603. 10.4049/jimmunol.177.12.8595 [DOI] [PubMed] [Google Scholar]
- 45.Hayworth JL, Mazzuca DM, Maleki Vareki S, Welch I, McCormick JK, Haeryfar SM. 2012. CD1d-independent activation of mouse and human iNKT cells by bacterial superantigens. Immunol. Cell Biol. 90:699–709. 10.1038/icb.2011.90 [DOI] [PubMed] [Google Scholar]
- 46.Yeung RS, Penninger JM, Kundig T, Khoo W, Ohashi PS, Kroemer G, Mak TW. 1996. Human CD4 and human major histocompatibility complex class II (DQ6) transgenic mice: supersensitivity to superantigen-induced septic shock. Eur. J. Immunol. 26:1074–1082. 10.1002/eji.1830260518 [DOI] [PubMed] [Google Scholar]
- 47.DaSilva L, Welcher BC, Ulrich RG, Aman MJ, David CS, Bavari S. 2002. Humanlike immune response of human leukocyte antigen-DR3 transgenic mice to staphylococcal enterotoxins: a novel model for superantigen vaccines. J. Infect. Dis. 185:1754–1760. 10.1086/340828 [DOI] [PubMed] [Google Scholar]
- 48.Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K. 2008. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J. Bacteriol. 190:300–310. 10.1128/JB.01000-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Borst DW, Betley MJ. 1993. Mutations in the promoter spacer region and early transcribed region increase expression of staphylococcal enterotoxin A. Infect. Immun. 61:5421–5425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Callahan JE, Herman A, Kappler JW, Marrack P. 1990. Stimulation of B10.BR T cells with superantigenic staphylococcal toxins. J. Immunol. 144:2473–2479 [PubMed] [Google Scholar]
- 51.Dohlsten M, Bjorklund M, Sundstedt A, Hedlund G, Samson D, Kalland T. 1993. Immunopharmacology of the superantigen staphylococcal enterotoxin A in T-cell receptor V beta 3 transgenic mice. Immunology 79:520–527 [PMC free article] [PubMed] [Google Scholar]
- 52.Leung DY, Gately M, Trumble A, Ferguson-Darnell B, Schlievert PM, Picker LJ. 1995. Bacterial superantigens induce T cell expression of the skin-selective homing receptor, the cutaneous lymphocyte-associated antigen, via stimulation of interleukin 12 production. J. Exp. Med. 181:747–753. 10.1084/jem.181.2.747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng AG, Kim HK, Burts ML, Krausz T, Schneewind O, Missiakas DM. 2009. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 23:3393–3404. 10.1096/fj.09-135467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng AG, DeDent AC, Schneewind O, Missiakas D. 2011. A play in four acts: Staphylococcus aureus abscess formation. Trends Microbiol. 19:225–232. 10.1016/j.tim.2011.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller LS, Cho JS. 2011. Immunity against Staphylococcus aureus cutaneous infections. Nat. Rev. Immunol. 11:505–518. 10.1038/nri3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ogston A. 1882. Micrococcus poisoning. J. Anat. Physiol. 16:526–567 [PMC free article] [PubMed] [Google Scholar]
- 57.Schmitz GR. 2011. How do you treat an abscess in the era of increased community-associated methicillin-resistant Staphylococcus aureus (MRSA)? J. Emerg. Med. 41:276–281. 10.1016/j.jemermed.2011.01.027 [DOI] [PubMed] [Google Scholar]
- 58.Robinson JL, Salvadori MI. 2011. Management of community-associated methicillin-resistant Staphylococcus aureus skin abscesses in children. Paediatr. Child Health 16:115–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kurts C, Panzer U, Anders HJ, Rees AJ. 2013. The immune system and kidney disease: basic concepts and clinical implications. Nat. Rev. Immunol. 13:738–753. 10.1038/nri3523 [DOI] [PubMed] [Google Scholar]
- 60.Chau TA, McCully ML, Brintnell W, An G, Kasper KJ, Vines ED, Kubes P, Haeryfar SM, McCormick JK, Cairns E, Heinrichs DE, Madrenas J. 2009. Toll-like receptor 2 ligands on the staphylococcal cell wall downregulate superantigen-induced T cell activation and prevent toxic shock syndrome. Nat. Med. 15:641–648. 10.1038/nm.1965 [DOI] [PubMed] [Google Scholar]
