

Abstract
The yeast phase of Histoplasma capsulatum is the virulent form of this thermally dimorphic fungal pathogen. Among the secreted proteome of Histoplasma, culture filtrate protein 4 (Cfp4) is a heavily glycosylated factor produced abundantly and specifically by Histoplasma yeast cells, suggesting its role in pathogenesis. We have generated three monoclonal antibodies as tools for characterization and detection of Cfp4 and determined the epitope each recognizes. Through site-directed mutagenesis of Cfp4, we identified three asparagines that function as the principal sites of N-linked glycan modification. To test the function of Cfp4 in Histoplasma pathogenesis, we generated Cfp4-deficient strains by insertional mutagenesis and by RNA interference. Cfp4-deficient strains are not attenuated in virulence in human macrophages or during lung infection in a murine model of histoplasmosis. Coinfection of differentially marked Cfp4-producing and Cfp4-deficient strains demonstrates that production of Cfp4 does not confer a fitness advantage to Histoplasma yeasts during murine lung infection. Despite no apparent role in acute virulence in mice, secretion of the Cfp4 glycoprotein by yeast cells is consistent across clinical and laboratory isolates of the North American type 1 and type 2 phylogenetic groups as well as a strain from Panama. In addition, human immune sera recognize the Histoplasma Cfp4 protein, confirming Cfp4 production during infection of human hosts. These results suggest the potential utility of Cfp4 as a diagnostic exoantigen for histoplasmosis.
INTRODUCTION
Histoplasma capsulatum is a member of the thermally dimorphic group of fungal pathogens that infect humans and other mammals, causing respiratory and systemic disease in these hosts (1–3). At ambient temperatures in the soil, Histoplasma grows as a saprobic conidium-producing mold. Disturbance of the mold form aerosolizes conidia, the inhalation of which initiates respiratory infection. Exposure to 37°C in the mammalian lung triggers a morphological and lifestyle change that results in the differentiation of conidia into yeast cells which parasitize host phagocytes. This conversion to the yeast form and expression of the yeast-phase regulon of genes are essential for the virulence of Histoplasma (4–6).
Unlike opportunistic fungi, Histoplasma survives the innate immune response largely by production of virulence factors that subvert or inactivate innate defenses (7). Histoplasma cells are efficiently taken up by phagocytes, chiefly alveolar macrophages which patrol the alveolar spaces. By expression of an α-linked glucan cell wall polysaccharide, Histoplasma yeasts conceal immunostimulatory cell wall β-glucans from detection by phagocytes (8). In addition, Histoplasma yeasts express an extracellular oxidative stress response system consisting of the extracellular superoxide dismutase, Sod3, and the extracellular catalase, CatB (9, 10). These secreted and cell surface-localized factors protect Histoplasma yeasts from the antimicrobial phagocyte-produced reactive oxygen during uptake by host phagocytes. Within phagocytes, Histoplasma yeasts grow and replicate, ultimately leading to lysis of the phagocyte and spread of the infection. A few factors facilitating the intramacrophage lifestyle of Histoplasma are beginning to be defined (11) and include Cbp1, a secreted factor of unknown function (12), production of siderophores and iron reductase systems that enable iron acquisition within the phagosome (13–15), de novo synthesis of essential vitamin cofactors (16), and thermotolerance (17).
Secretion is a hallmark of most Histoplasma virulence factors identified to date, positioning these factors to directly interact with host cells or molecules (18). As a foundation to better understand the secreted factors that contribute to Histoplasma's interaction with its host, we recently identified a core set of extracellular proteins produced by Histoplasma yeast cells (19). Five of the proteins secreted by yeasts lacked significant homology to other identified proteins from other organisms. These were designated culture filtrate proteins (Cfp), and the genes encoding three (Cfp1, Cfp4, and Cfp8) showed higher expression by pathogenic yeasts cells than by mycelia (19). At the protein level, Cfp4 was one of the most abundant extracellular proteins, second to the Cbp1 secreted factor.
In this study, we further characterize the Cfp4 protein and investigate its contribution to Histoplasma pathogenesis. We show that Cfp4 is heavily glycosylated and, through site-directed mutagenesis, identify which amino acids are attachment sites of N-linked glycan. Despite its abundant production, loss of Cfp4 does not reduce the virulence of two distinct strains of Histoplasma during acute respiratory infection, nor does Cfp4 provide a competitive advantage in coinfection experiments. Cfp4 is secreted by all strains tested from three different phylogenetic groups of Histoplasma. Cfp4 reacts with immune sera from histoplasmosis patients, providing evidence of production in vivo and suggesting that Cfp4 has potential as a diagnostic exoantigen.
MATERIALS AND METHODS
Culture of Histoplasma yeasts.
Histoplasma capsulatum strains (Table 1) included the laboratory strains G186A (ATCC 26027) and G217B (ATCC 26032), clinical isolates obtained from the Ohio State University Clinical Microbiology Lab, and mutants derived from the G186A and G217B backgrounds. Histoplasma cells were maintained as yeasts by growth at 37°C in Histoplasma-macrophage medium (HMM) (20). For the growth of uracil auxotroph strains, medium was supplemented with uracil (100 μg/ml). Liquid cultures were aerated by shaking (200 rpm) and grown until late exponential/early stationary phase. Growth rate and stage were determined by measuring liquid culture turbidity at 595 nm after addition of culture to 1 M NaOH. For experiments requiring defined yeast numbers, large clumps of yeasts of G186A background strains were removed by centrifugation (1 min at 50 × g), and the remaining dispersed cells were enumerated on a hemacytometer. For growth of fungal colonies, HMM was solidified with 0.6% agarose and supplemented with 25 μM FeSO4.
TABLE 1.
Histoplasma capsulatum strains
| Phylogenetic groupa | Strain | Genotype | Other designation |
|---|---|---|---|
| Pan | G186A | Wild type (ATCC 26027) | |
| WU8 | ura5-32Δ | CFP4 | |
| OSU6 | ura5-32Δ cfp4-1::T-DNA (hph) | cfp4 | |
| OSU15 | ura5-32Δ sod3-3Δ/pCR468 (URA5) | sod3Δ | |
| OSU18 | ura5-32Δ ags1-4Δ/pCR473 (URA5) | ags1Δ | |
| OSU44 | ura5-32Δ cfp4-1::T-DNA/pCR518 (URA5, PH2B-CFP4) | cfp4/CFP4 | |
| OSU45 | ura5-32Δ/pCR468 (URA5, gfp) | CFP4 | |
| OSU63 | ura5-32Δ/pEH09 (URA5, CFP4-RNAi) | CFP4-RNAi | |
| OSU77 | ura5-32Δ/pCR468 (URA5, gfp) | CFP4 | |
| OSU84 | ura5-32Δ cfp4-1::T-DNA/pCR468 (URA5, gfp) | cfp4 | |
| OSU85 | ura5-32Δ cfp4-1::T-DNA/pCR540 (URA5, rfp) | cfp4 | |
| OSU217 | ura5-32Δ/pCR628 (URA5, gfp) | Cfp4WT | |
| OSU218 | ura5-32Δ cfp4-1::T-DNA/pCR628 (URA5, gfp) | Cfp4− | |
| OSU219 | ura5-32Δ cfp4-1::T-DNA/pJA07 (URA5, CFP4) | Cfp4WT | |
| OSU220 | ura5-32Δ cfp4-1::T-DNA/pJA08 (URA5, CFP4N69A) | Cfp4N69A | |
| OSU221 | ura5-32Δ cfp4-1::T-DNA/pJA09 (URA5, CFP4N115A) | Cfp4N115A | |
| OSU222 | ura5-32Δ cfp4-1::T-DNA/pJA10 (URA5, CFP4N134A) | Cfp4N134A | |
| OSU223 | ura5-32Δ cfp4-1::T-DNA/pJA11 (URA5, CFP4N69A,N115A) | Cfp4N69A,N115A | |
| OSU224 | ura5-32Δ cfp4-1::T-DNA/pJA06 (URA5, CFP4N69A,N134A) | Cfp4N69A,N134A | |
| OSU225 | ura5-32Δ cfp4-1::T-DNA/pJA12 (URA5, CFP4N115A,N134A) | Cfp4N115A,N134A | |
| OSU226 | ura5-32Δ cfp4-1::T-DNA/pMK58 (URA5, CFP4N69A,N115A,N134A) | Cfp4N69A,N115A,N134A | |
| NAm2 | G217B | Wild type (ATCC26032) | |
| WU15 | ura5-42Δ | CFP4 | |
| OSU37 | ura5-42Δ/pCR473 (URA5, gfp-RNAi) | gfp-RNAi | |
| OSU87 | ura5-42Δ/pEH09 (URA5, CFP4-RNAi) | CFP4-RNAi | |
| Hc01 | Clinical isolate | ||
| Hc10 | Clinical isolate | ||
| Hc16 | Clinical isolate | ||
| Hc22 | Clinical isolate | ||
| Hc27 | Clinical isolate | ||
| NAm1 | Hc06 | Clinical isolate | |
| Hc20 | Clinical isolate |
Phylogenetic group classification as described by Kasuga et al. (48). Pan, Panama; NAm1, North American type 1; NAm2, North American type 2.
