

Abstract
The sepsis initial hyperinflammatory reaction, if not treated early, shifts to a protracted state of immunosuppression that alters both innate and adaptive immunity and is associated with elevated mortality. Myeloid-derived suppressor cells (MDSCs) are myeloid progenitors and precursors that fail to differentiate into mature innate-immunity cells and are known for their potent immunosuppressive activities. We previously reported that murine MDSCs expand dramatically in the bone marrow during late sepsis, induced by cecal ligation and puncture, and demonstrated that they contribute to late-sepsis immunosuppression. However, the molecular mechanism responsible for generating these immature Gr1+ CD11b+ myeloid cells during sepsis remains unknown. We show here that sepsis generates a microRNA (miRNA) signature that expands MDSCs. We found that miRNA 21 (miR-21) and miR-181b expression is upregulated in early sepsis and sustained in late sepsis. Importantly, we found that simultaneous in vivo blockade of both miRNAs via antagomiR (a chemically modified miRNA inhibitor) injection after sepsis initiation decreased the bone marrow Gr1+ CD11b+ myeloid progenitors, improved bacterial clearance, and reduced late-sepsis mortality by 74%. Gr1+ CD11b+ cells isolated from mice injected with antagomiRs were able to differentiate ex vivo into macrophages and dendritic cells and produced smaller amounts of the immunosuppressive interleukin 10 (IL-10) and transforming growth factor β (TGF-β) after stimulation with bacterial lipopolysaccharide, suggesting that immature myeloid cells regained their maturation potential and have lost their immunosuppressive activity. In addition, we found that the protein level of transcription factor NFI-A, which plays a role in myeloid cell differentiation, was increased during sepsis and that antagomiR injection reduced its expression. Moreover, knockdown of NFI-A in the Gr1+ CD11b+ cells isolated from late-septic mice increased their maturation potential and reduced their production of the immunosuppressive mediators, similar to antagomiR injection. These data support the hypothesis that sepsis reprograms myeloid cells and thus alters the innate immunity cell repertoire to promote immunosuppression, and they demonstrate that this process can be reversed by targeting miR-21 and miR-181b to improve late-sepsis survival.
INTRODUCTION
Recent studies in sepsis support the idea that immunosuppression, rather than excessive inflammation, contributes to most contemporary sepsis deaths (1–4). Sepsis, caused by infection or trauma, is initiated by a hyperinflammatory reaction, which shifts within a few days to a protracted state of anti-inflammation and immunosuppression (1–3, 5). This state of immunosuppression is associated with increased production of immunosuppressive cytokines, increased T cell and dendritic cell apoptosis, increased T regulatory cells, and enhanced local bacterial growth (1, 3, 5, 6). It has been postulated that although sepsis immunosuppression is considered an adaptive feedback mechanism to limit tissue damage during the onset of the early hyperinflammatory phase, its persistence increases the risk of secondary infections and predicts a poor outcome (1–3). In a postmortem study of sepsis, Torgersen et al. (7) reported that more than 70% of patients died after the first 3 days of sepsis onset, i.e., after the time in which most sepsis patients enter a state of hypoinflammation and immunosuppression (1). Using the clinically relevant model of polymicrobial sepsis in mice, we recently reported that most deaths occur after the first 5 days of sepsis induction (8). Thus, the rapid shift of sepsis response from early hyperinflammation to late immunosuppression may explain why more than 30 clinical trials using anti-inflammatory drugs failed to reduce overall sepsis mortality (9, 10). In large part, this is because these treatments are often given late, after the immunosuppressive state has already developed. Based on these and other studies, Hotchkiss and colleagues (1) have recently emphasized that sepsis should be considered an immunosuppressive disorder, and as such, sepsis patients should benefit from an immunostimulatory rather than anti-inflammatory therapy. This paradigm shift in the understanding of sepsis pathogenesis has renewed interest in immunosuppression as an underlying mechanism of late-sepsis pathogenesis.
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells that mainly include progenitors and precursors of monocytes, granulocytes, and dendritic cells, which expand under nearly all inflammatory conditions (11–13). The immature phenotypes and suppressive activities are the hallmark of these cells, as they potently suppress T cell and innate-immunity cell functions (13–15). Phenotypically, mouse MDSCs are characterized by the coexpression of the myeloid differentiation markers CD11b and Gr1 (12, 16). Because the Gr1 molecule comprises two epitopes, Ly6C and Ly6G, mouse MDSCs can be subdivided into monocytic (CD11b+ Ly6G− Ly6Chigh) and granulocytic (CD11b+ Ly6G+ Ly6Clow) subsets (12, 13, 16). Because human myeloid cells do not express Gr1, human MDSCs have been identified with the common myeloid marker CD33 in cancer patients (17). In addition, a CD11b+ CD14− CD15+ population, the equivalent of the mouse granulocytic MDSCs, has been identified in renal cell carcinoma patients (18), whereas a CD11b+ CD14+ HLA−/low population with monocytic morphology has been identified in melanoma patients (16). Despite this phenotypic heterogeneity of mouse and human MDSCs, both monocytic and granulocytic subpopulations are immunosuppressive (15), although they may suppress the immune response by different mechanisms (14, 16). Upon activation by tumor- or inflammation-driven factors, MDSCs express/produce immunosuppressive mediators, including arginase 1, inducible nitric oxide synthase (iNOS), reactive oxygen species (ROS), transforming growth factor β (TGF-β), interleukin 10 (IL-10), and cyclo-oxygenase 2 and prostaglandin E2 (12, 14, 16). These mediators then inhibit T cell proliferation and activation, induce T regulatory cells, and inhibit innate immune cell functions, thus suppressing both innate and adaptive immunity (12–14). Interestingly, recent studies reported that factors that eliminate MDSCs can overcome immune suppression and improve the immune response. For example, all-trans-retinoic acid, which promotes myeloid differentiation, has been shown to reduce MDSCs and to improve immunocompetence in tumor-bearing mice (19) and in mice with lipopolysaccharide (LPS)-induced immune suppression (20). We (8) and others (21, 22) have shown that MDSCs dramatically expand in the mouse bone marrow during the course of polymicrobial sepsis. In addition, we have shown that MDSCs from late sepsis are immunosuppressive, as they produced immunosuppressive cytokines and attenuated early sepsis hyperinflammation when transferred into mice undergoing sepsis response (8). Although several soluble factors, which can all be produced in the inflammatory microenvironment, such as vascular endothelial growth factor (VEGF), granulocyte colony-stimulating factor and monocyte colony-stimulating factor (M-CSF), and inflammatory cytokines, have been suggested to induce MDSC generation and expansion in the context of cancer (11–14), the molecular mechanism that generates MDSCs in sepsis remains unknown.
The hypothesis that MDSCs are activated myeloid cells that are trapped in the immature stages of their development (11, 12) supports the idea that dysregulation of the myeloid differentiation and maturation programs is primarily responsible for MDSC generation and expansion. Given that microRNAs (miRNAs) have been implicated in myeloid development and differentiation under normal and disease conditions (23), we employed an miRNA-based approach to investigate the mechanism of MDSC generation during murine sepsis. Our work demonstrates that miRNA 21 (miR-21) and miR-181b disrupt myeloid differentiation and maturation during the sepsis response and suggests that this negative path can be targeted to restore immunocompetency and improve survival.
MATERIALS AND METHODS
Mice.
Male BALB/c mice, 8 weeks old, were purchased from Harlan Sprague Dawley (Indianapolis, IN). The mice were housed in a pathogen-free facility and were acclimated to the new environment for a week before surgery. All experiments were conducted in accordance with National Institutes of Health guidelines and were approved by the East Tennessee State University Animal Care and Use Committee.
Sepsis model.
