

Abstract
Endodontic infections, in which oral bacteria access the tooth pulp chamber, are common and do not resolve once established. To investigate the effects of these infections on the innate immune response, we established a mouse subcutaneous chamber model, where a mixture of four oral pathogens commonly associated with these infections (endodontic pathogens [EP]), i.e., Fusobacterium nucleatum, Streptococcus intermedius, Parvimonas micra, and Prevotella intermedia, was inoculated into subcutaneously implanted titanium chambers. Cells that infiltrated the chamber after these infections were primarily neutrophils; however, these neutrophils were unable to control the infection. Infection with a nonpathogenic oral bacterial species, Streptococcus mitis, resulted in well-controlled infection, with bacterial numbers reduced by 4 to 5 log units after 7 days. Propidium iodide (PI) staining of the chamber neutrophils identified three distinct populations: neutrophils from EP-infected chambers were intermediate in PI staining, while cells in chambers from mice infected with S. mitis were PI positive (apoptotic) or negative (live). Strikingly, neutrophils from EP-infected chambers were severely impaired in their ability to phagocytose and to generate reactive oxygen species in vitro after removal from the chamber compared to cells from S. mitis-infected chambers. The mechanism of neutrophil impairment was necrotic cell death as determined by morphological analyses. P. intermedia alone could induce a similar neutrophil phenotype. We conclude that the endodontic pathogens, particularly P. intermedia, can efficiently disable and kill infiltrating neutrophils, allowing these infections to become established. These results can help explain the persistence of endodontic infections and demonstrate a new virulence mechanism associated with P. intermedia.
INTRODUCTION
Endodontic infections result from oral pathogenic bacteria reaching the dental pulp through cracks, unrepaired caries, and failed caries restorations and infecting the pulp and surrounding tissues (1). These infections invoke an intense inflammatory response causing tissue destruction and bone degradation, and they are the most common cause of tooth pain. Once established, these infections do not spontaneously resolve. The primary management of these infections is through root canal treatment involving removal of infected pulp tissue. It is estimated that more than 20 million of these procedures were performed in the United States in 2005 (2), with about 30% resulting from endodontic infection (3). These infections are polymicrobial, with more than 20 bacterial species associated with infections in the root canal or the surrounding periapical region (4). Understanding the physiology of both the infecting bacteria and the host response is essential to the development of new effective treatments for these infections.
A mouse model of endodontic infection that faithfully replicates many of the features of the human infections has provided substantial important information on the host response to these infections (5–11). In this model, the pulp chamber of the mouse molar is exposed and infected with a mixture of four bacterial species frequently found associated with human endodontic infections (9). While both adaptive and innate immune responses can contribute to the development of lesions in response to these infections, the primary mode of host defense in this model is innate immunity. Deletion of Th1 cytokines has no effect on lesion formation (10); however, late-stage infection and dissemination are controlled by T cells (11). The infections are controlled by interleukin-10 (IL-10), osteopontin, and IL-6, with a substantial role for macrophages through monocyte chemoattractant protein 1 (MCP-1) (5, 7, 8, 12). Taken together, these results suggest that at least at early stages, the host response to these infections is primarily through innate immunity. Neutrophils constitute the first-line innate immune defense in bacterial infections (13, 14), where they effect bacterial killing through several mechanisms, including phagocytosis and production of reactive oxygen species (15). Thus, it is likely that regulation of the accumulation or function of neutrophils can affect the course of these infections.
While analysis of many aspects of the host response is possible in the mouse model, detailed ex vivo analysis of the cellular infiltrates is challenging due to the small volume of the infected tissue in the mouse mandible and its location within a bony structure. To overcome this difficulty, we established a subcutaneous chamber model of infection, adapting an optimized model that has been used for the analysis of the host response to infection with periodontal bacteria (16–19). Substantial numbers of infiltrating host cells accumulate in these chambers in response to inoculation with bacterial species. We used these chambers to assess the initial host cellular response to infection with a combination of four bacterial species (Fusobacterium nucleatum, Streptococcus intermedius, Parvimonas micra, and Prevotella intermedia) to test the hypothesis that the infiltrating neutrophils function to control the infection. Unexpectedly, we found that the neutrophils infiltrating chambers infected with the endodontic species were unable to clear the infection, in contrast to a nonpathogenic species that was effectively destroyed. The persistence of the endodontic species was a result of bacterially caused severe damage to infiltrating neutrophils, which prevented the neutrophils from performing antibacterial functions.
MATERIALS AND METHODS
Mice.
Seven- to 8-week-old male C57BL/6 mice were purchased from the Jackson Laboratory and housed under pathogen-free conditions. All animal experiments were carried out according to the National Institutes of Health guidelines with the approval from the Institutional Animal Care and Use Committee at the Forsyth Institute (protocol 11-007), which is operating under the accreditation of the Association for Assessment and Accreditation of Laboratory Animal Care.
