

Abstract
Elongation factor P (EF-P) is a universally conserved bacterial translation factor. In many bacteria, EF-P is posttranslationally modified by PoxA, which covalently attaches a β-lysine to a conserved lysine residue of EF-P. Here we show that both EF-P and PoxA are necessary for virulence of the human diarrheal pathogen Shigella flexneri. Loss of either EF-P or PoxA leads to an impaired ability of S. flexneri to invade epithelial cells and form plaques in an epithelial cell monolayer. Proteomic analysis of efp and poxA deletion mutants revealed decreased levels of several virulence effector proteins, including IpaA, -B, and -C and IcsA. Additionally, mRNA levels of virB and virF, which encode master virulence regulators, were decreased in the efp mutant. The reduction in virF transcription was at least partially due to decreased levels of CpxA, which activates virF through the response regulator CpxR. The role of CpxAR in reduced synthesis of VirF and its downstream effectors was indicated by restoration of invasion when a mutation resulting in constitutively activated CpxR was introduced into the efp mutant. Thus, modified EF-P is required for appropriate synthesis of proteins involved in the virulence of this bacterial pathogen.
INTRODUCTION
Shigella flexneri is the causative agent of bacillary dysentery, a disease characterized by severe bloody diarrhea. S. flexneri invades the cells of the colonic mucosa, a process facilitated by the type III secretion system (T3SS) (1). Effector proteins injected into the host cell by the T3SS induce uptake of the bacterium (2). S. flexneri then replicates in the cytosol and spreads to adjacent cells via polymerization of host cell actin, a process mediated by the bacterial protein IcsA (3, 4). The resulting damage to the epithelial cell layer, in combination with the host inflammatory response, is responsible for the symptoms of dysentery.
All Shigella species possess a large virulence plasmid (pVir) that contains the majority of genes involved in Shigella virulence. A 31-kb section of this plasmid encodes the T3SS and many of the secreted effector proteins (5, 6). These include the Ipa (invasion plasmid antigen) proteins, which form a pore in the host membrane and facilitate bacterial invasion (7). Virulence in Shigella is controlled by two master virulence regulators, VirF and VirB (8, 9). Transcription of virF is activated by conditions that signal to the bacterium that it has arrived at the appropriate site of infection, including a temperature of 37°C, pH 7.4, and moderate osmolarity (10–12). The transcriptional regulation of virF is complex and requires the global regulators H-NS, Fis, and IHF, as well as the pH-responsive regulator CpxR (11, 13–16). VirF transcriptionally activates a second virulence regulator, VirB, which in turn facilitates transcription of the ipa genes and the genes encoding the T3SS components (8, 17). VirB also exhibits positive feedback on virF transcription (18). Independently of VirB, VirF directly regulates expression of icsA, which is needed for cell-to-cell spread (3, 19). In addition to factors encoded on the virulence plasmid, there are chromosomal genes that are necessary for successful host infection, including genes for iron regulation and carbon metabolism (20, 21).
Elongation factor P (EF-P) is a highly conserved bacterial translation factor that was recently shown to prevent ribosome stalling at consecutive proline residues in the nascent polypeptide chain (22, 23). Although the full specificity of EF-P is unclear, runs of three or more prolines, as well as PPG and certain APP motifs, have been identified as sequences that are usually translated more efficiently in the presence of EF-P (22–24). Crystal structures show that EF-P displays a striking structural similarity to a tRNA (25). In many bacteria, EF-P is posttranslationally modified by the addition of a β-lysine residue at a position that is structurally similar to the site of amino-acylation of a tRNA (26). This posttranslational modification is catalyzed by PoxA (also called EpmA, GenX, or YjeA), which attaches the β-lysine moiety to a conserved lysine residue of EF-P (26). In keeping with this scenario of molecular mimicry, PoxA has extensive structural similarity to a class II lysyl-tRNA synthetase (LysRS) and binds EF-P in a manner structurally similar to the binding of tRNALys by LysRS (26, 27). β-Lysine is synthesized by YjeK, and this modification of EF-P is the only known natural incorporation of β-lysine into a protein. The conserved lysine of EF-P is subsequently hydroxylated by an additional modifying enzyme, YfcM (28).
Properly modified EF-P is necessary for the virulence of several bacterial species, including Salmonella enterica and Agrobacterium tumefaciens (29, 30). Here we investigate the role of EF-P and its modification in the virulence of S. flexneri. We show that deletion of efp, poxA, or yjeK results in a virulence defect caused by reduced expression of master virulence regulators and a reduction in synthesis of the Ipa effector proteins. We also show that efp is needed for proper production of the histidine kinase CpxA and that altered activity of the CpxAR two-component system contributes to reduced virulence in an efp mutant.
MATERIALS AND METHODS
Media and growth conditions.
Liquid cultures of S. flexneri and Escherichia coli were grown in Luria-Bertani (LB) broth (1% tryptone, 0.5% yeast extract, 1% NaCl) at 30°C or 37°C, as indicated. S. flexneri was grown at 37°C on tryptic soy broth-agar (TSBA; Becton, Dickinson and Company, Franklin Lakes, NJ) plates containing 0.01% (wt/vol) Congo red. Antibiotics were used at the following concentrations: ampicillin, 25 μg/ml; kanamycin, 50 μg/ml; gentamicin, 20 μg/ml. Henle cells (intestinal 407; ATCC) were cultured in minimal essential medium (MEM; Gibco, Life Technologies, Carlsbad, CA) supplemented with 10% Bacto tryptone phosphate broth (Difco, Becton, Dickinson), 10% fetal bovine serum (Gibco), 2 mM glutamine, and nonessential amino acids (Gibco). Henle cells were cultured at 37°C with 95% air-5% CO2.
Bacterial strains and plasmid construction.