Generation of strains lacking Cfp4.
A transfer DNA (T-DNA) insertion at the CFP4 locus was generated using Agrobacterium tumefaciens-mediated transformation and recovery from mutant pools (21, 22). Briefly, A. tumefaciens strain LBA1100 containing plasmid pCM41 (encoding hygromycin resistance) was used to transform WU8 Histoplasma yeasts (23) and hygromycin-resistant transformants recovered by selection on HMM-uracil (100 μg/ml)-hygromycin (150 μg/ml). Pools containing 200 to 500 transformants each were made by flooding the transformation plates and collection of the yeast suspension. Total nucleic acids were isolated from an aliquot of each pool using mechanical disruption with 500-μm-diameter beads and ethanol precipitation, and the DNA was screened by PCR for insertions at the CFP4 locus using a right- or left-border primer specific to the T-DNA (21) and two nested CFP4 primers (ACGGCCGGTCCAGAACTGCTC and TCAATAATTGTATCCTGGCCCTC) located downstream of the CFP4 coding region. The individual insertion mutant (strain OSU6) was recovered from the mutant pool by successively subdividing the pool and screening by PCR until a single clone was obtained. The insertion site was determined by sequencing of the PCR product. The T-DNA insertion mutation in strain OSU6 was complemented by transformation of OSU6 with a linearized plasmid (pCR518) containing the CFP4 gene (amplified by PCR from G186A genomic DNA with primers AGGCGCGCCATGAAAACTGTTTGCTTGCCTACTTAC and CACTCGAGCAACTCGGGCGCTCTGTCAAAAAG) driven by the Histoplasma H2B promoter.
Depletion of Cfp4 in the G217B background was carried out using RNA interference (24). The CFP4 coding region was amplified from cDNA by PCR using CFP4-specific primers (GCAGCTGTTGACTCAATCAATGGC and GCGCTCTGTCAAAAAGACAGAAG), and inverse copies were cloned into the RNA interference (RNAi) vector, pCR427. The control green fluorescent protein (GFP)-RNAi and the CFP4-RNAi plasmids were linearized by digestion with PacI and transformed by electroporation (25) into the WU15 uracil auxotroph strain (23). Ura+ transformants were selected and screened for silencing of Cfp4 production by silver staining of culture filtrate proteins.
Determination of Cfp4 production and glycosylation.
The production of Cfp4 by Histoplasma yeasts was assessed by silver staining of polyacrylamide gels or by immunoblotting of yeast culture filtrates (19). Yeasts were grown in liquid medium to late exponential growth or stationary phase, the yeasts were removed by centrifugation (5 min at 2,000 × g), and the supernatants were passed through 0.45-μm-pore-size filters. Culture filtrate proteins were deglycosylated by overnight treatment with PNGase F (NEB), following reduction and denaturation of the culture filtrate proteins. The volumes of culture filtrate analyzed were normalized by culture density (absorbance at 595 nm) before separation by one-dimensional SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Cfp4 protein was visualized by silver staining of polyacrylamide gels or by immunoblotting after transfer of proteins to nitrocellulose. For immunoblotting, membranes were probed with monoclonal antibodies that recognize Cfp4 (see below), followed by a horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody (GenScript), and visualized by chemiluminescence (Pierce).
Mutation of putative N-linked glycosylation sites was done by PCR-based site-directed mutagenesis of the CFP4 gene using sense and antisense primers, which change the candidate asparagine codon to one encoding alanine. PCR amplicons representing the 5′ and 3′ regions of the CFP4 coding sequence flanking the target residue were generated from the cDNA template. Primers were engineered with the desired sequence change mutation. The two amplicons were subsequently annealed and extended in a second PCR by splicing by overlap extension (SOEing) (26, 27). The full-length, mutant CFP4 genes were cloned downstream of the H2B constitutive promoter in vector pCR628, which provides for Agrobacterium-mediated transformation of Histoplasma and chromosomal integration of the mutant CFP4 transgenes. For expression, constructs were transformed into the OSU6 strain background, which does not produce the native Cfp4 protein. Loss of N-linked glycosylation was determined by comparison of untreated and PNGase F-treated samples by immunoblotting of culture filtrates collected from transformants of each mutant construct.
Generation of anti-Cfp4 antibodies and epitope mapping.
Monoclonal antibodies to Histoplasma Cfp4 were generated by immunization of mice with a multiplexed library of seven Cfp4 peptides (SEAL; Abmart) and cloning of hybridoma fusions. Hybridomas were screened by immunoblotting of PNGase F-treated wild-type Histoplasma culture filtrates. Three positive clones were retained (2D20, 2F9, and 3G14). Hybridoma cell lines were expanded, the culture medium was collected, and the secreted antibody was concentrated by ultrafiltration of the medium using a 100-kDa-molecular-mass-cutoff membrane. To map the epitopes recognized, antibodies were screened for recognition of partial sequences of the Cfp4 protein (amino acids 70 to 113, 98 to 202, and 87 to 169) fused to glutathione S-transferase (GST) (in plasmid pGEX-2 [GE Healthcare Life Sciences]). The GST fusion proteins were expressed in Escherichia coli strain TG1 following a 3-h induction with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG). E. coli lysate proteins were prepared by boiling of bacteria in Laemmli sample buffer, separated by SDS-PAGE, and transferred to nitrocellulose for immunoblotting with the hybridoma supernatants.
Cfp4 recognition by human immune sera.
Histoplasma yeast strains WU8, WU15, OSU6, and OSU87 were grown in 100 ml liquid HMM until late exponential growth phase. Yeasts were removed by centrifugation (2,000 × g for 5 min) and then filtered through 0.22-μm-pore-size filters. Supernatants were concentrated using an Amicon ultrafiltration unit with a 10-kDa-molecular-mass-cutoff polyethersulfone (PES) membrane (Millipore). The protein concentrations of culture filtrates were determined using a DC protein assay with an ovalbumin protein standard (Bio-Rad). A total of 0.5 μg of untreated or PNGase F-treated deglycosylated culture filtrate proteins was separated by 10% SDS-PAGE, transferred to nitrocellulose, and probed with histoplasmosis patient immune serum at 1:500. The histoplasmosis sera were obtained from a 2012 outbreak investigation in Nebraska by the CDC (28). Characteristics of infected individuals are described in reference 29. Sera were previously confirmed positive by histoplasmin complement fixation and Histoplasma polysaccharide antigen testing. Being from an outbreak, the cases can be classified as probable acute or subacute pulmonary histoplasmosis. Immunoblots were also performed with pooled high-titer blastomycosis immune sera prepared from 5 individuals with high reactivity in a Blastomyces immunoassay (30). Control healthy human serum was from 5 residual sera from the Indiana Blood Bank chosen because Indiana is an area where Histoplasma is endemic. Protein recognition was visualized using biotinylated anti-human secondary antibody (0.15 μg/ml) and streptavidin-horseradish peroxidase (0.2 μg/ml) with Opti 4CN substrate color detection (Bio-Rad).
Macrophage infections.
To determine the role of Cfp4 in macrophage killing, we utilized the previously described P338D1-lacZ macrophage killing assay in which macrophage-expressed LacZ is used as an indicator of surviving macrophages (17). Macrophages were maintained in Ham's F-12 with 10% heat-inactivated fetal bovine serum (FBS). Macrophages were seeded at 2.5 × 104 macrophages per well of a 96-well plate and incubated at 37°C with 5% CO2-95% air for the duration of the assay. Yeasts of strains OSU45, OSU84, OSU18, OSU37, and OSU87 were grown to late exponential growth phase in HMM and counted on a hemacytometer. Yeasts were diluted and suspended in HMM-M (HMM buffered to pH 7.2 with 25 mM bicarbonate) with 10% FBS, and suspensions consisting of 2.5 × 104 yeasts were used to replace the medium on macrophages. Macrophage monolayers were examined visually daily and compared to wells containing uninfected macrophages. After 7 days, the number of surviving macrophages was determined by measuring the total β-galactosidase activity in each well (17). Briefly, culture media were removed, and remaining macrophages were lysed by the addition of phosphate-buffered saline (PBS) containing 0.5% Triton X-100, 2 mM MgCl2, and 2 mg/ml o-nitropenyl-β-d-galactopyranoside (ONPG). Substrate conversion was allowed to proceed for 30 min at room temperature before color change was read at 420 nm with correction at 595 nm.
Isolation and infection of human monocyte-derived macrophages.