Bacterial preparations.
The common human endodontic pathogens (EP) Prevotella intermedia (ATCC 25611), Streptococcus intermedius (ATCC 27335), Fusobacterium nucleatum (ATCC 25586), and Parvimonas micra (ATCC 33270) were grown on Trypticase soy agar with 5% sheep blood (TSA II plate; BBL) under anaerobic conditions (80% N2, 10% H2, and 10% CO2). Streptococcus mitis (ATCC 49456) was grown in Todd-Hewitt broth (THB) (BD) under aerobic conditions. Endodontic pathogens were harvested from the plate, resuspended in phosphate-buffered saline (PBS) (Invitrogen) and quantified spectrophotometrically. S. mitis was harvested from broth cultures by centrifugation at 3,000 × g for 10 min. For chamber infection, S. mitis was resuspended at 1 × 109 CFU/0.1 ml or 2.5 × 108 CFU of the four species (based on a previously determined CFU/optical density [OD] ratio) were mixed together for a total inoculum of 1 × 109 CFU/0.1 ml. The actual inoculum was determined just after infection by serial dilution using the drop plate method.
Chamber implantation.
Middorsal subcutaneous implantation of surgical-grade titanium coil chambers (length, 1.5 cm; diameter, 0.5 cm; prepared from tightly wound 0.37-in. titanium wire grade CP-1 [Dynamet, Washington PA]) was performed under isoflurane anesthesia (E-Z Anesthesia Classic System) as previously described (20). Before use, the free ends of the titanium wire were filed to remove sharp edges, and the chambers were sterilized by autoclaving. Each mouse received two chambers that were treated identically. Following a 10-day healing period, the chambers were used as a biological compartment for inducing inflammation. Bacterial suspensions were injected into the chambers of each mouse by using a 27-gauge needle.
Chamber fluid analysis.
Chamber exudates were harvested from uninfected chambers (0 h) or from chambers at 2, 12, 24, 72, and 168 h postinfection using a 27-gauge needle by flushing with 0.7 ml PBS with 5% fetal bovine serum (FBS). A 70-μm cell strainer (Falcon) was used to remove major clumps in the suspension, and passage through a 27-gauge needle was used to break further small clumps. Thirty microliters of exudates was collected for bacterial enumeration, and the remaining fluid was centrifuged for 5 min at 4°C and 240 × g. The supernatants were removed and stored at −80°C.
The pellets were immediately resuspended in RPMI medium (with 5% FBS), and the total host cell number was determined using acridine orange (AO)-propidium iodide (PI) staining (Cellometer Auto 2000; Nexcelom). Five hundred microliters of ACK buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA [Sigma]) was added to samples to lyse red blood cells for 5 min. The cells were then diluted in 2 ml of fluorescence-activated cell sorting (FACS) buffer (RPMI [Corning] with 5% FBS) and centrifuged. Cells were then resuspended in FACS buffer, and 1 × 106 cells were used for FACS. For S. mitis and EP experiments, 5 to 10 mice were used per point, except for at 0 and 2 h, where 5 mice per point were used for bacteria and cell counting, with the cells then pooled into two pools for FACS analysis. For individual species experiments, 5 mice per point were used. Bone marrow (BM) neutrophils were purified on Histopaque gradients as previously described (21).
Light microscopy.
Cells were placed onto cleaned round coverslips coated with fibronectin (100 μg/ml) or poly-d-lysine (100 μg/ml) in a 24-well plate; 1 × 105 cells in 500 μl of FACS buffer per well were seeded and incubated for 1 h in 5% CO2 at 37°C. After fixing with methanol, cells were stained with Wright-Giemsa staining buffer (HEMA3 Stat Pack; Fisher). Images were captured with a BX41 (Olympus) microscope.
Counting of bacteria.
For determining bacterial CFU, the drop plate method was used (22). Briefly, the collected suspension which contained cells and bacteria was serially diluted in prereduced anaerobically sterilized medium (for EP) (PRAS, Anaerobe Systems) or PBS (for S. mitis) and plated on TSA II or THB agar plates. Plates were cultured under appropriate conditions for 2 to 4 days, and colonies were counted. For determination of the identity of the colonies, colonies with different morphologies were suspended in water and subjected to PCR and sequencing using universal 16S rRNA gene primers 27F and 1492R (23).
Flow cytometric analysis.