Strains and plasmids used in this study are shown in Table 1. S. flexneri mutant strains HMS100, HMS110, HMS130, and HMS140 were generated by P1 bacteriophage transduction (31), using donor strains from the Keio collection (32). To create HMS120, the first 203 bp of yjeK, along with approximately 1.5 kb upstream of yjeK, were amplified from strain 2457T by using the AXyjeKfor2 and yjeKHI primers (see Table S1 in the supplemental material). The kanamycin cassette and approximately 1.5 kb downstream of the E. coli yjeK gene were amplified from JW4106 by using primers yjeKHIantiP1 and AXyjeKrev2. These two fragments were then combined using splice overlap extension PCR and ligated into pWSc1-Ts cut with EcoRV. After electroporation of this plasmid into 2457T, colonies containing the plasmid were grown overnight in the absence of antibiotics at 30°C. These cultures were then subcultured at a 1:200 dilution into cultures containing ampicillin and kanamycin. Cultures were grown for 7 to 10 h at 37°C to select for single crossovers. These cultures were then plated for single colonies on TSBA plus Congo red, ampicillin, and kanamycin and grown at 37°C. Colonies from these plates were used to inoculate overnight cultures grown at 37°C in the presence of 1% sucrose. These cultures were then plated for single colonies on TSBA plus Congo red, 10% sucrose, and kanamycin to select for double crossovers. The absence of the plasmid was confirmed by patching onto TSBA plates with or without ampicillin. For each mutant, replacement of the wild-type allele was confirmed by PCR analysis, using flanking primers listed in Table S1. All plasmid inserts were sequenced at the University of Texas at Austin DNA sequencing facility. HMS101 was created by site-directed mutagenesis of base 153 of cpxR from a C to an A, using primers cpxRc153a and cpxRc153a anti. The PCR product was then inserted into pWSc1-Ts cut with EcoRV and transferred to the chromosome by allelic exchange. This single base change was confirmed by sequencing. The pWSc1-Ts plasmid was created by a QuikChange (Agilent Technologies, Inc., Santa Clara, CA) PCR using pWSc-1 as the template. Primers were created using the QuikChange primer design program on the Agilent Technologies website and are listed in Table S1. After thermal cycling, the product was digested using the DpnI restriction enzyme (New England BioLabs). The purified product was then used to transform NEB high-efficiency competent DH5α cells (New England BioLabs) via the manufacturer's instructions. Plasmids pEfp, pPoxA, and pYjeK were constructed by amplifying the relevant genes from 2457T, using the efpfor/rev, poxAfor/rev, and yjeKfor/rev primer pairs. The PCR products were then ligated into pWKS30 cut with EcoRV.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| E. coli strains | ||
| JW4116 | ΔpoxA::Kan | Keio collection; 27 |
| JW4107 | Δefp::Kan | Keio collection |
| JW4106 | ΔyjeK::Kan | Keio collection |
| JW5381 | ΔyfcM::Kan | Keio collection |
| S. flexneri strains | ||
| 2457T | Wild type | Walter Reed Institute of Research |
| CFS100 | 2457T ΔpVir | C. Fisher |
| HMS100 | 2457T Δefp::Kan | This work |
| HMS110 | 2457T ΔpoxA::Kan | This work |
| HMS120 | 2457T yjeK Δ202-1007::Kan | This work |
| HMS130 | 2457T ΔyfcM::Kan | This work |
| HMS101 | HMS100 encoding CpxR D51E | This work |
| HMS140 | 2457T ΔcpxA::Kan | This work |
| Plasmids | ||
| pWKS30 | Low-copy-number vector; Ampr | 41 |
| pWSc-1 | sacB gene cloned into the SmaI site of pWKS30 | 42 |
| pWSc-1 Ts | Temperature-sensitive pWSc-1 | This work |
| pPoxA | poxA cloned into EcoRV site of pWKS30 | This work |
| pEfp | efp cloned into EcoRV site of pWKS30 | This work |
| pYjeK | yjeK cloned into EcoRV site of pWKS30 | This work |
Cell culture assays.
Invasion assays were performed using a modification of the method described by Gore and Payne (20). Briefly, a Congo red-positive colony was used to inoculate 3 ml of LB containing antibiotics, as appropriate, and was grown overnight at 30°C. This culture was diluted in LB without antibiotics and grown to mid-logarithmic phase at 37°C with aeration. Approximately 2 × 108 bacteria were added to a subconfluent monolayer of Henle cells in 35-mm six-well polystyrene plates (Corning Inc., Corning, NY) and centrifuged for 10 min at 700 × g. Plates were incubated for 30 min at 37°C and 5% CO2 and then washed with phosphate-buffered saline (PBS) four times to remove extracellular bacteria. Fresh MEM containing 40 μg/ml gentamicin was then added to the monolayers and incubated for an additional 45 min. Monolayers were washed twice with PBS and stained with Wright-Giemsa stain (Cambridge Diagnostic Products, Inc., Ft. Lauderdale, FL). Three hundred Henle cells from each well were visualized by microscopy, and those containing three or more bacteria were scored as invaded.
For plaque assays (33), bacteria were grown as described for the invasion assay. Confluent monolayers of Henle cells were inoculated with 105, 104, and 103 bacteria diluted in PBS by centrifugation of bacteria onto Henle cell monolayers for 10 min at 700 × g. Plates were incubated for 30 min at 37°C and 5% CO2 and then washed with PBS four times and overlaid with fresh MEM containing 20 μg/ml gentamicin. Infected monolayers were incubated for 72 h and then washed with PBS twice and stained with Wright-Giemsa stain.
2D-DIGE.
Bacteria were prepared according to instructions found on the Applied Biomics (Hayward, CA) website. To isolate total bacterial proteins, S. flexneri was grown to mid-logarithmic phase in LB. Cells were collected by centrifugation (7 min, 12,000 × g, 4°C). Cells were washed with PBS three times, and the final pellet was resuspended in 2D lysis buffer {7 M urea, 2 M thiourea, 4% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 30 mM Tris-HCl, pH 8.8} and sonicated briefly. The cell lysate was centrifuged at 16,300 × g for 2 min to remove unlysed cells. The supernatant was sent to Applied Biomics for two-dimensional differential in-gel electrophoresis (2D-DIGE), spot picking, and protein identification by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectroscopy.
Real-time PCR.
RNA was isolated from cells grown to mid-log phase at 37°C in LB medium by use of RNeasy minicolumns (Qiagen, Venlo, Netherlands). RNA was treated with DNase I (DNase I; Qiagen) on the column, and again after elution (DNase I; Invitrogen, Life Technologies, Grand Island, NY), according to the manufacturers' instructions. Two micrograms of RNA from each sample was reverse transcribed using a High Capacity cDNA Archive kit (Applied Biosystems, Life Technologies). Quantitative real-time reverse transcription-PCR (RT-PCR) was performed using TaqMan Universal PCR master mix (Applied Biosystems) and TaqMan probes (6-carboxyfluorescein labeled, minor-groove binding). All probes and target-specific primers were designed using Primer Express software (Applied Biosystems) and were synthesized by Applied Biosystems. cDNA preparations were used for RT-PCR, at a dilution of 1:1,000 for detection by the ipaC probe and at 1:100 for all other probes. Real-time PCR and analysis were performed using an Applied Biosystems 7300 real-time PCR system and software. Relative amounts of virF, virB, and ipaC cDNAs were normalized to the internal control dksA for each sample.
Immunoblotting.
Whole-cell preparations were prepared from approximately 109 cells, as determined by measuring the A650, grown to mid-log phase at 37°C in LB medium. Growth conditions and time of harvest were the same as for the invasion assay. Cells were collected by centrifugation and resuspended in SDS sample buffer (5% β-mercaptoethanol, 3% [wt/vol] SDS, 10% glycerol, 0.02% bromophenol blue, 63 mM Tris-Cl, pH 6.8), and samples were boiled for 10 min. Samples were electrophoresed in duplicate SDS-PAGE gels. After electrophoresis, proteins from one of the two gels were transferred to a 0.45-μm-pore-size nitrocellulose membrane (Hybond-ECL; GE Healthcare, Little Chalfont, United Kingdom) and incubated with the following antibodies: mouse monoclonal anti-IpaC, rabbit polyclonal anti-IcsA, monkey anti-S. flexneri convalescent-phase antiserum (E. V. Oaks, WRAIR), rabbit polyclonal anti-CpxA–maltose binding protein (MBP) (T. J. Silhavy, Princeton University), and rabbit polyclonal anti-VirF (E. Murphy, Ohio University). Proteins were detected using horseradish peroxidase (HRP)-conjugated goat anti-mouse, anti-human, or anti-rabbit antibody. Signals were detected by developing the blot with a Pierce ECL detection kit (Thermo-Fisher Scientific, Waltham, MA). The gels were scanned, and the relative intensities of the bands were compared using ImageJ software, version 1.48.
