

Abstract
Iron is essential for many cellular processes and is required by bacteria for replication. To acquire iron from the host, pathogenic Gram-negative bacteria secrete siderophores, including enterobactin (Ent). However, Ent is bound by the host protein lipocalin 2 (Lcn2), preventing bacterial reuptake of aferric or ferric Ent. Furthermore, the combination of Ent and Lcn2 (Ent+Lcn2) leads to enhanced secretion of interleukin-8 (IL-8) compared to that induced by either stimulus alone. Modified or structurally distinct siderophores, including yersiniabactin (Ybt) and glycosylated Ent (GlyEnt, or salmochelin), deliver iron to bacteria despite the presence of Lcn2. We hypothesized that the robust immune response to Ent and Lcn2 requires iron chelation rather than the Ent+Lcn2 complex itself and also can be stimulated by Lcn2-evasive siderophores. To test this hypothesis, cultured respiratory epithelial cells were stimulated with combinations of purified siderophores and Lcn2 and analyzed by gene expression microarrays, quantitative PCR, and cytokine immunoassays. Ent caused HIF-1α protein stabilization, induced the expression of genes regulated by hypoxia-inducible factor 1α (HIF-1α), and repressed genes involved in cell cycle and DNA replication, whereas Lcn2 induced expression of proinflammatory cytokines. Iron chelation by excess Ent or Ybt significantly increased Lcn2-induced secretion of IL-8, IL-6, and CCL20. Stabilization of HIF-1α was sufficient to enhance Lcn2-induced IL-6 secretion. These data indicate that respiratory epithelial cells can respond to bacterial siderophores that evade or overwhelm Lcn2 binding by increasing proinflammatory cytokine production.
INTRODUCTION
Due to its ability to assume multiple oxidative states, iron is an essential element in many human cellular processes, including DNA replication, oxygen metabolism, and electron transfer (1, 2). Iron homeostasis represents a unique challenge, since free ferric iron (Fe3+) is insoluble and ferrous iron (Fe2+) can be toxic to cells. Therefore, ferric iron is transported while complexed to transferrin, maintaining serum iron concentrations at ∼10−24 M (3–5). Bacteria require ∼10−6 M iron in their cytosol for cellular processes, a much higher concentration of iron than is readily available (3). To acquire the iron necessary for growth in the iron-limiting conditions of the human body, Gram-negative pathogens such as Escherichia coli and Klebsiella pneumoniae secrete the siderophore enterobactin (Ent). Ent is a prototypical catecholate siderophore with the highest known affinity for iron (3, 4, 6).
To counter the iron-scavenging effects of Ent, neutrophils and host mucosal cells secrete lipocalin 2 (Lcn2; neutrophil gelatinase-associated lipocalin [NGAL]; also called siderocalin or 24p3) (7). Lcn2 binds Ent in its binding pocket, either in its ferric (FeEnt) or aferric form, thereby disrupting bacterial iron acquisition and inhibiting bacterial replication (7–10). Lcn2 is critical for host defense, as Lcn2-deficient mice rapidly succumb to infection with E. coli and K. pneumoniae isolates that depend on Ent for iron acquisition (7, 11–13).
As an evasion mechanism, some strains of K. pneumoniae and other Gram-negative bacteria secrete siderophores that are not bound by Lcn2, including salmochelin and yersiniabactin (Ybt). Salmochelin is glycosylated Ent (GlyEnt), which cannot be bound by Lcn2 due to steric hindrance from added glucose groups (3). Additionally, the glucose groups decrease the membrane partitioning ability of Ent, potentially altering the ability of GlyEnt to access cellular iron (14). Ybt is a phenolate siderophore with high iron affinity that is structurally distinct from Ent and promotes pneumonia despite the presence of Lcn2 (3, 13, 15). Production of either GlyEnt or Ybt by strains of K. pneumoniae is sufficient for bacterial growth during nasal colonization and pneumonia (8, 13).
The interaction between siderophores and Lcn2 can modulate the inflammatory response to infection. Ent and Lcn2 each induce secretion of the neutrophil chemoattractant interleukin-8 (IL-8) by cultured respiratory epithelial cells (16). However, the combination of Ent and Lcn2 (Ent+Lcn2) is highly proinflammatory, increasing IL-8 production above the level of the combined effects of Ent and Lcn2 alone. During nasal colonization, Lcn2 enhances neutrophil influx in response to K. pneumoniae, producing both Ent and Ybt (8).
Certain siderophores have been shown to activate cytokine expression. For example, desferrioxamine (DFO), a nonpathogenic siderophore used therapeutically, induces IL-8 secretion through p38 mitogen-activated protein kinase (MAPK) signaling in a lung carcinoma cell line and an intestinal epithelial cell line (17, 18). DFO also stabilizes the global transcriptional regulator hypoxia inducible factor 1α (HIF-1α). Expression of HIF-1α protein is regulated through proline hydroxylation by prolyl hydroxylases (PHDs), a reaction that targets the protein for rapid proteasomal degradation and requires iron as a cofactor. Thus, HIF-1α stabilization can be induced by both oxygen and iron starvation (19). In turn, a wide variety of gene families can be activated, including genes involved in angiogenesis, iron metabolism, glycolysis, and inflammation (20–23). In contrast to DFO, the mechanism by which Ent induces cytokine production is unknown.
Whereas Lcn2 is known to induce IL-8 production and neutrophil recruitment in an Ent-dependent manner, Lcn2 is also an instrumental participant in the immune response to pathogens in an Ent-independent manner. During infection with Mycobacterium tuberculosis, Lcn2 induces alveolar macrophage expression of KC, a neutrophil chemoattractant, while inhibiting T cell accumulation and expression of the chemokine CXCL9 (24). Additionally, Lcn2 promotes proinflammatory IL-1β and gamma interferon (IFN-γ) secretion, as well as granulocyte recruitment, during malaria infection (25). These results indicate a role for Lcn2 in the inflammatory response to infections independent of its ability to bind Ent. Because iron chelation alone induces cytokine release, we hypothesized that the combined effects of siderophore-mediated iron starvation and the presence of Lcn2, rather than inherent properties of the Ent+Lcn2 complex, enhances inflammation in epithelial cells. The objective of this study was to determine the mechanism by which siderophores and Lcn2 combine to induce inflammatory responses in respiratory epithelial cells. To accomplish this, inflammatory gene expression pathways induced in response Ent, Lcn2, and Ent+Lcn2 were identified by microarray analysis of mRNA transcripts. To determine whether Lcn2 modulates inflammation specifically to Ent or more broadly in the context of iron starvation, respiratory epithelial cells were stimulated with the bacterial siderophores Ent, Ybt, and GlyEnt in combination with Lcn2, and iron starvation responses and cytokine secretion were measured.
MATERIALS AND METHODS
Cell culture.
A549 (ATCC CCL-185) cells, a human type II pneumocyte cell line, were cultured in F12K (Invitrogen, Carlsbad, CA) medium supplemented with 10% fetal bovine serum (Invitrogen) and 1:100 penicillin-streptomycin (Invitrogen).
Siderophore stimulation experiments.
Twenty-four-well plates were seeded with A549 cells at a concentration of 3.5 × 104 cells/well. After 2 days, cells were weaned from serum and antibiotics overnight. Cells then were stimulated overnight, as previously described (16), with the indicated combinations of 50 μM ferric ammonium citrate (FAC) (Sigma, St. Louis, MO), 50 or 100 μM Ent (Sigma or EMC Microcollections, Tubingen, Germany), 50 μM Ybt (EMC), 50 μM salmochelin S4 (EMC), 200 μM DFO (Sigma), 3 mM dimethyloxaloylglycine (DMOG; Sigma), or 25 μM lipocalin 2 (Lcn2) in F12K medium lacking serum or antibiotics. Prior to incubation with cells, siderophore-Lcn2 complexes were prepared by sequential incubation at room temperature of FAC and siderophore for 30 min, followed by addition of Lcn2 and incubation for an additional 30 min. Where indicated, complexes were spun through a 10,000-molecular-weight-cutoff (MWCO) centrifugal filter unit (EMD Millipore, Billerica, MA) prior to cell stimulation.
Cytokine measurement.
