

Abstract
Gastroesophageal reflux (GER) frequently occurs in patients with respiratory disease and is particularly prevalent in patients with cystic fibrosis. GER is a condition in which the duodenogastric contents of the stomach leak into the esophagus, in many cases resulting in aspiration into the respiratory tract. As such, the presence of GER-derived bile acids (BAs) has been confirmed in the bronchoalveolar lavage fluid and sputum of affected patients. We have recently shown that bile causes cystic fibrosis-associated bacterial pathogens to adopt a chronic lifestyle and may constitute a major host trigger underlying respiratory infection. The current study shows that BAs elicit a specific response in humans in which they repress hypoxia-inducible factor 1α (HIF-1α) protein, an emerging master regulator in response to infection and inflammation. HIF-1α repression was shown to occur through the 26S proteasome machinery via the prolyl hydroxylase domain (PHD) pathway. Further analysis of the downstream inflammatory response showed that HIF-1α repression by BAs can significantly modulate the immune response of airway epithelial cells, correlating with a decrease in interleukin-8 (IL-8) production, while IL-6 production was strongly increased. Importantly, the effects of BAs on cytokine production can also be more dominant than the bacterium-mediated effects. However, the effect of BAs on cytokine levels cannot be fully explained by their ability to repress HIF-1α, which is not surprising, given the complexity of the immune regulatory network. The suppression of HIF-1 signaling by bile acids may have a significant influence on the progression and outcome of respiratory disease, and the molecular mechanism underpinning this response warrants further investigation.
INTRODUCTION
Gastroesophageal reflux (GER) frequently occurs in patients with advanced lung diseases (1). This is particularly prevalent in patients with cystic fibrosis (CF) (2–8), chronic obstructive pulmonary disease (9), and ventilator-associated pneumonia (10). GER is caused by the leak of stomach contents into the esophagus, which may then lead to aspiration of duodenogastric contents into the respiratory tract. As such, GER-derived bile acids (BAs) have been detected in the bronchoalveolar lavage (BAL) fluid and sputum of affected patients at concentrations ranging from 0.4 μM (8) up to 32 μM (11).
Interestingly, BA aspiration seems to promote a predisposition to pulmonary infection (7, 11–14), including that with Pseudomonas aeruginosa, the predominant pathogen associated with respiratory diseases and morbidity and mortality in CF patients (15, 16). Recently, we have shown that bile modulates P. aeruginosa virulence behavior toward a chronic lifestyle (12), which is associated with a more severe form of disease. Moreover, BA aspiration has also been linked with airway inflammation, whereby a correlation between alveolar neutrophils (8) and interleukin-8 (IL-8) (11, 14) with BAL fluid BAs has been found. Moreover, in a rodent model of chronic aspiration, BA aspiration has also been associated with increased BAL fluid tumor necrosis factor alpha (TNF-α) (17).
The mechanisms underlying the impact of BA aspiration on lung inflammation and infection are still not known. It is conceivable that BAs in the airways may trigger host factors, such as transcription factors, which in turn modulate immune response pathways. One potential target is hypoxia-inducible factor 1 (HIF-1). HIF-1 has recently been characterized to be an emerging master regulator in response to infection (18–20) and inflammation (21). HIF-1 is composed of two protein subunits, HIF-1α and HIF-1β, but HIF-1 activity is predominantly dependent on HIF-1α protein stabilization (22). Indeed, while the HIF-1β subunit is constitutively expressed in cells, expression of HIF-1α protein is regulated at a posttranslational level by prolyl hydroxylase domain (PHD) proteins through the hydroxylation of HIF-1α and subsequent proteasomal degradation (23, 24). The signal first described to stabilize HIF-1α was hypoxia, which is defined as a decrease of oxygen availability. Hypoxia, by inhibiting PHD activity, consequently prevents HIF-1α degradation, leading to activation of HIF-1. Since then, several other signals have been described. More recently, infections with human pathogens (25–30), including P. aeruginosa (30–32), and chronic inflammation, e.g., rheumatoid arthritis (21), have been shown to stabilize HIF-1α in immune and/or epithelial cells. Consequently, HIF-1 induces genes involved in the host immune response, such as antimicrobial peptides, nitric oxide, and several cytokines, including TNF-α, which help in the fight against and limit the spread of infection (30). Interestingly, the Pseudomonas quinolone signal (PQS) from P. aeruginosa has been shown to modulate HIF-1, indicating that individual molecules can elicit a specific response from HIF-1 (33).
In light of recent reports describing the significant impact of BA aspiration and the role of HIF-1 in lung inflammation and infection, the effect of BAs on HIF-1 signaling in airway epithelial cells was investigated. As bile aspiration is predominant in CF patients and chronic lung infection/inflammation cycles still remain a major cause of mortality (15, 16), the effect of BAs on HIF-1 signaling was studied in CF airway epithelial cells.
MATERIALS AND METHODS
Chemicals.
All reagents were obtained from Sigma-Aldrich (Arklow, Ireland), unless stated otherwise.
Cell culture.
The IB3-1 (ATCC CRL-2777) cell line is a bronchial epithelial cell line derived from a CF patient with CFTR ΔF508/W1282X alleles, and S9 cells (ATCC CRL-2778) are IB3-1 cells corrected for CFTR expression by transfection with wild-type adeno-associated viral CFTR (AAVCFTR). The A549 (ATCC CCL-185) cell line is a lung adenocarcinoma epithelial cell line derived from a patient with a lung carcinoma. IB3-1 and S9 cells were cultured in bovine serum albumin-collagen-fibronectin-coated flasks using LHC-8 medium (Invitrogen, Bio-Science, Dublin, Ireland) supplemented with 10% (vol/vol) fetal bovine serum (FBS) and a combination of 100 units/ml penicillin and 100 μg/ml streptomycin (Invitrogen). A549 cells were cultured using M7278 minimal essential medium supplemented with 10% (vol/vol) FBS, 2 mM l-glutamine (Invitrogen), and a combination of 100 units/ml of penicillin and 100 μg/ml of streptomycin (Invitrogen). All cell lines were purchased from the American Type Culture Collection (ATCC; LGC Standards, Teddington, United Kingdom). Cells were maintained at 37°C in a humidified 5% CO2 atmosphere and used up to passage 20. All experiments were performed on cells that had reached about 80% confluence, after which FBS was removed.
Bile acid compounds.
All sodium salts of cholic acid (CA; catalog no. C9282; Sigma-Aldrich), chenodeoxycholic acid (CDCA; catalog no. C8261; Sigma-Aldrich), and deoxycholic acid (DCA; catalog no. D6750; Sigma-Aldrich) and their respective conjugates and sodium salts of taurocholic acid (TCA; catalog no. 86339; Sigma-Aldrich), glycolic acid (GCA; catalog no. G7132; Sigma-Aldrich), taurochenodeoxycholic acid (TCDCA; catalog no. T6260; Sigma-Aldrich), glycochenodeoxycholic acid (GCDCA; catalog no. G0759; Sigma-Aldrich), taurodeoxycholic acid (TDCA; catalog no. T0875; Sigma-Aldrich), and glycodeoxycholic acid (GDCA; catalog no. G9910; Sigma-Aldrich) were dissolved in H2O at a concentration of 20 mM. Acids of CA (catalog no., C1129; Sigma-Aldrich), CDCA (catalog no. C9377; Sigma-Aldrich), and DCA (catalog no. D2510; Sigma-Aldrich) were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 20 mM.
Enzyme-linked immunosorbent assay (ELISA) analysis of cytokine production.
The levels of the antigens IL-8, IL-6, and TNF-α in the supernatants (50 μl) of control IB3-1 cells and IB3-1 cells that had been treated with P. aeruginosa PAO1, heat-killed PAO1, lipopolysaccharide (LPS) extracted from PAO1, or serotype 10 LPS (Sigma-Aldrich) in the presence or absence of CDCA (50 μM) were measured using a human Mix-N-Match multianalyte ELISArray kit (CMEH0532A; Qiagen, West Sussex, United Kingdom), according to the manufacturer's instructions. Infections with live and heat-killed cells were performed at a multiplicity of infection (MOI) of 25:1, while LPS was used at a final concentration of 10 μg/ml. A SpectraMax Plus384 spectrophotometer (Molecular Devices, Sunnyvale, CA) was used to assay the absorbance, and A450 data were corrected using A530 readings per the manufacturer's guidelines.
Infection procedure.