RESULTS
S. flexneri efp, poxA, and yjeK mutants are defective in plaque formation.
The gene encoding the translation factor EF-P and genes responsible for its proper modification were each deleted from the genome of wild-type S. flexneri 2457T in order to investigate their roles in Shigella virulence. The efp, poxA, and yjeK mutants were tested for the ability to infect Henle cell monolayers in a plaque assay. To form plaques, Shigella must invade the cells of the monolayer, replicate intracellularly, and spread to adjacent cells, resulting in a zone of clearance in the monolayer that is visible upon staining (33). As shown in Fig. 1, deletion of efp completely abolished the ability of S. flexneri to form plaques, while both poxA and yjeK mutants formed tiny pinpoint plaques (Fig. 1). Although EF-P cannot be modified in the poxA and yjeK mutants, it appears that unmodified EF-P retains partial activity in these mutants. All three mutants formed plaques with wild-type morphology when the corresponding wild-type gene was provided on the low-copy-number vector pWKS30 (Fig. 1). In contrast to the virulence defect caused by loss of EF-P or its β-lysine modification, the hydroxyl modification of EF-P was not required for plaque formation, since the yfcM deletion mutant formed wild-type plaques (data not shown). The efp and poxA mutants were selected for further characterization to determine the cause of reduced plaque formation.
FIG 1.

Plaque formation is impaired in S. flexneri efp, poxA, and yjeK mutants. Henle cell monolayers were infected with approximately 105 CFU of bacteria. Plates were incubated for 3 days in the presence of gentamicin and then stained to visualize plaques.
S. flexneri efp and poxA mutants exhibit slightly impaired growth in vitro.
Poor growth of efp and poxA mutants could lead to poor plaque formation due to reduced replication inside host cells. To assess the effects of these mutations on growth of S. flexneri, the efp and poxA mutants were compared to the wild type for growth in LB. Both the efp and poxA mutants showed slight decreases in growth rate compared to the wild type, with the poxA mutant displaying a less severe growth defect than that of the efp mutant (Fig. 2). Both strains ultimately reached the same final density as the wild type. Wild-type growth rates were restored upon addition of the absent gene on a plasmid. This result suggests that the presence of EF-P, in either modified or nonmodified form, is required for wild-type fitness under these growth conditions, consistent with observations in Salmonella and E. coli (26, 29, 34).
FIG 2.
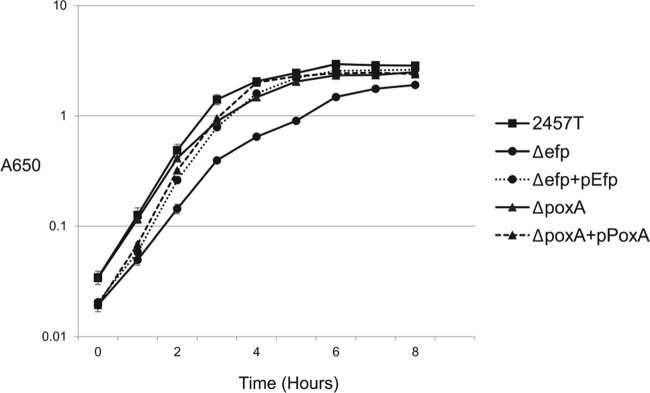
Growth of S. flexneri efp and poxA mutants in LB medium. Data shown are the averages of results from three biological replicates, and error bars represent 1 standard deviation.
The efp mutant shows minimal differences from the wild type in phenotypic microarray analysis.
Although the in vitro growth defect of the efp mutant in LB was relatively mild, it was possible that there might be a more severe growth defect within the host. This could be caused, for example, by a failure to acquire certain nutrients in the host or by increased susceptibility to stressors. To determine whether the efp mutation makes S. flexneri more sensitive to particular environmental stressors or affects the ability to use nutrients, we performed Biolog phenotype microarray analysis. Phenotype microarrays measure the ability of bacteria to respire under nearly 2,000 diverse conditions, including the presence of various nutrient sources, pHs, and osmolarities and the presence of antibiotics and other cellular stressors (35). Only a few differences were observed between the profiles of the wild type and the efp mutant. The primary difference observed was an increased sensitivity to alkaline pH under certain conditions in the efp mutant (see Fig. S1 in the supplemental material). No single defect or type of defect was identified in the Biolog analysis that would explain the inability of the mutant to form plaques.
Proteomic analysis of efp and poxA mutants by 2D-DIGE.
To look more directly at the effects of EF-P and PoxA in the cell, proteomic analysis by 2D-DIGE was performed. Bacteria grown to mid-logarithmic phase in LB were harvested, and whole-cell protein profiles were compared. A total of 45 spots were observed with expression ratios of ≥2.0 between either the poxA or efp mutant and the wild type. Additional proteins are likely affected by the mutations but may be missed in proteome analysis if they are expressed at very low levels in the wild type or if they comigrate with other proteins or are not well resolved on the gels.
Considerable overlap was observed between the protein profiles of the efp and poxA mutants (Fig. 3). The protein profile of the efp mutant was generally more divergent from the wild type than the poxA mutant profile. The 23 proteins that showed the greatest differences between the wild type and the mutants were identified by mass spectroscopy (Table 2). Among these proteins, 21 showed reduced levels in either the efp or poxA mutant, while 2 showed increased levels in at least one of the mutants. These changes in protein levels may represent both direct and indirect effects of the mutations. A direct effect would be suggested by the presence of polyproline motifs in affected proteins. Of the proteins with reduced levels in either mutant, none contained three or more sequential prolines, one of the motifs characterized as requiring EF-P (22). However, 10 proteins contained two sequential prolines, including 5 with a PPG motif, which has also been shown to contribute to ribosome stalling in the absence of EF-P (22). The APP motif is also frequently EF-P dependent (24), but APP was not observed in any of the identified proteins that did not also contain a PPG. Surprisingly, the two proteins found at increased levels in the efp mutant also contain sequential proline motifs. HslU contains PPA and PPG motifs, and IlvE contains PPA and PPF motifs. Thus, it is likely that these increases were due to indirect effects of the mutations.
FIG 3.