Peripheral blood monocytes were collected from healthy human volunteers, and monocytes were differentiated into macrophages as described previously (31). Human cells were obtained from healthy volunteers after obtaining written informed consent and Health Insurance Portability and Accountability Act (HIPAA) authorization. Human subject research was approved by the Biomedical Sciences Institutional Review Board at Ohio State University (protocol 2008H0242). Blood was collected by venipuncture into heparinized tubes. For monocyte collection, 20 ml of heparinized blood was mixed with 15 ml of saline. A total of 14 ml of Ficoll-Paque PLUS was underlayed to the blood suspension, and cells were separated by centrifugation (400 × g for 40 min at 18°C). The upper plasma layer was removed, the Ficoll and “buffy coats” were pooled, and the volumes were brought to 50 ml with saline. Monocytes were collected by centrifugation (200 × g for 15 min at 4°C) and transferred to Teflon wells in RPMI 1640 medium containing 20% autologous serum at a concentration of 2 × 106 cells/ml. Monocytes were incubated for 5 days at 37°C with 5% CO2-95% air for differentiation into macrophages. Cell suspensions were collected by centrifugation (200 × g for 15 min at 4°C), seeded at 8 × 104 total cells per well of a 96-well plate in RPMI 1640 medium with 10% autologous serum, and incubated for 2 h at 37°C before removal of nonadherent cells. For experiments involving activated macrophages, macrophages were treated with gamma interferon (IFN-γ) (1,000 U/ml; BioLegend) for 48 h.
For infection of monocyte-derived macrophages, Histoplasma yeasts were grown to late exponential growth phase in HMM and enumerated by a hemacytometer. Yeasts were added to the wells containing monocyte-derived macrophages at 2 × 103 yeasts/well, and infections were allowed to proceed for 4 h at 37°C with 5% CO2-95% air. To measure Histoplasma survival, the macrophages were lysed with water, and serial dilutions of the lysate were plated on HMM to determine viable CFU. The CFU counts from each infection were compared to wells containing yeast and complete media without macrophages to determine the relative yeast survival.
Bioinformatic analysis of Cfp4.
The CFP4 coding region was obtained from the sequenced genomes of G186A, Nam1, and G217B strains from The Genome Institute at Washington University (St. Louis) and from the Broad Institute (MIT). Translated sequences were examined using the ClustalW alignment algorithm within the BioEdit software package (http://www.mbio.ncsu.edu/bioedit). The presence of a secretion signal peptide was determined using SignalP v4.1 (32), and putative asparagine-linked glycosylation sites were predicted using NetNGlyc v1.0 (http://www.cbs.dtu.dk/services/NetNGlyc/) and GlycoEP (33).
Mouse infection assays.
To determine if Cfp4 is required for Histoplasma growth during infection, OSU45, OSU84, OSU37, and OSU87 yeasts were grown in HMM to late exponential growth, and suspensions were used to infect C57BL/6 mice (NCI) intranasally. Inocula were plated on solid HMM to determine the actual inoculum delivered. At incremental times, lung and spleen tissues were harvested and homogenized. Dilutions of the organ homogenates were plated on HMM to enumerate viable yeast CFU. Statistical analysis of data was performed using one-tailed Student's t test. All animal experiments were performed in compliance with the National Research Council's Guide for the Care and Use of Laboratory Animals (34) and were approved by the Institutional Animal Care and Use Committee at Ohio State University (protocol 2007A0241).
For competition assays, the WU8 wild-type strain was transformed with a green fluorescent protein expression plasmid (pCR468), and the cfp4::T-DNA mutant strain was transformed with a red fluorescent protein (td-Tomato RFP) expression plasmid (pCR540) to yield strains OSU77 and OSU85, respectively. Yeasts were grown in HMM to late exponential growth phase, enumerated by a hemacytometer, and GFP- and RFP-marked yeasts were mixed at a 1:1 ratio. The mixed suspension was used to inoculate liquid HMM at 5 × 106 total yeasts/ml and to infect mice (1 × 104 total yeasts per mouse). The original inoculum, broth culture after 4 days, and lung homogenates at 9 days postinfection were plated on solid HMM to determine total actual inoculum size and to determine the ratio of CFP4 (GFP fluorescent) and cfp4::T-DNA (RFP fluorescent) CFU. The GFP or RFP fluorescence of colonies was detected using a modified transilluminator (AlphaInnotech) with emission filters specific for GFP (530/15) or for RFP (620/20). The competition index was calculated as the ratio of OSU77 to OSU85 at the assay endpoint divided by the ratio of OSU77 to OSU85 in the initial inoculum.
RESULTS
Generation of Cfp4-depleted Histoplasma strains.
To probe the functional role of Cfp4 in the biology of Histoplasma, we created strains in which synthesis of the CFP4 gene product was prevented by insertional mutation or Cfp4 production was depleted by RNA interference. Cfp4 was originally identified from a proteomics analysis of the secreted proteome of the Panama lineage strain, G186A (19). Transcript sequencing confirmed the exon structure of the CFP4 gene (Fig. 1A). The gene encodes a 213-amino-acid protein of which notable features include a secretion signal at the N terminus and a proline/threonine-rich region (Fig. 1B). In addition, potential N-linked glycosylation sites were identified by bioinformatics analysis (Fig. 1B), and the verification of N-linked glycan modification of Cfp4 is detailed below. The Cfp4 protein is predicted to be 23 kDa in size, although the protein migrates as a broad 31- to 34-kDa band in one-dimensional acrylamide gels after enzymatic removal of N-linked glycosylation. To enable functional tests, we isolated mutants that were unable to produce the Cfp4 protein. We screened random T-DNA insertions to identify a mutant harboring a T-DNA insertion at the CFP4 locus (strain OSU6). The T-DNA insertion in OSU6 is located 209 bp upstream of the initiation codon (Fig. 1A). The insertion disrupts the CFP4 promoter, preventing expression of CFP4 as evidenced by lack of Cfp4 protein in deglycosylated culture filtrates derived from OSU6 yeasts (Fig. 1C). As an independent means of preventing Cfp4 function, we also created Cfp4-depleted lines through RNA interference (RNAi). Transduction of yeasts with the CFP4-RNAi plasmid, but not with an RNAi plasmid targeting gfp, prevented Cfp4 accumulation in yeast culture filtrates (Fig. 1C). In similar fashion, RNAi was used to suppress Cfp4 production by yeasts of the North American type 2 (NAm2) phylogenetic group (Fig. 1C). Examination of the proteins in the culture filtrate produced by these CFP4-RNAi yeasts showed loss of Cfp4 protein (Fig. 1C).
FIG 1.
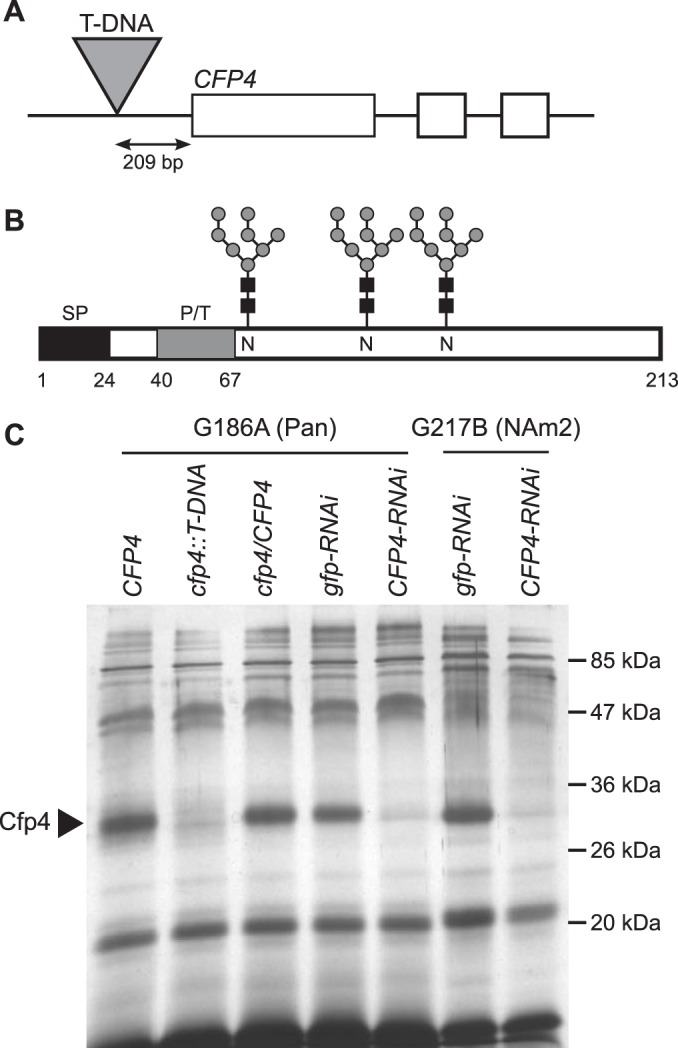
Generation of Cfp4-depleted Histoplasma strains. (A) Scaled depiction of the CFP4 gene, which consists of 3 exons (boxes), and the cfp4::T-DNA mutant (strain OSU6) in which a T-DNA insertion is located 209 bp upstream of the initiation codon. (B) Representation of the primary structure of the Cfp4 protein. Schematic indicates the secretion signal peptide (SP; black box), the proline/threonine-rich region (P/T; gray box), and three sites of N-linked glycosylation (glycan symbols) and the respective asparagines (N). Numbers indicate the amino acid residues. (C) Analysis of culture filtrates showing depletion of Cfp4 protein by T-DNA insertion and by RNAi. Culture filtrates were collected from G186A (Panama) and G217B (NAm2) background strains. Culture filtrate proteins were first treated with PNGase F to remove N-linked glycans, separated by one-dimensional SDS-PAGE, and visualized by silver staining. Strains analyzed include the Cfp4-producing parent strain (CFP4; strain WU8), the strain with a T-DNA insertion in the CFP4 gene promoter (cfp4::T-DNA; strain OSU6), and the complemented strain (cfp4/CFP4; strain OSU44). Cfp4 was also depleted by RNAi in both backgrounds, as shown by strains harboring plasmids for knockdown of the unrelated gfp gene (gfp-RNAi; strains OSU100 and OSU75) or CFP4 (CFP4-RNAi; strains OSU63 and OSU87).