Polymicrobial sepsis, which mimics general peritonitis occurring in humans (24–26), was induced by cecal ligation and puncture (CLP) as described previously (27). This model creates a prolonged infection with ∼90% mortality over 4 weeks. Briefly, mice were anesthetized via inhalation with 2.5% isoflurane (Abbott Laboratories, Abbott Park, IL). A midline abdominal incision was made, and the cecum was exteriorized, ligated distal to the ileocecal valve, and then punctured twice with a 21-gauge needle. A small amount of feces was extruded into the abdominal cavity. The abdominal wall and skin were sutured in layers with 3-0 silk. Sham-operated mice were treated identically, except that the cecum was neither ligated nor punctured. The mice received (intraperitoneally [i.p.]) 1 ml lactated Ringer's solution plus 5% dextrose for fluid resuscitation. To create the late-sepsis phenotype, mice were subcutaneously administered antibiotic (imipenem; 25 mg/kg body weight) or an equivalent volume of 0.9% saline. To establish intra-abdominal infection and approximate the clinical situation of early human sepsis, where there is a delay between the onset of sepsis and the delivery of therapy (28), injections of imipenem were given at 8 and 16 h after CLP. Based on our experience, these levels of injury and manipulation create prolonged infections with high mortality (∼60 to 70%) during the late/chronic phase (27).
The presence of early sepsis was confirmed by transient systemic bacteremia and elevated cytokine levels in the first 5 days after CLP. Late/chronic sepsis (after day 5) was confirmed by enhanced peritoneal bacterial overgrowth and reduced circulating proinflammatory cytokines.
Bacterial culture.
Immediately after mice were sacrificed, the peritoneal cavity was lavaged with 5 ml phosphate-buffered saline (PBS). The lavage fluid was cleared by centrifugation, diluted 6- to 8-fold, and plated on 5% sheep blood agar plates with Trypticase soy agar base (BD Biosciences, Sparks, MD). The plates were incubated for 24 h at 37°C under aerobic conditions. The plates were read by a microbiologist, and the numbers of CFU were determined and multiplied by the dilution factor.
Endotoxin challenge.
Septic mice that received miRNA antagomiRs (chemically modified miRNA inhibitors) were challenged (i.p.) with 10 μg of Gram-negative bacterial LPS (E. coli serotype 0111:B4; Sigma-Aldrich, St. Louis, MO). After 6 h, the mice were sacrificed, blood was collected via cardiac puncture, and sera were collected and used for cytokine measurements. In this experiment, mice were sampled between days 10 and 21 after CLP (i.e., during late sepsis) so that only moribund mice (i.e., those that it was determined would die within 24 h [the CLP plus mutant antagomiR group] based on our previous study [27]) and a corresponding number of surviving, healthy-appearing mice (the CLP plus antagomiR group) were challenged with LPS.
AntagomiR synthesis and injection.
AntagomiRs are chemically modified anti-sense single-stranded RNA molecules complementary to the mature miRNA. These miRNA inhibitors, unlike locked-nucleic acid (LNA)-based anti-miRNA oligonucleotides (which inhibit miRNA via sequestration), are stable in vivo for up to 2 weeks and inhibit the target miRNA via degradation (29). miR-21 and miR-181b sequences were downloaded from miRBase (http://mirbase.org), and antagomiRs were synthesized by GeneCopoeia (Rockville, MD). Mutant antagomiRs and scrambled anti-sense RNA sequences served as controls. Chemical modifications included cholesterol conjugation, 2′-O-methyl (2′-OMe) phosphoramidite, and phosphorothioate linkage. The antagomiR sequences were as follows: miR-21 antagomiR, 5′-mU*mC*mAmAmCmAmUmCmAmGmUmCmUmGmAmUmAmAmG*mC*mU*mA*-Chol-3′; miR-21 mutant antagomiR, 5′-mU*mC*mAmCmCmAmUmCmCmGmUmAmUmGmAmUmAmCmG*mC*mU*mA*-Chol-3′; miR-181b antagomiR, 5′-mA*mC*mCmCmAmCmCmGmAmCmAmGmCmAmAmUmGmAmAmU*mG*mU*mU*-Chol-3′; miR181b mutant antagomiR, 5′-mA*mC*mCmCmAmCmAmGmAmAmAmGmCmAmCmUmGmCmAmU*mG*mU*mU*-Chol-3′; and scrambled antagomiR, 5′-mGmCmGmUmAmUmUmAmUmAmGmCmCmGmAmUmUmAmAmCmGmAmC-3′, where m is 2′-OMe-modified phosphoramidite, * is phosphorothioate linkage, and Chol is hydroxypyrolinol-linked cholesterol. Mutated nucleotides are underlined.
AntagomiRs were injected via the tail vein at doses of 80 mg/kg in 100-μl volumes. Injection was performed 48 h after CLP (to allow initiation of sepsis), and target miRNA levels were measured 24 h after injection.
Isolation and purification of MDSCs.
Bone marrow and spleen Gr1+ CD11b+ MDSCs were harvested from sham-operated and septic mice using magnetically assisted cell sorting according to the manufacturer's protocol (Miltenyi Biotech, Auburn, CA). Briefly, the bone marrow cells were flushed out of the femurs with RPMI 1640 medium (without serum) under aseptic conditions (27). The spleens were minced in RPMI 1640 medium. A single-cell suspension of the bone marrow or spleen was made by pipetting and filtering through a 70-μm nylon strainer followed by erythrocyte lysis. To purify total Gr1+ CD11b+ MDSCs, bone marrow or spleen cells were first depleted of the Gr1− CD11b+ cells (mostly CD11c+ dendritic cells) using anti-CD11c microbeads and an LD column (Miltenyi). The CD11c+-depleted cell population was then subjected to positive selection of the Gr1+ CD11b+ cells by incubating with biotinylated mouse anti-Gr1 antibody (clone RB6-8C5; eBioscience, San Diego, CA) for 15 min at 4°C. After washing, the cells were incubated with anti-biotin magnetic beads for 20 min at 4°C and subsequently applied to an MS column. The purified Gr1+ CD11b+ cells were then washed and resuspended in sterile saline. Cell purity was determined by flow cytometry and was higher than 95%.
To isolate CD31+ MDSCs, total purified Gr1+ CD11b+ cells were incubated with anti-CD31 antibody (eBioscience), and CD31+ cells were subsequently selected with magnetic beads as described above.
miRNA array analysis.
To study miRNA expression during sepsis, we performed genome-wide miRNA expression profiling in whole bone marrow cells using the Mouse Genome miRNA PCR Array (Qiagen; catalog number MAM-200A). The array included miRNAs annotated by miRBase release 14 (www.mirbase.org). miRNA-enriched RNA was isolated from the bone marrow cells of naive and septic mice that were moribund and sacrificed during early (days 1 to 5) and late (days 10 to 21) sepsis and then hybridized to the array according to the manufacturer's protocol. Naive mice (day 0) served as a control. We considered an miRNA to be up- or downregulated in septic mice if its expression was modified at least 2-fold relative to naive mice.
Real-time PCR.
Quantitative real-time PCR was carried out to validate the array results. The PCRs were performed using the same RNA preparation used for the array and an assay primer set specific to each target miRNA according to the manufacturer's protocol (Qiagen). The relative expression of each miRNA was calculated using the 2−ΔΔCT cycle threshold method after normalization to the endogenous U6 RNA as an internal control.
miRNA Northern blotting.
Levels of miR-21 and miR-181b in whole bone marrow cells were measured using an miRNA Northern blot assay kit according to the manufacturer's instructions (Signosis, Santa Clara, CA). Briefly, 2-μg amounts of miRNA-enriched RNA were separated by using a 15% Tris-borate-EDTA (TBE) urea gel, transferred to a nylon membrane, and then immobilized by UV cross-linking. The membranes were hybridized overnight at 42°C with a biotin-labeled miRNA probe complementary to the target miRNA. The membranes were blocked for 30 min at room temperature and then incubated with streptavidin-horseradish peroxidase (HRP) for 45 min at room temperature. After washing, miRNAs were detected with a chemiluminescence substrate, the bands were visualized using the ChemiDoc XRS System (Bio-Rad), and the images were captured with Image Lab software V3.0 (Bio-Rad).
MDSC differentiation.
Total purified Gr1+ CD11b+ cells were cultured for 6 days with complete RPMI 1640 medium in the presence of 10 ng/ml of M-CSF (PeproTech Inc., Rocky Hill, NJ) plus 10 ng/ml recombinant IL-4 (rIL-4) (eBioscience, San Diego, CA). The cell phenotypes were analyzed by flow cytometry. In some experiments, a portion of the differentiated cells were washed and stimulated for 6 h with 100 ng/ml of LPS, and the culture supernatants were used for cytokine measurements by enzyme-linked immunosorbent assay (ELISA).
Flow cytometry.