Cytokine secretion was measured from A549 supernatants as previously described (16). Briefly, supernatants were collected from overnight A549 stimulations, cleared by centrifugation (1,000 × g, 5 min at 4°C), and assayed by enzyme-linked immunosorbent assay (ELISA) according to the manufacturers' directions for IL-8 (OptEIA; BD Biosciences, San Diego, CA) as well as CCL20 and IL-6 (Duoset; R&D Systems), using TMB substrate (Life Technologies, Carlsbad, CA) for color development, and measured using an Eon microplate spectrophotometer (BioTek, Winooski, VT).
RNA extraction, cDNA synthesis, and qPCR.
After collecting A549 supernatants, 700 μl TRIzol reagent (Ambion, Carlsbad, CA) was added to the cells, and RNA was extracted according to the manufacturer's protocol. cDNA was synthesized using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Carlsbad, CA) and a 2720 thermal cycler (Applied Biosystems). Quantitative real-time PCR (qPCR) was conducted with TaqMan gene expression master mix (Applied Biosystems) or SYBR green Power master mix (Applied Biosystems). TaqMan assays (Invitrogen) for IL-8 (Hs00174103_m1), VEGFA (Hs00900055_m1), CCL20 (Hs01011368_m1), and IL-6 (Hs00985639_m1) were analyzed using a Realplex2 machine (Eppendorf, Hauppauge, NY) with gene expression measured relative to the housekeeping gene GAPDH (Hs99999905_m1) or GUSB (Hs00939627_m1) using the comparative threshold cycle (CT) method (26). Relative expression of NDRG1 (Hs.Pt.56a.4224283.g; Integrated DNA Technologies, Coralville, IA) and IL1R1 (Hs.PT.58.15515070) was analyzed using a Realplex2 machine with gene expression measured by SYBR green relative to the housekeeping gene GAPDH (Hs.PT.39a.22214836) using the comparative CT method (26).
Lipocalin 2 purification.
Recombinant human Lcn2 was purified based on methods previously described (8, 27). Briefly, E. coli strain BL-21, containing a plasmid encoding a human Lcn2-glutathione S-transferase (Lcn2-GST) fusion protein, was grown to mid-logarithmic phase in terrific broth supplemented with 50 μM ferrous sulfate, and Lcn2-GST expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 3 h at 37°C. Cells were pelleted by centrifugation; treated with lysozyme (0.5 mg/ml; incubated on ice for 1 h; Sigma), deoxycholate (2.8 mg/ml; incubated at 37°C for 20 min; Fisher Scientific, Pittsburgh, PA), and DNase (4.5 μg/ml; incubated at room temperature for 10 min; Roche, Branchburg, NJ) in lysis buffer (50 mM Tris, 100 mM NaCl, pH 7.5) supplemented with protease inhibitor (Complete EDTA-free cocktail tablets, Roche); and disrupted by sonication using a model 505 sonic dismembrator (four 30-s pulses at 40% amplitude with a 30-s pause between pulses; Fisher Scientific). Lcn2-GST was purified from the lysate using a glutathione Sepharose 4B bead column (GE Amersham, Piscataway, NJ) followed by elution with glutathione elution buffer (50 mM Tris, 40 mM reduced glutathione [Sigma], pH 8.5) and overnight cleavage using human thrombin (25 U per liter of E. coli; Sigma) during dialysis through a 10,000-MWCO membrane (Thermo Fisher Scientific) in buffered solution (50 mM Tris, 100 mM NaCl, pH 7.5). Digested protein then was sterilized using a 0.22-μm filter (EMD Millipore) and gel filtered using a Superdex 75 column attached to an AKTA fast-performance liquid chromatography (FPLC) system (GE Healthcare) using buffer containing phosphate-buffered saline (PBS) to remove GST. The biological activity of purified Lcn2 was confirmed by retention with Fe-Ent after centrifugation over a 10,000-MWCO column as measured by absorbance at 340 nm and growth inhibition of lipocalin-sensitive K. pneumoniae strain KP20 when added to human serum, as previously described (13).
CAS assay.
The chrome azurol S (CAS) assay was performed to determine the iron-chelating capabilities of Ent, GlyEnt (salmochelin S4), and Ybt at concentrations between 1 and 200 μM as previously described (28).
Microarray analysis.
A549 cells were stimulated overnight as described above. RNA was purified using the miRNeasy kit (Qiagen) and submitted to the University of Pennsylvania microarray facility for hybridization on the Affymetrix human gene 1.0ST gene chip (University of Pennsylvania microarray facility). Transcript abundance was estimated with the robust multiarray average (RMA) algorithm and log transformed (29). A cutoff for a significant difference in gene expression between experimental groups of a fold change of >1.3 with a P value of 0.01 was used. Gene sets with significant changes were used for enrichment analysis by comparison to the Broad Institute Molecular Signatures Database (http://www.broadinstitute.org/gsea/msigdb/index.jsp) (30). Gene ontology terms for each gene were obtained through downloads of annotation files from the Affymetrix web site.
Calcein treatment.
A549 lung epithelial cells were seeded and serum starved as described above. Cells were washed twice with RPMI without phenol red (Invitrogen) and pretreated with 1 μM calcein (Sigma) for 30 min in a standard cell culture incubator. Cells then were washed twice with RPMI without phenol red and treated overnight with siderophores with or without FAC. Fluorescence imaging was performed with an Olympus IX52 inverted microscope (Center Valley, PA), and images were analyzed with cellSens Entry imaging software (Olympus).
Western blotting.
A549 lung epithelial cells were seeded, serum starved, and stimulated as stated above. Following overnight stimulation, cellular fractionation was performed to collect nuclear proteins as previously described (31) or with radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA, 1% NP-40, 0.1% SDS) with protease inhibitors. Fractions were denatured in 6× sample buffer and separated on a 7.5% or 10% SDS-PAGE gel (Bio-Rad, Hercules, CA) at 120 V for 1.5 h. Protein was transferred onto a polyvinylidene difluoride (PVDF) or nitrocellulose membrane (Millipore) at 100 V for 1 h and immediately blocked in 5% nonfat milk in Tris-buffered saline with 0.1% Tween (TBST). Membranes were probed with α-HIF-1α (1:2,000 [R&D Systems] or 1:1,000 [Santa Cruz Inc., Dallas, TX]) and α-β-actin (1:5,000; Novus Biologicals, Littleton, CO) or with α-glyceraldehyde-3-phosphate dehydrogenase (α-GAPDH) (1:1,000; Santa Cruz Inc.) as a loading control overnight, followed by mouse IgG-horseradish peroxidase (HRP) secondary antibody (1:1,000 [R&D Systems or Cell Signaling Technologies, Danvers, MA]), in 5% bovine serum albumin (BSA) in TBST or 5% nonfat milk in TBST. Protein expression was detected using ECL substrate (Pierce) and visualized on a ChemiDoc/RS+ imaging system (Bio-Rad). Protein concentration was determined with a microbicinchoninic acid (BCA) assay kit (Pierce).
Statistical analysis.
Array, qPCR, and ELISA data were log transformed and fit to analysis of variance (ANOVA) models with one mean per group, and pairs of treatments were compared. Additionally, some comparisons of the differences in two pairs of treatments were made. Where indicated, unpaired, two-tailed t tests were performed using Prism 6 software (GraphPad Software, Inc.).
Microarray data accession number.
The array data and our statistical analysis have been deposited in NCBI's Gene Expression Omnibus (GEO) and are accessible through GEO series accession number GSE54962 (32).
RESULTS
Enterobactin and Ent+Lcn2 induce distinct gene expression and cytokine secretion responses in respiratory epithelial cells.