P. aeruginosa strain PAO1, originally obtained from B. Iglewski, was cultured aerobically for 16 h in LHC-8 medium at 37°C under agitation. Heat-killed PAO1 cells were obtained by incubation of phosphate-buffered saline (PBS)-washed cells at 95°C for 1 h. Aliquots were spread plated on nonselective agar and incubated at 37°C to confirm the heat-killed treatment. IB3-1 cells were infected at an MOI of 25 PAO1 cells to 1 IB3-1 cell for 3 h. The MOI was confirmed by plate counting of serial dilutions of strain PAO1 onto Luria broth (LB) plates.
LPS extraction and visualization.
LPS was extracted using the method described by Hitchcock and Brown (34). PAO1 cells were grown overnight in LB, and the pellet of 1.5 ml was collected by centrifugation at 13,000 rpm for 2 min. The cell pellet was resuspended in 1.5 ml PBS, after which the optical density at 600 nm was adjusted to 1.0 in a total volume of 5 ml PBS. The suspension was centrifuged at 13,000 rpm, and the supernatant was discarded. The pellet was resuspended in 100 μl of lysis buffer (10% SDS [100 μl], 60% glycerol [83 μl], 0.1 M Tris-HCl, pH 6.8 [100 μl], sterile H2O [197 μl], β-mercaptoethanol [20 μl]) and heated at 100°C for 10 min. The sample was then stored at −20°C overnight. Proteinase K (12.5 μl of a 2-mg/ml solution) was added to the thawed suspension and incubated at 60°C for 1 h. Following this, 10 μl RNase (1 mg/ml) and 5 μl DNase (1 mg/ml) were added, and the mixture was incubated at 37°C for 1 h. LPS was subsequently dialyzed extensively in sterile distilled water using a 12- to 14-kDa-cutoff membrane (Medicell International Ltd., London, United Kingdom). Purified LPS was solubilized in NuPAGE LDS sample buffer (Invitrogen, Bio-Science, Dublin, Ireland), and 16 μl from each sample was separated on a 15% SDS-gel with a 5% stacking gel under reducing conditions at 150 mA for 2 h. LPS was visualized by silver staining, performed using a PlusOne kit (Amersham GE Healthcare, Dublin, Ireland). Coomassie staining was performed to confirm the absence of bacterial proteins in the LPS extract.
Western blot analysis.
Total proteins were isolated from airway epithelial cells by sonication in a lysis buffer, as previously described (33). Twenty micrograms of proteins was resolved on an 8% (vol/vol) SDS-polyacrylamide gel and transferred to a nitrocellulose membrane (pore size, 0.45 μm; Amersham GE Healthcare, Dublin, Ireland). The following antibodies were used: mouse anti-human HIF-1α (catalog no. 610958; clone 54; BD Transduction Laboratories, Dublin, Ireland), mouse anti-human HIF-1β (catalog no. 611078; clone 29; BD Transduction Laboratories), and mouse anti-human heat shock cognate 70 (HSC70; catalog no. sc-7298; B-6; Santa Cruz Biotechnology, Heidelberg, Germany). These antibodies were diluted at ratios of 1:1,000, 1:2,000, and 1:10,000, respectively, according to the manufacturer's instructions. The secondary mouse antibody (Dako Diagnostics Ireland) was used at a dilution of 1:1,000. Detection was performed using enhanced chemiluminescence (Fisher Scientific, Pierce, Dublin, Ireland).
RNA isolation, cDNA synthesis, and quantitative real-time PCR.
Total RNAs were extracted from IB3-1 airway epithelial cells using an RNeasy minikit (Qiagen, West Sussex, United Kingdom). After DNase treatment using RQ1 RNase-free DNase (Promega, Southampton, United Kingdom), 1 μg of RNA was reverse transcribed into cDNA using oligo(dT) primer and avian myeloblastosis reverse transcriptase (Promega) according to the manufacturer's recommendations. Quantitative real-time PCR was performed using a FastStart TaqMan probe master (Roche Diagnostics, West Sussex, United Kingdom) and a PTC-200 thermocycler (MJ Research, Bio-Rad, United Kingdom) according to the manufacturer's instructions. The primer pairs (Table 1) used for each transcript were specifically designed using the Universal Probe Library Assay Design Center (Roche). The relative quantification of mRNA levels was calculated by the 2−ΔΔCT method using hypoxanthine phosphoribosyltransferase 1 (HPRT-1) threshold cycle (CT) values for normalization. In order to address the core biological questions, normalized values for IB3-1 cells alone were assigned an arbitrary value of 1 (30).
TABLE 1.
Primer pairs used for this studya
| GenBank accession no. | Gene name | Orientationb | Primer sequence | Probe no. from Roche |
|---|---|---|---|---|
| NM_000194.2 | HPRT-1 | F | TGACCTTGATTTATTTTGCATACC | 73 |
| HPRT-1 | R | CGAGCAAGACGTTCAGTCCT | ||
| NM_000600.3 | IL-6 | F | GATGAGTACAAAAGTCCTGATCCA | 40 |
| IL-6 | R | CTGCAGCCACTGGTTCTGT | ||
| NM_000584.2 | IL-8 | F | AGACAGCAGAGCACACAAGC | 72 |
| IL-8 | R | ATGGTTCCTTCCGGTGGT | ||
| NM_000594.2 | TNF-α | F | CAGCCTCTTCTCCTTCCTGAT | 29 |
| TNF-α | R | GCCAGAGGGCTGATTAGAGA | ||
| NM_000758.2 | GM-CSF | F | TCTCAGAAATGTTTGACCTCCA | 1 |
| GM-CSF | R | GCCCTTGAGCTTGGTGAG | ||
| NM_000759.3 | G-CSF | F | ACTTTGCCACCACCATCTG | 48 |
| G-CSF | R | TGGAAAGCAGAGGCGAAG | ||
| NM_000625.4 | NOS2 | F | ACCAGTACGTTTGGCAATGG | 37 |
| NOS2 | R | TCAGCATGAAGAGCGATTTCT |
Gene accession numbers, gene names, primer sequences, and the corresponding Roche probes used for quantitative real-time PCR are shown.
F, forward; R, reverse.
Cytotoxicity assay.
The release of lactate dehydrogenase (LDH) into cell culture supernatants was measured using a LDH cytotoxicity detection kit (Roche) according to the manufacturer's instructions. Briefly, IB3-1 cells were seeded onto 96-well plates and treated with BAs at 50 and 100 μM. Following 16 h of incubation at 37°C in 5% CO2, the supernatants were removed, added to a catalyst reaction mixture in a fresh plate, and further incubated at 37°C in 5% CO2 for 30 min to allow color development. After this period, the plate was analyzed on an ELISA plate reader at 490 nm. Cytotoxicity was expressed as a percentage of the total amount of LDH released from untreated cells and cells treated with DMSO or with BAs in comparison with the total amount of LDH released from cells treated with 0.1% Triton X-100 (which was considered 100% cytotoxicity).
Statistical analysis.
Three independent biological replicates were performed for all experiments described in this report. Statistical analyses were performed using a one- or two-way analysis of variance (ANOVA) test, followed by Tukey multiple-comparisons analysis. Statistical analysis of the ELISA data was performed by an unpaired, two-tailed Student t test, followed by the use of Welch's correction. Differences were considered significant if the P value was ≤0.05.
RESULTS
BAs repress HIF-1α protein in airway epithelial cells.
Hypoxia-inducible factor 1 (HIF-1) has recently been characterized to be an emerging master regulator in response to infection. As gastroesophageal reflux (GER)-derived bile aspiration has also been linked to lung infection, the effects of bile acids (BAs) on HIF-1 were studied. Clinical manifestations of GER disease, in particular, esophageal mucosal damage, are often attributed to BAs; however, evidence suggests that the pathophysiology of GER is far more complex (35). In order to ensure that the impact of bile on HIF-1 is due to a specific response rather than a general response, BAs were first tested for cytotoxicity toward the epithelial cell lines under investigation. For this, the four major human BAs (36), trihydroxylated cholic acid (CA), dihydroxylated chenodeoxycholic acid (CDCA) and deoxycholic acid (DCA), and monohydroxylated lithocholic acid (LCA), were incubated with IB3-1 airway epithelial cells. Concentrations of 50 and 100 μM were selected, as these are similar to the highest concentrations found in airway secretions (11). As BAs may be present in humans as sodium salts or acids, both versions were tested, except for LCA, where only acid was commercially available.