MDSCs were analyzed by flow cytometry. Bone marrow cells and total or differentiated Gr1+ CD11b+ MDSCs were labeled by incubation for 30 min on ice in staining buffer (PBS plus 2% fetal bovine serum [FBS]) with the appropriate fluorochrome-conjugated antibodies. After washing, the samples were analyzed with a FACSCaliber flow cytometer (BD Biosciences, Sparks, MD). About 15,000 events were acquired and analyzed using CellQuest Pro software (BD Biosciences). The following antibodies were used: anti-Gr1 conjugated to fluorescein isothiocyanate (FITC), anti-CD11b conjugated to phycoerythrin (PE), anti-F4/80 conjugated to allophycocyanin (APC), anti-CD11c conjugated to PE, anti-major histocompatibility complex class II (MHC-II) conjugated to FITC, and anti-CD31 conjugated to PE57 (eBioscience). An appropriate isotype-matched control was used for each antibody.
siRNA-mediated knockdown.
Total purified Gr1+ CD11b+ MDSCs were transfected with pools of NFI-A-specific or scrambled (control) small interfering RNAs (siRNAs) at a 0.5 μM final concentration (Santa Cruz Biotechnology, Santa Cruz, CA) using HiPerFect transfection reagent according to the manufacturer's instructions (Qiagen, Valencia, CA). After 24 h, cells were either differentiated for 6 days with M-CSF plus rIL-4 (as described above) or stimulated for 6 h with LPS to assess cytokine production.
Western blot analysis.
Equal amounts of protein extracts were mixed with 5× Laemmli sample buffer, separated on an SDS-10% polyacrylamide gel (Bio-Rad), and subsequently transferred to nitrocellulose membranes (Thermo Fisher Scientific, Waltham, MA). After blocking with 5% milk in Tris-buffered saline–Tween 20 for 1 h at room temperature, the membranes were probed overnight at 4°C with the appropriate primary antibody. After washing, the blots were incubated with the appropriate HRP-conjugated secondary antibody (Life Technologies, Grand Island, NY) for 2 h at room temperature. Proteins were detected with the enhanced chemiluminescence detection system (Thermo Fisher Scientific, Waltham, MA), the bands were visualized using the ChemiDoc XRS System (Bio-Rad), and the images were captured with Image Lab Software V3.0. The membranes were stripped and reprobed with actin (Sigma-Aldrich) antibody as a loading control.
ELISA and nitrite assay.
Cytokine concentrations were measured by ELISA. Circulating (serum) levels of tumor necrosis factor alpha (TNF-α), IL-6, IL-10, and TGF-β in septic mice injected with antagomiRs or their levels in the culture supernatants of LPS-stimulated MDSCs were determined using specific ELISA kits (eBioscience) according to the manufacturer's instructions. Blood was collected via cardiac puncture at the time of sacrifice (8). A nitrite assay was performed as described previously (8) to determine NO production by LPS-stimulated MDSCs. Each sample was run in triplicate.
Statistical analysis.
The Kaplan-Meier survival curve was plotted using GraphPad Prism version 5.0 (GraphPad Software), and survival significance was determined by a log-rank test. Other data were analyzed using Microsoft Excel V3.0, and statistical significance was determined by an unpaired Student's t test. All values are expressed as means and standard deviations (SD); P values of ≤0.05 were considered statistically significant.
RESULTS
Expression of miR-21 and miR-181b is induced and maintained during sepsis.
Because miRNAs have been shown to participate in myeloid cell development and differentiation under various conditions (23, 30), we investigated whether miRNAs play a role in myeloid differentiation and MDSC expansion during sepsis. To do this, we performed genome-wide miRNA expression profiling in mixed bone marrow cells using the Mouse Genome miRNA PCR array (Qiagen, catalog number MAM-200A), which includes miRNAs annotated by miRBase release 14 (www.miRBase.org). We considered an miRNA to be up- or downregulated if its expression was modified at least 2-fold relative to naive mice. We detected changes in expression of 29 miRNAs, which fell into 3 categories: (i) 17 miRNAs were increased (∼2- to 6-fold) in early sepsis and returned to baseline levels in late sepsis (including miR-125b, miR-126-3p, miR-146a, miR-147, and miR-155), (ii) 9 miRNAs were decreased (∼2- to 4.1-fold) in early sepsis and returned to baseline levels in late sepsis (including miR-17-5p, miR-20a, miR-106a, miR-150, and miR-223), and (iii) 3 miRNAs were increased (∼3.2- to 18.5-fold) in early sepsis (miR-21, miR-181b, and miR-196b). Expression of two miRNAs (miR-21 and miR-181b) had sustained increases in late sepsis, whereas one miRNA (miR-196b) diminished but remained (1.2-fold) above the baseline level in late sepsis. Because these three miRNAs were elevated in both early and late sepsis, we hypothesized that they might be involved in MDSC expansion. Using reverse transcription (RT)-PCR, we confirmed that miR-21 expression was increased from 18.5-fold in early sepsis to 27-fold in late sepsis, whereas miR-181b was increased from 15-fold in early sepsis to 22.6-fold in late sepsis. These results were confirmed by Northern blotting (Fig. 1). We could not confirm the 1.2-fold increase in miR-196b.
FIG 1.
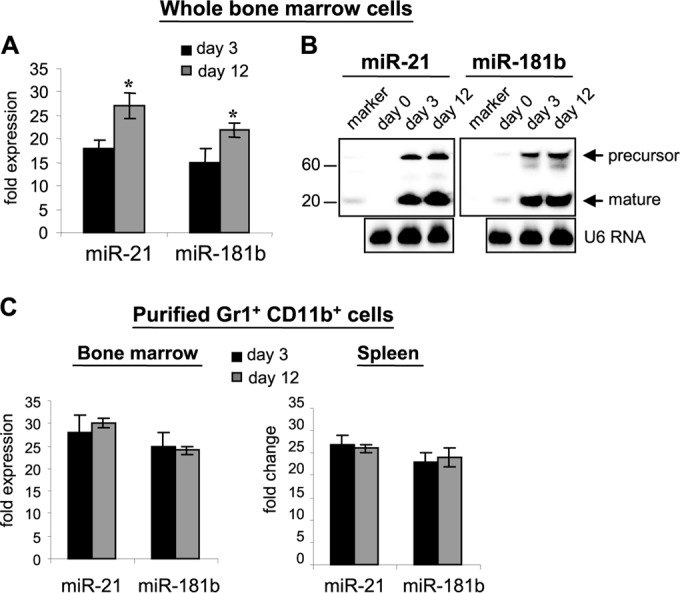
miR-21 and miR-181b expression is increased during sepsis. Sepsis was induced by CLP. Whole bone marrow cells were pooled (n = 5 per group) from septic mice that were moribund and were sacrificed at day 3 (representing early sepsis) and day 12 (representing late sepsis). A group of naive mice (day 0) served as a control. (A) miRNA-enriched RNA was isolated from whole bone marrow cells and analyzed by real-time PCR using miR-21- and miR-181b-specific assay primer sets. Values were normalized to U6 RNA as an internal control. The data are expressed as means ± SD of three assays (*, P < 0.05) and are presented as fold increases relative to naive mice (day 0), set at 1-fold. (B) The miRNA-enriched RNA was analyzed for miR-21 and miR-181b expression using a Northern blot assay kit. The results are representative of three assays. (C) miRNA-enriched RNA was isolated from Gr1+ CD11b+ cells that were purified and pooled (n = 6 mice per group) from bone marrow cells from normal and septic mice and then analyzed as for panel A.
To determine whether miR-21 and miR-181b are linked to MDSC expansion during sepsis, we measured their levels in Gr1+ CD11b+ cells purified from the bone marrow of septic mice. As shown in Fig. 1C, the miR-21 level was increased 28-fold in early sepsis (compared to naive/day 0, set at 1-fold), whereas the miR-181b level was increased 26-fold, and these increases were maintained at similar levels in late sepsis. In contrast to their levels in whole bone marrow cells (Fig. 1A and B), there were no significant changes in miR-21 and miR-181b levels in purified Gr1+ CD11b+ cells as mice entered the late sepsis phase. Such a difference may be due to the presence of fewer Gr1+ CD11b+ cells within the whole bone marrow cell population in early sepsis than in late sepsis, which was eliminated when we used equal numbers of purified Gr1+ CD11b+ cells. We also observed closely similar patterns of increases in the levels of miR-21 and miR-181b in Gr1+ CD11b+ cells purified from the spleen (Fig. 1C).