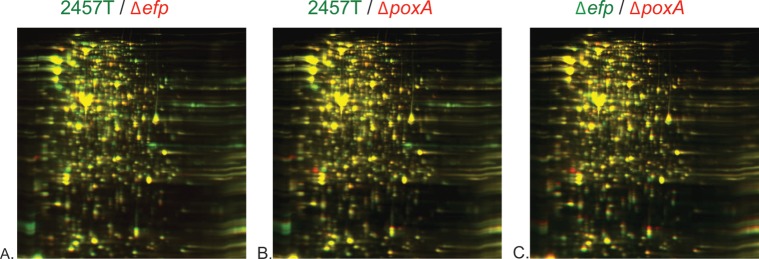
2D-DIGE profiles of wild-type S. flexneri 2457T versus the Δefp mutant (HMS100) (A), 2457T versus the ΔpoxA mutant (HMS110) (B), and the Δefp mutant (HMS100) versus the ΔpoxA mutant (HMS110) (C). For each panel, the strain listed in green is labeled with green fluorescence, while the strain listed in red is labeled with red fluorescence. Whole cells were prepared in lysis buffer and sonicated.
TABLE 2.
Proteins differentially expressed between wild-type 2457T and the efp or poxA mutanta
| Gene name | efp mutant/2457T expression ratiob | poxA mutant/2457T expression ratiob | Annotated function | Polyproline motif(s) | Identifier |
|---|---|---|---|---|---|
| yhbS | −5.1 | −2.2 | Predicted acetyltransferase | GI:24114447 | |
| yieF | −4.8 | −4.5 | Chromate reductase, flavoprotein | GI:332764088 | |
| fkpA | −4.5 | −3.8 | Heat shock peptidyl-prolyl isomerase; also has a chaperone function | PPE, PPN | GI:24114611 |
| atpD | −3.8 | −3 | Beta subunit of ATP synthase | PPG | GI:112791342 |
| fabG | −3.1 | −1.9 | Reductase involved in fatty acid biosynthesis | GI:333004903 | |
| ipaC | −2.9 | −2.5 | Secreted invasion protein | GI:281603882 | |
| rplI | −2.9 | 1 | Ribosomal subunit protein L9 | GI:15804792 | |
| acnB | −2.7 | −1.2 | Bifunctional aconitate hydratase 2/2-methylisocitrate dehydratase | PPA, PPG, APP | GI:56479606 |
| typA (bipA) | −2.5 | −1.8 | Ribosome binding GTPase | GI:61248993 | |
| ipaA | −2.5 | −1.7 | Secreted virulence effector protein | GI:31983589 | |
| mreB | −2.5 | −1.3 | Rod shape determination | PPE, PPG | GI:332752454 |
| manX | −2.4 | −1.2 | Mannose-specific PTS component | GI:333003654 | |
| ospC2 | −2.3 | −1.4 | Unknown function; secreted by T3SS | GI:31983556 | |
| apy/phoN2 | −2.1 | −2.1 | Periplasmic phosphatase, apyrase | GI:31983594 | |
| infC | −2.1 | −1.9 | Translation initiation factor IF-3 | PPV | GI:39931271 |
| iucB | −2 | −1.6 | Siderophore biosynthesis | PPRP | GI:74313968 |
| iucD | −2 | −1.6 | Siderophore biosynthesis | PPC, PPA | GI:332764119 |
| mrp | −2 | −1.6 | ATP-binding protein involved in chromosome partitioning | PPG | GI:30063550 |
| rpe | −1.5 | 6.1 | Ribulose phosphate-3-epimerase | GI:332749517 | |
| afeA | −1.2 | −2 | Protein folding catalyst | GI:333003122 | |
| ilvE | 2.7 | −1.2 | Branched-chain amino acid aminotransferase | PPA, PPF | GI:332751003 |
| hslU | 8.4 | 8.1 | ATP-dependent protease ATP-binding subunit | PPA, PPG | GI:82778893 |
Spots were identified using MALDI-TOF mass spectroscopy. The identifier listed corresponds to the protein ID in GenBank. Proteins encoded on the virulence plasmid are shown in bold.
The relative amount of each spot was determined, and ratios are shown for the mutants compared to the wild type by 2D-DIGE. Negative numbers indicate that smaller amounts of the protein were present in the mutant.
The proteins identified in the proteome analysis as being affected in the efp mutant had a variety of functions. Although most of these did not fall into identifiable groups, a large group of proteins represented in the analysis were virulence proteins.
efp and poxA mutants have reduced levels of several virulence proteins.
Of the 21 proteins identified from 2D-DIGE analysis that showed reduced levels in the efp mutant, 4 were encoded on the virulence plasmid of S. flexneri: Apy, OspC2, IpaC, and IpaA. The level of the effector protein IpaC was decreased approximately 3-fold in the efp mutant (2.5-fold in the poxA mutant) (Table 2). To determine whether the efp mutations affected other virulence effectors in addition to those detected by 2D-DIGE, immunoblotting with antisera against the virulence proteins was used. Convalescent-phase antiserum from a monkey infected with wild-type S. flexneri recognized several of the major virulence proteins, including IpaA, IpaB, IpaC, and IcsA, as well as the major outer membrane protein of S. flexneri (Fig. 4A). There were reduced amounts of all of the virulence proteins in the efp mutant. The amount of the major outer membrane protein was unaffected, indicating that this was not a general effect on protein synthesis. Specific antisera against IpaC and IcsA (Fig. 4B) were used to confirm the identities of these proteins. The Ipa proteins are the primary effectors involved in the invasion of epithelial cells and are essential for entry of S. flexneri into the host cell, as well as subsequent lysis of the phagocytic vacuole and spread to the adjacent cells. The reduced levels of these Ipa proteins prompted us to further investigate how the absence of efp and poxA affects the ability of S. flexneri to invade host cells.
FIG 4.

The efp mutant has reduced levels of virulence proteins. (A) Virulence proteins were detected by Western blotting using a convalescent-phase monkey antiserum to S. flexneri proteins. Whole-cell lysates of equivalent numbers of cells of wild-type 2457T (WT), the CFS100 strain, which lacked the virulence plasmid and served as the negative control (ΔpVir), the efp mutant HMS100 (Δefp), and the Δefp mutant complemented with wild-type efp (Δefp/pEfp) were used. The positions of the major virulence antigens and the major outer membrane protein are shown on the right. (B) The same samples as in panel A were probed with a monoclonal antibody against IpaC or a polyclonal antiserum against IcsA to confirm the reduced amounts of the proteins.
efp and poxA mutants have reduced invasion.
The efp and poxA mutants were tested for the ability to invade epithelial cell monolayers. The efp mutant exhibited 4- to 5-fold less invasion of Henle cells than wild-type S. flexneri, while the poxA mutant invaded at slightly less than half the level of the wild type (Fig. 5). This suggests that the primary virulence defect of these mutants occurs at the point of invasion, although there may be additional defects in subsequent steps of virulence. These results are consistent with the observed decreases in the amounts of IpaC and other invasion proteins (Table 2; Fig. 4).
FIG 5.

efp and poxA mutants invade Henle cells at reduced levels compared to those of the wild type. Henle cell monolayers were infected with 2 × 108 CFU of bacteria. Plates were incubated for 30 min, treated with medium containing gentamicin, and stained. Henle cells containing S. flexneri were scored by microscopy. Data shown are the averages of results from three biological replicates, and error bars represent 1 standard deviation. Invasion values were compared to those of the WT by using the Student t test. *, P < 0.01.
Virulence is affected upstream of virF transcription.