Treatment of IB3-1 cells by Triton X-100 at 0.1% (vol/vol) was chosen as a positive control of cytotoxicity, and the cytotoxicity that it caused was given an arbitrary value of 100%. LCA, the monohydroxylated BA, was found to be extremely toxic (close to 100%) for airway cells (Fig. 1A). Treatment with CDCA and DCA, the two dihydroxylated BAs, led to the moderate release of LDH (14 to 27%) at both concentrations used (Fig. 1A), although only the acid version was found to be significantly toxic in comparison to the effect of no treatment. The sodium salts versions used at 50 μM were not cytotoxic. CA, the trihydroxylated BA, was not found to be toxic for the airway epithelial cells, except for the acid version of CA used at 100 μM (7%) (Fig. 1A). LDH release was correlated with cell morphology, as illustrated with IB3-1 cell imaging (Fig. 1B). The acid version of LCA at 100 μM led to the destruction of all airway epithelial cells, whereas acid versions of CA, CDCA, and DCA at 100 μM did not significantly impact cell morphology in comparison with that of untreated control cells (Fig. 1B). Taken together, toxicity for airway cells was inversely proportional to the status of hydroxylation, which is similar to the toxicity found in other human tissues (36, 37).
FIG 1.
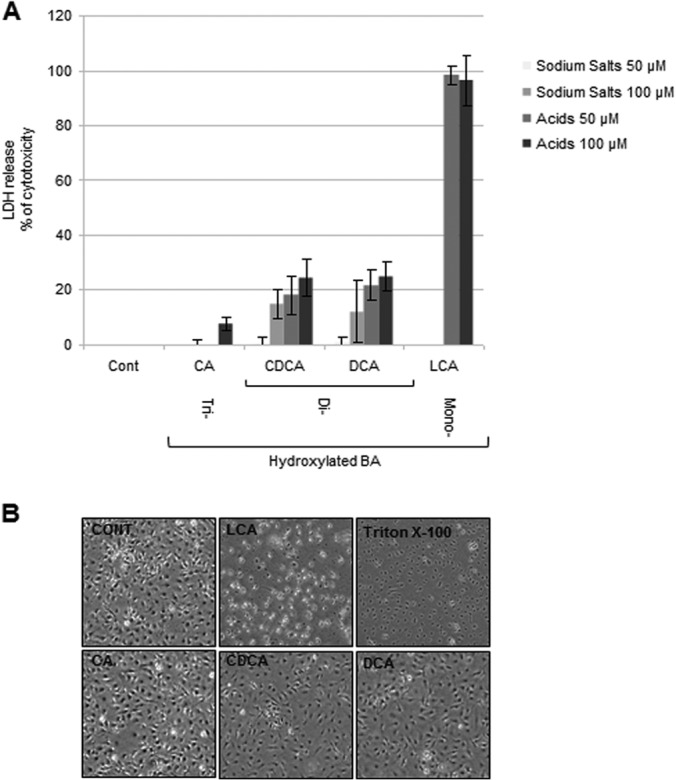
The toxicity of bile acids is inversely correlated to the degree of hydroxylation. (A) Release of LDH in the culture medium of IB3-1 airway epithelial cells that were untreated (control [Cont]) or treated with sodium salts of the trihydroxylated BA CA, the two dihydroxylated BAs (CDCA and DCA), acids of CA, CDCA, and DCA, or the monohydroxylated BA (LCA) at concentrations of 50 and 100 μM for 16 h. Cytotoxicity is expressed as the mean percentage ± standard deviation (SD) (n = 3) of the total amount of LDH released from IB3-1 cells treated with 0.1% Triton X-100, which was given an arbitrary value of 100%. A one-way ANOVA test was performed for comparison of treated cells with control untreated cells, followed by Tukey multiple-comparisons analysis. **, P ≤ 0.01; ***, P ≤ 0.001. (B) Phase-contrast microscopy of IB3-1 cells that were untreated (control) or treated with acids of LCA, CA, CDCA, and DCA at a concentration of 100 μM for 16 h or with Triton X-100 at 0.1%. Phase-contrast microscopy data represent those from one of three independent experiments with comparable results. Magnification, ×40.
The effects of three individual bile acids, CA, CDCA, and DCA, on HIF-1 signaling were assessed, as the toxicity of LCA was too high. BAs were tested in their noncytotoxic sodium salt forms, as these are the most prevalent forms under physiological conditions (38). Interestingly, incubation of CDCA and DCA with airway epithelial cells led to a dose-dependent downregulation of HIF-1α protein levels with a starting concentration of 25 μM for CDCA and 1 μM for DCA (Fig. 2A and B). In contrast, CA had no effect on HIF-1α protein levels even at the highest concentration of 100 μM (Fig. 2C). Importantly, similar results were obtained for the acid versions of CA, CDCA, and DCA (Fig. 2), suggesting that HIF-1α protein repression was not specific to acid or sodium salt versions of the BAs.
FIG 2.
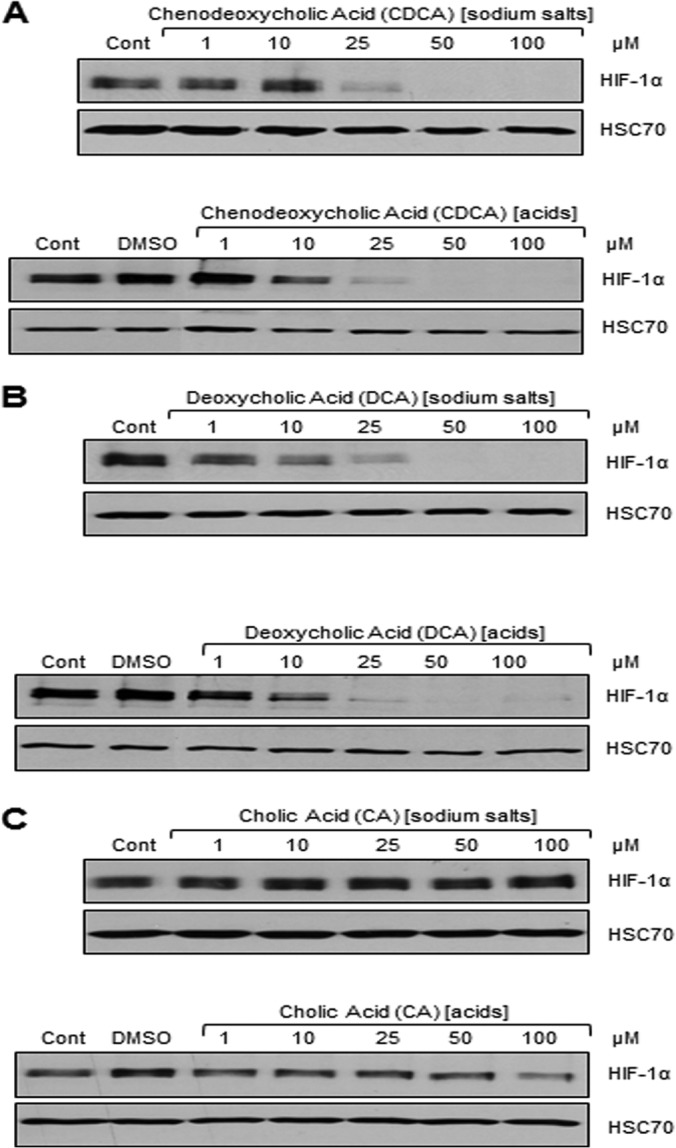
The two dihydroxylated BAs CDCA and DCA repress HIF-1α protein, whereas the monohydroxylated CA does not. The levels of HIF-1α and HSC70 (loading control) proteins in IB3-1 airway epithelial cells untreated (control) or treated with DMSO or with the sodium salt or acid version of CDCA (A), DCA (B), and CA (C) at a concentration of 1, 10, 25, 50, and 100 μM for 16 h are shown. Western blot data represent those from one of three independent experiments with comparable results.
In order to check if HIF-1α downregulation was linked to the CFTR mutation(s) and/or CF disease, the nontoxic concentration of 50 μM of the sodium salts of CDCA and DCA, which led to the complete repression of HIF-1α protein in IB3-1 cells, was tested in CFTR-corrected IB3-1 airway epithelial cells (S9 cells) and lung adenocarcinoma cells (A549 cells). Addition of both CDCA and DCA led to the repression of HIF-1α protein in both cell lines (Fig. 3), suggesting that HIF-1α protein repression is not related to the CF status but appears to be a general effect of dihydroxylated BAs. Finally, the effect of BAs appeared to be specific for HIF-1α protein repression, as the protein levels of the second HIF-1 protein subunit, HIF-1β, were not affected by either CDCA or DCA in any of the airway cell lines tested (Fig. 3).