- 61.Neumann B, Emmanuilidis K, Stadler M, Holzmann B. 1998. Distinct functions of interferon-gamma for chemokine expression in models of acute lung inflammation. Immunology 95:512–521. 10.1046/j.1365-2567.1998.00643.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rajagopalan G, Tilahun AY, Asmann YW, David CS. 2009. Early gene expression changes induced by the bacterial superantigen staphylococcal enterotoxin B and its modulation by a proteasome inhibitor. Physiol. Genomics 37:279–293. 10.1152/physiolgenomics.90385.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muraille E, Pajak B, Urbain J, Moser M, Leo O. 1999. Role and regulation of IL-12 in the in vivo response to staphylococcal enterotoxin B. Int. Immunol. 11:1403–1410. 10.1093/intimm/11.9.1403 [DOI] [PubMed] [Google Scholar]
- 64.Donohue JH, Rosenberg SA. 1983. The fate of interleukin-2 after in vivo administration. J. Immunol. 130:2203–2208 [PubMed] [Google Scholar]
- 65.Muhlradt PF, Opitz HG. 1982. Clearance of interleukin 2 from the blood of normal and T cell-depleted mice. Eur. J. Immunol. 12:983–985. 10.1002/eji.1830121117 [DOI] [PubMed] [Google Scholar]
- 66.McLoughlin RM, Lee JC, Kasper DL, Tzianabos AO. 2008. IFN-gamma regulated chemokine production determines the outcome of Staphylococcus aureus infection. J. Immunol. 181:1323–1332. 10.4049/jimmunol.181.2.1323 [DOI] [PubMed] [Google Scholar]
- 67.Tessier PA, Naccache PH, Diener KR, Gladue RP, Neote KS, Clark-Lewis I, McColl SR. 1998. Induction of acute inflammation in vivo by staphylococcal superantigens. II. Critical role for chemokines, ICAM-1, and TNF-alpha. J. Immunol. 161:1204–1211 [PubMed] [Google Scholar]
- 68.Verdrengh M, Tarkowski A. 1997. Role of neutrophils in experimental septicemia and septic arthritis induced by Staphylococcus aureus. Infect. Immun. 65:2517–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Robertson CM, Perrone EE, McConnell KW, Dunne WM, Boody B, Brahmbhatt T, Diacovo MJ, Van Rooijen N, Hogue LA, Cannon CL, Buchman TG, Hotchkiss RS, Coopersmith CM. 2008. Neutrophil depletion causes a fatal defect in murine pulmonary Staphylococcus aureus clearance. J. Surg. Res. 150:278–285. 10.1016/j.jss.2008.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. 2000. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J. Immunol. 164:3713–3722. 10.4049/jimmunol.164.7.3713 [DOI] [PubMed] [Google Scholar]
- 71.Zeng X, Moore TA, Newstead MW, Deng JC, Lukacs NW, Standiford TJ. 2005. IP-10 mediates selective mononuclear cell accumulation and activation in response to intrapulmonary transgenic expression and during adenovirus-induced pulmonary inflammation. J. Interferon Cytokine Res. 25:103–112. 10.1089/jir.2005.25.103 [DOI] [PubMed] [Google Scholar]
- 72.van Wamel WJ, Rooijakkers SH, Ruyken M, van Kessel KP, van Strijp JA. 2006. The innate immune modulators staphylococcal complement inhibitor and chemotaxis inhibitory protein of Staphylococcus aureus are located on beta-hemolysin-converting bacteriophages. J. Bacteriol. 188:1310–1315. 10.1128/JB.188.4.1310-1315.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rooijakkers SH, Ruyken M, van Roon J, van Kessel KP, van Strijp JA, van Wamel WJ. 2006. Early expression of SCIN and CHIPS drives instant immune evasion by Staphylococcus aureus. Cell Microbiol. 8:1282–1293. 10.1111/j.1462-5822.2006.00709.x [DOI] [PubMed] [Google Scholar]
- 74.Postma B, Poppelier MJ, van Galen JC, Prossnitz ER, van Strijp JA, de Haas CJ, van Kessel KP. 2004. Chemotaxis inhibitory protein of Staphylococcus aureus binds specifically to the C5a and formylated peptide receptor. J. Immunol. 172:6994–7001. 10.4049/jimmunol.172.11.6994 [DOI] [PubMed] [Google Scholar]
- 75.Rooijakkers SH, Ruyken M, Roos A, Daha MR, Presanis JS, Sim RB, van Wamel WJ, van Kessel KP, van Strijp JA. 2005. Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat. Immunol. 6:920–927. 10.1038/ni1235 [DOI] [PubMed] [Google Scholar]