Characterization of Cfp4 N-linked glycosylation.
We generated custom monoclonal antibodies to Cfp4 as a tool to track production and posttranslational modification of the Cfp4 protein. Mice were immunized with a chimeric protein containing seven selected Cfp4 epitopes (Fig. 2A). Following hybridoma establishment, the hybridoma culture filtrates were screened for recognition of Histoplasma Cfp4. Three hybridoma lines producing antibodies that detect Histoplasma Cfp4 were obtained: 2D20, 2F9, and 3G14. The antibodies specifically recognize the Histoplasma Cfp4 protein since immunoblots of wild-type Histoplasma yeast culture filtrate proteins react with a 33-kDa band but fail to detect anything in culture filtrates derived from mutant yeasts unable to produce Cfp4 (Fig. 2B). To map the epitope recognized by each of the monoclonal antibodies, hybridoma supernatants were screened against lysates from E. coli expressing different overlapping fragments of Cfp4 fused to glutathione S-transferase (Fig. 2C; see also Fig. S1 in the supplemental material). Immunoblotting results (Fig. 2D) indicate that 2D20 and 2F9 both recognize epitope 2 (Cfp4 fragments contained in pCR617 and pCR619 but not pCR618), whereas the 3G14 hybridoma produces an antibody that is specific for epitope 5 (Cfp4 fragments contained in pCR618 but not pCR617 and pCR619). The recognition of epitope 2 by 2D20 and 2F9 is qualitatively different, suggesting that the antibodies produced by these two hybridomas are not identical.
FIG 2.

Mapping of epitopes recognized by monoclonal antibodies to Cfp4. (A) Primary amino acid sequence of the Cfp4 protein showing the location of the epitopes used (boxed amino acids) for immunization of mice (epitopes 0 to 6). (B) Specificity of Cfp4 recognition by monoclonal antibodies as demonstrated by immunoblots of Histoplasma culture filtrates from Cfp4-producing and Cfp4-lacking yeasts. Culture filtrates were collected from Cfp4-producing (CFP4; strain OSU45) and cfp4 mutant yeasts (cfp4::T-DNA; strain OSU84). PNGase F-treated samples were immunoblotted with hybridoma supernatants 2D20, 2F9, and 3G14. (C) Schematic of the Cfp4 protein and the constructs used to map the recognized Cfp4 epitopes. Shaded boxes represent epitopes 0 to 6, and horizontal bars below show the Cfp4 fragments produced in E. coli as fusions to glutathione S-transferase (GST) from plasmids pCR617, pCR618, and pCR619. (D) Delineation of the epitopes recognized by each monoclonal antibody. Crude E. coli lysates from GST-expressing or GST::Cfp4 fusion-expressing bacteria were immunoblotted with hybridoma supernatants.
Previously, we determined that the Cfp4 protein was highly modified by N-linked glycosylation (19). Glycosylation is characteristic of many extracellular proteins that transit through the general secretory pathway. In particular, N-linked glycosylation substantially alters the Cfp4 protein migration in one-dimensional denaturing acrylamide gels; glycosylated Cfp4 migrates variably as a high-molecular-mass diffuse smear, whereas deglycosylation of Cfp4 by PNGase F treatment increases the mobility of Cfp4 as a specific band near 33 kDa (see Fig. S2 in the supplemental material). Even after removal of N-linked glycans through the N-linked-specific deglycosylase PNGase F, Cfp4 does not migrate with the predicted molecular mass, implying additional posttranslational modifications (e.g., O-linked glycosylation).
To determine which asparagine residues serve as sites for N-linked glycosylation, we used site-directed mutagenesis to eliminate candidate positions for glycan attachment. Between the three major phylogenetic lineages (NAm1, NAm2, and Panama), the respective Cfp4 proteins have 80 to 85% amino acid identity, with most variation occurring in the proline/threonine-rich region (amino acids 40 to 67) that follows the signal peptide (Fig. 3A). The threonine residues in the proline/threonine-rich region of amino acids may serve as the site for O-linked glycosylation. Bioinformatic examination of the Cfp4 amino acid sequence N-glycan attachment prediction algorithms identified 7 asparagine residues for potential N-linked glycan modification, including four with the consensus N-linked glycosylation sequon Asn-X-Ser or Asn-X-Thr (Fig. 3A). The sequon beginning with the asparagine at amino acid 147 was not conserved in all strains, missing either the asparagine (NAm1) or the serine/threonine residue of the consensus site (NAm2), and was not considered further. The remaining three sites, N69, N115, and N134, were considered potential sites for N-linked glycan attachment to Cfp4.
FIG 3.
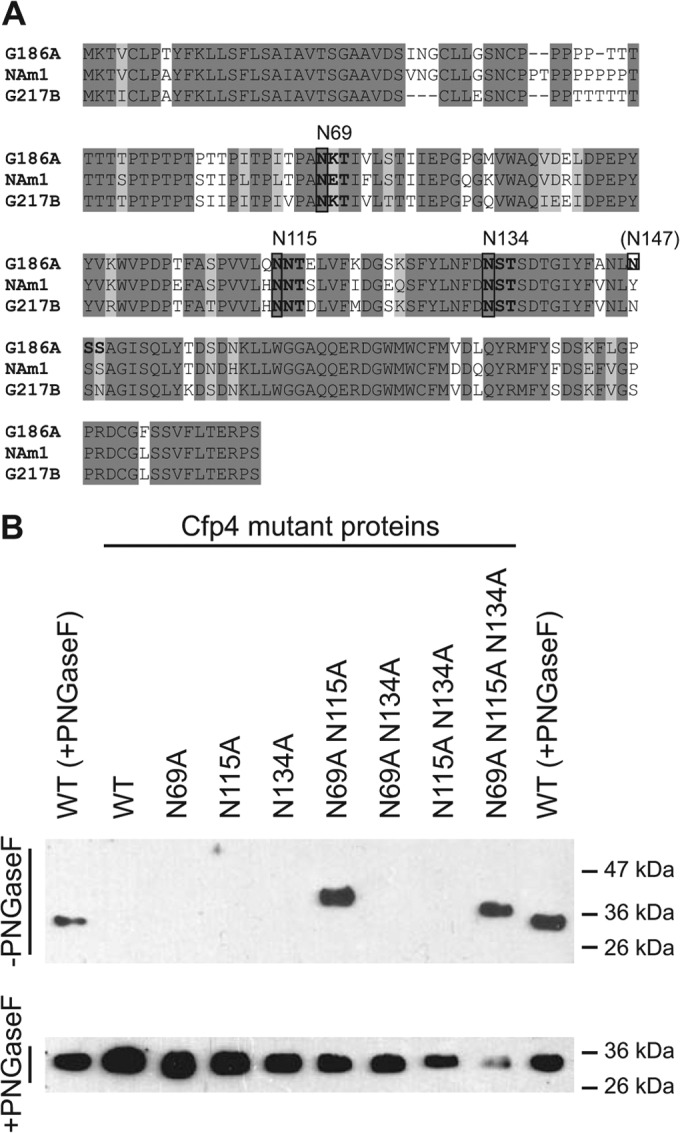
Identification of N-linked glycosylation modification sites on Cfp4. (A) Amino acid sequence of Cfp4 from three phylogenetic groups of Histoplasma indicating sites of potential N-linked glycosylation. Shading of amino acids represents completely conserved residues (dark shading) or conservation in 2 of 3 lineages (light shading). Consensus N-linked glycosylation sequons (Asn-X-Ser/Thr) are indicated in bold, with the corresponding asparagine residue above the sequence. (B) Delineation of N-linked glycosylation sites on Cfp4 through mutation of candidate sites. Culture filtrates from Histoplasma yeasts expressing mutant Cfp4 proteins (strains OSU217 to OSU226) were immunoblotted using the 2D20 antibody. Culture filtrate proteins were electrophoresed without prior treatment with PNGase F (−PNGaseF; top) or with PNGase F treatment (+PNGaseF; bottom). The leftmost and rightmost lanes in both panels contained PNGase F-treated culture filtrate proteins from the wild type [WT (+PNGaseF); G186A strain]. Labels above the panels indicate which specific asparagines were mutated to alanines in the mutant proteins.