Cells (1 × 106/sample) were incubated with anti-Ly6G (1A8), -CD11b (M1/70), or -F4/80 monoclonal antibodies (BioLegend). TruStain fcX (anti-mouse CD16/32) antibody (BioLegend) was added 10 min prior to the antibody staining to prevent nonspecific binding of Fc receptor on neutrophils. Isotype control antibodies (BioLegend) were used at the same concentration as experimental antibodies. Analysis was performed on a FACS Aria II (BD Bioscience). All data files were analyzed using FlowJo software (TreeStar Inc.). Propidium iodide (4 μg/ml final concentration) was added just before analysis. In some cases, cells were incubated at 4°C in the dark after staining with the LIVE/DEAD fixable green dead cell stain kit (Invitrogen) for 30 min according to the manufacturer's instructions. The fluorescein isothiocyanate (FITC) annexin V apoptosis detection kit I (BD Pharmingen) was used according to the manufacturer's instructions, with incubation for 15 min prior to FACS.
Phagocytosis.
pH-sensitive pHrodo green Escherichia coli bioparticles conjugate (Invitrogen) was used after opsonization. Briefly, mouse serum (Sigma) was passively adsorbed onto particles for 30 min at 37°C. The particles were washed twice with PBS to remove excess serum, and 1 × 106 chamber cells/500 μl of FACS buffer were incubated with 100 μl (1 mg/ml) of bioparticles at 37°C for 2 h according to the manufacturer's instructions. As a control for attached but not phagocytic ingested particles, parallel reaction mixtures were incubated on ice. Cells were then stained with Ly6G, and phagocytosis of the E. coli particles by neutrophils (Ly6G+) was assessed by flow cytometry. Specific phagocytosis was calculated by subtracting the geometric median fluorescence intensity (MFI) of phagocytized bioparticles in the on-ice treatment groups from that of the same sample incubated at 37°C.
Detection of ROS.
To measure generation of reactive oxygen species (ROS), CellROX Deep Red reagent (Invitrogen) was used according to the manufacturer's instructions. Briefly, 1 × 106 chamber cells in FACS buffer were untreated (ROS production) or treated with 400 μM tert-butyl hydroperoxide (TBHP) (maximal ROS production) for 30 min and then incubated with 250 μM CellROX Deep Red reagent for 1 h at 37°C in FACS buffer. Cells were then stained with Ly6G and analyzed by FACS. The geometric median fluorescence intensity of red staining was used to determine ROS production.
Electron microscopy.
Cells were fixed with paraformaldehyde and glutaraldehyde, postfixed with 1% osmium tetroxide, contrasted with uranyl acetate, dehydrated in ethanol, and embedded in LR White resin (Electron Microscopy Sciences). Resin blocks were cut with an ultramicrotome (PowerTome XL; RMC Products) into 70- to 100-nm-thick ultrathin sections, which were immediately placed on 400-mesh carbon-coated copper grids (Electron Microscopy Sciences). The morphology of neutrophils was observed by transmission electron microscopy (TEM) (1200EX; JEOL) using the bright-field mode operated at 100 kV and captured by an AMT charge-coupled device (CCD) camera. For biofilm analysis, straight titanium wires, 0.037 in. by 1 to 1.3 cm long, were placed inside the chambers at the time of implantation. Chambers were then infected with EP or S. mitis as described above. Three days later, the wires were removed from the chambers, fixed with 2% formaldehyde–3% glutaraldehyde in 0.1 M cacodylic acid, and then treated for 1 h with 2% osmium tetroxide in 0.1 M S-collidine buffer. After dehydration, wire samples were sputter coated with gold (Desk V; Denton Vacuum) and then examined with a scanning electron microscope (SEM) (Evo LS10; Carl Zeiss).
MPO activity.
Stored supernatants were used for myeloperoxidase (MPO) activity assay (Cayman). After thawing, samples were centrifuged at 18,000 × g for 15 min at 4°C, and proteinase inhibitor (Roche) was added to each sample prior to the assay. Protein concentrations in the samples were determined with the bicinchoninic acid protein assay kit (Pierce). Data are expressed as units MPO activity/μg protein.
Statistical analyses.
The significance of differences was calculated by one-way analysis of variance (ANOVA) or a two-tailed t test using GraphPad Prism. Groups compared are indicated by brackets in the figures or in the figure legends. Data shown are means ± standard deviations (SD).
RESULTS
Chamber model of infection.
In order to quantify and characterize in detail the cellular response to infection with the endodontic pathogens, we adapted the subcutaneous chamber model. In this model, titanium wire (0.037-in. diameter) is wound into a tight coil of 1.5 cm by 0.5 cm (Fig. 1A). These coils are implanted under the dorsal skin of mice (Fig. 1B), where they become fluid filled and then become encapsulated in a thin fibrous tissue after 10 days (Fig. 1C). At this time, they are infected with bacterial suspensions by direct injection into the chamber through the skin.
FIG 1.