FIG 3.
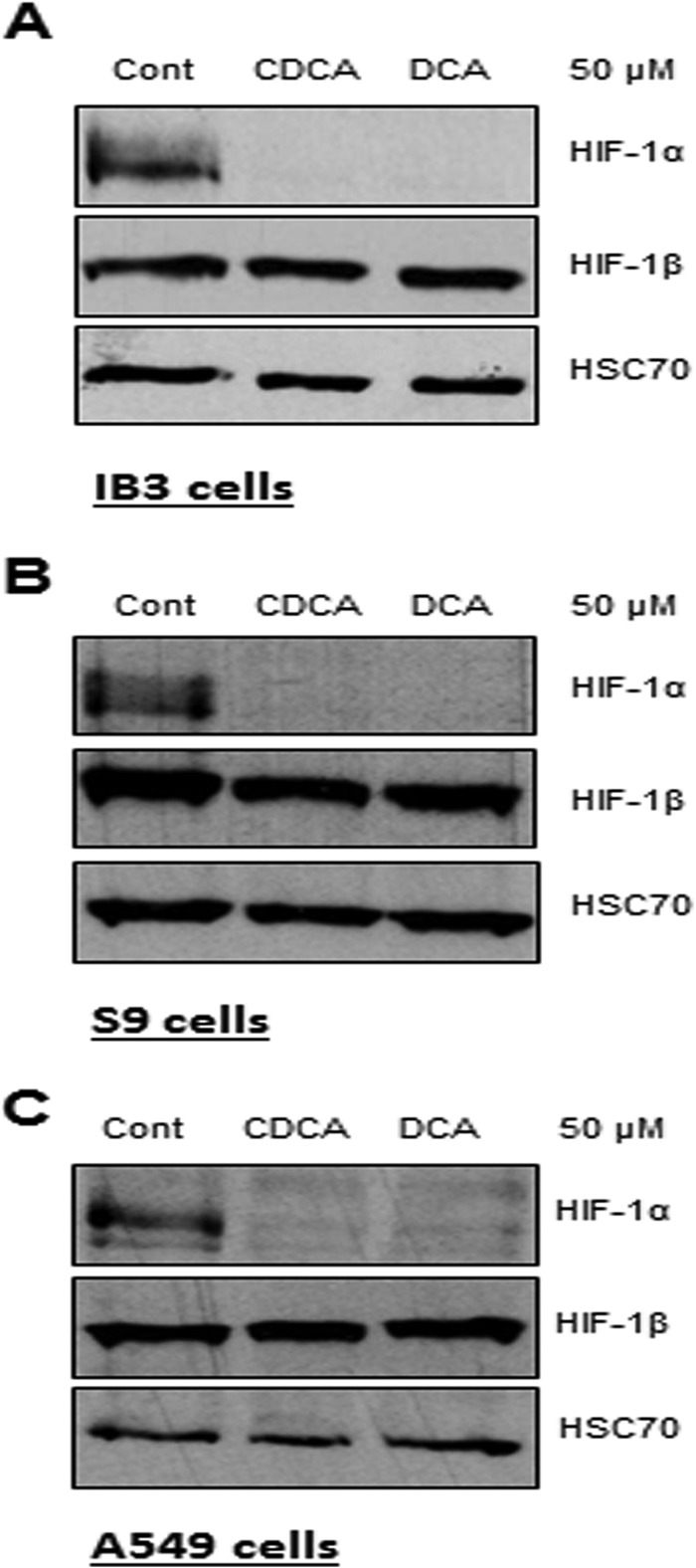
CDCA and DCA repress HIF-1α protein in CF and non-CF airway cells without affecting HIF-1β protein levels. The levels of the HIF-1α, HIF-1β, and HSC70 (loading control) proteins in CF-affected IB3-1 cells (A), CFTR-corrected S9 cells (B), and A549 airway epithelial cells (C) that were untreated (control) or treated with sodium salts of CDCA or DCA at a concentration of 50 μM for 16 h are shown. Western blot data represent those from one of three independent experiments with comparable results.
As both conjugated and unconjugated BAs have been detected in airway secretions (10), the impact of BAs conjugated with either taurine or glycine on HIF-1α protein levels was studied. Conjugation of CA had no effect on HIF-1α protein levels (Fig. 4A), whereas conjugation of CDCA and DCA by either taurine (TCDCA and TDCA, respectively) or glycine (GCDCA or GDCA, respectively) totally abolished HIF-1α protein downregulation (Fig. 4B and C). Altogether, unconjugated forms of the dihydroxylated BAs found in airway secretions, CDCA and DCA, led to the dose-dependent downregulation of HIF-1α protein.
FIG 4.

Conjugation of CDCA and DCA by either taurine or glycine abolishes HIF-1α protein repression. The levels of HIF-1α and HSC70 (loading control) proteins in IB3-1 airway epithelial cells untreated (control) or treated with sodium salts of nonconjugated CA (A), CDCA (B), and DCA (C) or CA, CDCA, and DCA conjugated with taurine (TCA, TCDCA, and TDCA, respectively) or glycine (GCA, GCDCA, and GDCA, respectively) at a concentration of 50 μM for 16 h are shown. Western blot data represent those from one of three independent experiments with comparable results.
CDCA and DCA led to HIF-1α protein degradation through the 26S proteasome machinery via the PHD pathway.
In order to investigate the mechanism by which CDCA and DCA repressed the HIF-1α protein, it was first investigated if HIF-1α was modulated at the mRNA level in IB3-1 cells. HIF-1α mRNA levels were not affected in response to CDCA and DCA compared to the levels in control untreated cells (data not shown), indicating that repression of the HIF-1α protein by CDCA and DCA was not due to modulation of its transcriptional expression.
HIF-1α protein is known to be strongly regulated at the posttranslational level. Prolyl hydroxylase domain (PHD) enzymes are major regulators of HIF-1α protein levels through an iron-oxygen-dependent hydroxylation mechanism, in turn leading to HIF-1α degradation by the 26S proteasome machinery (23, 24). Therefore, it was hypothesized that unconjugated dihydroxylated BA-mediated HIF-1α protein repression might occur at this posttranslational level. In order to study the potential involvement of this pathway, chemical inhibitors of PHD enzymes and the 26S proteasome machinery were used. Dimethyloxaloylglycine (DMOG; 2 mM), 2,2′-dipyridyl (DP; 100 μM), and cobalt chloride (CoCl2; 150 μM), all of which are known to be PHD inhibitors, and Z-Leu-Leu-Leu-al (MG132; 1 μM) and N-acetyl-Leu-Leu-norleucinal (MG101; 25 μM), both of which are 26S proteasome inhibitors, were tested. All of the inhibitors, alone or in the presence of CDCA and DCA, led to significant HIF-1α stabilization, in comparison to untreated control cells (Fig. 5). Therefore, in the presence of the PHD or 26S proteasome inhibitors, CDCA and DCA were no longer able to repress HIF-1α protein. Thus, CDCA and DCA degrade HIF-1α protein via the 26S proteasome machinery through the PHD enzymes, which are involved in the most characterized and major pathway involved in HIF-1 regulation.
FIG 5.

CDCA and DCA lead to concentration-dependent HIF-1α protein degradation through the PHD pathway and 26S proteasome machinery. The levels of HIF-1α and HSC70 (loading control) proteins in IB3-1 airway epithelial cells that were untreated (control) or treated with the PHD inhibitor DMOG (2 mM and 0.2 mM) (A), DP (100 μM and 25 μM) (B), or CoCl2 (150 μM and 25 μM) (C) or with the 26S proteasome inhibitor MG132 (1 μM) (D) or MG101 (25 μM) (E), alone or in association with treatment with the sodium salts of CDCA or DCA at a concentration of 50 μM for 16 h, are shown. Western blot data represent those from one of three independent experiments with comparable results.
However, importantly, when PHD inhibitors were used at a lower concentration but a concentration sufficient for HIF-1α stabilization, i.e., 200 μM for DMOG (Fig. 5A) and 25 μM for DP and CoCl2 (Fig. 5B and C), both CDCA and DCA were still able to repress HIF-1α protein, suggesting that a minimal concentration of PHD inhibitors is required to block dihydroxylated BA-mediated HIF-1α protein degradation.
CDCA represses HIF-1α protein and modulates immune-related cytokine levels during infection with P. aeruginosa.