In vivo blockade of miR-21 and miR-181b by antagomiRs improves late-sepsis survival.
Because miR-21 and miR-181b were the only miRNAs that were induced and maintained throughout the sepsis response, we asked whether reducing their levels can impact the sepsis outcome. We used chemically modified miRNA inhibitors known as antagomiRs against miR-21 and miR-181b. AntagomiRs are biostable (in vivo) and can inhibit the target miRNA for up to 2 weeks (29) by inducing its degradation, unlike LAN-based anti-miRNAs, which inhibit their target by sequestration (31). miR-21 and miR-181b antagomiRs or their mutants were injected (i.p) via the tail vein at doses of 80 mg/kg body weight. To allow the initiation of sepsis first, antagomiRs were administered 48 h after CLP, and survival was monitored for 28 days.
Administration of miR-21 antagomiRs alone had no effect, whereas miR-181b antagomiRs alone improved survival by 5% (data not shown). Importantly, when mice received a mixture of both antagomiRs, survival was improved by 74% compared with mutant/control antagomiRs (Fig. 2A). The effect of antagomiRs on mortality was not observed until day 6 after CLP (i.e., 4 days after antagomiR injection). It should be noted that our CLP model is manipulated to develop into early and late sepsis phases. We define late sepsis in mice as the period after day 5 (8). We also measured peritoneal bacteria (which are associated with a high mortality rate) in mice that survived or died during the late sepsis phase. Peritoneal bacteria were diminished in mice that received a mixture of miR-21 and miR-181b antagomiRs but remained significantly high in mice that received mutant antagomiRs (Fig. 2B). These results suggest that miR-21 and miR-181b synergize to promote sepsis immunosuppression.
FIG 2.
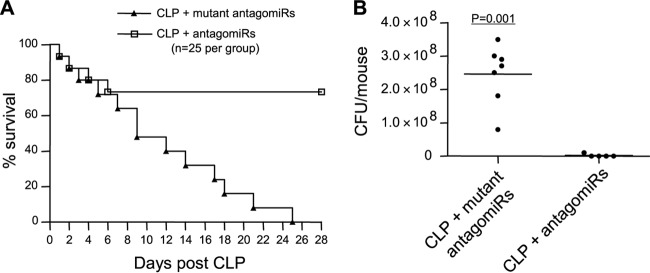
In vivo blockade of miR-21 and miR-181b by antagomiRs improves late-sepsis survival. (A) Kaplan-Meier survival curve. Sepsis was induced by CLP. After 48 h (to allow sepsis initiation), mice were injected via the tail vein with a mixture of miR-21 and miR-181b antagomiRs or their mutants at doses of 80 mg/kg in 100 μl saline, and survival was reported for 28 days. (B) Diminished peritoneal bacteria in late-septic mice that received antagomiRs. Mice that were moribund at days 8 to 21 after CLP (i.e., during late sepsis) were subjected to peritoneal lavage immediately after killing. A corresponding number of surviving, healthy-appearing mice (the mutant antgomiR group) were sacrificed and analyzed at the same time and are reported as “surviving.” The lavage fluid was cultured on 5% sheep blood agar plates, and the CFU were determined 24 h later.
Blockade of miR-21 and miR-181b reduces the numbers of Gr1+ CD11b+ MDSCs by promoting their differentiation and maturation.
We set out to determine if the increases in bacterial clearance and survival observed during late sepsis are due to changes in MDSC numbers. Because we observed significant increases in the number of bone marrow MDSCs only after day 6 of CLP (i.e., during late sepsis) and because late-sepsis MDSCs are immunosuppressive (8), we focused our analyses on the late sepsis phase (the period after day 5). Flow cytometry analysis revealed that the numbers of Gr+ CD11b+ cells in the bone marrow samples were significantly high (89%) in the control mice (the mutant antagomiR group) that were moribund and were sacrificed at days 10 to 21. This number was decreased to 20% in mice that received miR-21 and miR-181b antagomiRs, survived, and were sampled during the same period (Fig. 3A and B).
FIG 3.

miR-21 and miR-181b antagomiRs reduce the numbers of Gr1+ CD11b+ cells in late sepsis. Bone marrow cells were harvested from moribund (the antagomiR group) and surviving (the mutant antagomiR group) mice that were sacrificed at days 10 to 21 after CLP (i.e., during late sepsis). (A) Example of flow cytometry of cells gated on Gr1+ CD11b+ staining. (B) Quantitative analysis of total Gr1+ CD11b+ cells. The data are expressed as means and SD of 5 mice per group (*, P < 0.05). (C) Percentages of CD31+ cells within the total Gr1+ CD11b+ cell population. The data are expressed as means and SD of 7 mice per group (*, P < 0.05). (D) Differentiation of Gr1+ CD11b+ cells. Gr1+ CD11b+ cells were differentiated for 6 days with M-CSF and rIL-4 (10 ng/ml/each). The percentages of F4/80+ CD11b+ (macrophage) and CD11c+ MHC-II+ (dendritic) cells were determined by flow cytometry. The data are expressed as means and SD of 8 mice per group (*, P < 0.05).
We have previously shown that most late-septic Gr1+ CD11b+ cells coexpress the early and more immature myeloid cell marker CD31 (8), which is lost with further myeloid differentiation and maturation (32). Flow cytometry analysis revealed that the numbers of Gr1+ CD11b+ cells that coexpressed CD31 marker represented 75% of the total Gr1+ CD11b+ cell population in the bone marrow of the septic mice that received mutant antagomiRs and were moribund (Fig. 3C). Interestingly, the numbers in this CD31+ subset declined to 16% in the mice that received miR-21 and miR-181b antagomiRs and survived. We also examined whether this decline in the CD31+ subset within the Gr1+ CD11b+ population was associated with changes in the Gr1+ CD11b+ differentiation and maturation potential. Purified Gr1+ CD11b+ cells were incubated with M-CSF plus rIL-4 for 6 days, and the numbers of differentiated F4/80+ CD11b+ (macrophage) and CD11c+ MHC-II+ (dendritic) cells were determined by flow cytometry. The results showed that the level of differentiation of Gr1+ CD11b+ cells from mice that received antagomiRs into macrophages and dendritic cells was significantly higher than that of cells from the mice that received mutant antagomiRs (Fig. 3D). Together, these results demonstrate that miR-21 and miR-181b generate and expand MDSCs in the bone marrow during sepsis and suggest that their blockade reduces MDSCs by allowing the differentiation and maturation of the immature Gr1+ CD11b+ myeloid progenitors.
Mice that survive late sepsis due to blockade of miR-21 and miR-181b are immunoreactive.
Late-septic mice produce less proinflammatory but more anti-inflammatory/immunosuppressive cytokines despite the presence of an ongoing infection, an indication of immunosuppression (8). We hypothesized that the mice that survived late sepsis due to miR-21 and miR-181b blockade should be immunoreactive, since they cleared peritoneal bacteria (Fig. 2). If this was true, then they should respond to a challenge with the Gram-negative bacterial LPS. Mice that survived (the antagomiR group) or were moribund (the mutant antagomiR group) were pooled at days 12 to 21 after CLP (i.e., during late sepsis) and then challenged (i.p) with a sublethal dose (10 μg) of LPS. A group of sham-operated mice also received LPS and served as a positive control. After 6 h, the systemic response to LPS challenge was determined by measuring the circulating levels of the proinflammatory (TNF-α and IL-6) and anti-inflammatory/immunosuppressive (IL-10 and TGF-β) cytokines. As shown in Fig. 4, the antagomiR mouse group produced significantly larger amounts of TNF-α and IL-6 but significantly smaller amounts of IL-10 and TGF-β than the mutant antagomiR group. In addition, the cytokine profiles in the surviving antagomiR mouse group were similar to those in sham-operated/control mice, suggesting that they had immunoreactive innate immune systems, whereas mice without miR-21 and miR-181b inhibition remained immunosuppressed.
FIG 4.