Candidate asparagines were mutated to alanine singly and in combination, and the transgenes were expressed in the cfp4 mutant background (strain OSU6). N-linked glycosylation of Cfp4 prevents the wild-type protein from efficiently entering and migrating through acrylamide gels, whereas Cfp4 without N-linked glycans migrates around 33 kDa. Thus, absence of the Cfp4 mutant protein on immunoblots of culture filtrates without PNGase F treatment was used as an indicator that the mutant Cfp4 proteins still had substantial N-linked glycans. None of the single asparagine-to-alanine mutations permitted migration of Cfp4 through acrylamide gels, consistent with N-linked glycans present at multiple sites (Fig. 3B). Consequently, mutated asparagines were combined as pairs and as a triple mutant protein lacking all three candidate asparagines. Expression of the N69A N115 N134A triple mutant enabled migration of Cfp4 through the acrylamide gel nearly as well as fully deglycosylated Cfp4, indicating that at least two of these residues are sites of substantial glycosylation. The N69A N134A protein as well as the N115A N134A protein did not enter the gel, indicating substantial glycan modification still remaining in these double mutant proteins. On the other hand, the N69A N115A Cfp4 protein migrated into the gel, albeit with slightly slower mobility than the triple N69A N115A N134A mutant protein. Together, these results are consistent with N69 and N115 being sites of large N-glycan modification, and the slower mobility of the N69A N115A mutant protein that retains the N134 site suggests some smaller oligosaccharide attachment to N134.
Cfp4 is a conserved exoantigen.
To test for conservation of Cfp4 protein production and secretion, culture filtrates from laboratory and clinical isolates were screened by immunoblotting for Cfp4. Clinical isolates of Histoplasma were typed as either NAm1 or NAm2 by exploiting PCR size polymorphisms of the YPS3 gene and PCR-restriction fragment length polymorphisms (RFLP) in the SOD3 gene (see Fig. S3 in the supplemental material). Culture filtrates from two NAm1 isolates (Hc06 and Hc20) were tested, as well as six NAm2 isolates (Hc01, Hc10, Hc16, Hc22, Hc27, and G217B) and the G186A Panama lineage strain. Immunoblots of the culture filtrate proteins showed that all strains secreted Cfp4 protein (Fig. 4), indicating that Cfp4 is a conserved extracellular factor of Histoplasma yeasts. The Cfp4 protein from NAm1 isolates showed slightly slower mobility than the Cfp4 of NAm2 isolates after removal of N-linked glycans. Although the NAm1 Cfp4 protein is 5 amino acids larger than the NAm2 Cfp4 protein, this difference is unlikely to be the sole contributor to the greater-than-1-kDa shift observed. This may suggest the Cfp4 of NAm1 Histoplasma has a higher degree of O-linked glycan modification than that of NAm2, since the O-linked glycans are not affected by PNGase F treatment.
FIG 4.
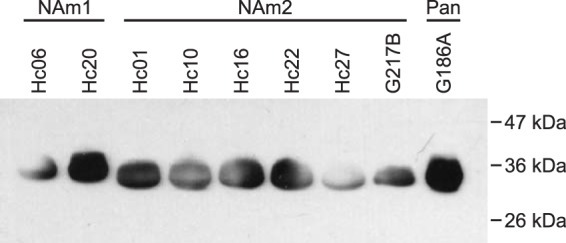
Secretion of Cfp4 protein by three divergent phylogenetic lineages of Histoplasma yeasts. Culture filtrates collected from NAm1 strains (Hc06 and Hc20), NAm2 strains (Hc01, Hc10, Hc16, Hc22, Hc27, G217B), and the Panama strain (G186A) were treated with PNGase, and the proteins were separated by one-dimensional SDS-PAGE followed by immunoblotting with 2D20 and 2F9 antibodies.
Given the conserved production of Cfp4 by Histoplasma strains, we evaluated the possible utility of Cfp4 as a diagnostic immunoreactive antigen. Previous results indicated that the CFP4 gene is transcribed during mouse lung infection (19). To test for Cfp4 production during human infection, culture filtrates derived from G186A and G217B background wild-type and cfp4 mutant strains were probed with immune sera from eight human patients with confirmed Histoplasma exposure by complement fixation tests. As the sera were obtained from an outbreak investigation, the cases were likely acute or subacute pulmonary histoplasmosis. Without PNGase F treatment, a few background bands were observed, but few differences were noted between Cfp4-proficient and Cfp4-lacking culture filtrates. However, after PNGase F treatment of the Histoplasma-secreted proteins, human immune sera strongly recognized a Histoplasma protein around 33 kDa (Fig. 5). The identity of this immunoreactive protein was Cfp4, since no 33-kDa protein was detected in culture filtrates harvested from Cfp4-deficient strains. The human immune sera recognized both the G217B (NAm2) Cfp4 and the G186A (Panama) Cfp4 despite all patients being from the United States and not exposed to the Panama Histoplasma lineage (data not shown). At a qualitative level, no particular correlation was observed between Cfp4 reactivity by immunoblotting and complement fixation titers (Fig. 5). Normal (healthy) human sera did not react with the Cfp4 protein, nor did pooled immune sera from patients infected with the related fungus Blastomyces dermatitidis (Fig. 5). The strong immunoreactivity of the Cfp4 protein with histoplasmosis immune sera indicates Cfp4 has potential as a diagnostic antigen for human histoplasmosis.
FIG 5.
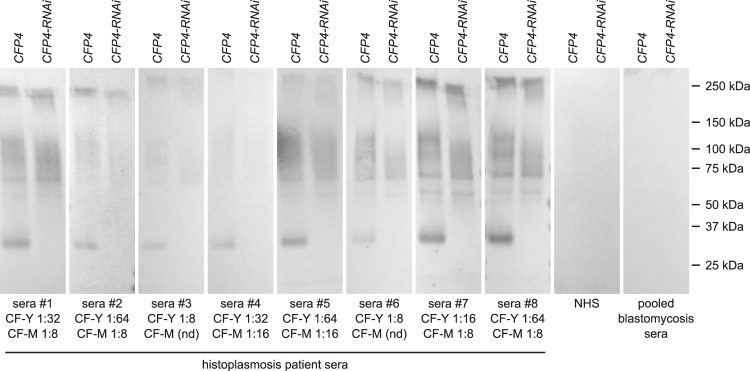
Immunoreactivity of Cfp4 in humans with histoplasmosis. Immunoblots of culture filtrates collected from Cfp4-producing (CFP4; OSU37) and Cfp4-depleted (CFP4-RNAi; OSU87) Histoplasma yeasts using sera from patients with histoplasmosis, healthy individuals (normal human sera [NHS]from residents of Indiana), and pooled high-titer sera from patients with blastomycosis. Culture filtrate proteins (500 ng total protein) were treated with PNGase F and separated by electrophoresis for immunoblotting. The yeast-histoplasmin and mycelial-histoplasmin complement fixation titers (CF-Y and CF-M, respectively) of individual histoplasmosis patient serum are listed below each immunoblot. “nd” indicates no detection by complement fixation tests.
Cfp4 is not essential for Histoplasma pathogenesis.
The conserved production, abundant secretion, and pathogenic yeast-phase-specific expression of Cfp4 suggests that Cfp4 could contribute to Histoplasma pathogenesis. Since macrophages are the primary host cell for Histoplasma yeasts, we investigated whether Cfp4 was required for macrophage infection. To test for Cfp4 function in macrophage infection and lysis, wild-type and Cfp4-deficient Histoplasma yeasts were used to infect an lacZ-transgenic murine macrophage cell line which enables rapid quantification of surviving macrophages (17). Lack of Cfp4 did not reduce the ability of Histoplasma yeast to kill macrophages (Fig. 6A), as both the Cfp4-producing (OSU45 and OSU37) and Cfp4-lacking (OSU84 and OSU87) yeasts caused lysis of 70 to 75% of the macrophages relative to macrophage monolayers without Histoplasma infection. In contrast, macrophages infected with a virulence-attenuated strain due to loss of α-(1,3)-glucan caused lysis of only 30% of the macrophages. No deficiency in macrophage killing was found between wild-type and cfp4 mutant yeast from either the G186A (Panama) or the G217B (NAm 2) background.
FIG 6.