HIF-1 is an important regulator of several key host response networks, and key targets include the inflammation-related cytokines IL-6, IL-8, and TNF-α (39, 40). Therefore, to establish the downstream significance of HIF-1α modulation by BAs, ELISA analysis for all three cytokines was performed in IB3-1 cells in the presence and absence of CDCA, a BA that has been specifically found in the airway secretions of patients with ventilator-associated pneumonia (10). CDCA treatment led to a reduction in IL-8 production (with the levels in CDCA-treated cells falling below the limit of detection of the ELISA), while the level of IL-6 was significantly increased 7.3-fold (±0.86-fold) compared to that in IB3-1 cells in the absence of CDCA (Fig. 6A). The expression of TNF-α in the absence of CDCA was below the limit of detection for the assay, and while the level of expression was increased by CDCA, the increase did not reach significance (data not shown). Thus, CDCA appears to influence immune response cytokines, in addition to HIF-1α, and warrants further investigation.
FIG 6.

Under conditions of infection with P. aeruginosa, CDCA represses HIF-1α protein levels and affects proinflammatory cytokine expression profiles. (A) ELISA analysis of cytokine production by IB3-1 cells in the presence or absence of 50 μM CDCA for uninfected cells or for cells at 3 h after treatment with PAO1, heat-killed (HK) PAO1, or LPS extracted from PAO1 cells. #, values that were below the limit of detection of the ELISA kit. Statistical significance was assessed via an unpaired, two-tailed Student t test with the Welch correction. *, P < 0.05; **, P < 0.005. The data presented represent those from three independent replicates. (B) Western blot analysis of HIF-1α and HSC70 (loading control) proteins in IB3-1 airway epithelial cells in the presence or absence of 50 μM CDCA for uninfected cells or for cells at 3 h after treatment with PAO1, heat-killed PAO1, or LPS extracted from PAO1 cells. Data represent those from one of three independent experiments with comparable results. (C) Relative expression of IL-6, IL-8, TNF-α, GM-CSF, and G-CSF mRNA in IB3-1 airway epithelial cells that were uninfected (control), infected with P. aeruginosa PAO1 at an MOI of 25:1 for 3 h (PAO1) in combination with preincubation with sodium salts of CDCA (PAO1 + CDCA), or treated with CDCA only (CDCA) at a concentration of 50 μM for 19 h. The fold change is expressed as the mean (n = 3) and is relative to the level for untreated and uninfected control cells, for which expression was given an arbitrary value of 1, except for TNF-α, where the fold change is relative to that for cells infected with PAO1, which was given an arbitrary value of 1. ND, not determined. A two-way ANOVA test was performed. *, P ≤ 0.05 in comparison with IB3-1 alone (control); **, P ≤ 0.01 in comparison with IB3-1 alone (control); ***, P ≤ 0.001 in comparison with IB3-1 alone (control); ∧, P ≤ 0.05 in comparison with PAO1-treated cells; ∧∧∧, P ≤ 0.001 in comparison with PAO1-treated cells; ##, P ≤ 0.01 in comparison with CDCA-treated cells; ###, P ≤ 0.001 in comparison with CDCA-treated cells.
GER and respiratory infections often co-occur in individuals, particularly in CF patients. As shown in this study, BAs are able to repress HIF-1α protein and modulate cytokine production. On the other hand, bacterial cell contact is known to induce HIF-1α (31, 33), though there is also emerging evidence that secreted signal molecules can selectively suppress this major regulator (33). Therefore, the effect of BAs on HIF-1α levels during infection was investigated. For this, IB3-1 airway epithelial cells were infected with P. aeruginosa strain PAO1 in the absence or the presence of CDCA. Infection by PAO1 for 3 h at an MOI of 25:1 led to HIF-1α protein stabilization in IB3-1 airway epithelial cells, as previously published (31) (Fig. 6B). As demonstrated here, the addition of CDCA alone led to HIF-1α protein degradation. Interestingly, pretreatment with CDCA at 50 μM for 16 h prevented HIF-1α stabilization in response to PAO1 infection. This impact of CDCA on HIF-1α protein levels in response to infection was not due to a decrease in bacterial growth, as the number of PAO1 cells in the cell culture supernatant or attached to the airway cells was found to be the same under all conditions (data not shown). ELISA analysis of cytokine production in infected cells revealed significant suppression of IL-8 by CDCA (P = 0.0124), while IL-6 production was significantly increased (P = 0.0021) upon addition of the BAs compared to that with infection alone (Fig. 6A).
To further investigate the role of BAs during infection, the mRNA levels of the cytokines IL-6, IL-8, and TNF-α plus those of granulocyte macrophage colony-stimulating factor (GM-CSF) and granulocyte colony-stimulating factor (G-CSF) (all of which are found in the airway secretions of CF patients [41–43]) were investigated in the presence and absence of CDCA and in response to infection with P. aeruginosa strain PAO1. All cytokines except TNF-α were detected in untreated controls, and their levels were given an arbitrary value of 1. TNF-α mRNA was detected only upon infection, and its level was then given an arbitrary value of 1 (Fig. 6C). All cytokines were significantly induced by PAO1 infection in comparison with their levels in uninfected and untreated control cells (Fig. 6C). When cells pretreated with CDCA were infected with P. aeruginosa, this pattern of cytokine expression was altered. PAO1 induction of IL-8 and GM-CSF mRNA expression was significantly reduced in the presence of CDCA, whereas PAO1 induction of IL-6, TNF-α, and G-CSF mRNA expression was significantly enhanced by pretreatment with CDCA (Fig. 6C). Interestingly, in agreement with the ELISA data, treatment with CDCA alone significantly induced IL-6 mRNA expression in the presence and absence of infection and had no significant effect on TNF-α expression. While the reduction in IL-8 protein levels by CDCA was not reflected at the level of mRNA expression, it was interesting to note the significant reduction in IL-8 induction by PAO1 in the presence of CDCA. Moreover, CDCA significantly repressed GM-CSF and G-CSF expression in comparison with that in uninfected and untreated control cells.
The cytokines IL-8, IL-6, and TNF-α have all been characterized to be HIF-1 targets (39, 40). Therefore, while it is clear that BAs can significantly modulate the immune response of airway epithelial cells to infection, the effect of CDCA on cytokine levels cannot be fully explained by its ability to repress HIF-1α protein. This is not surprising, given the complexity of the immune regulatory network. Expression of the mRNA of another important HIF-1 target gene, the gene for inducible nitric oxide synthase (iNOS), was not detected in the IB3-1 cell line under untreated/treated and/or infected conditions. Moreover, in this cell line, iNOS mRNA was also not expressed after hypoxic exposure (1% O2 for 16 h in a Coy Scientific hypoxia chamber) or following treatment with human TNF-α (HumanKine; Sigma-Aldrich) at 5 or 50 ng/ml after 4 h or 16 h (data not shown).
Importantly, bacterial LPS can affect HIF-1 signaling and downstream immune responses, and hence, it is possible that BAs could mediate their effect during infection through modulation of LPS. Therefore, LPS (serotype O5) was extracted from PAO1 and used to treat IB3-1 cells as described above. Upon exposure to PAO1 LPS, HIF-1α was found to be weakly stabilized (Fig. 6B) and cytokine production was enhanced (the level of IL-8 production increased 7.09-fold [±2.16-fold], and the level of IL-6 production increased 2.34-fold [±0.025-fold]) (Fig. 6A). This was in contrast to the results for serotype 10 LPS (Sigma-Aldrich) used at 0.1 or 10 μg/ml for 4 h or 16 h, which did not impact cytokine levels (Fig. 7A and data not shown) or HIF-1α protein levels (Fig. 7B) in IB3-1 cells, which is supported by the observations made in a previous study (44). Moreover, PAO1 LPS and serotype 10 LPS showed distinct profiles (data not shown). It was also interesting to note that heat-killed PAO1 cells were capable of stabilizing HIF-1α (Fig. 6B), as well as significantly inducing cytokine production, with IL-8 levels increasing 6.74-fold (±2.01-fold) and IL-6 levels increasing 2.01-fold (±0.164-fold) at 3 h postinfection (Fig. 6A). As with live cells, addition of CDCA led to a reduction in IL-8 levels and increased IL-6 production compared to the IL-8 levels and levels of IL-6 production obtained by infection with heat-killed cells or LPS treatment alone. However, in the case of IL-8, the reduction was not significant. Taken together, these findings indicate a complex interaction between pathogen and host involving bile acids, HIF-1, and cytokine production.
FIG 7.