The combination of Ent and Lcn2 strongly induces secretion of IL-8, a proinflammatory cytokine responsible for neutrophil chemotaxis (16). However, the mechanism of this inflammation and whether Ent+Lcn2 triggers additional cellular responses is unknown. To identify cellular pathways induced or repressed by combinations of Fe, Ent, Lcn2, or Ent+Lcn2, microarray analysis was performed on mRNA transcripts from stimulated A549 respiratory epithelial cells. We calculated the number of significant probe set differences for many possible comparisons for 29,096 probe sets from our two experiments (see Table S1 in the supplemental material). For comparisons that are the focus of this study, we collapsed the probe sets to 19,419 distinct genes (see Table S2). Here, we report results in terms of distinct genes. To identify gene expression specific to the iron status of Ent, cells were stimulated with a combination of Fe and Ent, and an interaction test was used to determine if the Ent-versus-PBS difference was significantly larger or smaller than the Fe-Ent-versus-Fe difference by a factor of at least 1.3 and with P < 0.01 [(Ent/PBS)/(Fe-Ent/Fe)]. This interaction test demonstrated induction of 1,152 genes and repression of 812 genes in response to aferric Ent. Gene ontology analysis indicated significant induction of genes related to MAPK phosphatase activity, apoptosis, and response to cytokine stimulus and repression of genes related to the cell cycle, DNA replication, mitosis, and DNA repair (see Table S3 and Fig. S1). Consistent with iron starvation, aferric Ent specifically upregulated expression of NDRG1, a tumor metastasis gene that is induced in response to cell-permeable iron chelators (Fig. 1A) (33). Aferric Ent also significantly upregulated expression of IL-8. In addition to downregulation of cell cycle genes, Ent strongly reduced expression of the IL-1 receptor gene IL1R1. To confirm microarray findings, A549 cells were stimulated in an independent experiment with combinations of Fe, Ent, and Lcn2, and gene expression was measured by qPCR (Fig. 1C and D). NDRG1 was significantly induced by Ent compared to induction in the presence of PBS (21.5-fold; P = 1.1E−11) and met the selection criteria described above where the increase in induction from PBS to Ent was significantly more than the increase from Fe to Fe-Ent (35.8-fold more; P = 1.4E−10). Similarly, IL-8 was induced by Ent more than by PBS (17-fold; P = 3.4E−9) and met the interaction selection criteria used in the microarray (3-fold more; P = 0.003) (Fig. 1F). Ent treatment repressed IL1R1 expression significantly compared to that of PBS treatment (0.29-fold; P = 1.6E−5) (Fig. 1D), although it narrowly missed the interaction selection criteria (P = 0.054).
FIG 1.

Enterobactin and Ent+Lcn2 induce distinct gene expression patterns. Heat maps of relative gene expression by A549 respiratory cells in response to combinations of 50 μM enterobactin (Ent) and 50 μM ferric ammonium citrate (Fe) alone (A) or with 25 μM lipocalin 2 (Lcn2) (B), as measured by microarray, are shown. Red indicates upregulation of gene expression and green indicates downregulation of gene expression relative to the median for each experiment. Stimulations were repeated independently, and gene expression was validated by qPCR of NDRG1 (C), IL1R1 (D), VEGFA (E), IL-8 (F), CCL20 (G), and IL-6 (H) in response to combinations of Fe, Ent, and Lcn2. Data are shown as means ± standard errors of the means (SEM) from 3 replicate samples and are representative of at least 2 independent experiments. Statistics were calculated using ANOVA (#, P < 0.001 for the indicated comparisons).
To identify gene expression uniquely altered by Ent+Lcn2, a second experiment was performed comparing responses to this stimulus to the response to both Lcn2 alone and Fe-Ent+Lcn2. Lcn2 alone significantly induced 56 genes and repressed 80 genes (selection criteria of P < 0.01; fold change, >1.3), and gene ontology analysis demonstrated induction of genes involved in the immune response, innate immune response, and chemokine and cytokine activity (see Table S3 and Fig. S2 in the supplemental material). The set of repressed genes did not significantly overlap a gene ontology group. Induced genes included the cytokine genes IL-8, IL-6, CCL20, CXCL1, CXCL5, complement component C3, and LCN2 itself.
Ent+Lcn2 significantly induced expression of 239 genes and repressed 36 genes compared to Lcn2 and Fe-Ent+Lcn2 (P < 0.01 and a fold change of >1.3 for both Ent+Lcn2 versus Lcn2 and Ent+Lcn2 versus Fe-Ent+Lcn2) (see Fig. S3 in the supplemental material). The intersection of this gene set and the set induced by Ent described above contained 137 induced and 21 repressed genes (P < 1E−200 by Mantel-Haenszel chi-square statistic for association), indicating that the iron status of Ent conferred a strong effect on gene expression regardless of the presence of Lcn2. Accordingly, Ent+Lcn2 significantly induced NDRG1 expression compared to both Lcn2 and Fe-Ent+Lcn2 (Fig. 1C).
Gene ontology analysis of Ent+Lcn2-induced genes indicated significant induction of genes involved in glycolysis, response to hypoxia, and the endoplasmic reticulum unfolded protein response and repression of genes related to the mitotic prometaphase (see Table S3 in the supplemental material). Induced genes that are associated with the response to hypoxia included VEGFA, ADM, TFRC, and ELGN3 (Fig. 1A and B; also see Fig. S3 in the supplemental material). Independent stimulations of A549 cells indicated that Ent induced VEGFA relative to PBS and met the interaction selection criteria compared to Fe and Fe-Ent (P = 4.4E−5), whereas Ent+Lcn2 induced significantly more expression than Lcn2 or Fe-Ent+Lcn2, as measured by qPCR (Fig. 1E). VEGFA is an angiogenesis gene regulated by HIF-1α, indicating that Ent and Ent+Lcn2 activate HIF-1α, and ELGN3 is a prolyl hydroxylase that regulates HIF function (20, 34). Indeed, enrichment analysis for motif gene sets indicated Ent+Lcn2 induced HIF-1-responsive genes (see Table S2).
Two cytokine genes showed strong induction in response to Ent+Lcn2 compared to both Lcn2 and Fe-Ent+Lcn2: IL-6 and CCL20 (Fig. 1B). In contrast, neither cytokine was induced significantly by aferric Ent based on the interaction test (Fig. 1A). Separate stimulation of A549 cells with combinations of Fe, Ent, and Lcn2 confirmed induction by Ent+Lcn2 compared to both Lcn2 and Fe-Ent+Lcn2, as measured by qPCR (Fig. 1G to H). Based on the PBS control, basal transcription of CCL20 and IL-6 was very low. Gene expression in response to combinations of Fe and Ent were similarly low and could not be reliably determined. Therefore, relative expression of CCL20 and IL-6 was calculated by comparing each stimulus's transcript level to that of Lcn2, rather than PBS, as baseline expression. IL-8 also was significantly induced by Ent+Lcn2 compared to Lcn2 and Fe-Ent+Lcn2 as measured by qPCR (P < 0.0001). In contrast to the expression pattern of IL-6 and CCL20, aferric Ent strongly induced IL-8 expression as described above.
To correlate changes in gene expression with cytokine secretion, A549 cells were stimulated with combinations of Fe, Ent, and Lcn2, and IL-6, IL-8, and CCL20 were measured by ELISA (Fig. 2A to C). As previously reported, Ent and Lcn2 individually induced IL-8 secretion, and the combination of Ent and Lcn2 induced IL-8 secretion that was greater than the response to either Lcn2 or Fe-Ent+Lcn2 (Fig. 2A) (16). However, this was in contrast to the expression pattern where Ent induced significantly more IL-8 expression than Lcn2 (P = 1.3E−7) (Fig. 1F). This suggests that Lcn2 acts posttranscriptionally to increase IL-8 production or secretion. The patterns of CCL20 and IL-6 secretion differ from the pattern of IL-8 secretion. CCL20 was secreted at lower levels than the PBS control in response to Ent or Fe-Ent (P < 0.001) (Fig. 2B), whereas IL-6 was not detectable in response to either stimulus (Fig. 2C). Similar to the IL-8 response, Lcn2 induced secretion of CCL20, and IL-6 and Ent+Lcn2 induced greater secretion than Lcn2 or Fe-Ent+Lcn2. Together, the microarray data identify new gene targets induced and repressed by Fe, Ent, and Lcn2, and independent qPCR confirmation and ELISA data indicate IL-8, CCL20, and IL-6 are gene targets strongly activated in respiratory epithelial cells in response to the combination of Ent and Lcn2.
FIG 2.