Chamber model of infection. (A) Titanium coiled-wire chambers (scale is mm) were implanted under the dorsal skin of C57BL/6 mice. (B) Dashed boxes indicate the positions of the chambers. (C) Ten days after infection, the chambers became encapsulated in a thin fibrous tissue.
S. mitis infection is cleared efficiently in this model, but EP are not.
We infected these chambers with a nonpathogenic oral species Streptococcus mitis, or with a mixture of four endodontic pathogens (EP): Prevotella intermedia, Fusobacterium nucleatum, Streptococcus intermedius, and Parvimonas micra. The chamber contents were collected at different times after infection, and live bacterial numbers were measured. While S. mitis infection in these chambers could be controlled, with bacterial counts dropping by 4 to 5 log units over 7 days (168 h), the EP persisted in the chambers over this time frame (Fig. 2A), with even a slight increase in CFU at later times. Bacterial colonies grown from chamber contents at all time points had differing morphologies, consistent with recovery of all four species. Sequencing of 16S rRNA gene alleles of several colonies confirmed that all species were recovered from the chambers (data not shown).
FIG 2.
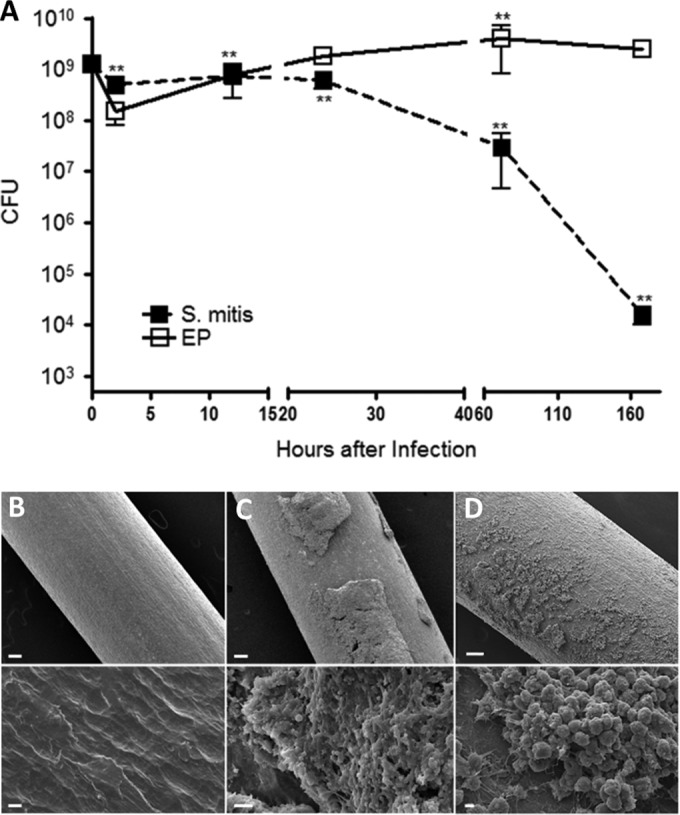
S. mitis but not EP bacterial numbers are controlled in the chambers: association with biofilm formation. (A) Bacterial suspensions were inoculated at 1 × 109 to 2 × 109 cells/chamber at 0 h, and chamber contents were collected at different times after infection as indicated. Live bacteria were quantified as described in Materials and Methods. **, P < 0.01 compared to 0 h by 1-way ANOVA. (B to D) Titanium wires were collected from infected chambers at 72 h after infection and analyzed by scanning electron microscopy. (B) Wire only, no bacteria. (C) Wire from EP-infected chamber. (D) Wire from S. mitis-infected chamber. Bars, 100 μm (upper panels) or 2 μm (lower panels).
To determine if biofilms formed inside the chambers, a length of straight titanium wire (the same wire used to form the chamber) was placed inside the chamber during infection, recovered at 72 h after infection, and processed for scanning electron microscopy. The results suggest that thick biofilms formed the on the chamber material during EP infection (Fig. 2C). Wires recovered from S. mitis-infected chambers were coated with a thinner layer of material that was comprised largely of neutrophils (Fig. 2D). Whether these neutrophils are adherent to areas of biofilm is difficult to discern in these analyses.
Neutrophils in EP-infected chambers are abnormal.