- 76.Bokarewa MI, Jin T, Tarkowski A. 2006. Staphylococcus aureus: staphylokinase. Int. J. Biochem. Cell Biol. 38:504–509. 10.1016/j.biocel.2005.07.005 [DOI] [PubMed] [Google Scholar]
- 77.Llewelyn M, Cohen J. 2002. Superantigens: microbial agents that corrupt immunity. Lancet Infect. Dis. 2:156–162. 10.1016/S1473-3099(02)00222-0 [DOI] [PubMed] [Google Scholar]
- 78.Fraser J, Arcus V, Kong P, Baker E, Proft T. 2000. Superantigens–powerful modifiers of the immune system. Mol. Med. Today 6:125–132. 10.1016/S1357-4310(99)01657-3 [DOI] [PubMed] [Google Scholar]
- 79.Vergeront JM, Stolz SJ, Crass BA, Nelson DB, Davis JP, Bergdoll MS. 1983. Prevalence of serum antibody to staphylococcal enterotoxin F among Wisconsin residents: implications for toxic-shock syndrome. J. Infect. Dis. 148:692–698. 10.1093/infdis/148.4.692 [DOI] [PubMed] [Google Scholar]
- 80.Grumann D, Ruotsalainen E, Kolata J, Kuusela P, Jarvinen A, Kontinen VP, Broker BM, Holtfreter S. 2011. Characterization of infecting strains and superantigen-neutralizing antibodies in Staphylococcus aureus bacteremia. Clin. Vaccine Immunol. 18:487–493. 10.1128/CVI.00329-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rellahan BL, Jones LA, Kruisbeek AM, Fry AM, Matis LA. 1990. In vivo induction of anergy in peripheral V beta 8+ T cells by staphylococcal enterotoxin B. J. Exp. Med. 172:1091–1100. 10.1084/jem.172.4.1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Miller C, Ragheb JA, Schwartz RH. 1999. Anergy and cytokine-mediated suppression as distinct superantigen-induced tolerance mechanisms in vivo. J. Exp. Med. 190:53–64. 10.1084/jem.190.1.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sundstedt A, Dohlsten M. 1998. In vivo anergized CD4+ T cells have defective expression and function of the activating protein-1 transcription factor. J. Immunol. 161:5930–5936 [PubMed] [Google Scholar]
- 84.Watson AR, Janik DK, Lee WT. 2012. Superantigen-induced CD4 memory T cell anergy. I. Staphylococcal enterotoxin B induces Fyn-mediated negative signaling. Cell Immunol. 276:16–25. 10.1016/j.cellimm.2012.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.MacDonald HR, Baschieri S, Lees RK. 1991. Clonal expansion precedes anergy and death of V beta 8+ peripheral T cells responding to staphylococcal enterotoxin B in vivo. Eur. J. Immunol. 21:1963–1966. 10.1002/eji.1830210827 [DOI] [PubMed] [Google Scholar]
- 86.Renno T, Hahne M, MacDonald HR. 1995. Proliferation is a prerequisite for bacterial superantigen-induced T cell apoptosis in vivo. J. Exp. Med. 181:2283–2287. 10.1084/jem.181.6.2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Niedergang F, Hemar A, Hewitt CR, Owen MJ, Dautry-Varsat A, Alcover A. 1995. The Staphylococcus aureus enterotoxin B superantigen induces specific T cell receptor down-regulation by increasing its internalization. J. Biol. Chem. 270:12839–12845. 10.1074/jbc.270.21.12839 [DOI] [PubMed] [Google Scholar]
- 88.Makida R, Hofer MF, Takase K, Cambier JC, Leung DY. 1996. Bacterial superantigens induce V beta-specific T cell receptor internalization. Mol. Immunol. 33:891–900. 10.1016/0161-5890(96)84615-3 [DOI] [PubMed] [Google Scholar]
- 89.Cauley LS, Miller EE, Yen M, Swain SL. 2000. Superantigen-induced CD4 T cell tolerance mediated by myeloid cells and IFN-gamma. J. Immunol. 165:6056–6066. 10.4049/jimmunol.165.11.6056 [DOI] [PubMed] [Google Scholar]
- 90.Duthie ES, Lorenz LL. 1952. Staphylococcal coagulase; mode of action and antigenicity. J. Gen. Microbiol. 6:95–107. 10.1099/00221287-6-1-2-95 [DOI] [PubMed] [Google Scholar]
- 91.Novick R. 1967. Properties of a cryptic high-frequency transducing phage in Staphylococcus aureus. Virology 33:155–166. 10.1016/0042-6822(67)90105-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.