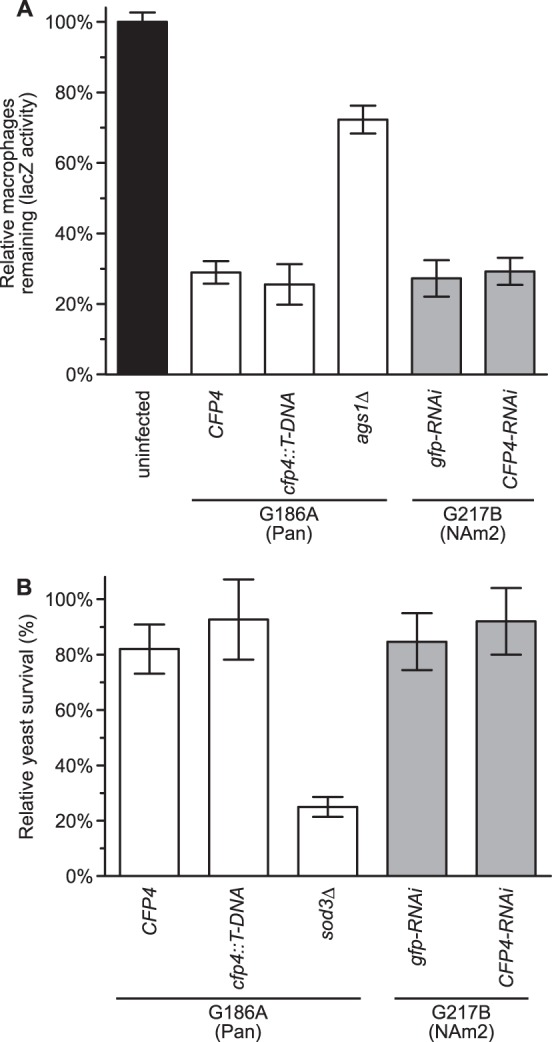
Cfp4 production is not essential for Panama or NAm2 lineage Histoplasma virulence in macrophages. (A) Determination of macrophage killing ability of yeasts with and without Cfp4. P388D1 macrophages expressing LacZ were infected with Histoplasma yeasts at a multiplicity of infection (MOI) of 1:1. After 7 days of coculture, the relative survival of macrophages was determined by quantifying the remaining macrophage-produced LacZ activity. As controls, macrophages were left uninfected or were infected with the attenuated Histoplasma strain lacking α-glucan (ags1Δ; strain OSU18), which is deficient in macrophage killing. (B) Histoplasma yeast survival in IFN-γ-activated human macrophages with and without Cfp4. Human macrophages were derived from peripheral blood monocytes, activated with IFN-γ, and infected with Histoplasma yeasts at an MOI of 1:50. Yeast survival was determined by enumeration of viable CFU after 4 h of coincubation at 37°C. Results are plotted as survival relative to yeasts incubated in the absence of macrophages. In all experiments, the data represent the means ± standard deviations (n = 3 replicates). All macrophage infections were conducted using Cfp4-producing yeasts (CFP4; strain OSU45) or cfp4 mutant yeasts (cfp4::T-DNA; strain OSU84) of the G186A (Panama) background or Cfp4-producing yeasts (gfp-RNAi; strain OSU37) or Cfp4-depleted yeasts (CFP4-RNAi; strain OSU87) of the G217B (NAm2) background. The superoxide dismutase-deficient sod3Δ strain (OSU15) was used as a control for attenuated survival in macrophages. No statistically significant differences were detected (Student's t test) between infections with Cfp4-producing and Cfp4-deficient strains.
Although Histoplasma-dependent lysis of host cells was equivalent with and without Cfp4, Cfp4 could function in enhancing Histoplasma's ability to infect and survive within macrophages. To test this, Cfp4-producing and Cfp4-deficient yeasts were used to infect human primary macrophages derived from peripheral blood monocytes. We observed no reduced survival of Cfp4-deficient yeasts in nonactivated macrophages (data not shown). To increase the stringency of this test, experiments were repeated with macrophages that were activated by treatment with IFN-γ prior to infection. Enumeration of intracellular yeasts showed no differences in survival and replication between the Cfp4-producing (OSU45 and OSU37) and Cfp4-deficient (OSU84 and OSU87) strains (Fig. 6B). This survival of Cfp4-deficient yeasts is in contrast to yeasts that lack the Sod3 extracellular superoxide dismutase, which has reduced survival in activated macrophages (Fig. 6B). Thus, Cfp4 is not essential for infection of and survival within human macrophages.
Although Cfp4 does not appear to function in the interaction of Histoplasma with macrophages in culture, we used Cfp4-producing (OSU45) and Cfp4-deficient (OSU84) strains to establish sublethal respiratory infections in mice to address any potential role of Cfp4 in Histoplasma virulence in vivo. After intranasal infection, both the wild-type and cfp4 mutant Histoplasma yeasts exhibited similar infection kinetics in lung tissue with rising fungal burdens through day 12 and afterward declining with the onset of cell-mediated immunity (Fig. 7A and B). The kinetics and fungal burden in spleen tissue were also identical for mice infected with wild-type and cfp4 mutant strains, indicating that Cfp4 was not required for extrapulmonary dissemination of the infection (Fig. 7C and D). As differences exist between the virulence factor production and requirement between Histoplasma phylogenetic groups (35–37), we tested whether Cfp4 was required by NAm2 Histoplasma using the gfp-RNAi and cfp4-RNAi lines of the G217B background (OSU37 and OSU87, respectively). Similar to the results with the G186A background, lung infection kinetics and fungal burdens were similar regardless of the production of Cfp4 (Fig. 8A and B). In the G217B background, a 5-fold reduction in spleen fungal burden was observed at day 8 and was also decreased at day 12 in the absence of Cfp4 (Fig. 8C and D). This was consistent through multiple mouse infections. Although statistically significant, this Cfp4-dependent difference in virulence is transient, and after day 16, identical fungal burdens are found in the spleens of mice infected with Cfp4-producing and Cfp4-lacking Histoplasma yeasts.
FIG 7.

Cfp4 is not essential for Panama lineage Histoplasma yeasts' ability to cause acute histoplasmosis in mice. Virulence of G186A background yeasts in vivo as assessed by fungal burdens in lungs (A and B) and spleens (C and D) following sublethal infection. Mice were infected intranasally with Cfp4-producing (CFP4; strain OSU45) or Cfp4-lacking (cfp4::T-DNA; strain OSU84) yeasts, and the fungal burden in tissues was determined by plating of the tissue homogenates and enumeration of viable CFU at 4-day increments postinfection. Data represent the fungal burdens in individual mice, and horizontal bars indicate the average burden. Dashed horizontal line in lung graphs indicates the inoculum delivered. Fungal burdens at each time point between Cfp4-producing and cfp4 mutant strains were compared by Student's t test, but no data points had statistical significance (P < 0.05).
FIG 8.

Cfp4 is not essential for NAm2 lineage Histoplasma yeasts' ability to cause acute histoplasmosis in mice. Virulence of G217B background yeasts in vivo as assessed by fungal burdens in lungs (A and B) and spleens (C and D) following sublethal infection. Mice were infected intranasally with Cfp4-producing (gfp-RNAi; strain OSU37) or Cfp4-lacking (CFP4-RNAi; strain OSU87) yeasts, and the fungal burden in tissues was determined by plating of the tissue homogenates and enumeration of viable CFU at 4-day increments postinfection. Data represent the fungal burdens in individual mice, and horizontal bars indicate the average burden. Dashed horizontal line in lung graphs indicates the inoculum delivered. Fungal burdens at each time point between Cfp4-producing and cfp4 mutant strains were compared by Student's t test, and statistical significance is indicated by asterisks (*, P < 0.05; **, P < 0.01).
As a more sensitive test of the potential contribution of Cfp4 to Histoplasma pathogenesis, we competed Cfp4-producing and Cfp4-deficient strains in vivo. To enable separate enumeration of Cfp4-producing yeast and Cfp4-deficient yeast after coinfection of the same mouse, we marked the Cfp4-producing yeast with the td-Tomato red fluorescent protein (RFP) and Cfp4-deficient yeasts with green fluorescent protein (GFP). To compute the relative fitness (i.e., competitive index), equal amounts of the two strains (OSU77 and OSU85) were used to inoculate mice and liquid broth, and the resulting proliferation in both environments was determined. The growths of both Cfp4-producing and Cfp4-deficient yeasts were nearly identical in liquid broth culture (competitive index of 0.8) (Fig. 9), indicating no role for Cfp4 during growth in vitro. After 9 days of lung infection, the competitive index was 0.8, also indicating Cfp4 confers no significant fitness advantage in vivo. These in vivo fitness data indicate that Cfp4 is dispensable for acute histoplasmosis.
FIG 9.
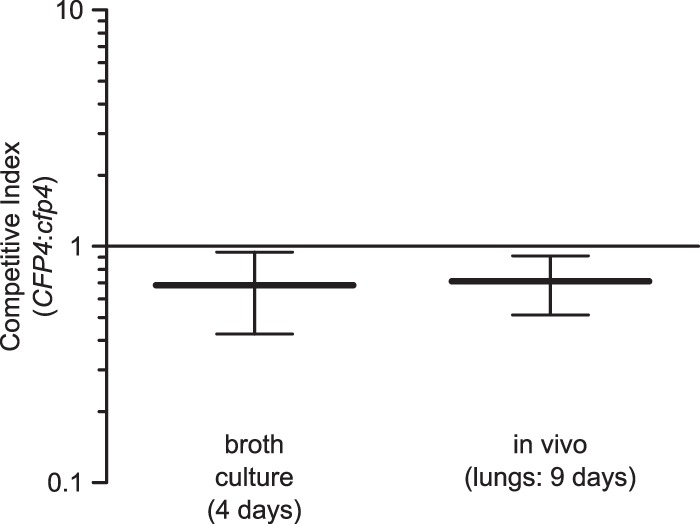
Cfp4 does not confer a competitive advantage to Histoplasma during murine infection. Competitive index determination of Cfp4-producing and Cfp4-deficient yeasts in vitro and in vivo. Equivalent proportions of Cfp4-producing yeasts marked with GFP (strain OSU77) and Cfp4-lacking yeasts marked with RFP (strain OSU85) were combined and used to inoculate liquid medium (broth culture) and for intranasal infection of mice (inoculum of 104 total yeast cells). After 4 days of growth in broth culture and 9 days of lung infection, dilutions of the yeast suspensions or the lung homogenates were plated and compared to platings of the inoculum mixture. The proportions of Cfp4-producing and Cfp4-deficient yeasts were determined as green and red fluorescent colonies, respectively. The competitive index was calculated as the GFP/RFP ratio at the endpoint divided by the GFP/RFP ratio of the inoculum. Horizontal lines and error bars represent the mean competitive index ± standard deviation (n = 4 replicates [in vitro] or infected mice [in vivo]). The competitive index did not differ significantly from 1.0 for in vitro (P = 0.51) or in vivo (P = 0.58) growth, as determined by Student's t test. Additionally, no significant difference (P = 0.88) was found between in vivo and in vitro growth.