miR-21 and miR-181b antagomiRs restore responsiveness of late-septic mice to bacterial LPS. At days 12 to 21 after CLP, mice that were moribund (the antagomiR group) and a corresponding number of surviving, healthy-appearing mice (the mutant antagomiR group) were challenged (i.p.) with 10 μg of bacterial LPS. A group of sham-operated mice also received LPS and served as a positive control. After 6 h, blood was collected and sera were obtained and used to determine the circulating levels of pro- and anti-inflammatory cytokines by ELISA. The data are expressed as means and SD of 8 mice per group. *, P < 0.05 compared with CLP plus mutant antagomiRs; **, P < 0.05 compared with sham operated.
The transcription factor NFI-A is increased in sepsis and is a downstream effector of miR-21 and miR-181b, and its knockdown in vitro promotes Gr1+ CD11b+ cell differentiation.
To gain further insights into the mechanism by which miR-21 and miR-181b generate MDSCs, we profiled the expression of proteins implicated in myeloid cell differentiation and maturation and those known to be a target for regulation by miRNAs (23, 33, 34). These proteins included transcription factors belonging to the CCAAT/enhancer-binding protein (C/EBP) family, the NFI family, and SP1. Western blot analysis of bone marrow cells showed that only the expression of C/EBPβ and NFI-A proteins was markedly induced during early sepsis and further increased and sustained in late sepsis (Fig. 5A).
FIG 5.
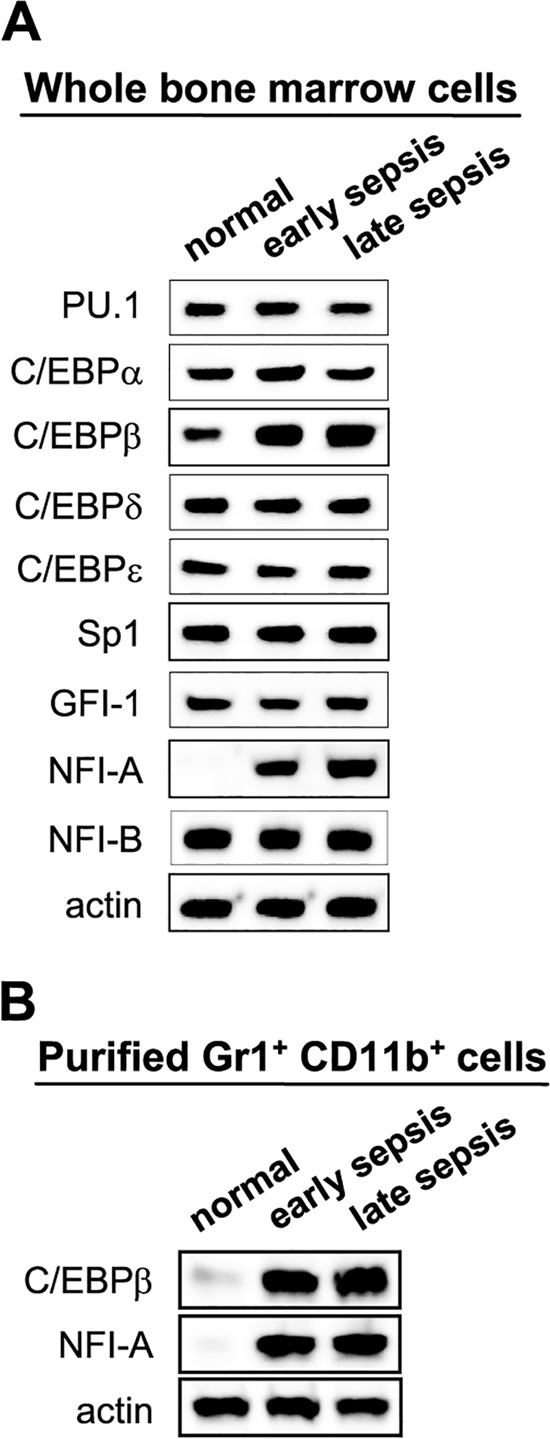
Expression of transcription factors C/EBPβ and NFI-A is upregulated during sepsis. Shown is Western blot analysis of proteins known to affect myeloid cell differentiation and/or maturation. (A) Whole bone marrow cells were pooled (n = 5 per group) from normal and septic mice that were moribund and sacrificed at days 3 and 4 (representing early sepsis) and days 10 to 14 (representing late sepsis). (B) Purified bone marrow cells Gr1+ CD11b+ cells from 5 healthy, 7 early-septic, and 5 late-septic mice were pooled. Cell extracts were prepared and probed for the indicated proteins. Only a sample of β-actin is shown.
The induction of C/EBPβ and NFI-A proteins in the bone marrow compartment during sepsis was confirmed using purified Gr1+ CD11b+ cells. The results (Fig. 5B) suggest that, because of the marked increases in their expression, one or both proteins may play a role in Gr1+ CD11b+ MDSC expansion in sepsis. Next, we investigated whether these two transcription factors are downstream targets of miR-21 and/or miR-181b. Gr1+ CD11b+ cells were purified from the bone marrow of late-septic mice that received antagomiRs or their mutants and then analyzed by Western blotting to measure C/EBPβ and NFI-A proteins. Figure 6A shows that miR-21 and miR-181b antagomiRs markedly reduced NFI-A protein levels but did not affect C/EBPβ, suggesting that NFI-A is a downstream target for induction by miR-21 and miR-181. It should be mentioned that inhibition of either miRNA alone had no noticeable effect (data not shown).
FIG 6.
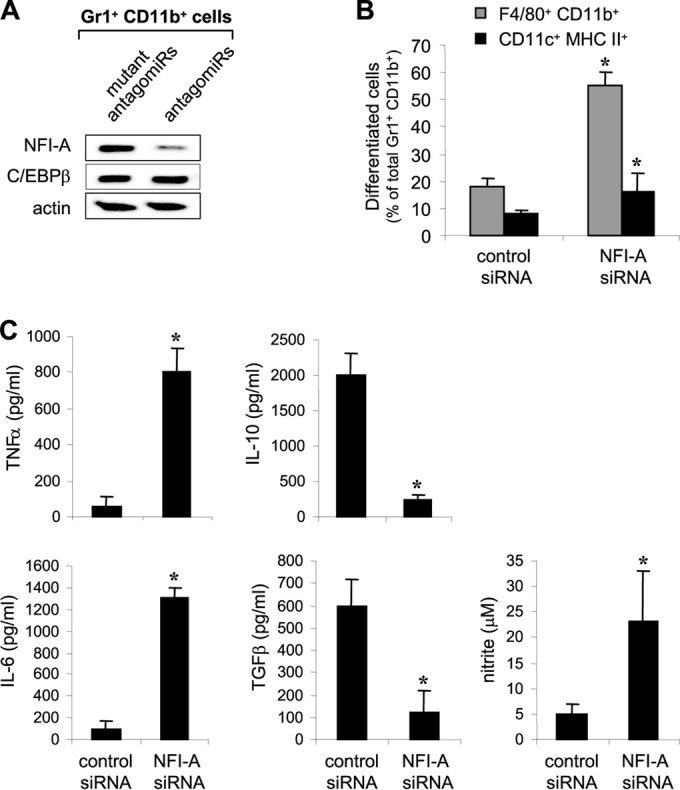
NFI-A is a downstream target of miR-21 and miR-181b, and its knockdown in Gr1+ CD11b+ cells from late-septic mice increases their differentiation. (A) AntagomiRs inhibit NFI-A expression in bone marrow of septic mice. Sepsis was induced by CLP. After 48 h (to allow sepsis initiation), mice were injected via the tail vein with a mixture of miR-21 and miR-181b antagomiRs or their mutants at doses of 80 mg/kg in 100 μl saline. Bone marrow cells were harvested from the moribund mice that were sacrificed at days 8 to 21 after CLP (i.e., during late sepsis). Gr1+ CD11b+ cells from 6 mice were then purified and pooled. Proteins were isolated and analyzed by Western blotting. (B) Knockdown of NFI-A ex vivo in late-septic Gr1+ CD11b+ cells increases their differentiation potential. Gr1+ CD11b+ cells were purified from moribund mice (without antagomiR injection) that were sacrificed at days 8 to 21 and then transfected with NFI-A-specific or scrambled siRNA. After 24 h, the cells were differentiated for 6 days with M-CSF and rIL-4 (10 ng/ml each) and analyzed by flow cytometry for the expression of macrophage (F4/80 and CD11b) and dendritic cell (CD11c MHC-II) markers. The data are expressed as means and SD of 7 mice (*, P < 0.05). (C) Knockdown of NFI-A ex vivo in late-septic Gr1+ CD11b+ cells restores their reactivity to bacterial LPS. Gr1+ CD11b+ cells were purified and transfected as for panel B. After 24 h, the cells were differentiated for 6 days into macrophages with 10 ng/ml of M-CSF. The cells were then washed and stimulated for 6 h with 1 μg/ml of LPS. The culture supernatants were collected and analyzed by ELISA for cytokine production. Production of NO was measured by a nitrite assay using the same supernatants. The data are expressed as means and SD of 8 mice (*, P < 0.05).