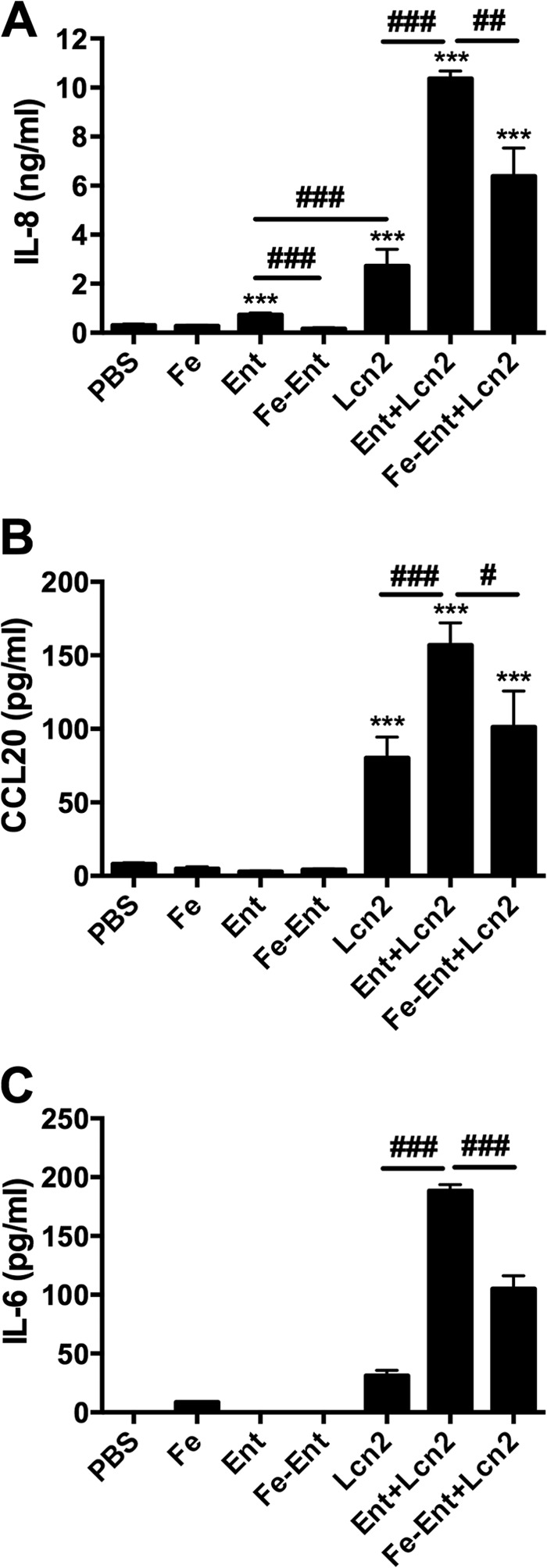
In combination, Ent and Lcn2 strongly induce cytokine production in A549 respiratory cells. Cells were stimulated for 16 h with combinations of 50 μM FAC (Fe), 50 μM Ent, or 25 μM Lcn2. IL-8 (A), CCL20 (B), and IL-6 (C) secretion were measured by ELISA. Values shown are means ± SEM from 3 replicate samples and are representative of at least 2 independent experiments. Statistics were calculated using one-way ANOVA (***, P < 0.0001 induction relative to PBS; #, P < 0.05; ##, P < 0.01; ###, P < 0.001 for the indicated comparison).
Unbound enterobactin enhances lipocalin 2 induction of proinflammatory cytokines.
The enhanced cytokine production induced by Ent+Lcn2 compared to those of Lcn2 and Fe-Ent+Lcn2 indicates that secretion is triggered by siderophore-dependent iron chelation (Fig. 2A to C). This could be mediated either by Ent bound to Lcn2 in the binding calyx (16) or unbound Ent. To distinguish these possibilities, Ent and Lcn2 were incubated in a 2:1 ratio to allow binding and then spun through a 10,000-MWCO column to retain Lcn2 (20.5 kDa) and Ent+Lcn2 complexes but not Ent alone (669 Da). Consistent with the loss of unbound Ent, spinning Ent+Lcn2 in an MWCO column diminished its ability to stimulate IL-8 and IL-6 secretion (P < 0.0001) (Fig. 3), reducing IL-8 and IL-6 secretion to levels comparable to that of Lcn2 alone (P = 0.5 and 0.1, respectively). Addition of Ent to retained Ent+Lcn2 following spinning through an MWCO column restored its ability to stimulate IL-8 and IL-6 secretion, indicating that the MWCO column has no effect on the stimulatory effect of Lcn2, and that unbound Ent is required to enhance secretion of IL-8 and IL-6. Additionally, stimulation of respiratory epithelial cells with Ent and Lcn2 at 4:1 and 2:1 ratios induced increased IL-8 secretion, whereas stimulation with Ent and Lcn2 at a 1:1 ratio did not (data not shown). These data indicate that the combination of unbound Ent and Lcn2, rather than Ent+Lcn2 complexes themselves, stimulates robust IL-8 and IL-6 secretion.
FIG 3.
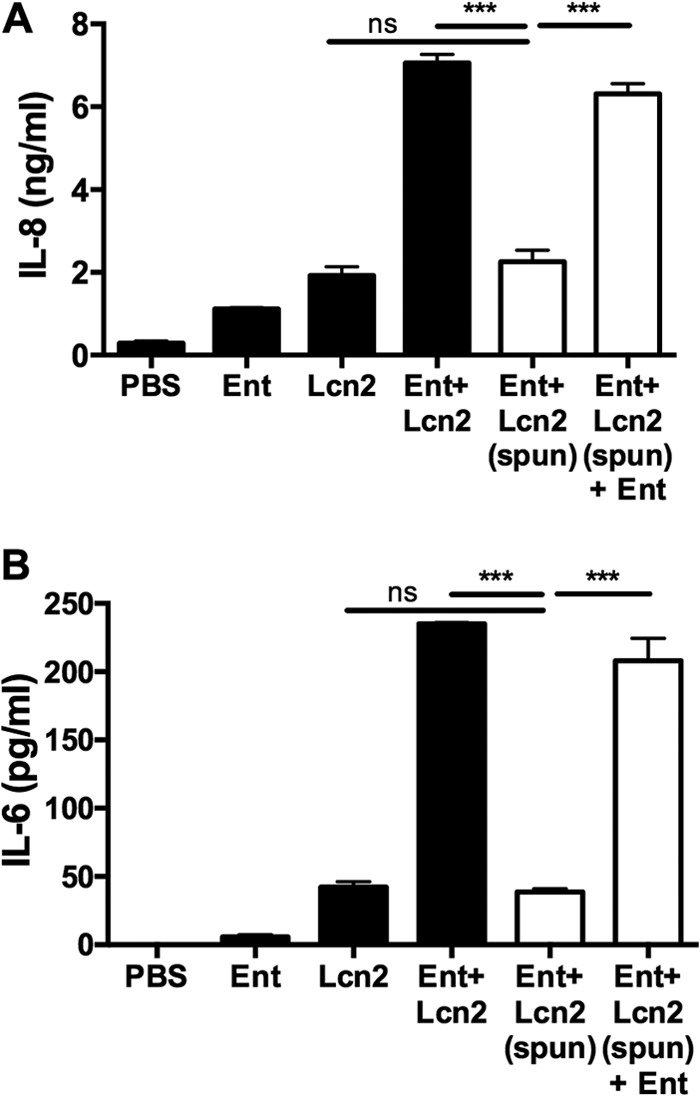
Unbound Ent in combination with Lcn2 is required for synergistic IL-8 and IL-6 secretion in A549 cells. Combinations of 50 μM Ent (669 Da) and 25 μM Lcn2 (20.5 kDa) were spun, as indicated, through a 10,000-MWCO column, and cells were stimulated with the retentate, containing Lcn2 or Ent bound by Lcn2, for 16 h. IL-8 (A) and IL-6 (B) secretion were measured by ELISA. Values shown are means ± SEM from 3 replicate samples and are representative of at least 2 independent experiments. Statistics were calculated using one-way ANOVA (***, P < 0.0001; ns, P > 0.05).
Iron chelation by yersiniabactin in combination with lipocalin 2 strongly induces cytokine secretion.