In addition to IpaC, several virulence proteins were reduced in the mutants. This could be a direct effect on the synthesis of each of these proteins, or it could be indirect, resulting from reduced synthesis of an upstream regulatory protein. We sought to determine where EF-P exerts its effect in this regulatory cascade. Transcription of the ipa operon is facilitated by the regulator VirB, whose transcription is activated by the master virulence regulator VirF, as summarized in Fig. 6. There are no polyproline motifs in VirF, VirB, or any of the downstream virulence proteins we identified, suggesting that EF-P does not act directly on translation of these proteins. By performing quantitative real-time PCR analysis, we found that ipaC mRNA levels were reduced approximately 3-fold in the efp mutant, pointing to an effect upstream of transcription of the ipa operon (Fig. 7A). Consistent with this hypothesis, mRNA levels of the transcriptional regulator VirB were reduced between 2- and 3-fold, and virF mRNA was reduced approximately 2-fold in the efp mutant. VirF protein levels were also decreased in the efp mutant and could be rescued by complementation of the efp mutant by supplying efp on a low-copy-number plasmid (Fig. 7B). The reduced levels of both virF and virB mRNAs, as well as VirF protein, suggest an effect of EF-P on Shigella virulence that occurs upstream of VirF transcription.
FIG 6.
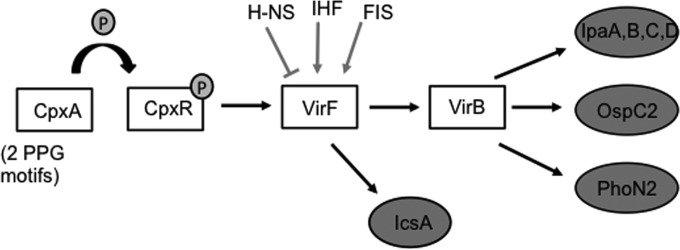
Shigella virulence gene regulation. The diagram shows the simplified regulatory pathway of Shigella virulence and regulation of virF, with emphasis on the CpxAR two-component system, which plays a role in the virulence defect of the efp mutant. Proteins in circles are virulence effector proteins, which were found at decreased levels in the efp mutant. Note that although the proteins are depicted in the figure, the regulation shown occurs at the level of transcription.
FIG 7.
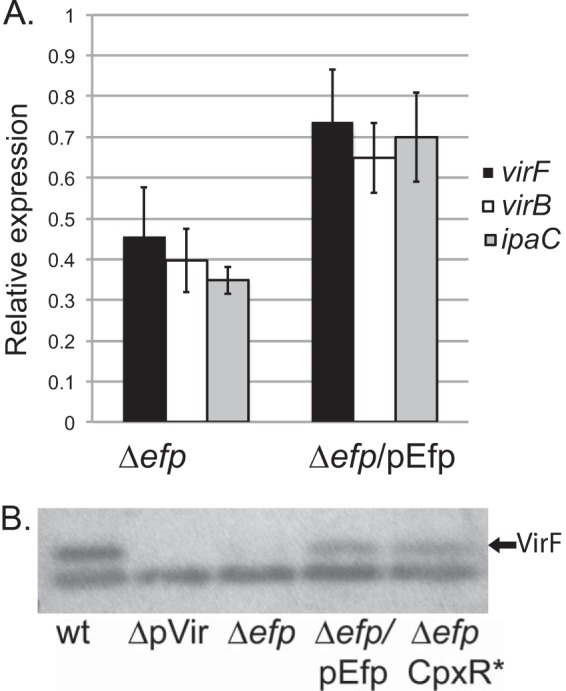
Effect of efp mutation on virulence gene expression. (A) mRNA levels of virF, virB, and ipaC were reduced in the efp mutant, as determined by quantitative real-time PCR analysis. Values were normalized to the amount of dksA mRNA in each sample, and results shown are relative to the wild-type level, which was set to 1. Values shown are averages of results from three independent experiments. Error bars depict 1 standard deviation. mRNA levels of virF, virB, and ipaC were compared to those of the wild type by using the Student t test. *, P < 0.01; **, P < 0.001. (B) VirF levels were reduced in the S. flexneri efp mutant. VirF was detected by Western blotting using antisera to VirF. Whole-cell lysates of equivalent numbers of cells of wild-type 2457T, 2457T cured of the virulence plasmid (CFS100), and the Δefp, Δefp/pEfp, and Δefp CpxR D51E mutants were used. The lower band is a protein detected in all samples, which served as a loading control.
CpxA levels are reduced in an efp mutant.
Transcription of virF is affected by four upstream regulators: H-NS, IHF, FIS, and CpxR (Fig. 6). None of these regulators contain polyproline motifs of any length. However, the response regulator CpxR, which stimulates virF transcription, is activated by the sensor kinase CpxA, which does contain two PPG motifs. While PPG motifs do not correlate absolutely with a requirement for Efp, their presence in CpxA suggested that it was a candidate for further analysis. The Cpx two-component system is activated in response to conditions that create membrane stress, including alkaline pH and changes in the lipid composition of the outer membrane (11, 16, 36). At physiological pH, phosphorylated CpxR transcriptionally activates virF, and almost no VirF is made in the absence of cpxR (16). In the efp mutant, CpxA protein levels as determined by immunoblotting were approximately one-third the level in wild-type S. flexneri, as determined by ImageJ analysis of the scanned image (Fig. 8A). To confirm that CpxA levels were lower due to reduced translation of the message rather than reduced transcription of cpxA, quantitative RT-PCR was used to detect cpxA mRNA. mRNA levels of cpxA were not significantly different from those of the wild type (Fig. 8B). This result confirms that CpxA levels are affected at the level of translation, likely due to the role of EF-P in the translation of this protein. This reduction in CpxA protein may reduce the amount of active CpxR-P available to induce virF transcription.
FIG 8.
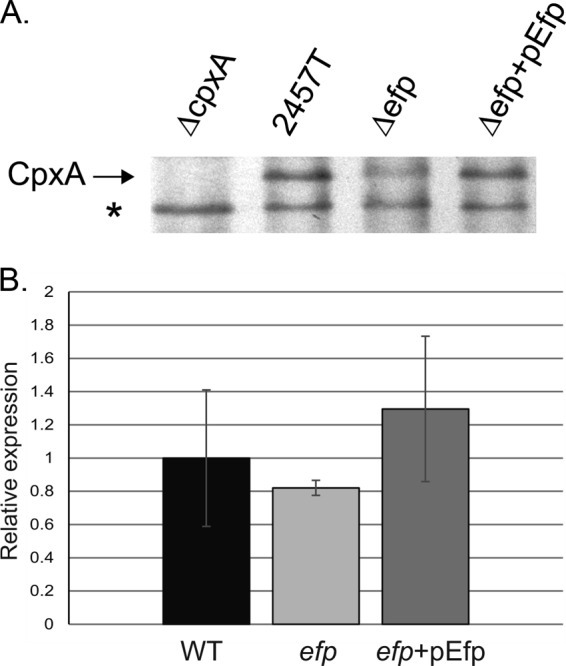
CpxA protein levels, but not mRNA levels, are lower in the efp mutant. (A) CpxA was detected by Western blotting using antisera to CpxA-MBP. Whole-cell lysates of equivalent numbers of cells of the wild type (2457T) and the ΔcpxA (HMS140), Δefp (HMS100), and Δefp/pEfp mutants were used. The star indicates a protein detected in all samples, which served as a loading control. (B) cpxA mRNA was measured using quantitative real-time PCR analysis. Values were normalized to the amount of dksA mRNA in each sample, and results shown are relative to the wild-type level, which was set to 1. Values shown are averages of results from three independent experiments. Error bars depict 1 standard deviation. mRNA levels of cpxA were compared to those in the wild type by using the Student t test, and differences were not significant (P > 0.5).