DISCUSSION
Virulence is a unique feature of the yeast phase of Histoplasma, and nearly all demonstrated virulence factors exhibit preferential production by yeast cells instead of mycelia (Cbp1, α-1,3-glucan, Sod3, CatB, and Yps3) (9, 10, 19, 24, 37, 38). The Cfp4 protein shares many characteristics with established Histoplasma virulence factors, including yeast-phase-specific production, extracellular localization, and conserved production among three separate phylogenetic groups. Despite these indicators, our data with Cfp4-depleted strains indicate that Cfp4 is dispensable for acute infection of mice as well as survival and replication in macrophages. Even a highly sensitive competitive index test failed to show any fitness advantage conferred by expression of Cfp4. One possible explanation for why no role in virulence for Cfp4 was found is that a low level of Cfp4 could still be produced in the mutant and the RNAi strains which is sufficient for pathogenesis. However, we were unable to detect any Cfp4 produced either through silver staining of protein gels or by immunoblotting, indicating little, if any, production of Cfp4 by the mutant strains. In contrast, Cfp4 is one of the most abundantly secreted proteins by wild-type strains. Alternatively, Cfp4's role in pathogenesis may be masked by another factor with redundant function, as was observed with CatB and an intracellular catalase, CatP (10). As a third possibility, Cfp4's pathogenesis function may be specific for human but not murine hosts. This explanation seems unlikely, as our experiments with cultured human macrophages also did not indicate a role for Cfp4, even with cytokine activation of the macrophages.
Regardless of any function in pathogenesis, as a secreted factor, Cfp4 has potential as a diagnostic marker for histoplasmosis. Serological tests for host antibodies to Histoplasma antigens play an important role in diagnosis of acute, subacute, and chronic pulmonary infections (39–43). In a multicenter U.S. study, Hage et al. determined that current serology tests are positive in 90% of pulmonary cases but only 75% in disseminated cases, likely due to the effect of suppressed antibody production in immunocompromised and immunosuppressed hosts (42). Our experiments with human immune sera demonstrate that Cfp4 is produced during human infection and that it is an immunoreactive antigen. The good specificity of anti-Cfp4 antibodies for Histoplasma Cfp4 indicates a possible role for this protein as a diagnostic tool for Histoplasma infection with little cross-reactivity to related fungi. Future investigations will be needed to determine if antibody responses to Cfp4 are more sensitive than current serological markers. However, development of antibodies by the host requires time and an efficient immune response, both of which histoplasmosis patients may lack. Antigen, rather than antibody, detection is one way to avoid reliance on B-cell responses, and the level of antigenemia often correlates with the fungal burden in active disease (42, 44–47). As an abundantly secreted protein, Cfp4 could potentially be a target for antigen detection-based diagnostics, but further studies to establish its utility as a biomarker will be required.
Determination of the precise positions at which Cfp4 is modified by N-linked glycosylation identified protein regions that could be exploited for detection of the Cfp4 protein. By focusing on regions of the Cfp4 protein that are not modified, detection schemes can be designed that could recognize a variety of different Cfp4 glycoforms. Using site-directed mutagenesis, we mapped the sites of N-glycan attachment to the Cfp4 polypeptide backbone at positions 69, 115, and 134. Cfp4 detection by immunoblotting requires prior treatment with PNGase F to facilitate electrophoretic mobility and transfer of the protein. We have not determined if the monoclonal antibodies can detect native Cfp4 in solution; however, the 2D20 and 2F9 antibodies recognize an epitope lacking sites for N-linked glycans, suggesting the possibility of recognition of native Cfp4. These monoclonal antibodies sufficiently detected Cfp4 produced by divergent clinical strains. The Cfp4 protein thus represents an exoantigen with conserved production across both North American lineages as well as the Panama lineage. This makes the Cfp4 glycoprotein a Histoplasma-specific antigen that could be potentially developed and employed in multiple geographic regions for histoplasmosis diagnosis.
Supplementary Material
ACKNOWLEDGMENTS
We thank J.-M. Balada-Llasat at the Ohio State University Clinical Microbiology Lab for supplying clinical Histoplasma isolates used in this study. We thank E. K. Dickerhoof for classification of clinical isolates, J. C. Angle for construction of Cfp4 expression vectors, and J. A. Edwards for assistance with hybridoma cultures.
This research was supported by research grant AI083335 from the National Institutes of Health and a predoctoral fellowship to E. D. Holbrook from the Public Health Preparedness for Infectious Diseases Program at Ohio State University.
L. J. Wheat is owner of MiraVista Diagnostics, and S. M. Richer and E. D. Holbrook are currently employees of MiraVista Diagnostics.
Footnotes
Published ahead of print 11 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01893-14.
REFERENCES
- 1. Ajello L. 1971. Distribution of Histoplasma capsulatum in the United States, p 103–122 In Ajello L, Chick EW, Furcolow MF. (ed), Histoplasmosis. Charles C. Thomas Publisher, Springfield, IL [Google Scholar]
- 2. Rippon J. 1988. Histoplasmosis, p 381–423 In Medical mycology: the pathogenic fungi and the pathogenic actinomycetes, 3rd ed. W.B. Saunders Co., Philadelphia, PA [Google Scholar]
- 3. Kauffman CA. 2009. Histoplasmosis. Clin. Chest Med. 30:217–225. 10.1016/j.ccm.2009.02.002 [DOI] [PubMed] [Google Scholar]
- 4. Medoff G, Sacco M, Maresca B, Schlessinger D, Painter A, Kobayashi GS, Carratu L. 1986. Irreversible block of the mycelial-to-yeast phase transition of Histoplasma capsulatum. Science 231:476–479. 10.1126/science.3001938 [DOI] [PubMed] [Google Scholar]
- 5. Nemecek JC, Wüthrich M, Klein BS. 2006. Global control of dimorphism and virulence in fungi. Science 312:583–588. 10.1126/science.1124105 [DOI] [PubMed] [Google Scholar]
- 6. Nguyen VQ, Sil A. 2008. Temperature-induced switch to the pathogenic yeast form of Histoplasma capsulatum requires Ryp1, a conserved transcriptional regulator. Proc. Natl. Acad. Sci. U. S. A. 105:4880–4885. 10.1073/pnas.0710448105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rappleye CA. 2014. Pathogenesis mechanisms of Histoplasma capsulatum, p 251–269 In Sullivan DJ, Moran GP. (ed), Human pathogenic fungi: molecular biology and pathogenic mechanisms. Caister Academic Press, Poole, United Kingdom [Google Scholar]
- 8. Rappleye CA, Eissenberg LG, Goldman WE. 2007. Histoplasma capsulatum alpha-(1,3)-glucan blocks innate immune recognition by the beta-glucan receptor. Proc. Natl. Acad. Sci. U. S. A. 104:1366–1370. 10.1073/pnas.0609848104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Youseff BH, Holbrook ED, Smolnycki KA, Rappleye CA. 2012. Extracellular superoxide dismutase protects Histoplasma yeast cells from host-derived oxidative stress. PLoS Pathog. 8:e1002713. 10.1371/journal.ppat.1002713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Holbrook ED, Smolnycki KA, Youseff BH, Rappleye CA. 2013. Redundant catalases detoxify phagocyte reactive oxygen and facilitate Histoplasma capsulatum pathogenesis. Infect. Immun. 81:2334–2346. 10.1128/IAI.00173-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rappleye CA. 2014. Molecular mechanisms of Histoplasma pathogenesis, p 129–140 In Kurzai O. (ed), Human fungal pathogens, 2nd ed. Springer Press, New York, NY [Google Scholar]
- 12. Sebghati TS, Engle JT, Goldman WE. 2000. Intracellular parasitism by Histoplasma capsulatum: fungal virulence and calcium dependence. Science 290:1368–1372. 10.1126/science.290.5495.1368 [DOI] [PubMed] [Google Scholar]
- 13. Hilty J, George Smulian A, Newman SL. 2011. Histoplasma capsulatum utilizes siderophores for intracellular iron acquisition in macrophages. Med. Mycol. 49:633–642. 10.3109/13693786.2011.558930 [DOI] [PubMed] [Google Scholar]