The fact that unbound Ent enhances cytokine responses to Lcn2 suggests that this cellular response also occurs in response to iron chelation by siderophores to which Lcn2 cannot bind. To test this hypothesis, respiratory epithelial cells were stimulated with combinations of Fe and the Lcn2-evasive siderophores Ybt and GlyEnt, and qPCR for the iron starvation gene NDRG1 was performed (Fig. 4A). Similar to Ent, Ybt strongly induced gene expression of NDRG1, as measured by qPCR, which was reversed by Fe (P < 0.0001). In contrast, GlyEnt did not induce NDRG1 (P = 0.6). To verify the iron chelation ability of the siderophores, A549 cells were treated with calcein, a membrane-permeable ester that is cleaved upon entering a cell, causing fluorescence that is quenched by the cellular labile iron pool (35). Addition of Ent and Ybt chelated iron away from calcein, increasing fluorescence, whereas addition of GlyEnt did not (Fig. 4B). Preloading the siderophores with Fe prevented induction of calcein fluorescence. Because GlyEnt has different membrane-partitioning activities than Ent that could confer differing abilities to chelate intracellular iron, iron chelation in solution was measured by the chromogenic CAS assay (28). Ent and Ybt rapidly and efficiently induced a color change in the CAS reagent, whereas GlyEnt did not (data not shown). Combined, these data indicate the ability of Ent and Ybt to disrupt cellular iron homeostasis.
FIG 4.
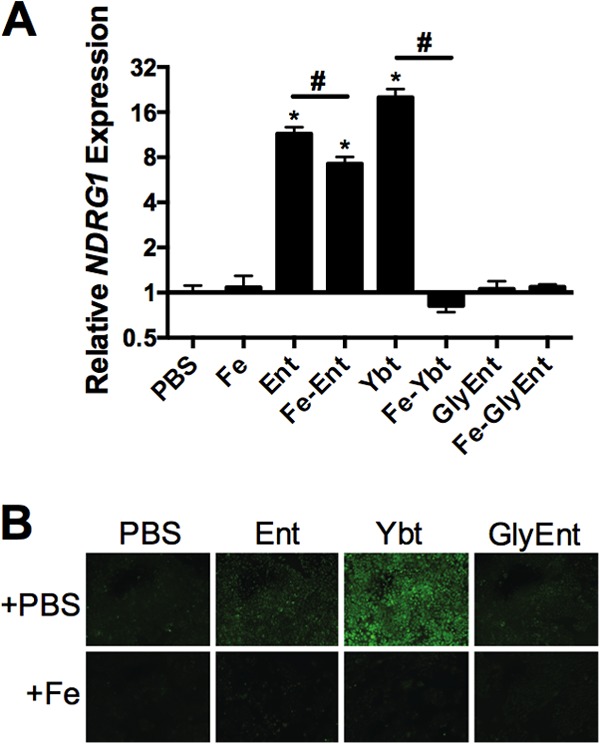
Ybt and Ent, but not GlyEnt, induce expression of the iron starvation gene NDRG1 and calcein fluorescence. (A) Cells were stimulated for 16 h with combinations of 50 μM FAC (Fe), 50 μM Ent, 50 μM Ybt, or 50 μM GlyEnt. NDRG1 expression, a marker associated with iron chelation, was measured by qPCR. (B) Cells were stimulated for 16 h with combinations of 100 μM FAC (Fe), 100 μM Ent, 100 μM Ybt, or 100 μM GlyEnt, and calcein fluorescence was examined. Values shown are means ± SEM from 3 replicate samples and are representative of at least 2 independent experiments. Statistics were calculated using one-way ANOVA (*, P < 0.0001 for induction relative to PBS; #, P < 0.0001 for the indicated comparison).
To determine if host iron chelation by nonligand siderophores can induce increased cytokine release in the presence of Lcn2, respiratory epithelial cells were stimulated with Ybt or GlyEnt and Lcn2 (Fig. 5). Ybt alone significantly increased IL-8 and IL-6 secretion and induced CCL20 secretion, whereas levels were undetectable in the control. Furthermore, Ybt+Lcn2 induced significantly more IL-8 (Fig. 5A), IL-6 (Fig. 5B), and CCL20 (Fig. 5C) secretion than Lcn2 alone. Induction of cytokine secretion by Ybt and Ybt+Lcn2 correlated with host iron chelation, as measured by increased NDRG1 gene expression (Fig. 5D). Lcn2 alone had no effect on NDRG1 expression. Neither GlyEnt nor GlyEnt+Lcn2 induced NDRG1 expression. Additionally, GlyEnt+Lcn2 did not enhance IL-8, IL-6, or CCL20 secretion compared to Lcn2 alone, consistent with the inability of GlyEnt to perturb intracellular iron levels (Fig. 4). To determine if a pharmacologic iron chelator could induce increased cytokine release, we stimulated respiratory epithelial cells with DFO in the presence of Lcn2. DFO+Lcn2 induced secretion of IL-8, IL-6, and CCL20 that correlated with expression of NDRG1 (Fig. 5E and F; also see Fig. S4 in the supplemental material.) These data indicate that iron chelation by a siderophore other than Ent enhances Lcn2-dependent proinflammatory cytokine release in respiratory epithelial cells.
FIG 5.

Ybt+Lcn2 and DFO+Lcn2 induce chemokine release by A549 respiratory cells. Cells were stimulated for 16 h with combinations of 50 μM Ybt, 50 μM GlyEnt, 200 μM DFO, or 25 μM Lcn2, and ELISA was used to measure IL-8 (A), IL-6 (B and E), and CCL20 (C) secretion. Relative NDRG1 expression (D and F) was assayed using qPCR. Values shown are means ± SEM from 3 replicate samples and are representative of at least 2 independent experiments. Statistics were calculated using one-way ANOVA (**, P < 0.01 relative to PBS; ##, P < 0.01; ###, P < 0.001 for the indicated comparison; ns, P > 0.05).
Induction of HIF-1α stabilization in the presence of lipocalin 2 is sufficient to enhance inflammation.
Gene expression analysis indicated that Ent and Ent+Lcn2 induced HIF-regulated genes, including VEGFA (Fig. 1A, B, and E). HIF-1α has been shown to regulate inflammation and enhance expression of cytokines, including IL-6 (36, 37). HIF-1α is rapidly targeted for degradation by prolyl hydroxylases (PHDs) but is stabilized through inactivation of PHDs by iron limitation, hypoxia, or the dioxygenase inhibitor DMOG (38). To determine if HIF-1α is stabilized by stimulation with Ent, Western blotting of nuclear fractions was performed. Stimulation with Ent induced nuclear stabilization of HIF-1α, similar to the stabilization of HIF-1α observed in response to DMOG (Fig. 6A). Additionally, stimulation with Ent+Lcn2, but not Lcn2 alone, induced nuclear stabilization of HIF-1α (Fig. 6B and C). To determine if stabilization of HIF-1α through inactivation of prolyl hydroxylases is sufficient to enhance Lcn2-dependent inflammation, A549 cells were treated with DMOG alone or in combination with Lcn2. DMOG in combination with Lcn2 did not increase secretion of IL-8 compared to Lcn2 alone (P = 0.2) (Fig. 6D) or CCL20 (data not shown); however, DMOG+Lcn2 stimulation induced IL-6 expression significantly above the level of Lcn2 alone (P < 0.01) (Fig. 6E). These data indicate that Ent induces stabilization of HIF-1α that, in combination with Lcn2, is sufficient to induce a proinflammatory immune response.
FIG 6.

Ent stabilizes HIF-1α in A549 respiratory epithelial cells, which is sufficient to enhance Lcn2-dependent IL-6 secretion. Cells were stimulated for 16 h with combinations of 50 μM Ent, 3 mM DMOG, or 25 μM Lcn2, and Western blotting or ELISA was used to measure HIF-1α stabilization (A, B, and C), IL-8 secretion (D), or IL-6 secretion (E). Western blot data are representative of 2 independent experiments. ELISA values shown are means ± SEM from 3 replicate samples and are representative of at least 2 independent experiments. Statistics were calculated using unpaired two-tailed t tests (**, P < 0.01; ns, P > 0.05).