(A) ELISA analysis of IB3-1 cytokine production at 3 h postinfection with LPS from P. aeruginosa serotype 10 (Sigma-Aldrich). #, values that were below the limit of detection of the ELISA kit. Statistical significance was assessed via an unpaired, two-tailed Student t test with the Welch correction. *, P < 0.05. Data presented represent those from three independent replicates. (B) Levels of HIF-1α and HSC70 (loading control) proteins in IB3-1 airway epithelial cells untreated (control) or treated with LPS (Sigma-Aldrich) at 0.1 or 10 μg/ml after 4 h or 16 h. Western blot data represent those from one of three independent experiments with comparable results.
DISCUSSION
The aim of this study was to investigate the impact of BAs on the airway response, particularly the effect on HIF-1, a key master regulator of the immune and infection response. BAs are emerging as important factors contributing to respiratory infection and inflammation, although to date the mechanism(s) of action has not been elucidated. Here we demonstrate for the first time that certain BAs, both in their acid and in their salt forms, can specifically repress HIF-1α stabilization and that this is mediated at least in part via the PHD/26S proteasome pathway (see Fig. 8 for an overview).
FIG 8.

Impact of bile acids on HIF-1 signaling. GER-associated bile aspiration frequently occurs in patients with CF, leading to the presence of physiologically active concentrations of BAs in the lower respiratory tract of these patients. Although aspirated BAs are known to modulate components of the immune response, the host regulators underpinning this remain largely unknown. In this study, we demonstrate that (i) BAs and particularly unconjugated CDCA and DCA destabilize HIF-1α protein levels through the 26S proteasome machinery via the PHD pathway. Moreover, we demonstrate that (ii) CDCA represses the HIF-1α protein induced upon infection with P. aeruginosa, one of the main pathogens associated with CF disease. Finally, we show that (iii) CDCA modulates the proinflammatory cytokine gene expression profile under basal conditions and in response to infection. These findings are highly significant in light of two recent reports showing that BAs influence respiratory pathogens toward a chronic lifestyle (12) and that the P. aeruginosa PQS signal molecule, triggered by bile, can also directly repress HIF-1α protein (33). Therefore, (iv) bile aspiration and PQS may adversely impact the immune response through dual or synergistic repression of HIF-1α. Taken together, repression of HIF-1 signaling by BAs and/or PQS and the resultant perturbation of the cytokine profile may have a significant effect on the progression of CF disease by promoting and/or maintaining infections.
It was interesting to note that conjugation of the BAs eliminated the repression of HIF-1α stabilization. However, it has been reported previously that conjugation of BAs leads to the loss of the biological effect (45, 46). Although, in general, most of the BAs are present in humans in their conjugated forms (36), GER treatment, including treatment with proton pump inhibitors and acid suppression therapy, has been shown to result in deconjugation of BAs (47). Therefore, the ratio of unconjugated BAs/conjugated BAs is increased in these patients (47), and indeed, the unconjugated forms of CDCA, DCA, and LCA have all been found in airway secretions, whereas only the glycoconjugate form of CDCA (GCDCA) has been detected (10). Thus, it is conceivable that the effect of unconjugated BAs on HIF-1α stabilization may be the most clinically relevant in GER patients.
Both HIF-1 and BAs are known to modulate the immune response. More than 30 years ago, BAs were described for their immunosuppressive activities in cholestasis (48), which is defined by the arrest of bile flow and associated with elevated serum BA levels. Indeed, cholestasis has frequently been linked to the impaired function of macrophages (48) and lymphocytes (49). In vitro experiments have shown that BAs are able to directly inhibit the production of proinflammatory cytokines (50, 51). As such, the immunosuppressive properties of BAs could occur through the HIF-1 pathway. Indeed, in our study, we show that the most prevalent unconjugated BA found in airway secretions (10), CDCA, modulates cytokine expression under basal conditions but also in response to infection with P. aeruginosa (Fig. 6 and 8). This could be partially explained by the ability of CDCA to cause the degradation of HIF-1α, which, in particular, would effect IL-8 expression, which is known to be controlled by the HIF-1 pathway (26, 39). Interestingly, the expression of GM-CSF is also decreased by CDCA, but GM-CSF has not yet been described to be a direct HIF-1 target gene. However, IL-6 and TNF-α expression, which has been shown to be controlled by HIF-1 (30, 40), was strongly potentiated in epithelial cells in the presence of CDCA, a condition under which HIF-1α is suppressed, reflecting the different levels of regulation and the complexity of the immune system. It has recently been reported that IL-6 can compromise tissue repair, shifting the inflammatory response from an acute to a chronic profibrotic stage (52). In addition, HIF-1 stabilization has been shown to be central to effective inflammatory resolution in intestinal cells, with HIF-1 interference leading to chronic inflammation (53). Taken together, our data strongly support the hypothesis that BA modulation of cytokines and HIF-1α may contribute to the chronic inflammation and reduced lung function associated with aspiration in patients with respiratory disease.
It has been established that HIF-1α is stabilized in response to bacterial infection (23). Therefore, the suppression of HIF-1α by BAs during coculture experiments is a significant development. Although the pathway underpinning this suppression remains to be elucidated, there is evidence to suggest both direct and indirect mechanisms. Previously, it was shown that the effects of BAs on LPS- or endotoxin-stimulated TNF-α secretion were not a result of a direct interaction between BAs and LPS (54) or inactivation of endotoxin (51) but occurred through a direct inhibitory effect of BAs on the cells. It has been postulated that the immunosuppression caused by BAs might be a result of cell membrane damage. However, in this study and in a previous study (54), no cytotoxicity was observed at the concentrations used. Additionally, our results show that CDCA did not affect P. aeruginosa growth. PAO1 LPS was specifically found to be involved in the modulation of HIF-1α by BAs in our model, while heat-killed PAO1 also induced both IL-6 and IL-8, consistent with the findings presented in previous reports (55, 56). However, this does not rule out the involvement of bacterial secreted factors in BA-mediated suppression of HIF-1α and modulation of the immune response. We have recently shown that the P. aeruginosa signaling molecule PQS is capable of repressing HIF-1α at physiologically relevant concentrations through a PHD-independent mechanism (33). We have also shown that PQS production in P. aeruginosa is strongly stimulated by BAs (12), suggesting that synergistic suppression of HIF-1α may occur during infection of CF patients (Fig. 8), up to 80% of whom are GER and P. aeruginosa positive (4, 8).
Taken together, these new BA/HIF-1 interaction data tend to suggest that the disruption in HIF-1 signaling, coupled with the resulting modulation in cytokine levels, may actually promote infection(s) (7, 11, 13, 14) and/or maintain chronic infections (12), thus further contributing to the process of chronic inflammation in patients suffering from GER and pulmonary diseases (Fig. 8).
ACKNOWLEDGMENTS
We thank Pat Higgins and Emma Hennessy for excellent technical assistance.
This research was supported in part by grants awarded to F.O. by the Science Foundation of Ireland (SSPC2 12/RC/2275; 13-TIDA-B2625, 07/IN.1/B948; 12/TIDA/B2411; 12/TIDA/B2405,09/RFP/BMT 2350); the Department of Agriculture, Fisheries and Food (DAFF11/F/009 MabS; FIRM/RSF/CoFoRD; FIRM 08/RDC/629); the Environmental Protection Agency (EPA 2008-PhD/S-2); the Irish Research Council for Science, Engineering and Technology (PD/2011/2414; RS/2010/2413); the European Commission (FP7-PEOPLE-2013-ITN 607786; OCEAN2012 287589; FP7-KBBE-2012-6 CP-TP 311975; FP7-KBBE-2012-6 CP-TP-312184; Marie Curie 256596); the Marine Institute (Beaufort award C2CRA 2007/082); Teagasc (Walsh Fellowship 2013); and the Health Research Board (HRA/2009/146).