Because antagomiR administration also enhanced Gr1+ CD11b+ cell differentiation and maturation (Fig. 3), we reasoned that the increase in NFI-A protein during sepsis may prevent myeloid cell differentiation and maturation and thus expands the immature Gr1+ CD11b+ cell population. To test this possibility, we knocked down NFI-A in Gr1+ CD11b+ cells purified from the late-septic mice (without antagomiRs) and then differentiated them with M-CSF plus rIL-4 for 6 days. Flow cytometry showed that Gr1+ CD11b+ cells with NFI-A knockdown differentiated readily and gave rise to significantly more macrophages and dendritic cells than the control knockdown cells (Fig. 6B), suggesting that NFI-A upregulation during sepsis is primarily responsible for MDSC expansion. Finally, we tested the immunocompetence of these differentiated cells by measuring their cytokine production after stimulation with LPS for 6 h. Differentiated cells lacking NFI-A produced significantly larger amounts of the proinflammatory cytokines TNF-α and IL-6 than the control knockdown cells, which retained an immunosuppressive phenotype, producing significantly larger amounts of IL-10 and TGF-β. These cells also produced less NO than NFI-A knockdown cells (Fig. 6C). Together, these results demonstrate that NFI-A is induced downstream due to upregulation of miR-21 and miR-181b and suggest that NFI-A plays a main role in MDSC expansion in sepsis by blocking Gr1+ CD11b+ cell differentiation and maturation.
DISCUSSION
Although the immunosuppressive role of MDSCs in sepsis pathogenesis is not completely understood, it is evident that their generation and immature phenotypes underscore dysregulation of the myeloid cell differentiation and maturation programs (11, 35). The differentiation of innate-immunity cells from myeloid progenitors is controlled by sequential changes in the expression of myeloid-specific transcription factors, which often are targets of regulation by miRNAs (23, 36, 37). Our recent findings showing that MDSCs expand during polymicrobial sepsis in mice and that they play a role in late-sepsis immunosuppression (8) encouraged us to investigate the path leading to their generation. In the current study, we uncovered a molecular path that leads to MDSC expansion in the bone marrow during sepsis. We show that miR-21 and miR-181b are induced by the sepsis inflammatory reaction to drive MDSC expansion and that they upregulate the expression of NFI-A, a transcription factor implicated in myeloid differentiation. Importantly, we show that this negative path can be reversed in vivo by inhibiting miR-21 and miR-181b, which reduced MDSCs and improved late-sepsis survival, thus providing insight into how changes in miRNA expression can impact sepsis pathogenesis.
miRNAs are short (20- to 23-nucleotide) noncoding, single-stranded RNA molecules that are transcribed from introns of protein-coding genes or exons of unique noncoding genes (38, 39). miRNAs bind to complementary sequences within the 3′ untranslated region (UTR) of their target mRNA as part of a protein complex known as the RNA-induced silencing complex (RISC), which induces degradation and/or inhibits translation of the target mRNA, resulting in downregulation of the target protein (38, 40). Several miRNAs have been implicated in the regulation of innate and adaptive immune systems under normal and pathological conditions by targeting myeloid cell proliferation, differentiation, and maturation (reviewed in references 23 and 37), but none has been linked to MDSC generation in sepsis. In most cases, miRNA expression is regulated by signals and transcription factors induced by inflammatory changes (39). Our study demonstrated that miR-21 and miR-181b are induced in Gr1+ CD11b+ cells during sepsis, and their in vivo inhibition by antagomiRs significantly improved late-sepsis survival. Importantly, we showed that simultaneous inhibition of both miRNAs eliminated Gr1+ CD11b+ MDSCs and enhanced peritoneal bacterial clearance, indicating restoration of the innate immune response.
We previously showed that the majority of Gr1+ CD11b+ MDSCs detected in the bone marrow of late-septic mice were immature myeloid progenitors that also coexpressed the CD31+ marker and were unable to differentiate ex vivo into macrophages and dendritic cells (8). We attributed the expansion in Gr1+ CD11b+ MDSCs to the increases in this CD31+ cell subset (8). The current study showed that the CD31+ subset within the total Gr1+ CD11b+ cell population in the bone marrow of late-septic mice was reduced from 75% to 16% after miR-21 and miR-181b inhibition (Fig. 3). In particular, the decline in this CD31+ progenitor subset was associated with increased ability of the remaining Gr1+ CD11b+ cells to differentiate ex vivo into macrophages and dendritic cells. These results clearly demonstrate that miR-21 and miR-181b disrupt myeloid progenitor differentiation and maturation and thus suggest that they underlie the Gr1+ CD11b+ expansion we observe during late sepsis.
The finding that NFI-A expression was induced in Gr1+ CD11b+ cells during sepsis with a pattern similar to that of miR-21 and miR-181b is particularly important. NFI-A maintains the myeloid progenitors in an undifferentiated state, and its downregulation is required for normal monocyte and granulocyte differentiation and maturation (33, 34, 36). Recent studies have reported that ectopic expression of NFI-A in human myeloid progenitors counteracts their monocytic differentiation (34), whereas silencing its expression promotes their granulocytic differentiation (41). Interestingly, we showed that in vivo inhibition of miR-21 and miR-181b reduced NFI-A expression and Gr1+ CD11b+ cell numbers and that the Gr1+ CD11b+ cells with NFI-A knockdown by siRNA were able to differentiate readily into macrophages and dendritic cells (Fig. 6). While these findings clearly demonstrate that NFI-A is a downstream effector of miR-21 and miR-181b upregulation, they do not explain how the blockade of miR-21 and miR-181b could inhibit NFI-A expression. Bioinformatic analysis revealed that both miR-21 and miR-181 have no complementary sequences (i.e., binding sites) within the 3′ UTR of NFI-A, making it unlikely that they directly induce NFI-A expression. However, our results indicate that both miRNAs converge on NFI-A, as its expression was reduced only by the concurrent inhibition of both miRNAs. In addition, the notion that the vast majority of miRNAs repress rather than activate expression of their targets suggests that miR-21 and miR-181b induce NFI-A expression during sepsis indirectly. In this regard, a recent study reported that the transcription factor PU.1, which is also implicated in myeloid differentiation, induces miR-424, which in turn inhibits NFI-A expression in myeloid cells (34). However, in the context of sepsis, we did not observe changes in PU.1 protein (data not shown). It has also been reported that miR-223, whose expression is increased with myeloid differentiation and maturation, represses NFI-A in human myeloid progenitors, allowing their differentiation (33, 41). It is possible that miR-21 and miR-181b can induce NFI-A during sepsis by targeting other factors, like miR-223. In addition, it is unclear from our results how NFI-A blocks Gr1+ CD11b+ progenitor differentiation in sepsis. NFI-A has been shown to inhibit the cyclin-dependent kinase inhibitor p21 (42). It has been suggested that absence of p21 in conjunction with p27 (another cyclin-dependent kinase inhibitor) increases hematopoietic stem cell and myeloid progenitor cell proliferation and numbers in response to growth in cytokine stimulation (43). Because these proteins inhibit cyclin-dependent kinases, they may ultimately activate NF-κB. Persistent activation of NF-κB has been associated with myeloproliferation disorder and accumulation of the Gr1+ CD11b+ myeloid progenitors in mice (44). Although the exact mechanism by which NFI-A generates MDSCs in sepsis remains unknown, our study places NFI-A as an important target of miR-21 and miR-181b in the molecular path that generates Gr1+ CD11b+ MDSCs and subsequent immunosuppression in sepsis. In support of this, the in vivo inhibition of miR-21 and miR-181b, similarly to the in vitro knockdown of NFI-A, enhanced Gr1+ CD11b+ progenitor differentiation and maturation.