Host cells accumulating in uninfected chambers and in chambers at different times after infection were collected and counted, and the proportion of neutrophils was determined by flow cytometry using an antibody for the neutrophil marker Ly6G. Uninfected chambers contained fewer than 106 cells, of which about 50% were neutrophils (Fig. 3A, hatched bar). We showed previously that in uninfected chambers, the cell number and composition remain unchanged over 7 days (20). Infection with either S. mitis or EP resulted in a 10-fold increase in total cell numbers by 12 h after infection (Fig. 3A, points, left axis). At 2, 12, and 24 h, there were significantly more cells in the S. mitis-infected chambers than in those infected with EP. This was reversed by 7 days, when the total cell number in EP-infected chambers was significantly higher than that in S. mitis-infected chambers. The proportion of neutrophils was 70 to 80% from 12 to 72 h and was similar in both S. mitis- and EP-infected chambers (Fig. 3A, bars, right axis). An apparent decrease in the proportion of neutrophils at 168 h in EP-infected chambers is likely the result of a lower level of expression of Ly6G on the neutrophil population at this time point rather than a change in the cell population (Fig. 3C) (see below). At 2 h the proportion of neutrophils was lower in both types of chambers than at later times, suggesting that the initial cellular infiltrate comprises primarily nonneutrophils, although the identity of these cells was not determined. Interestingly, we were unable to detect a clear population of macrophages (F4/80+) or monocytes (Cd11b+/Ly6G−) (24) in these chambers (Fig. 3B). While the results are shown for 7 days after infection, similar results were obtained at each time point (data not shown). It is not clear why these cell types are absent from the chamber, but we hypothesize that macrophages may be adherent to the perichamber tissue and are not released during collection of the chamber exudates.
FIG 3.

Cellular infiltrates in EP-infected and S. mitis-infected chambers are distinct. (A) Total infiltrating cell numbers (left axis, points) and percent neutrophils (right axis, bars) in chambers infected with S. mitis (S.m.) or EP as indicated; 0 h represents uninfected chambers. *, P < 0.05 for EP compared to S. mitis, by t test. (B) Top panel, representative histogram of F4/80 staining of 7-day cells (black or dotted line) compared to isotype control (shaded histogram). Bottom panels, representative CD11b Ly6G double staining of 7-day chamber cells. Monocytes (CD11b+ Ly6g−, Q1) are absent in both populations. (C) PI/Ly6G staining of chamber cells from uninfected (0 h) S. mitis (top row)- and EP (bottom row)-infected chambers. Cells from chambers were collected at different times as indicated and analyzed by FACS for Ly6G and PI staining as described in Materials and Methods. Dashed boxes represent the R1, R2, and R3 gates as shown for the 0-h sample. Quantification of the percent neutrophils in the different populations (n = 5) is depicted by the pie charts below each dot plot. *, P < 0.001 by one-way ANOVA with Bonferroni's multiple-comparison test comparing each population from EP-infected chambers with that from S. mitis-infected chambers.
Despite the similar proportions of infiltrating cells, there were dramatic differences in the characteristics of the cells in chambers infected with S. mitis and EP. Propidium iodide (PI) was used to determine the proportions of live and dead neutrophils in the chambers at different times after infection, since neutrophils are short-lived cells and neutrophil death can regulate the host response to infection (25). Neutrophils in uninfected chambers were predominantly live (PI negative) and were designated the R1 population (Fig. 3C, top row). S. mitis-infected chambers contained up to 36% R1 cells at early times after infection (2, 12, and 24 h), along with a population of strongly PI-positive cells (R3) that comprised the majority of the neutrophil population (Fig. 3C, middle row). In EP-infected chambers, on the other hand, very few PI-negative live cells were seen at any time, with a maximum of 14% at 2 h after infection. There was also a population of R3 cells in these chambers, but it was not distinct and decreased over time. Rather, 47 to 84% of the population in these chambers was stained to an intermediate level with PI (R2 population) (Fig. 3C, bottom row). Quantification of these populations revealed significant differences between the neutrophil populations in S. mitis- and EP-infected chambers (Fig. 3C, pie charts). The distinct difference in these populations was also evident by analysis of forward scatter and side scatter, where cells in the EP-infected chambers become continually smaller and less granular (data not shown). None of the PI-positive populations reacted with isotype control antibodies or with other antibodies such as F4/80, and CD11b staining was lower in the PI-positive population than in PI-negative cells (data not shown), indicating that the Ly6G staining of these populations was specific.
Changes in the EP-infected chambers by day 7.
By 7 days after infection, there were striking differences in the appearance of the tissue surrounding the EP- and S. mitis-infected chambers. While S. mitis-infected chambers retained their preinfection size, substantial swelling and edema had occurred around the EP-infected chambers (Fig. 4A and B) so that the diameter of the EP-infected chambers was significantly larger than that of the S. mitis-infected chambers (Fig. 4C). The fluid collected from S. mitis-infected chambers was clear, while EP-infected chamber fluid was white and cloudy (Fig. 4D). This appearance probably reflects the increase in dead cells and bacteria accumulated in these chambers at this time; although the proportion of neutrophils remains the same as at earlier times, their characteristics have changed, with lower Ly6G expression and lower forward scatter (Fig. 3 and data not shown).
FIG 4.