Footnotes
Published ahead of print 9 June 2014
REFERENCES
- 1.Sweet MP, Patti MG, Hoopes C, Hays SR, Golden JA. 2009. Gastro-oesophageal reflux and aspiration in patients with advanced lung disease. Thorax 64:167–173. 10.1136/thx.2007.082719 [DOI] [PubMed] [Google Scholar]
- 2.Blondeau K, Dupont LJ, Mertens V, Verleden G, Malfroot A, Vandenplas Y, Hauser B, Sifrim D. 2008. Gastro-oesophageal reflux and aspiration of gastric contents in adult patients with cystic fibrosis. Gut 57:1049–1055. 10.1136/gut.2007.146134 [DOI] [PubMed] [Google Scholar]
- 3.Blondeau K, Pauwels A, Dupont L, Mertens V, Proesmans M, Orel R, Brecelj J, Lopez-Alonso M, Moya M, Malfroot A, De Wachter E, Vandenplas Y, Hauser B, Sifrim D. 2010. Characteristics of gastroesophageal reflux and potential risk of gastric content aspiration in children with cystic fibrosis. J. Pediatr. Gastroenterol. Nutr. 50:161–166. 10.1097/MPG.0b013e3181acae98 [DOI] [PubMed] [Google Scholar]
- 4.Hallberg K, Fandriks L, Strandvik B. 2004. Duodenogastric bile reflux is common in cystic fibrosis. J. Pediatr. Gastroenterol. Nutr. 38:312–316. 10.1097/00005176-200403000-00016 [DOI] [PubMed] [Google Scholar]
- 5.Ledson MJ, Tran J, Walshaw MJ. 1998. Prevalence and mechanisms of gastro-oesophageal reflux in adult cystic fibrosis patients. J. R. Soc. Med. 91:7–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mousa HM, Woodley FW. 2012. Gastroesophageal reflux in cystic fibrosis: current understandings of mechanisms and management. Curr. Gastroenterol. Rep. 14:226–235. 10.1007/s11894-012-0261-9 [DOI] [PubMed] [Google Scholar]
- 7.Palm K, Sawicki G, Rosen R. 2012. The impact of reflux burden on Pseudomonas positivity in children with cystic fibrosis. Pediatr. Pulmonol. 47:582–587. 10.1002/ppul.21598 [DOI] [PubMed] [Google Scholar]
- 8.Pauwels A, Decraene A, Blondeau K, Mertens V, Farre R, Proesmans M, Van Bleyenbergh P, Sifrim D, Dupont LJ. 2012. Bile acids in sputum and increased airway inflammation in patients with cystic fibrosis. Chest 141:1568–1574. 10.1378/chest.11-1573 [DOI] [PubMed] [Google Scholar]
- 9.Rascon-Aguilar IE, Pamer M, Wludyka P, Cury J, Coultas D, Lambiase LR, Nahman NS, Vega KJ. 2006. Role of gastroesophageal reflux symptoms in exacerbations of COPD. Chest 130:1096–1101. 10.1378/chest.130.4.1096 [DOI] [PubMed] [Google Scholar]
- 10.Wu YC, Hsu PK, Su KC, Liu LY, Tsai CC, Tsai SH, Hsu WH, Lee YC, Perng DW. 2009. Bile acid aspiration in suspected ventilator-associated pneumonia. Chest 136:118–124. 10.1378/chest.08-2668 [DOI] [PubMed] [Google Scholar]
- 11.D'Ovidio F, Mura M, Tsang M, Waddell TK, Hutcheon MA, Singer LG, Hadjiliadis D, Chaparro C, Gutierrez C, Pierre A, Darling G, Liu M, Keshavjee S. 2005. Bile acid aspiration and the development of bronchiolitis obliterans after lung transplantation. J. Thorac. Cardiovasc. Surg. 129:1144–1152. 10.1016/j.jtcvs.2004.10.035 [DOI] [PubMed] [Google Scholar]
- 12.Reen FJ, Woods DF, Mooij MJ, Adams C, O'Gara F. 2012. Respiratory pathogens adopt a chronic lifestyle in response to bile. PLoS One 7:e45978. 10.1371/journal.pone.0045978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Doef HP, Arets HG, Froeling SP, Westers P, Houwen RH. 2009. Gastric acid inhibition for fat malabsorption or gastroesophageal reflux disease in cystic fibrosis: longitudinal effect on bacterial colonization and pulmonary function. J. Pediatr. 155:629–633. 10.1016/j.jpeds.2009.06.040 [DOI] [PubMed] [Google Scholar]
- 14.Vos R, Blondeau K, Vanaudenaerde BM, Mertens V, Van Raemdonck DE, Sifrim D, Dupont LJ, Verleden GM. 2008. Airway colonization and gastric aspiration after lung transplantation: do birds of a feather flock together? J. Heart Lung Transplant. 27:843–849. 10.1016/j.healun.2008.05.022 [DOI] [PubMed] [Google Scholar]
- 15.Gibson RL, Burns JL, Ramsey BW. 2003. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 168:918–951. 10.1164/rccm.200304-505SO [DOI] [PubMed] [Google Scholar]
- 16.O'Sullivan BP, Freedman SD. 2009. Cystic fibrosis. Lancet 373:1891–1904. 10.1016/S0140-6736(09)60327-5 [DOI] [PubMed] [Google Scholar]
- 17.Appel JZ, III, Lee SM, Hartwig MG, Li B, Hsieh CC, Cantu E, III, Yoon Y, Lin SS, Parker W, Davis RD. 2007. Characterization of the innate immune response to chronic aspiration in a novel rodent model. Respir. Res. 8:87. 10.1186/1465-9921-8-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eltzschig HK, Carmeliet P. 2011. Hypoxia and inflammation. N. Engl. J. Med. 364:656–665. 10.1056/NEJMra0910283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nizet V, Johnson RS. 2009. Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 9:609–617. 10.1038/nri2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zinkernagel AS, Johnson RS, Nizet V. 2007. Hypoxia inducible factor (HIF) function in innate immunity and infection. J. Mol. Med. (Berl.) 85:1339–1346. 10.1007/s00109-007-0282-2 [DOI] [PubMed] [Google Scholar]
- 21.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. 2003. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112:645–657. 10.1016/S0092-8674(03)00154-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang GL, Jiang BH, Rue EA, Semenza GL. 1995. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. U. S. A. 92:5510–5514. 10.1073/pnas.92.12.5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. 2001. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292:468–472. 10.1126/science.1059796 [DOI] [PubMed] [Google Scholar]
- 24.Schofield CJ, Ratcliffe PJ. 2005. Signalling hypoxia by HIF hydroxylases. Biochem. Biophys. Res. Commun. 338:617–626. 10.1016/j.bbrc.2005.08.111 [DOI] [PubMed] [Google Scholar]
- 25.Werth N, Beerlage C, Rosenberger C, Yazdi AS, Edelmann M, Amr A, Bernhardt W, von Eiff C, Becker K, Schafer A, Peschel A, Kempf VAJ. 2010. Activation of hypoxia inducible factor 1 is a general phenomenon in infections with human pathogens. PLoS One 5:e11576. 10.1371/journal.pone.0011576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cane G, Ginouves A, Marchetti S, Busca R, Pouyssegur J, Berra E, Hofman P, Vouret-Craviari V. 2010. HIF-1alpha mediates the induction of IL-8 and VEGF expression on infection with Afa/Dr diffusely adhering E. coli and promotes EMT-like behaviour. Cell. Microbiol. 12:640–653. 10.1111/j.1462-5822.2009.01422.x [DOI] [PubMed] [Google Scholar]
- 27.Rupp J, Gieffers J, Klinger M, van Zandbergen G, Wrase R, Maass M, Solbach W, Deiwick J, Hellwig-Burgel T. 2007. Chlamydia pneumoniae directly interferes with HIF-1alpha stabilization in human host cells. Cell. Microbiol. 9:2181–2191. 10.1111/j.1462-5822.2007.00948.x [DOI] [PubMed] [Google Scholar]
- 28.Sharma M, Machuy N, Bohme L, Karunakaran K, Maurer AP, Meyer TF, Rudel T. 2011. HIF-1alpha is involved in mediating apoptosis resistance to Chlamydia trachomatis-infected cells. Cell. Microbiol. 13:1573–1585. 10.1111/j.1462-5822.2011.01642.x [DOI] [PubMed] [Google Scholar]
- 29.Peyssonnaux C, Boutin AT, Zinkernagel AS, Datta V, Nizet V, Johnson RS. 2008. Critical role of HIF-1alpha in keratinocyte defense against bacterial infection. J. Investig. Dermatol. 128:1964–1968. 10.1038/jid.2008.27 [DOI] [PubMed] [Google Scholar]