DISCUSSION
In addition to disrupting bacterial iron acquisition, Lcn2 enhances inflammation in vitro and in vivo in response to Ent (8, 16). In this way, Lcn2 may tailor inflammation based on microbial iron metabolism. To determine the mechanism of inflammation induced by Ent and Lcn2, we performed a microarray analysis to identify genes modulated in response to Fe, Ent, and Lcn2 and confirmed changes in gene expression using qPCR and ELISA. We then determined whether the strong induction of cellular immune responses by Ent+Lcn2 was due to the ligand-protein complex or, more broadly, to iron chelation. We found that the host immune response is activated in response to Lcn2 and amplified through iron chelation by siderophores rather than in response to the Ent+Lcn2 complex itself. Furthermore, Ent induces HIF-1α stabilization alone and in the presence of Lcn2, and HIF-1α stabilization is sufficient to enhance Lcn2-dependent secretion of the cytokine IL-6. These findings indicate a novel host response to microbial iron metabolism in which cellular stress induced by siderophore-mediated iron chelation and the presence of Lcn2 leads to activation of a limited set of cytokines, namely, IL-6, IL-8, and CCL20. These findings also indicate a novel mechanism for siderophore-induced cytokine secretion, linking HIF-1α stabilization by pathogen-associated siderophores to IL-6 secretion.
Without its ligand, Lcn2 has been shown to modulate cytokine expression. In cells of the central nervous system, Lcn2 modulates lipopolysaccharide-induced cytokine production, including IL-6 and CCL20, as well as adipokine production in adipocytes (39, 40). In models of ischemia and reperfusion, Lcn2 controls neutrophil recruitment by regulating expression of chemokines, including IL-6, and their cell surface receptors (41). Consistent with these studies, our findings indicate that Lcn2 induces IL-6 and CCL20 secretion from respiratory epithelial cells. IL-6 is an inflammatory cytokine active in the regulation of the acute-phase response in hepatocytes and is capable of upregulating expression of hepcidin (42). Hepcidin regulates plasma iron concentrations by inhibiting enterocyte uptake of iron and iron recycling by macrophages and is upregulated during infection and inflammation (43). IL-6 is also a differentiation factor for Th17 lymphocytes that mediate protective immunity against siderophore-producing pathogens, such as K. pneumoniae (44). In turn, CCL20 is a lymphocyte chemoattractant whose expression is amplified by IL-6 production, recruiting Th17 cells to sites of inflammation by binding to its cognate receptor, CCR6. Thus, it is possible that expression of CCL20 initiates an adaptive immune response (45–47).
Lcn2-induced cytokines also are induced in response to disruptions in iron homeostasis. Iron chelation by DFO induces IL-8 and CCL20 production in intestinal epithelial cells (17, 48). In respiratory epithelial cells, the combination of siderophores and Lcn2 induces robust expression of IL-6 and CCL20. Therefore, the cytokine response to bacterial siderophores and Lcn2 could serve as a multifaceted failsafe mechanism. First, IL-8 can recruit neutrophils to the site of infection. Second, IL-6 can upregulate hepcidin to limit further iron availability for invading bacteria. Finally, IL-6 and CCL20 can act in concert to attract mature Th17 to sites of infection and commit naive T cells to the Th17 pathway.
The presence or absence of siderophores likely is crucial to the effect of Lcn2 on inflammation. In recent work, stimulation of macrophages with Streptococcus pneumoniae induced IL-10 production in an Lcn2-dependent manner, which skewed macrophages toward a deactivated phenotype (49). In human and animal models, increased Lcn2 correlated with worsening of pneumococcal pneumonia. These findings contrast with the results of this work, which demonstrate proinflammatory effects of Lcn2, and previous work by our group and others, demonstrating that Lcn2 is a crucial antimicrobial peptide that enhances survival during infection, particularly with K. pneumoniae (7, 8, 11, 13). In addition, our microarray analysis did not indicate any change in the gene expression of IL-10 in response to Lcn2. We hypothesize that the difference in outcome is because Streptococcus pneumoniae does not require siderophores for its pathogenesis, and Lcn2 cannot properly modulate inflammation during infection without siderophore-mediated iron chelation. In fact, patient survival from Gram-negative pneumonia correlated with increased Lcn2 in the bronchoalveolar lavage fluid (49).
Iron homeostasis and metabolism are tightly regulated systems that require the expression and function of many proteins, including transferrin, transferrin receptor, and ferritin. Disruption of these systems due to iron chelation exerts a wide range of pathological effects on cells, including disruption of DNA replication, apoptosis, and cell cycle arrest (33, 50, 51). Although these properties of iron chelators show promise as anti-cancer therapies, our data suggest that bacterial siderophores act as cytotoxins during infection. Clinical isolates of K. pneumoniae produce 50 to 100 μM Ent in pure culture (data not shown), quantities sufficient to induce the hypoxia and iron starvation responses described here. The induction of cellular stresses in response to siderophores and Lcn2 during infection may lead to significant pathological effects during infection. However, our results indicate that Lcn2 can cooperate with these cellular stress responses to induce robust cytokine release and recruit inflammatory cells to combat the bacterial source of toxic siderophores.
Although the inflammatory response to siderophores and Lcn2 is activated in response to iron chelation rather than a siderophore-Lcn2 complex, the cellular responses to Ent, Ybt, and GlyEnt are distinct. Stimulation with Ybt or Ybt+Lcn2 induces more IL-8, IL-6, and CCL20 secretion and NDRG1 gene expression than equimolar stimulation with Ent or Ent+Lcn2. This is surprising, because Ent has the highest known affinity for iron. In fact, stimulation of A549 cells with increasing molar concentrations of siderophores illustrates a higher threshold concentration to induce IL-8 secretion by Ybt than that by Ent (data not shown). This is consistent with the pattern shown in Fig. 4A, in which Fe-Ent induces more NDRG1 gene expression than Fe-Ybt. Despite equimolar addition of Fe to Ent, trace free Ent is capable of chelating cellular iron and inducing NDRG1 expression. GlyEnt may not induce cellular iron chelation or proinflammatory cytokine secretion because of its decreased membrane partitioning abilities (14). Addition of GlyEnt to an entirely siderophore-deficient strain of K. pneumoniae restores bacterial growth, indicating that GlyEnt is able to acquire iron for bacterial growth (52). Differential secretion of Ent, Ybt, and GlyEnt during infection may cause dissimilar pathological effects through triggering varied levels of cytokine production.
Expression of HIF-1α protein is regulated through hydroxylation by prolyl hydroxylases (PHDs), a modification that targets the protein for rapid proteasomal degradation (19). Since PHDs require iron as a cofactor, HIF-1α stabilization can be induced by both oxygen and iron starvation (53). Indeed, siderophores previously have been shown to induce HIF-1α stabilization (54, 55). In a previous study, Ybt was shown to stabilize HIF-1α, but effects on inflammation were not assessed. GlyEnt also was reported to induce HIF-1α, but this required high concentrations of siderophores (≥200 μM) (54). The current study demonstrates induction of HIF-1α stabilization by the prototypical siderophore Ent and Ent+Lcn2 at physiologic concentrations. Additionally, we illustrate that HIF-1α stabilization in combination with Lcn2 is sufficient for IL-6 secretion, linking HIF-1α-regulated genes with inflammatory pathways. HIF-1α stabilization in combination with Lcn2 is not sufficient to induce IL-8 or CCL20 secretion, suggesting that additional pathways are activated in response to siderophore-Lcn2 stimulation that enhance inflammation. IL-8 production by epithelial cells is regulated by a combination of MAPK and NF-κB signaling pathways (18). Microarray analysis in response to treatment with Ent indicated upregulation of dual-specificity phosphatases (DUSPs), indicating that MAPK signaling is involved in IL-8 secretion in response to siderophore-Lcn2.
In summary, our results introduce a novel role for Lcn2 as a rheostat that modulates the response to iron chelation by bacterial siderophores. We propose a model in which a small amount of Ent can be bound and neutralized by Lcn2 (Fig. 7A), resulting in low levels of Lcn2-induced cytokine secretion. However, high levels of Ent (Fig. 7B) or Ybt (Fig. 7C) can overwhelm Lcn2 binding capacity, causing the accumulation of unbound siderophores. These siderophores chelate host cellular iron and, in combination with Lcn2, induce robust secretion of IL-6, CCL20, and IL-8 in airway epithelial cells. Ent induces HIF-1α stabilization, and HIF-1α in combination with Lcn2 is sufficient to induce IL-6. In contrast, HIF-independent pathways likely are required to augment IL-8 and CCL20 expression. DFO and Ybt also combine with Lcn2 to induce inflammation, indicating this is a generalized response to siderophore-mediated iron starvation. In this way, Lcn2 can prevent iron sequestration by Ent without triggering a substantial immune response but can potently upregulate inflammation when overwhelmed by siderophores that perturb epithelial cell iron homeostasis.