(A and B) Appearance of S. mitis (A)- and EP (B)-infected chambers at 7 days after infection, indicated by arrows, showing swelling around EP-infected chambers. (C) Quantification of width of chamber tissue. (D) Appearance of fluid recovered from S. mitis-infected chambers (left) and EP-infected chambers (right). ***, P < 0.001 by t test.
Neutrophils from EP-infected chambers are functionally impaired.
In order to understand the nature of the cells in the different chamber populations, we tested their ability to perform bacterial killing functions, including phagocytosis and production of reactive oxygen species. These assays were designed to be evaluated ex vivo by flow cytometry, so we could gate on the neutrophil population. Phagocytosis was measured using pH-sensitive fluorochrome-labeled E. coli (pHrhodo). The fluorescence intensity of these bacteria increases 8- to 10-fold at the pH of the lysosome compared to the extracellular pH, distinguishing phagocytosed bacteria in lysosomes from nonspecifically bound cells. Specific phagocytosis was defined as the mean fluorescence intensity (MFI) of control samples minus the MFI of samples incubated on ice, where phagocytosis is inhibited. Neutrophils from S. mitis-infected chambers performed substantial phagocytosis at 12 and 24 h after infection, and lower levels of phagocytosis ability persisted until 7 days after infection. Compared to these neutrophils, the phagocytic ability of cells from the EP-infected chambers was severely impaired. While neutrophils from EP-infected chambers were able to perform phagocytosis at 12 h after infection similarly to cells from S. mitis-infected chambers, beyond this time there was a steep drop-off in total phagocytic ability of the EP cells from that of the S. mitis-infected chambers (Fig. 5A). From 24 h to 7 days after infection, cells from EP-infected chambers had only about 1/3 of the phagocytic capacity as cells from S. mitis-infected chambers. Similarly, the generation of reactive oxygen species measured directly (Fig. 5B) or maximal potential ROS generation measured in the presence of the ROS activator TBHP (Fig. 5C) was significantly lower in cells from the EP-infected chambers than in those from S. mitis-infected chambers at early time points. By 7 days after infection however, the ability of the cells from EP-infected chambers to generate reactive oxygen species was substantially higher than that of cells from the S. mitis-infected chambers, suggesting that at later times, the EP induce an altered cellular response in the chamber neutrophils. In an in vitro bacterial killing assay, significantly lower killing ability of neutrophils from EP-infected chambers than of those from S. mitis-infected chambers was also observed (data not shown). On the other hand, myeloperoxidase activity was significantly higher in fluid collected from the EP-infected chambers than in that from chambers those infected with S. mitis at most time points (Fig. 5D), indicating increased release of myeloperoxidase from cells in EP-infected chambers.
FIG 5.

Neutrophil function is suppressed in EP-infected chambers. Phagocytosis and ROS production were measured by FACS in Ly6G+ cells. (A) Phagocytosis was determined using pHrhodo-labeled E. coli, and specific phagocytosis was calculated as described in Materials and Methods. (B) Production of reactive oxygen species was detected using CellROX Deep Red. The geometric median fluorescence intensity (MFI) determined after 1 h of incubation at 37°C is shown. (C) Maximal ROS production in each sample was determined by treatment with CellROX Deep Red in the presence of the ROS stimulator TBHP. (D) Myeloperoxidase (MPO) activity was measured in total chamber fluid. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (by 1-way ANOVA).
R2 and R3 populations have different characteristics.
To elucidate the difference between the R2 and R3 populations, we investigated their mechanisms of cell death. We used an alternative cell-impermeative fluorochrome (LIVE/DEAD fixable; Invitrogen) that reacts with amines on the cell surface of intact cells, to give dim staining, or with intracellular amines in cells with compromised membranes, resulting in strong staining. PI-positive cells from both S. mitis- and EP-infected chambers stained strongly with this reagent, indicating that both had compromised membranes (Fig. 6A). However, while PI-positive cells from the S. mitis-infected chambers stained with annexin V, suggesting that they were late apoptotic, the R2 population was predominantly annexin V negative (Fig. 6B). Annexin V-negative, PI-positive neutrophils have been previously characterized as primary necrotic (26). We further tested the ability of the different cell populations to adhere to fibronectin, a process that requires integrin binding followed by focal adhesion formation and actin polymerization, or to poly-d-lysine, where adherence results from passive interaction of cell surface proteins with the positively charged surface. Cells isolated from S. mitis-infected chambers at 24 h, representing predominantly live R1 cells, or at 72 h, comprising mostly PI-positive R3 cells, adhered equally well to fibronectin and to poly-d-lysine, suggesting that integrin-based cell adhesion function was retained in these cells, although adhesion to fibronectin was significantly reduced at 72 h. In contrast, at 24 h, the adhesion of EP cells to fibronectin was only about 25% of that of S. mitis cells, and cells from EP-infected chambers (R2) adhered significantly less well to fibronectin than to poly-d-lysine at both time points (Fig. 6C). Together, these results demonstrate that the R3 and R2 populations represent neutrophils approaching cell death through different pathways, likely apoptosis and necrosis, respectively.