Our previous studies (8) have shown that MDSCs are potently immunosuppressive, as they produced high levels of the immunosuppressive mediators IL-10, TGF-β, and arginase when adoptively transferred into mice undergoing the early sepsis hyperinflammatory response. This finding is complemented by our results showing that late-septic mice did not respond to LPS challenge and instead produced higher levels of IL-10 and TGF-β (Fig. 4). Of note, the miR-21 and miR-181b blockade restored the immunoreactivity of these mice to the LPS challenge, as demonstrated by the significant increases in the levels of circulating TNF-α and IL-6, a typical inflammatory response that is observed after infection or bacterial challenge (24, 45, 46). These results indicate that eliminating MDSCs is necessary for restoring immune cell functions and responses during late sepsis. Consistent with this, our in vitro experiments showed that Gr1+ CD11b+ cells with NFI-A knockdown not only differentiated into mature cells, but also responded to LPS stimulation by producing more TNF-α and IL-6 and less IL-10 and TGF-β, similar to the pattern seen in vivo after the miRNA blockade, suggesting they were immunoreactive. IL-10 and TGF-β suppress many innate- and adaptive-immunity cell functions and can activate MDSCs themselves (13, 14, 47), thus providing an autocrine loop for amplifying the immunosuppression we observe in late sepsis.
A few other studies have also shown that MDSCs expand during polymicrobial sepsis (21, 22, 48) and that their depletion with anti-Gr1 antibody (although nonspecific) improves the immune response (21, 22). In addition, Sander et al. (22) and Derive et al. (48) reported that adoptive transfer of late-septic MDSCs into mice undergoing early sepsis improved survival and thus described MDSCs as “immunoprotective.” This description, however, may be confusing, given that early sepsis is a hyperinflammatory response and is expected to be attenuated by these “immunosuppressive” MDSCs. Also, in these two studies, the mice were followed for only 5 to 8 days after the transfer, i.e., around the time the late septic phase ensues. Thus, it is reasonable to describe MDSCs as immunoprotective only in the context of early sepsis. We also observe this immunoprotective effect on early sepsis, but because our model also develops into a late septic phase, we did not observe any protective effect from the MDSC transfer once the mice entered the late immunosuppressive state (8).
Finally, while our study clearly implicates miR-21 and miR-181b in sepsis immunosuppression, an important question remains: what signal/factor drives their upregulation during sepsis? It has been suggested that mediators, such as IL-6, IL-10, VEGF, M-CSF, and granulocyte-macrophage colony-stimulating factor (GM-CSF), some of which are produced in the tumor or inflammatory microenvironment, may generate and expand MDSCs (14, 16). IL-6 and IL-10 can activate STAT3, which has been linked to dysregulation of myeloid cell differentiation and increases in immature progenitors (49, 50). Indeed, we observed an increase in STAT3 activation/phosphorylation during sepsis (data not shown). Also, IL-6 increases in early sepsis while IL-10 increases in late sepsis. Whether IL-6 and IL-10 alternately activate STAT3 and whether STAT3 induces miR-21 and miR-181b during sepsis warrant further investigation.
In summary, we have uncovered the molecular path that generates Gr1+ CD11b+ MDSCs during sepsis. This anti-inflammatory path involves miR-21 and miR-181b, which synergize to activate NFI-A expression downstream. As a negative factor for myeloid cell differentiation, NFI-A increases the immature-progenitor pool, thus expanding the Gr1+ CD11b+ MDSC population. Because it is becoming evident that most sepsis mortality is due to late-sepsis immunosuppression, eliminating MDSCs may hold promise for restoring the immune response during sepsis. Targeting MDSCs has recently been shown to reduce immune suppression and improve antitumor immunity in tumor-bearing animals. Our results suggest that inhibiting miR-21 and miR-181b eliminates MDSCs and their immunosuppressive effects. Thus, this study provides an attractive therapeutic approach for the treatment of sepsis immunosuppression.
ACKNOWLEDGMENT
This work was supported by National Institutes of Health Grant R01 GM103887 (to M.E.).
Footnotes
Published ahead of print 30 June 2014
REFERENCES
- 1.Hotchkiss RS, Monneret G, Payen D. 2013. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 13:260–268. 10.1016/S1473-3099(13)70001-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCall CE, Yoza B, Liu T, El Gazzar M. 2010. Gene-specific epigenetic regulation in serious infections with systemic inflammation. J. Innate Immun. 2:395–405. 10.1159/000314077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Remick DG. 2007. Pathophysiology of sepsis. Am. J. Pathol. 170:1435–1444. 10.2353/ajpath.2007.060872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shelley O, Murphy T, Paterson H, Mannick JA, Lederer JA. 2003. Interaction between the innate and adaptive immune systems is required to survive sepsis and control inflammation after injury. Shock 20:123–129. 10.1097/01.shk.0000079426.52617.00 [DOI] [PubMed] [Google Scholar]
- 5.Shubin NJ, Monaghan SF, Ayala A. 2011. Anti-inflammatory mechanisms of sepsis. Contrib. Microbiol. 17:108–124. 10.1159/000324024 [DOI] [PubMed] [Google Scholar]
- 6.Perl M, Chung CS, Garber M, Huang X, Ayala A. 2006. Contribution of anti-inflammatory/immune suppressive processes to the pathology of sepsis. Front. Biosci. 11:272–299. 10.2741/1797 [DOI] [PubMed] [Google Scholar]
- 7.Torgersen C, Moser P, Luckner G, Mayr V, Jochberger S, Hasibeder WR, Dunser MW. 2009. Macroscopic postmortem findings in 235 surgical intensive care patients with sepsis. Anesth. Analg. 108:1841–1847. 10.1213/ane.0b013e318195e11d [DOI] [PubMed] [Google Scholar]
- 8.Brudecki L, Ferguson DA, McCall CE, El Gazzar M. 2012. Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect. Immun. 80:2026–2034. 10.1128/IAI.00239-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen J, Opal S, Calandra T. 2012. Sepsis studies need new direction. Lancet Infect. Dis. 12:503–505. 10.1016/S1473-3099(12)70136-6 [DOI] [PubMed] [Google Scholar]
- 10.Wenzel RP, Edmond MB. 2012. Septic shock-evaluating another failed treatment. N. Engl. J. Med. 366:2122–2124. 10.1056/NEJMe1203412 [DOI] [PubMed] [Google Scholar]
- 11.Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moreno C, Scumpia PO, Laface DM, Heyworth PG, Efron PA, Moldawer LL. 2011. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol. Med. 17:281–292. 10.1007/s00894-010-0723-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gabrilovich DI, Nagaraj S. 2009. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 9:162–174. 10.1038/nri2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ostrand-Rosenberg S, Sinha P. 2009. Myeloid-derived suppressor cells: linking inflammation and cancer. J. Immunol. 182:4499–4506. 10.4049/jimmunol.0802740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kong YY, Fuchsberger M, Xiang SD, Apostolopoulos V, Plebanski M. 2013. Myeloid derived suppressor cells and their role in diseases. Curr. Med. Chem. 20:1437–1444. 10.2174/0929867311320110006 [DOI] [PubMed] [Google Scholar]
- 15.Lees JR, Azimzadeh AM, Bromberg JS. 2011. Myeloid derived suppressor cells in transplantation. Curr. Opin. Immunol. 23:692–697. 10.1016/j.coi.2011.07.004 [DOI] [PubMed] [Google Scholar]
- 16.Condamine T, Gabrilovich DI. 2011. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 32:19–25. 10.1016/j.it.2010.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. 2001. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J. Immunol. 166:678–689. 10.4049/jimmunol.166.1.678 [DOI] [PubMed] [Google Scholar]
- 18.Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, McDermott D, Quiceno D, Youmans A, O'Neill A, Mier J, Ochoa AC. 2005. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 65:3044–30481 [DOI] [PubMed] [Google Scholar]