- 14. Hwang LH, Mayfield JA, Rine J, Sil A. 2008. Histoplasma requires SID1, a member of an iron-regulated siderophore gene cluster, for host colonization. PLoS Pathog. 4:e1000044. 10.1371/journal.ppat.1000044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zarnowski R, Cooper KG, Brunold LS, Calaycay J, Woods JP. 2008. Histoplasma capsulatum secreted γ-glutamyltransferase reduces iron by generating an efficient ferric reductant. Mol. Microbiol. 70:352–368. 10.1111/j.1365-2958.2008.06410.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garfoot AL, Zemska O, Rappleye CA. 2014. Histoplasma capsulatum depends on de novo vitamin biosynthesis for intraphagosomal proliferation. Infect. Immun. 82:393–404. 10.1128/IAI.00824-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Edwards JA, Zemska O, Rappleye CA. 2011. Discovery of a role for Hsp82 in Histoplasma virulence through a quantitative screen for macrophage lethality. Infect. Immun. 79:3348–3357. 10.1128/IAI.05124-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holbrook ED, Rappleye CA. 2008. Histoplasma capsulatum pathogenesis: making a lifestyle switch. Curr. Opin. Microbiol. 11:318–324. 10.1016/j.mib.2008.05.010 [DOI] [PubMed] [Google Scholar]
- 19. Holbrook ED, Edwards JA, Youseff BH, Rappleye CA. 2011. Definition of the extracellular proteome of pathogenic-phase Histoplasma capsulatum. J. Proteome Res. 10:1929–1943. 10.1021/pr1011697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Worsham PL, Goldman WE. 1988. Quantitative plating of Histoplasma capsulatum without addition of conditioned medium or siderophores. Med. Mycol. 26:137–143. 10.1080/02681218880000211 [DOI] [PubMed] [Google Scholar]
- 21. Youseff BH, Dougherty JA, Rappleye CA. 2009. Reverse genetics through random mutagenesis in Histoplasma capsulatum. BMC Microbiol. 9:236. 10.1186/1471-2180-9-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zemska O, Rappleye CA. 2012. Agrobacterium-mediated insertional mutagenesis in Histoplasma capsulatum, p 51–66 In Brand AC, MacCallum DM. (ed), Host-fungus interactions. Humana Press, New York, NY: [DOI] [PubMed] [Google Scholar]
- 23. Marion CL, Rappleye CA, Engle JT, Goldman WE. 2006. An alpha-(1,4)-amylase is essential for alpha-(1,3)-glucan production and virulence in Histoplasma capsulatum. Mol. Microbiol. 62:970–983. 10.1111/j.1365-2958.2006.05436.x [DOI] [PubMed] [Google Scholar]
- 24. Rappleye CA, Engle JT, Goldman WE. 2004. RNA interference in Histoplasma capsulatum demonstrates a role for alpha-(1,3)-glucan in virulence. Mol. Microbiol. 53:153–165. 10.1111/j.1365-2958.2004.04131.x [DOI] [PubMed] [Google Scholar]
- 25. Woods JP, Heinecke EL, Goldman WE. 1998. Electrotransformation and expression of bacterial genes encoding hygromycin phosphotransferase and beta-galactosidase in the pathogenic fungus Histoplasma capsulatum. Infect. Immun. 66:1697–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Horton RM, Cai ZL, Ho SN, Pease LR. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8:528–535 [PubMed] [Google Scholar]
- 27. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. 10.1016/0378-1119(89)90358-2 [DOI] [PubMed] [Google Scholar]
- 28. Centers for Disease Control and Prevention (CDC). 2012. Histoplasmosis outbreak among day camp attendees—Nebraska, June 2012. MMWR Morb. Mortal. Wkly. Rep. 61:747–748 [PubMed] [Google Scholar]
- 29. O'Keefe A, Frederick J, Harmon B, Safranek T, Buss B, Park B, Yeoman K. 2012. Notes from the field: histoplasmosis outbreak among day camp attendees—Nebraska, June 2012. JAMA 308:1853–1854. 10.1001/jama.2012.14070 [DOI] [Google Scholar]
- 30. Richer SM, Smedema ML, Durkin MM, Brandhorst TT, Hage CA, Connolly PA, Leland DS, Davis TE, Klein BS, Wheat LJ. 2014. Development of a highly sensitive and specific blastomycosis antibody enzyme immunoassay using Blastomyces dermatitidis surface protein BAD-1. Clin. Vaccine Immunol. 21:143–146. 10.1128/CVI.00597-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schlesinger LS. 1993. Macrophage phagocytosis of virulent but not attenuated strains of Mycobacterium tuberculosis is mediated by mannose receptors in addition to complement receptors. J. Immunol. 150:2920–2930 [PubMed] [Google Scholar]
- 32. Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8:785–786. 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- 33. Chauhan JS, Rao A, Raghava GPS. 2013. In silico platform for prediction of N-, O- and C-glycosites in eukaryotic protein sequences. PLoS One 8:e67008. 10.1371/journal.pone.0067008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC [Google Scholar]
- 35. Edwards JA, Alore EA, Rappleye CA. 2011. The yeast-phase virulence requirement for α-glucan synthase differs among Histoplasma capsulatum chemotypes. Eukaryot. Cell 10:87–97. 10.1128/EC.00214-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Edwards JA, Rappleye CA. 2011. Histoplasma mechanisms of pathogenesis—one portfolio doesn't fit all. FEMS Microbiol. Lett. 324:1–9. 10.1111/j.1574-6968.2011.02363.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bohse ML, Woods JP. 2007. Expression and interstrain variability of the YPS3 gene of Histoplasma capsulatum. Eukaryot. Cell 6:609–615. 10.1128/EC.00010-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Patel JB, Batanghari JW, Goldman WE. 1998. Probing the yeast phase-specific expression of the CBP1 gene in Histoplasma capsulatum. J. Bacteriol. 180:1786–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hamilton AJ, Bartholomew MA, Figueroa J, Fenelon LE, Hay RJ. 1990. Evidence that the M antigen of Histoplasma capsulatum var. capsulatum is a catalase which exhibits cross-reactivity with other dimorphic fungi. J. Med. Vet. Mycol. 28:479–485 [PubMed] [Google Scholar]
- 40. Edwards LB, Acquaviva FA, Livesay VT, Cross FW, Palmer CE. 1969. An atlas of sensitivity to tuberculin, PPD-B, and histoplasmin in the United States. Am. Rev. Respir. Dis. 99(Suppl):1–132 [PubMed] [Google Scholar]
- 41. Guimarães AJ, Nosanchuk JD, Zancopé-Oliveira RM. 2006. Diagnosis of histoplasmosis. Braz. J. Microbiol. 37:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hage CA, Ribes JA, Wengenack NL, Baddour LM, Assi M, McKinsey DS, Hammoud K, Alapat D, Babady NE, Parker M, Fuller D, Noor A, Davis TE, Rodgers M, Connolly PA, El Haddad B, Wheat LJ. 2011. A multicenter evaluation of tests for diagnosis of histoplasmosis. Clin. Infect. Dis. 53:448–454. 10.1093/cid/cir435 [DOI] [PubMed] [Google Scholar]
- 43. Wheat LJ. 2006. Improvements in diagnosis of histoplasmosis. Expert Opin. Biol. Ther. 6:1207–1221. 10.1517/14712598.6.11.1207 [DOI] [PubMed] [Google Scholar]
- 44. Wheat LJ, Garringer T, Brizendine E, Connolly P. 2002. Diagnosis of histoplasmosis by antigen detection based upon experience at the histoplasmosis reference laboratory. Diagn. Microbiol. Infect. Dis. 43:29–37. 10.1016/S0732-8893(02)00367-X [DOI] [PubMed] [Google Scholar]
- 45. Gómez BL, Figueroa JI, Hamilton AJ, Diez S, Rojas M, Tobón A, Restrepo A, Hay RJ. 1999. Detection of the 70-kilodalton histoplasma capsulatum antigen in serum of histoplasmosis patients: correlation between antigenemia and therapy during follow-up. J. Clin. Microbiol. 37:675–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wheat LJ, Cloud G, Johnson PC, Connolly P, Goldman M, Le Monte A, Fuller DE, Davis TE, Hafner R, Clinical Trials Group AIDS, Mycoses Study Group of NIAID 2001. Clearance of fungal burden during treatment of disseminated histoplasmosis with liposomal amphotericin B versus itraconazole. Antimicrob. Agents Chemother. 45:2354–2357. 10.1128/AAC.45.8.2354-2357.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Williams BJ, Connolly-Stringfield P, Bartlett M, Durkin M, Garringer T, Blair R, Connolly K, Tewari RP, Wheat LJ. 1991. Correlation of Histoplasma capsulatum polysaccharide antigen with the severity of infection in murine histoplasmosis. J. Clin. Lab. Anal. 5:121–126. 10.1002/jcla.1860050209 [DOI] [PubMed] [Google Scholar]
- 48. Kasuga T, White TJ, Koenig G, McEwen J, Restrepo A, Castañeda E, Da Silva Lacaz C, Heins-Vaccari EM, De Freitas RS, Zancopé-Oliveira RM, Qin Z, Negroni R, Carter DA, Mikami Y, Tamura M, Taylor ML, Miller GF, Poonwan N, Taylor JW. 2003. Phylogeography of the fungal pathogen Histoplasma capsulatum. Mol. Ecol. 12:3383–3401. 10.1046/j.1365-294X.2003.01995.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.