FIG 7.

Lcn2 acts as a sensor by modulating airway epithelial cell inflammatory cytokine secretion in response to iron chelation by unbound Ent and Ybt. (A) Small amounts of Ent can be bound and neutralized by Lcn2, leading to a low level of Lcn2-induced cytokine secretion in the airway. Large amounts of Ent (B) or Ybt (C) evade Lcn2 binding, leading to altered host iron status and HIF-1α stabilization. The combination of cellular iron depletion and Lcn2 signaling increases production of inflammatory cytokines, such as IL-8, IL-6, and CCL20.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by Public Health Service grant GM085612 from the National Institute of General Medical Sciences (M.A.B.), CA148828 from the National Cancer Institute (Y.M.S.), and a University of Michigan Rackham predoctoral grant (V.I.H.), and it was partially supported by University of Michigan Cancer Center Support grant P30 CA046592 from the National Institutes of Health (R.K.).
We thank Harry Mobley, Marc Hershenson, and Beth Moore for guidance and manuscript revisions.
We have no conflicting financial interests to report.
Footnotes
Published ahead of print 30 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01849-14.
REFERENCES
- 1.Pantopoulos K, Porwal SK, Tartakoff A, Devireddy L. 2012. Mechanisms of mammalian iron homeostasis. Biochemistry 51:5705–5724. 10.1021/bi300752r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gkouvatsos K, Papanikolaou G, Pantopoulos K. 2012. Regulation of iron transport and the role of transferrin. Biochim. Biophys. Acta 1820:188–202. 10.1016/j.bbagen.2011.10.013 [DOI] [PubMed] [Google Scholar]
- 3.Fischbach MA, Lin H, Liu DR, Walsh CT. 2006. How pathogenic bacteria evade mammalian sabotage in the battle for iron. Nat. Chem. Biol. 2:132–138. 10.1038/nchembio771 [DOI] [PubMed] [Google Scholar]
- 4.Raymond KN, Dertz EA, Kim SS. 2003. Enterobactin: an archetype for microbial iron transport. Proc. Natl. Acad. Sci. U. S. A. 100:3584–3588. 10.1073/pnas.0630018100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Correnti C, Strong RK. 2012. Mammalian siderophores, siderophore-binding lipocalins, and the labile iron pool. J. Biol. Chem. 287:13524–13531. 10.1074/jbc.R111.311829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abergel RJ, Warner JA, Shuh DK, Raymond KN. 2006. Enterobactin protonation and iron release: structural characterization of the salicylate coordination shift in ferric enterobactin. J. Am. Chem. Soc. 128:8920–8931. 10.1021/ja062046j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. 2004. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432:917–921. 10.1038/nature03104 [DOI] [PubMed] [Google Scholar]
- 8.Bachman MA, Miller VL, Weiser JN. 2009. Mucosal lipocalin 2 has pro-inflammatory and iron-sequestering effects in response to bacterial enterobactin. PLoS Pathog. 5:e1000622. 10.1371/journal.ppat.1000622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. 2002. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell 10:1033–1043. 10.1016/S1097-2765(02)00708-6 [DOI] [PubMed] [Google Scholar]
- 10.Abergel RJ, Moore EG, Strong RK, Raymond KN. 2006. Microbial evasion of the immune system: structural modifications of enterobactin impair siderocalin recognition. J. Am. Chem. Soc. 128:10998–10999. 10.1021/ja062476+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan YR, Liu JS, Pociask DA, Zheng M, Mietzner TA, Berger T, Mak TW, Clifton MC, Strong RK, Ray P, Kolls JK. 2009. Lipocalin 2 is required for pulmonary host defense against Klebsiella infection. J. Immunol. 182:4947–4956. 10.4049/jimmunol.0803282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berger T, Togawa A, Duncan GS, Elia AJ, You-Ten A, Wakeham A, Fong HE, Cheung CC, Mak TW. 2006. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U. S. A. 103:1834–1839. 10.1073/pnas.0510847103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bachman MA, Oyler JE, Burns SH, Caza M, Lepine F, Dozois CM, Weiser JN. 2011. Klebsiella pneumoniae yersiniabactin promotes respiratory tract infection through evasion of lipocalin 2. Infect. Immun. 79:3309–3316. 10.1128/IAI.05114-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo M, Lin H, Fischbach MA, Liu DR, Walsh CT, Groves JT. 2006. Enzymatic tailoring of enterobactin alters membrane partitioning and iron acquisition. ACS Chem. Biol. 1:29–32. 10.1021/cb0500034 [DOI] [PubMed] [Google Scholar]
- 15.Perry RD, Balbo PB, Jones HA, Fetherston JD, DeMoll E. 1999. Yersiniabactin from Yersinia pestis: biochemical characterization of the siderophore and its role in iron transport and regulation. Microbiology 145(Part 5):1181–1190. 10.1099/13500872-145-5-1181 [DOI] [PubMed] [Google Scholar]
- 16.Nelson AL, Ratner AJ, Barasch J, Weiser JN. 2007. Interleukin-8 secretion in response to aferric enterobactin is potentiated by siderocalin. Infect. Immun. 75:3160–3168. 10.1128/IAI.01719-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi EY, Kim EC, Oh HM, Kim S, Lee HJ, Cho EY, Yoon KH, Kim EA, Han WC, Choi SC, Hwang JY, Park C, Oh BS, Kim Y, Kimm KC, Park KI, Chung HT, Jun CD. 2004. Iron chelator triggers inflammatory signals in human intestinal epithelial cells: involvement of p38 and extracellular signal-regulated kinase signaling pathways. J. Immunol. 172:7069–7077. 10.4049/jimmunol.172.11.7069 [DOI] [PubMed] [Google Scholar]
- 18.Li J, Kartha S, Iasvovskaia S, Tan A, Bhat RK, Manaligod JM, Page K, Brasier AR, Hershenson MB. 2002. Regulation of human airway epithelial cell IL-8 expression by MAP kinases. Am. J. Physiol. Lung Cell. Mol. Physiol. 283:L690–L699 [DOI] [PubMed] [Google Scholar]
- 19.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr 2001. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292:464–468. 10.1126/science.1059817 [DOI] [PubMed] [Google Scholar]
- 20.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. 1996. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16:4604–4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tacchini L, Bianchi L, Bernelli-Zazzera A, Cairo G. 1999. Transferrin receptor induction by hypoxia. HIF-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. J. Biol. Chem. 274:24142–24146 [DOI] [PubMed] [Google Scholar]
- 22.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O'Neill LA. 2013. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496:238–242. 10.1038/nature11986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. 2003. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112:645–657. 10.1016/S0092-8674(03)00154-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guglani L, Gopal R, Rangel-Moreno J, Junecko BF, Lin Y, Berger T, Mak TW, Alcorn JF, Randall TD, Reinhart TA, Chan YR, Khader SA. 2012. Lipocalin 2 regulates inflammation during pulmonary mycobacterial infections. PLoS One 7:e50052. 10.1371/journal.pone.0050052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao H, Konishi A, Fujita Y, Yagi M, Ohata K, Aoshi T, Itagaki S, Sato S, Narita H, Abdelgelil NH, Inoue M, Culleton R, Kaneko O, Nakagawa A, Horii T, Akira S, Ishii KJ, Coban C. 2012. Lipocalin 2 bolsters innate and adaptive immune responses to blood-stage malaria infection by reinforcing host iron metabolism. Cell Host Microbe 12:705–716. 10.1016/j.chom.2012.10.010 [DOI] [PubMed] [Google Scholar]