- 19.Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, Gabrilovich D. 2003. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 63:4441–4449 [PubMed] [Google Scholar]
- 20.Martire-Greco D, Landoni VI, Chiarella P, Rodriguez-Rodrigues N, Schierloh P, Rearte B, Isturiz MA, Fernandez GC. 2014. All-trans-retinoic acid improves immunocompetence in a murine model of lipopolysaccharide-induced immunosuppression. Clin. Sci. 126:355–365. 10.1042/CS20130236 [DOI] [PubMed] [Google Scholar]
- 21.Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, O'Malley KA, Wynn JL, Antonenko S, Al-Quran SZ, Swan R, Chung CS, Atkinson MA, Ramphal R, Gabrilovich DI, Reeves WH, Ayala A, Phillips J, Laface D, Heyworth PG, Clare-Salzler M, Moldawer LL. 2007. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J. Exp. Med. 204:1463–1474. 10.1084/jem.20062602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sander LE, Sackett SD, Dierssen U, Beraza N, Linke RP, Muller M, Blander JM, Tacke F, Trautwein C. 2010. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J. Exp. Med. 207:1453–1464. 10.1084/jem.20091474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Connell RM, Zhao JL, Rao DS. 2011. MicroRNA function in myeloid biology. Blood 118:2960–2969. 10.1182/blood-2011-03-291971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belikoff B, Hatfield S, Sitkovsky M, Remick DG. 2011. Adenosine negative feedback on A2A adenosine receptors mediates hyporesponsiveness in chronically septic mice. Shock 35:382–387. 10.1097/SHK.0b013e3182085f12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osuchowski MF, Welch K, Siddiqui J, Remick DG. 2006. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J. Immunol. 177:1967–1974. 10.4049/jimmunol.177.3.1967 [DOI] [PubMed] [Google Scholar]
- 26.Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA. 2002. Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock 17:463–467. 10.1097/00024382-200206000-00004 [DOI] [PubMed] [Google Scholar]
- 27.Brudecki L, Ferguson DA, Yin D, Lesage GD, McCall CE, El Gazzar M. 2012. Hematopoietic stem-progenitor cells restore immunoreactivity and improve survival in late sepsis. Infect. Immun. 80:602–611. 10.1128/IAI.05480-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mazuski JE, Sawyer RG, Nathens AB, DiPiro JT, Schein M, Kudsk KA, Yowler C. 2002. The Surgical Infection Society guidelines on antimicrobial therapy for intra-abdominal infections: an executive summary. Surg. Infect. 3:161–173. 10.1089/109629602761624171 [DOI] [PubMed] [Google Scholar]
- 29.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. 2005. Silencing of microRNAs in vivo with ‘antagomirs'. Nature 438:685–689. 10.1038/nature04303 [DOI] [PubMed] [Google Scholar]
- 30.Roy S, Sen CK. 2011. MiRNA in innate immune responses: novel players in wound inflammation. Physiol. Genomics. 43:557–565. 10.1152/physiolgenomics.00160.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stenvang J, Lindow M, Kauppinen S. 2008. Targeting of microRNAs for therapeutics. Biochem. Soc. Trans. 36:1197–1200. 10.1042/BST0361197 [DOI] [PubMed] [Google Scholar]
- 32.Ling V, Luxenberg D, Wang J, Nickbarg E, Leenen PJ, Neben S, Kobayashi M. 1997. Structural identification of the hematopoietic progenitor antigen ER-MP12 as the vascular endothelial adhesion molecule PECAM-1 (CD31). Eur. J. Immunol. 27:509–514. 10.1002/eji.1830270223 [DOI] [PubMed] [Google Scholar]
- 33.Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, Bozzoni I. 2005. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 123:819–831. 10.1016/j.cell.2005.09.023 [DOI] [PubMed] [Google Scholar]
- 34.Rosa A, Ballarino M, Sorrentino A, Sthandier O, De Angelis FG, Marchioni M, Masella B, Guarini A, Fatica A, Peschle C, Bozzoni I. 2007. The interplay between the master transcription factor PU.1 and miR-424 regulates human monocyte/macrophage differentiation. Proc. Natl. Acad. Sci. U. S. A. 104:19849–19854. 10.1073/pnas.0706963104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cuenca AG, Moldawer LL. 2012. Myeloid-derived suppressor cells in sepsis: friend or foe? Intensive Care Med. 38:928–930. 10.1007/s00134-012-2575-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laslo P, Pongubala JM, Lancki DW, Singh H. 2008. Gene regulatory networks directing myeloid and lymphoid cell fates within the immune system. Semin. Immunol. 20:228–235. 10.1016/j.smim.2008.08.003 [DOI] [PubMed] [Google Scholar]
- 37.Tsitsiou E, Lindsay MA. 2009. microRNAs and the immune response. Curr. Opin. Pharmacol. 9:514–520. 10.1016/j.coph.2009.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baltimore D, Boldin MP, O'Connell RM, Rao DS, Taganov KD. 2008. MicroRNAs: new regulators of immune cell development and function. Nat. Immunol. 9:839–845. 10.1038/ni.f.209 [DOI] [PubMed] [Google Scholar]
- 39.O'Connell RM, Rao DS, Chaudhuri AA, Baltimore D. 2010. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 10:111–122. 10.1038/nri2708 [DOI] [PubMed] [Google Scholar]
- 40.Kim VN, Han J, Siomi MC. 2009. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell. Biol. 10:126–139. 10.1038/nrm2632 [DOI] [PubMed] [Google Scholar]
- 41.Zardo G, Ciolfi A, Vian L, Starnes LM, Billi M, Racanicchi S, Maresca C, Fazi F, Travaglini L, Noguera N, Mancini M, Nanni M, Cimino G, Lo-Coco F, Grignani F, Nervi C. 2012. Polycombs and microRNA-223 regulate human granulopoiesis by transcriptional control of target gene expression. Blood 119:4034–4046. 10.1182/blood-2011-08-371344 [DOI] [PubMed] [Google Scholar]
- 42.Ouellet S, Vigneault F, Lessard M, Leclerc S, Drouin R, Guerin SL. 2006. Transcriptional regulation of the cyclin-dependent kinase inhibitor 1A (p21) gene by NFI in proliferating human cells. Nucleic Acids Res. 34:6472–6487. 10.1093/nar/gkl861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Broxmeyer HE, Franklin DS, Cooper S, Hangoc G, Mantel C. 2012. Cyclin dependent kinase inhibitors differentially modulate synergistic cytokine responsiveness of hematopoietic progenitor cells. Stem Cells Dev. 21:1597–1603. 10.1089/scd.2011.0476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao JL, Rao DS, Boldin MP, Taganov KD, O'Connell RM, Baltimore D. 2011. NF-kappaB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc. Natl. Acad. Sci. U. S. A. 108:9184–9189. 10.1073/pnas.1105398108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Belikoff BG, Hatfield S, Georgiev P, Ohta A, Lukashev D, Buras JA, Remick DG, Sitkovsky M. 2011. A2B adenosine receptor blockade enhances macrophage-mediated bacterial phagocytosis and improves polymicrobial sepsis survival in mice. J. Immunol. 186:2444–2453. 10.4049/jimmunol.1001567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muenzer JT, Davis CG, Chang K, Schmidt RE, Dunne WM, Coopersmith CM, Hotchkiss RS. 2010. Characterization and modulation of the immunosuppressive phase of sepsis. Infect. Immun. 78:1582–1592. 10.1128/IAI.01213-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y, Lai L, Chen Q, Song Y, Xu S, Ma F, Wang X, Wang J, Yu H, Cao X, Wang Q. 2012. MicroRNA-494 is required for the accumulation and functions of tumor-expanded myeloid-derived suppressor cells via targeting of PTEN. J. Immunol. 188:5500–5510. 10.4049/jimmunol.1103505 [DOI] [PubMed] [Google Scholar]
- 48.Derive M, Bouazza Y, Alauzet C, Gibot S. 2012. Myeloid-derived suppressor cells control microbial sepsis. Intensive Care Med. 38:1040–1049. 10.1007/s00134-012-2574-4 [DOI] [PubMed] [Google Scholar]
- 49.Kohanbash G, Okada H. 2012. MicroRNAs and STAT interplay. Semin. Cancer Biol. 22:70–75. 10.1016/j.semcancer.2011.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang H, Nguyen-Jackson H, Panopoulos AD, Li HS, Murray PJ, Watowich SS. 2010. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood 116:2462–2471. 10.1182/blood-2009-12-259630 [DOI] [PMC free article] [PubMed] [Google Scholar]