FIG 6.
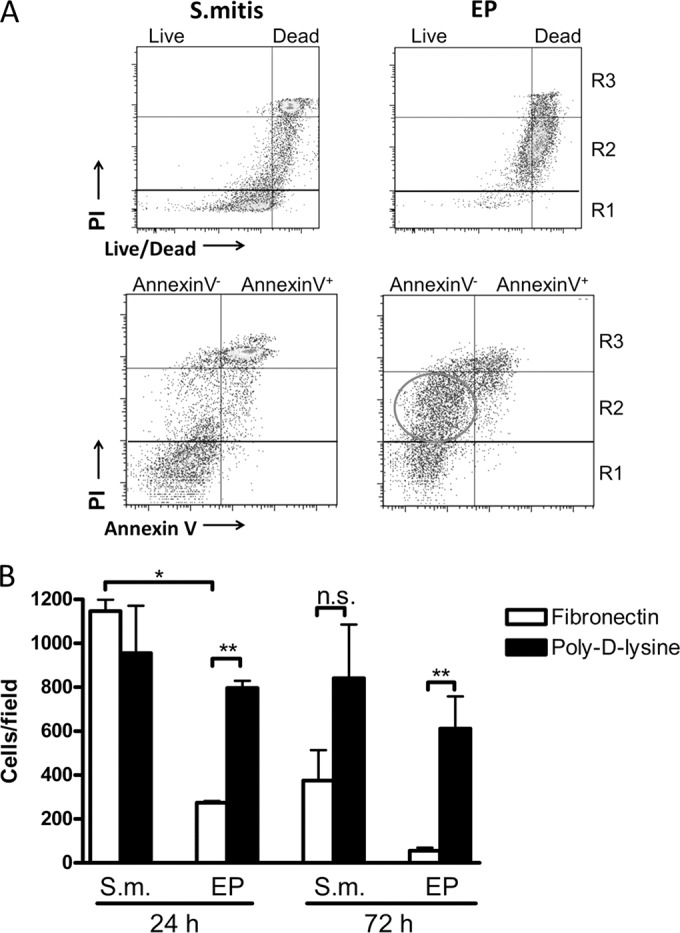
Neutrophils from EP-infected chambers are not apoptotic but have features of dead cells. (A) Top panels, neutrophils were collected from S. mitis- or EP-infected chambers at 24 h after infection and stained with PI together with LIVE/DEAD stain (Invitrogen). A representative image of 4 determinations is shown. Bottom panels, annexin V/PI staining of neutrophils from S. mitis- or EP-infected chambers at 24 h after infection. Regions representing R1, R2, and R3 populations as well as annexin V-positive and -negative populations are indicated. The circled area indicates the annexin V-negative R2 population. A representative image of 4 determinations is shown. (B) Neutrophils from S. mitis (S.m.)- or EP-infected chambers were collected at the indicated times. Equal numbers of cells were applied to coverslips coated with fibronectin or poly-d-lysine and allowed to adhere for 1 h. After washing, the cells were stained and the number of cells per field determined by counting. **, P < 0.01; *, P < 0.05 (by t test). n.s., not significant.
R2 and R3 neutrophils differ morphologically.
We then assessed the morphology of the different cell populations by Wright staining and transmission electron microscopy (TEM). We were unable to isolate the R1 population by cell sorting, probably because these cells have a short life and are converted to R3 during the sorting process. Therefore, we examined cells isolated from chambers with a prominent population of each of the three PI staining types. R1 cells were represented by isolated bone marrow neutrophils (Fig. 7, row 1) or by cells from S. mitis-infected chambers at 24 h, where 25 to 30% of the cells are R1 (Fig. 7, row 2). R2 cells comprise 85 to 90% of the population in cells from EP-infected chambers at 72 h (Fig. 7, row 3). At 72 h after infection, 85 to 90% of cells from the S. mitis-infected chambers are R3 (Fig. 7, row 4). The morphologies of these three populations seen by Wright staining were very different, with neutrophils from S. mitis-infected chambers showing ring-shaped or multilobed nuclei (similar to bone marrow neutrophils) (Fig. 7a, row 1) and numerous phagocytosed bacteria at 24 h (Fig. 7a, row 2). By TEM, these cells also had a structure similar to that of BM neutrophils (Fig. 7b, rows 1 and 2), except that R1 cells from S. mitis-infected chambers had phagocytosed bacteria and more convoluted nuclei. R3 cells from these chambers had a distinct morphology by Wright staining: the nuclear structure was less distinct, and the cells were surrounded by fibrous exudates consistent with extracellular DNA representing neutrophil extracellular traps (NETs) (Fig. 7a, row 4), although we have not confirmed that these structures are NETs. TEM showed that these cells had less-dense nuclei and an amorphous cell structure, with membrane blebbing consistent with apoptosis. R2 cells, on the other hand, showed a morphology very different from those of the other cell types. By Wright staining, the cells were present in groups connected by amorphous substance, with dense small nuclei and no visible nuclear structure (Fig. 7a, row 3). TEM revealed a dramatic loss of cell structure (Fig. 7b, row 3). The plasma membrane of these cells appeared disrupted, and while granules were still present, a normal cellular structure was absent. Strikingly, the nuclei of these cells were fragmented or absent. Together, these results support the idea that R3 neutrophils are late apoptotic, while R2 neutrophils from the EP-infected chambers have features more similar to those of necrosis.