- 26.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3:1101–1108. 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- 27.Yang J, Goetz D, Li JY, Wang W, Mori K, Setlik D, Du T, Erdjument-Bromage H, Tempst P, Strong R, Barasch J. 2002. An iron delivery pathway mediated by a lipocalin. Mol. Cell 10:1045–1056. 10.1016/S1097-2765(02)00710-4 [DOI] [PubMed] [Google Scholar]
- 28.Schwyn B, Neilands JB. 1987. Universal chemical assay for the detection and determination of siderophores. Anal. Biochem. 160:47–56. 10.1016/0003-2697(87)90612-9 [DOI] [PubMed] [Google Scholar]
- 29.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249–264. 10.1093/biostatistics/4.2.249 [DOI] [PubMed] [Google Scholar]
- 30.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. 2011. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27:1739–1740. 10.1093/bioinformatics/btr260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sunters A, Armstrong VJ, Zaman G, Kypta RM, Kawano Y, Lanyon LE, Price JS. 2010. Mechano-transduction in osteoblastic cells involves strain-regulated estrogen receptor alpha-mediated control of insulin-like growth factor (IGF) I receptor sensitivity to Ambient IGF, leading to phosphatidylinositol 3-kinase/AKT-dependent Wnt/LRP5 receptor-independent activation of beta-catenin signaling. J. Biol. Chem. 285:8743–8758. 10.1074/jbc.M109.027086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A. 2013. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 41:D991–D995. 10.1093/nar/gks1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le NT, Richardson DR. 2004. Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene: a link between iron metabolism and proliferation. Blood 104:2967–2975. 10.1182/blood-2004-05-1866 [DOI] [PubMed] [Google Scholar]
- 34.del Peso L, Castellanos MC, Temes E, Martin-Puig S, Cuevas Y, Olmos G, Landazuri MO. 2003. The von Hippel Lindau/hypoxia-inducible factor (HIF) pathway regulates the transcription of the HIF-proline hydroxylase genes in response to low oxygen. J. Biol. Chem. 278:48690–48695. 10.1074/jbc.M308862200 [DOI] [PubMed] [Google Scholar]
- 35.Thomas F, Serratrice G, Beguin C, Aman ES, Pierre JL, Fontecave M, Laulhere JP. 1999. Calcein as a fluorescent probe for ferric iron. Application to iron nutrition in plant cells. J. Biol. Chem. 274:13375–13383 [DOI] [PubMed] [Google Scholar]
- 36.Jeong HJ, Hong SH, Park RK, Shin T, An NH, Kim HM. 2005. Hypoxia-induced IL-6 production is associated with activation of MAP kinase, HIF-1, and NF-kappaB on HEI-OC1 cells. Hear. Res. 207:59–67. 10.1016/j.heares.2005.04.003 [DOI] [PubMed] [Google Scholar]
- 37.Eltzschig HK, Carmeliet P. 2011. Hypoxia and inflammation. N. Engl. J. Med. 364:656–665. 10.1056/NEJMra0910283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM. 2006. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: the role of HIF-1alpha, HIF-2alpha, and other pathways. J. Biol. Chem. 281:15215–15226. 10.1074/jbc.M511408200 [DOI] [PubMed] [Google Scholar]
- 39.Zhang J, Wu Y, Zhang Y, Leroith D, Bernlohr DA, Chen X. 2008. The role of lipocalin 2 in the regulation of inflammation in adipocytes and macrophages. Mol. Endocrinol. 22:1416–1426. 10.1210/me.2007-0420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee S, Kim JH, Seo JW, Han HS, Lee WH, Mori K, Nakao K, Barasch J, Suk K. 2011. Lipocalin-2 is a chemokine inducer in the central nervous system: role of chemokine ligand 10 (CXCL10) in lipocalin-2-induced cell migration. J. Biol. Chem. 286:43855–43870. 10.1074/jbc.M111.299248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sickinger S, Maier H, Konig S, Vallant N, Kofler M, Schumpp P, Schwelberger H, Hermann M, Obrist P, Schneeberger S, Margreiter R, Troppmair J, Pratschke J, Aigner F. 2013. Lipocalin-2 as mediator of chemokine expression and granulocyte infiltration during ischemia and reperfusion. Transpl. Int. 26:761–769. 10.1111/tri.12116 [DOI] [PubMed] [Google Scholar]
- 42.Wrighting DM, Andrews NC. 2006. Interleukin-6 induces hepcidin expression through STAT3. Blood 108:3204–3209. 10.1182/blood-2006-06-027631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nemeth E, Ganz T. 2006. Regulation of iron metabolism by hepcidin. Annu. Rev. Nutr. 26:323–342. 10.1146/annurev.nutr.26.061505.111303 [DOI] [PubMed] [Google Scholar]
- 44.Chen K, McAleer JP, Lin Y, Paterson DL, Zheng M, Alcorn JF, Weaver CT, Kolls JK. 2011. Th17 cells mediate clade-specific, serotype-independent mucosal immunity. Immunity 35:997–1009. 10.1016/j.immuni.2011.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murakami M, Hirano T. 2012. The pathological and physiological roles of IL-6 amplifier activation. Int. J. Biol. Sci. 8:1267–1280. 10.7150/ijbs.4828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miossec P, Korn T, Kuchroo VK. 2009. Interleukin-17 and type 17 helper T cells. N. Engl. J. Med. 361:888–898. 10.1056/NEJMra0707449 [DOI] [PubMed] [Google Scholar]
- 47.Li Q, Laumonnier Y, Syrovets T, Simmet T. 2013. Recruitment of CCR6-expressing Th17 cells by CCL20 secreted from plasmin-stimulated macrophages. Acta Biochim. Biophys. Sin (Shanghai) 45:593–600. 10.1093/abbs/gmt049 [DOI] [PubMed] [Google Scholar]
- 48.Lee HJ, Choi SC, Choi EY, Lee MH, Seo GS, Kim EC, Yang BJ, Lee MS, Shin YI, Park KI, Jun CD. 2005. Iron chelator induces MIP-alpha/CCL20 in human intestinal epithelial cells: implication for triggering mucosal adaptive immunity. Exp. Mol. Med. 37:297–310. 10.1038/emm.2005.40 [DOI] [PubMed] [Google Scholar]
- 49.Warszawska JM, Gawish R, Sharif O, Sigel S, Doninger B, Lakovits K, Mesteri I, Nairz M, Boon L, Spiel A, Fuhrmann V, Strobl B, Muller M, Schenk P, Weiss G, Knapp S. 2013. Lipocalin 2 deactivates macrophages and worsens pneumococcal pneumonia outcomes. J. Clin. Investig. 123:3363–3372. 10.1172/JCI67911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fu D, Richardson DR. 2007. Iron chelation and regulation of the cell cycle: 2 mechanisms of posttranscriptional regulation of the universal cyclin-dependent kinase inhibitor p21CIP1/WAF1 by iron depletion. Blood 110:752–761. 10.1182/blood-2007-03-076737 [DOI] [PubMed] [Google Scholar]
- 51.Nurtjahja-Tjendraputra E, Fu D, Phang JM, Richardson DR. 2007. Iron chelation regulates cyclin D1 expression via the proteasome: a link to iron deficiency-mediated growth suppression. Blood 109:4045–4054. 10.1182/blood-2006-10-047753 [DOI] [PubMed] [Google Scholar]
- 52.Bachman MA, Lenio S, Schmidt L, Oyler JE, Weiser JN. 2012. Interaction of lipocalin 2, transferrin, and siderophores determines the replicative niche of Klebsiella pneumoniae during pneumonia. mBio 3:e00224–11. 10.1128/mBio.00224-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chepelev NL, Willmore WG. 2011. Regulation of iron pathways in response to hypoxia. Free Radic. Biol. Med. 50:645–666. 10.1016/j.freeradbiomed.2010.12.023 [DOI] [PubMed] [Google Scholar]
- 54.Hartmann H, Eltzschig HK, Wurz H, Hantke K, Rakin A, Yazdi AS, Matteoli G, Bohn E, Autenrieth IB, Karhausen J, Neumann D, Colgan SP, Kempf VA. 2008. Hypoxia-independent activation of HIF-1 by enterobacteriaceae and their siderophores. Gastroenterology 134:756–767. 10.1053/j.gastro.2007.12.008 [DOI] [PubMed] [Google Scholar]
- 55.Asikainen TM, Ahmad A, Schneider BK, Ho WB, Arend M, Brenner M, Gunzler V, White CW. 2005. Stimulation of HIF-1alpha, HIF-2alpha, and VEGF by prolyl 4-hydroxylase inhibition in human lung endothelial and epithelial cells. Free Radic. Biol. Med. 38:1002–1013. 10.1016/j.freeradbiomed.2004.12.004 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.