To compensate for the reduced level of CpxA, we constructed a mutant (HMS101) with constitutively activated CpxR in the efp mutant background. Aspartate 51 of CpxR is phosphorylated by CpxA, leading to active CpxR, which can then stimulate transcription. Changing the aspartate residue to glutamate, which mimics aspartyl-phosphate, can result in constitutive activation (37, 38). The S. flexneri efp mutant producing CpxR D51E was tested for invasion and plaque formation to determine whether CpxA-independent activation of CpxR would suppress the virulence defect of the efp mutant. The CpxR D51E mutation completely restored the ability of the efp mutant to invade (Fig. 9). Thus, activation of CpxR is sufficient to rescue the invasive ability of the efp mutant, likely by increasing levels of the virulence regulators. VirF protein levels were measured by immunoblotting, and activation of CpxR largely restored VirF levels in the efp mutant (Fig. 7B).
FIG 9.

The CpxR D51E mutation restores invasion of the Δefp mutant. Data shown are averages of results from three biological replicates, and error bars represent 1 standard deviation. Invasion values for the Δefp and Δefp CpxR D51E (HMS101) strains were compared to that of the WT by using the Student t test. *, P < 0.001.
To confirm that the increased levels of VirF resulted in increased synthesis of the virulence proteins, Western blots of cell lysates were probed with the monkey convalescent-phase antiserum (Fig. 10A) and with an antibody against IpaC or IcsA (Fig. 10B). The presence of CpxR D51E in the efp mutant resulted in nearly wild-type levels of the Ipa proteins, although IcsA levels were not fully restored. Thus, CpxR D51E suppressed the effect of the efp mutation on the invasion proteins. Although levels of VirF and the virulence proteins in the efp mutant were increased by the activation of CpxR, this mutation did not restore wild-type plaque morphology. Instead, small areas of cell death with turbid centers were observed in the cell monolayer (Fig. 11). This altered plaque morphology suggests that the bacteria invaded the Henle cells but were defective in intracellular replication or cell-to-cell spread. Thus, while the reduced level of virF expression as a consequence of the loss of signaling through CpxAR is likely responsible for the invasion defect of the efp mutant, the mutant must have additional defects that affect plaque formation subsequent to invasion. IcsA is required for actin polymerization and intercellular spread of the bacteria. Although IcsA levels were partially restored in vitro by the CpxR mutation (Fig. 10), IcsA may not be expressed appropriately and processed in the mutant in the cytoplasm of epithelial cells. Additionally, other virulence or intracellular metabolism proteins required for plaque formation may be affected by EF-P, independent of its effect on the Cpx pathway.
FIG 10.

The CpxR D51E mutation results in increased synthesis of virulence proteins in the efp mutant. (A) Virulence proteins were detected by Western blotting using monkey convalescent-phase antiserum to S. flexneri proteins. Whole-cell lysates of equivalent numbers of cells of wild-type 2457T (WT), CFS100, which lacks the virulence plasmid (ΔpVir), the efp mutant HMS100 (Δefp), and the Δefp cpxR D51E mutant (Δefp, cpxR*) were used. The positions of the major virulence antigens and the major outer membrane protein are shown on the right. (B) The same samples as in panel A were probed with a monoclonal antibody against IpaC or a polyclonal antiserum against IcsA to confirm the reduced amounts of the proteins.
FIG 11.

Plaques formed by S. flexneri strains in Henle cell monolayers. (A) 2457T; (B) Δefp mutant (HMS100); (C) Δefp CpxR D51E mutant (HMS101); (D) uninfected.
DISCUSSION
EF-P and its β-lysine modification catalyzed by PoxA are necessary for the optimal virulence of S. flexneri (Fig. 1 and 4). The virulence defect in efp and poxA mutants is due in part to reduced synthesis of Ipa and IcsA proteins. IpaA, -B, and -C, which were observed at reduced levels in efp and poxA mutants, are necessary for facilitating bacterial uptake by the host cell (39, 40). It is likely that the few bacteria that are able to enter host cells are impaired in subsequent steps of infection that result in cell-to-cell spread. IcsA, which is required for actin-based motility of Shigella in the host cell cytoplasm, was also produced at lower levels in the efp mutant, and a lack of IcsA results in a loss of plaque formation (3). Apy and OspC2 are proteins encoded on the virulence plasmid that were also observed at reduced levels in the efp and poxA mutants. Although the exact roles of these proteins in Shigella virulence are unknown, both proteins may play a role in host cell death, and their absence might contribute to reduced plaque formation (41, 42). The reduction of the invasion proteins as well as other virulence effectors is a consequence of reduced transcription of the master regulators of virulence, VirF and VirB. The amount of VirF protein was lower in the efp mutant than in the wild type, and the transcription of virB, which is activated by VirF, was also reduced.
Transcription of Shigella virulence regulators is subject to complex control to ensure proper expression of virulence factors, increasing the chance of successful infection. The CpxAR two-component system is an important part of this regulatory network, leading to activation of virF transcription only at the appropriate pH (16). In addition, CpxA may be necessary for posttranscriptional processing of VirB (43). The amount of the sensor kinase CpxA was reduced in the efp mutant, most likely due to reduced translation of its mRNA due to polyproline motifs. We did not observe CpxA in our proteomic analysis; however, only a limited number of proteins were identified, and CpxA is frequently not detected in 2D-PAGE analysis (44). The reduction in CpxA may explain several of the phenotypes observed in the efp mutant. The increased sensitivity of the efp mutant to alkaline pH observed under certain conditions in the phenotype microarray analysis may have resulted from a lack of pH sensing and signaling through CpxAR. Similarly, the low levels of VirF and subsequent loss of virulence gene expression and invasion of cultured cells are consistent with a reduction in CpxA. An alteration of CpxR (CpxR D51E) that renders it active, and thus no longer dependent on CpxA, restored the ability of the efp mutant to invade Henle cells. The increased level of active CpxR in this strain restored synthesis of VirF, leading to greater production of virulence proteins. Although the CpxR D51E mutation allowed the efp mutant to invade at wild-type levels, it did not rescue the plaque defect of the efp mutant. It is unclear if the inability to form plaques is due to inappropriate regulation of expression of virulence genes or if EF-P is needed for optimal translation of other proteins required for intracellular growth or spread.