- 30.Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS. 2005. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J. Clin. Invest. 115:1806–1815. 10.1172/JCI23865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Legendre C, Mooij MJ, Adams C, O'Gara F. 2011. Impaired expression of hypoxia-inducible factor-1alpha in cystic fibrosis airway epithelial cells—a role for HIF-1 in the pathophysiology of CF? J. Cyst. Fibros. 10:286–290. 10.1016/j.jcf.2011.02.005 [DOI] [PubMed] [Google Scholar]
- 32.Shao Z, Zhang Y, Ye Q, Saldanha JN, Powell-Coffman JA. 2010. C. elegans SWAN-1 binds to EGL-9 and regulates HIF-1-mediated resistance to the bacterial pathogen Pseudomonas aeruginosa PAO1. PLoS Pathog. 6:e1001075. 10.1371/journal.ppat.1001075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Legendre C, Reen FJ, Mooij MJ, McGlacken GP, Adams C, O'Gara F. 2012. Pseudomonas aeruginosa alkyl quinolones repress hypoxia-inducible factor 1 (HIF-1) signaling through HIF-1alpha degradation. Infect. Immun. 80:3985–3992. 10.1128/IAI.00554-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hitchcock PJ, Brown TM. 1983. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J. Bacteriol. 154:269–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tack J. 2006. Review article: the role of bile and pepsin in the pathophysiology and treatment of gastro-oesophageal reflux disease. Aliment. Pharmacol. Ther. 24(Suppl 2):S10–S16. 10.1111/j.1365-2036.2006.03040.x [DOI] [PubMed] [Google Scholar]
- 36.Hofmann AF. 1999. The continuing importance of bile acids in liver and intestinal disease. Arch. Intern. Med. 159:2647–2658. 10.1001/archinte.159.22.2647 [DOI] [PubMed] [Google Scholar]
- 37.Ferreira M, Coxito PM, Sardao VA, Palmeira CM, Oliveira PJ. 2005. Bile acids are toxic for isolated cardiac mitochondria: a possible cause for hepatic-derived cardiomyopathies? Cardiovasc. Toxicol. 5:63–73. 10.1385/CT:5:1:063 [DOI] [PubMed] [Google Scholar]
- 38.Begley M, Gahan CG, Hill C. 2002. Bile stress response in Listeria monocytogenes LO28: adaptation, cross-protection, and identification of genetic loci involved in bile resistance. Appl. Environ. Microbiol. 68:6005–6012. 10.1128/AEM.68.12.6005-6012.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim KS, Rajagopal V, Gonsalves C, Johnson C, Kalra VK. 2006. A novel role of hypoxia-inducible factor in cobalt chloride- and hypoxia-mediated expression of IL-8 chemokine in human endothelial cells. J. Immunol. 177:7211–7224. 10.4049/jimmunol.177.10.7211 [DOI] [PubMed] [Google Scholar]
- 40.Werno C, Menrad H, Weigert A, Dehne N, Goerdt S, Schledzewski K, Kzhyshkowska J, Brune B. 2010. Knockout of HIF-1alpha in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis 31:1863–1872. 10.1093/carcin/bgq088 [DOI] [PubMed] [Google Scholar]
- 41.Liou TG, Adler FR, Keogh RH, Li Y, Jensen JL, Walsh W, Packer K, Clark T, Carveth H, Chen J, Rogers SL, Lane C, Moore J, Sturrock A, Paine R, III, Cox DR, Hoidal JR. 2012. Sputum biomarkers and the prediction of clinical outcomes in patients with cystic fibrosis. PLoS One 7:e42748. 10.1371/journal.pone.0042748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McAllister F, Henry A, Kreindler JL, Dubin PJ, Ulrich L, Steele C, Finder JD, Pilewski JM, Carreno BM, Goldman SJ, Pirhonen J, Kolls JK. 2005. Role of IL-17A, IL-17F, and the IL-17 receptor in regulating growth-related oncogene-alpha and granulocyte colony-stimulating factor in bronchial epithelium: implications for airway inflammation in cystic fibrosis. J. Immunol. 175:404–412. 10.4049/jimmunol.175.1.404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Osika E, Cavaillon JM, Chadelat K, Boule M, Fitting C, Tournier G, Clement A. 1999. Distinct sputum cytokine profiles in cystic fibrosis and other chronic inflammatory airway disease. Eur. Respir. J. 14:339–346 [DOI] [PubMed] [Google Scholar]
- 44.Blau H, Klein K, Shalit I, Halperin D, Fabian I. 2007. Moxifloxacin but not ciprofloxacin or azithromycin selectively inhibits IL-8, IL-6, ERK1/2, JNK, and NF-kappaB activation in a cystic fibrosis epithelial cell line. Am. J. Physiol. Lung Cell. Mol. Physiol. 292:L343–L352. 10.1152/ajplung.00030.2006 [DOI] [PubMed] [Google Scholar]
- 45.Song S, Guha S, Liu K, Buttar NS, Bresalier RS. 2007. COX-2 induction by unconjugated bile acids involves reactive oxygen species-mediated signalling pathways in Barrett's oesophagus and oesophageal adenocarcinoma. Gut 56:1512–1521. 10.1136/gut.2007.121244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Debruyne PR, Bruyneel EA, Karaguni IM, Li X, Flatau G, Muller O, Zimber A, Gespach C, Mareel MM. 2002. Bile acids stimulate invasion and haptotaxis in human colorectal cancer cells through activation of multiple oncogenic signaling pathways. Oncogene 21:6740–6750. 10.1038/sj.onc.1205729 [DOI] [PubMed] [Google Scholar]
- 47.Theisen J, Nehra D, Citron D, Johansson J, Hagen JA, Crookes PF, DeMeester SR, Bremner CG, DeMeester TR, Peters JH. 2000. Suppression of gastric acid secretion in patients with gastroesophageal reflux disease results in gastric bacterial overgrowth and deconjugation of bile acids. J. Gastrointest. Surg. 4:50–54. 10.1016/S1091-255X(00)80032-3 [DOI] [PubMed] [Google Scholar]
- 48.Drivas G, James O, Wardle N. 1976. Study of reticuloendothelial phagocytic capacity in patients with cholestasis. Br. Med. J. 1:1568–1569. 10.1136/bmj.1.6025.1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keane RM, Gadacz TR, Munster AM, Birmingham W, Winchurch RA. 1984. Impairment of human lymphocyte function by bile salts. Surgery 95:439–443 [PubMed] [Google Scholar]
- 50.Calmus Y, Guechot J, Podevin P, Bonnefis MT, Giboudeau J, Poupon R. 1992. Differential effects of chenodeoxycholic and ursodeoxycholic acids on interleukin 1, interleukin 6 and tumor necrosis factor-alpha production by monocytes. Hepatology 16:719–723. 10.1002/hep.1840160317 [DOI] [PubMed] [Google Scholar]
- 51.Greve JW, Gouma DJ, Buurman WA. 1989. Bile acids inhibit endotoxin-induced release of tumor necrosis factor by monocytes: an in vitro study. Hepatology 10:454–458. 10.1002/hep.1840100409 [DOI] [PubMed] [Google Scholar]
- 52.Fielding CA, Jones GW, McLoughlin RM, McLeod L, Hammond VJ, Uceda J, Williams AS, Lambie M, Foster TL, Liao CT, Rice CM, Greenhill CJ, Colmont CS, Hams E, Coles B, Kift-Morgan A, Newton Z, Craig KJ, Williams JD, Williams GT, Davies SJ, Humphreys IR, O'Donnell VB, Taylor PR, Jenkins BJ, Topley N, Jones SA. 2014. Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity 40:40–50. 10.1016/j.immuni.2013.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, Bayless AJ, Scully M, Saeedi BJ, Golden-Mason L, Ehrentraut SF, Curtis VF, Burgess A, Garvey JF, Sorensen A, Nemenoff R, Jedlicka P, Taylor CT, Kominsky DJ, Colgan SP. 2014. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 40:66–77. 10.1016/j.immuni.2013.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, Hinuma S, Fujisawa Y, Fujino M. 2003. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 278:9435–9440. 10.1074/jbc.M209706200 [DOI] [PubMed] [Google Scholar]
- 55.Gomez MI, Sokol SH, Muir AB, Soong G, Bastien J, Prince AS. 2005. Bacterial induction of TNF-alpha converting enzyme expression and IL-6 receptor alpha shedding regulates airway inflammatory signaling. J. Immunol. 175:1930–1936. 10.4049/jimmunol.175.3.1930 [DOI] [PubMed] [Google Scholar]
- 56.Tang A, Sharma A, Jen R, Hirschfeld AF, Chilvers MA, Lavoie PM, Turvey SE. 2012. Inflammasome-mediated IL-1beta production in humans with cystic fibrosis. PLoS One 7:e37689. 10.1371/journal.pone.0037689 [DOI] [PMC free article] [PubMed] [Google Scholar]