Park et al. (45) showed that all of the EF-P purified from E. coli cells possesses the β-lysine modification, leading to the hypothesis that this modification is necessary or highly advantageous for the function of EF-P. Compared to the efp mutant, the poxA mutant of S. flexneri displayed an intermediate reduction in virulence, as well as a higher growth rate and less severe decreases in many of the affected proteins identified by proteomic analysis. Our results indicate that unmodified EF-P is more beneficial for S. flexneri virulence than a complete lack of EF-P. This is consistent with in vitro data showing that unmodified EF-P does have residual activity in preventing ribosome stalling (22).
Modification of EF-P is necessary for the virulence of Salmonella enterica, as mutation of either poxA or yjeK leads to attenuation of virulence in mice (29). Like S. flexneri, S. enterica uses a T3SS to invade host epithelial cells. Navarre et al. found virulence effector proteins to be a major group with altered levels in a proteomic analysis of a poxA mutant (29). Unlike the case for S. flexneri, these virulence proteins were present in larger amounts in the poxA mutant, including high levels of the S. enterica virulence regulatory protein HilA. It is likely that the in vivo virulence of S. enterica was affected due to inappropriate expression of the virulence effector proteins. Zou et al. performed Biolog phenotype microarray analysis on an efp mutant of S. enterica and found a large number of differences between the wild type and the efp mutant, including increased sensitivity to a multitude of cellular stressors (34). This is in contrast to the relatively few differences we observed between wild-type S. flexneri and the efp mutant. Collectively, these studies show that while both of these intracellular pathogens require modified EF-P for wild-type virulence, EF-P plays different roles in the virulence of these two bacterial species.
Significant progress has been made recently in uncovering the function and significance of EF-P and its modification. The presence of polyproline motifs within proteins has been shown to be associated with a requirement for EF-P for their efficient translation. However, Hersch et al. showed that not all proteins containing polyproline motifs depend on EF-P for their translation, while other, unknown peptide motifs may confer EF-P dependence, illustrating that our understanding of EF-P function is still limited (24). This study extends our knowledge of the roles that EF-P plays in bacterial physiology and virulence.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ed Oaks, Erin Murphy, and Tom Silhavy for generously providing antisera. We are indebted to Carolyn Fisher for technical assistance and for sharing strains. Elizabeth Wyckoff provided a thorough review and editing of the manuscript.
This work was funded by Public Health Service grant AI16935 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Published ahead of print 16 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01532-13.
REFERENCES
- 1.Jennison AV, Verma NK. 2004. Shigella flexneri infection: pathogenesis and vaccine development. FEMS Microbiol. Rev. 28:43–58. 10.1016/j.femsre.2003.07.002 [DOI] [PubMed] [Google Scholar]
- 2.Sansonetti PJ, Egile C. 1998. Molecular bases of epithelial cell invasion by Shigella flexneri. Antonie Van Leeuwenhoek 74:191–197. 10.1023/A:1001519806727 [DOI] [PubMed] [Google Scholar]
- 3.Bernardini ML, Mounier J, d'Hauteville H, Coquis-Rondon M, Sansonetti PJ. 1989. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc. Natl. Acad. Sci. U. S. A. 86:3867–3871. 10.1073/pnas.86.10.3867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monack DM, Theriot JA. 2001. Actin-based motility is sufficient for bacterial membrane protrusion formation and host cell uptake. Cell. Microbiol. 3:633–647. 10.1046/j.1462-5822.2001.00143.x [DOI] [PubMed] [Google Scholar]
- 5.Maurelli AT, Baudry B, d'Hauteville H, Hale TL, Sansonetti PJ. 1985. Cloning of plasmid DNA sequences involved in invasion of HeLa cells by Shigella flexneri. Infect. Immun. 49:164–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sasakawa C, Kamata K, Sakai T, Makino S, Yamada M, Okada N, Yoshikawa M. 1988. Virulence-associated genetic regions comprising 31 kilobases of the 230-kilobase plasmid in Shigella flexneri 2a. J. Bacteriol. 170:2480–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menard R, Prevost MC, Gounon P, Sansonetti P, Dehio C. 1996. The secreted Ipa complex of Shigella flexneri promotes entry into mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 93:1254–1258. 10.1073/pnas.93.3.1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adler B, Sasakawa C, Tobe T, Makino S, Komatsu K, Yoshikawa M. 1989. A dual transcriptional activation system for the 230 kb plasmid genes coding for virulence-associated antigens of Shigella flexneri. Mol. Microbiol. 3:627–635. 10.1111/j.1365-2958.1989.tb00210.x [DOI] [PubMed] [Google Scholar]
- 9.Sakai T, Sasakawa C, Yoshikawa M. 1988. Expression of four virulence antigens of Shigella flexneri is positively regulated at the transcriptional level by the 30 kilodalton VirF protein. Mol. Microbiol. 2:589–597. 10.1111/j.1365-2958.1988.tb00067.x [DOI] [PubMed] [Google Scholar]
- 10.Maurelli AT, Blackmon B, Curtiss R. 1984. Temperature-dependent expression of virulence genes in Shigella species. Infect. Immun. 43:195–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakayama S, Watanabe H. 1995. Involvement of cpxA, a sensor of a two-component regulatory system, in the pH-dependent regulation of expression of Shigella sonnei virF gene. J. Bacteriol. 177:5062–5069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernardini ML, Fontaine A, Sansonetti PJ. 1990. The two-component regulatory system ompR-envZ controls the virulence of Shigella flexneri. J. Bacteriol. 172:6274–6281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hromockyj AE, Tucker SC, Maurelli AT. 1992. Temperature regulation of Shigella virulence: identification of the repressor gene virR, an analogue of hns, and partial complementation by tyrosyl transfer RNA (tRNA1(Tyr)). Mol. Microbiol. 6:2113–2124. 10.1111/j.1365-2958.1992.tb01385.x [DOI] [PubMed] [Google Scholar]
- 14.Porter ME, Dorman CJ. 1994. A role for H-NS in the thermo-osmotic regulation of virulence gene expression in Shigella flexneri. J. Bacteriol. 176:4187–4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falconi M, Prosseda G, Giangrossi M, Beghetto E, Colonna B. 2001. Involvement of FIS in the H-NS-mediated regulation of virF gene of Shigella and enteroinvasive Escherichia coli. Mol. Microbiol. 42:439–452. 10.1046/j.1365-2958.2001.02646.x [DOI] [PubMed] [Google Scholar]
- 16.Nakayama S, Watanabe H. 1998. Identification of cpxR as a positive regulator essential for expression of the Shigella sonnei virF gene. J. Bacteriol. 180:3522–3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tobe T, Yoshikawa M, Mizuno T, Sasakawa C. 1993. Transcriptional control of the invasion regulatory gene virB of Shigella flexneri: activation by VirF and repression by H-NS. J. Bacteriol. 175:6142–6149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kane KA, Dorman CJ. 2012. VirB-mediated positive feedback control of the virulence gene regulatory cascade of Shigella flexneri. J. Bacteriol. 194:5264–5273. 10.1128/JB.00800-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tran CN, Giangrossi M, Prosseda G, Brandi A, Di Martino ML, Colonna B, Falconi M. 2011. A multifactor regulatory circuit involving H-NS, VirF and an antisense RNA modulates transcription of the virulence gene icsA of Shigella flexneri. Nucleic Acids Res. 39:8122–8134. 10.1093/nar/gkr521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gore AL, Payne SM. 2010. CsrA and Cra influence Shigella flexneri pathogenesis. Infect. Immun. 78:4674–4682. 10.1128/IAI.00589-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy ER, Payne SM. 2007. RyhB, an iron-responsive small RNA molecule, regulates Shigella dysenteriae virulence. Infect. Immun. 75:3470–3477. 10.1128/IAI.00112-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doerfel LK, Wohlgemuth I, Kothe C, Peske F, Urlaub H, Rodnina MV. 2013. EF-P is essential for rapid synthesis of proteins containing consecutive proline residues. Science 339:85–88. 10.1126/science.1229017 [DOI] [PubMed] [Google Scholar]
- 23.Woolstenhulme CJ, Parajuli S, Healey DW, Valverde DP, Petersen EN, Starosta AL, Guydosh NR, Johnson WE, Wilson DN, Buskirk AR. 2013. Nascent peptides that block protein synthesis in bacteria. Proc. Natl. Acad. Sci. U. S. A. 110:E878–E887. 10.1073/pnas.1219536110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hersch SJ, Wang M, Zou SB, Moon K-M, Foster LJ, Ibba M, Navarre WW. 2013. Divergent protein motifs direct elongation factor P-mediated translational regulation in Salmonella enterica and Escherichia coli. mBio 4:e00180–13. 10.1128/mBio.00180-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blaha G, Stanley RE, Steitz TA. 2009. Formation of the first peptide bond: the structure of EF-P bound to the 70S ribosome. Science 325:966–970. 10.1126/science.1175800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yanagisawa T, Sumida T, Ishii R, Takemoto C, Yokoyama S. 2010. A paralog of lysyl-tRNA synthetase aminoacylates a conserved lysine residue in translation elongation factor P. Nat. Struct. Mol. Biol. 17:1136–1143. 10.1038/nsmb.1889 [DOI] [PubMed] [Google Scholar]
- 27.Bailly M, de Crécy-Lagard V. 2010. Predicting the pathway involved in post-translational modification of elongation factor P in a subset of bacterial species. Biol. Direct 5:3. 10.1186/1745-6150-5-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peil L, Starosta AL, Virumäe K, Atkinson GC, Tenson T, Remme J, Wilson DN. 2012. Lys34 of translation elongation factor EF-P is hydroxylated by YfcM. Nat. Chem. Biol. 8:695–697. 10.1038/nchembio.1001 [DOI] [PubMed] [Google Scholar]
- 29.Navarre WW, Zou SB, Roy H, Xie JL, Savchenko A, Singer A, Edvokimova E, Prost LR, Kumar R, Ibba M, Fang FC. 2010. PoxA, yjeK, and elongation factor P coordinately modulate virulence and drug resistance in Salmonella enterica. Mol. Cell 39:209–221. 10.1016/j.molcel.2010.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng WT, Banta LM, Charles TC, Nester EW. 2001. The chvH locus of Agrobacterium encodes a homologue of an elongation factor involved in protein synthesis. J. Bacteriol. 183:36–45. 10.1128/JB.183.1.36-45.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 32.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oaks EV, Wingfield ME, Formal SB. 1985. Plaque formation by virulent Shigella flexneri. Infect. Immun. 48:124–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zou SB, Hersch SJ, Roy H, Wiggers JB, Leung AS, Buranyi S, Xie JL, Dare K, Ibba M, Navarre WW. 2012. Loss of elongation factor P disrupts bacterial outer membrane integrity. J. Bacteriol. 194:413–425. 10.1128/JB.05864-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bochner BR, Gadzinski P, Panomitros E. 2001. Phenotype microarrays for high-throughput phenotypic testing and assay of gene function. Genome Res. 11:1246–1255. 10.1101/gr.186501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mileykovskaya E, Dowhan W. 1997. The Cpx two-component signal transduction pathway is activated in Escherichia coli mutant strains lacking phosphatidylethanolamine. J. Bacteriol. 179:1029–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klose KE, Weiss DS, Kustu S. 1993. Glutamate at the site of phosphorylation of nitrogen-regulatory protein NTRC mimics aspartyl-phosphate and activates the protein. J. Mol. Biol. 232:67–78. 10.1006/jmbi.1993.1370 [DOI] [PubMed] [Google Scholar]
- 38.Thanikkal EJ, Mangu JCK, Francis MS. 2012. Interactions of the CpxA sensor kinase and cognate CpxR response regulator from Yersinia pseudotuberculosis. BMC Res. Notes 5:536. 10.1186/1756-0500-5-536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tran Van Nhieu G, Ben-Ze'ev A, Sansonetti PJ. 1997. Modulation of bacterial entry into epithelial cells by association between vinculin and the Shigella IpaA invasin. EMBO J. 16:2717–2729. 10.1093/emboj/16.10.2717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blocker A, Gounon P, Larquet E, Niebuhr K, Cabiaux V, Parsot C, Sansonetti P. 1999. The tripartite type III secreton of Shigella flexneri inserts IpaB and IpaC into host membranes. J. Cell Biol. 147:683–693. 10.1083/jcb.147.3.683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santapaola D, Del Chierico F, Petrucca A, Uzzau S, Casalino M, Colonna B, Sessa R, Berlutti F, Nicoletti M. 2006. Apyrase, the product of the virulence plasmid-encoded phoN2 (apy) gene of Shigella flexneri, is necessary for proper unipolar IcsA localization and for efficient intercellular spread. J. Bacteriol. 188:1620–1627. 10.1128/JB.188.4.1620-1627.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buchrieser C, Glaser P, Rusniok C, Nedjari H, D'Hauteville H, Kunst F, Sansonetti P, Parsot C. 2000. The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella flexneri. Mol. Microbiol. 38:760–771. 10.1046/j.1365-2958.2000.02179.x [DOI] [PubMed] [Google Scholar]
- 43.Mitobe J, Arakawa E, Watanabe H. 2005. A sensor of the two-component system CpxA affects expression of the type III secretion system through posttranscriptional processing of InvE. J. Bacteriol. 187:107–113. 10.1128/JB.187.1.107-113.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pasquali C, Frutiger S, Wilkins MR, Hughes GJ, Appel RD, Bairoch A, Schaller D, Sanchez JC, Hochstrasser DF. 1996. Two-dimensional gel electrophoresis of Escherichia coli homogenates: the Escherichia coli SWISS-2DPAGE database. Electrophoresis 17:547–555. 10.1002/elps.1150170325 [DOI] [PubMed] [Google Scholar]
- 45.Park J-H, Johansson HE, Aoki H, Huang BX, Kim H-Y, Ganoza MC, Park MH. 2012. Post-translational modification by β-lysylation is required for activity of Escherichia coli elongation factor P (EF-P). J. Biol. Chem. 287:2579–2590. 10.1074/jbc.M111.309633 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.