FIG 7.

Neutrophils from S. mitis-infected chambers either are healthy or have apoptotic features, while those from EP-infected chambers appear necrotic. Cells were collected from S. mitis- or EP-infected chambers at time points when a large proportion of the cells are identified as R1, R2, or R3 by PI/Ly6G FACS analysis. These cells were then applied to coverslips and stained with Wright stain or fixed and processed for TEM. (a) Wright-Giemsa staining; (b) TEM; (c) representative PI/Lyg6 staining of analyzed populations. Rows: 1, partially purified bone marrow neutrophils represent R1 cells; 2, cells collected from S. mitis-infected chambers at 24 h after infection are both R1 and R3; 3, cells collected from EP-infected chambers at 24 h are predominantly R2; 4, cells collected from S. mitis-infected chambers at 72 h represent the R3 population. R1 cells in row 2 were identified as distinct from cells with morphology similar to R3 cells identified in row 4. N, nucleus; arrows (a) and open arrowheads (b), phagocytosed bacteria; dashed arrows (a), extracellular strands consistent with NETs. Bars, 20 μm (a) and 5 μm (b).
P. intermedia alone can generate R2 neutrophils.
Our observations suggested that the four endodontic pathogens are able to damage and kill infiltrating neutrophils, but it is not clear if one of these species is responsible or if all four are necessary. Therefore, we infected chambers with each of the four species individually at 1 × 109 CFU/chamber and evaluated the bacterial load and cellular infiltrates at 24, 72, and 168 h after infection. Analysis of the bacterial load at different times showed that there was an initial drop in CFU of all four species by 24 h, with a further decline at 72 h. However, the number of P. intermedia cells recovered did not further decline at 7 days, unlike for the other species, which all had significantly lower bacterial numbers at this time point. All four species were killed less effectively than S. mitis, however (Fig. 8A). The number of infiltrating host cells was variable between the species and for most species was variable at the different time points. Only in P. intermedia-infected chambers was there a significant increase in infiltrating cell numbers at 168 h (Fig. 8B), similar to the case for the EP-infected chambers. Importantly, at 24 h after infection, only P. intermedia caused the appearance of the R2 PI-intermediate population (Fig. 8C, top panel); quantification of the data from five different animals (Fig. 8C, bottom panel) indicated a highly significant increase in the R2 population and decrease in the R3 population in cells from P. intermedia-infected chambers compared to those from the other three species. The ability of neutrophils from chambers infected with the different species to perform phagocytosis was tested at the different time points, and variable results were obtained. At all time points, however, the phagocytosis ability of neutrophils from P. intermedia-infected chambers was very low. This is best appreciated by adding together the specific phagocytosis levels at all three time points (Fig. 8D, hatched bars); this value is significantly lower for neutrophils from P. intermedia-infected chambers than for those from chambers infected with the other three species (Fig. 8D). The generation of reactive oxygen species, on the other hand, was similar in all species, with the exception of F. nucleatum, where high-level ROS production was seen at 24 h after infection (Fig. 8E). The sum of the ROS production at all time points was highest for F. nucleatum but was not significantly different between the other species. Taken together, these results suggest that while all four endodontic pathogens are killed less efficiently than S. mitis, P. intermedia is most responsible for the damage to the neutrophil population.
FIG 8.

P. intermedia alone causes neutrophil damage. Each of the four endodontic species was injected individually into chambers at 1 × 109 cells/chamber. (A) Live bacteria were enumerated as for Fig. 2. ***, P < 0.001 for P. intermedia versus all other species by 1-way ANOVA. (B) Infiltrating host cells recovered from infected chambers at different times were quantified as for Fig. 3. ***, P < 0.001; *, P < 0.05 (by 1-way ANOVA). (C) Upper panels, the PI staining pattern of chamber neutrophils (Ly6G+) was determined in cells collected at 24 h after infection as for Fig. 3. Lower panel, quantification of different PI-stained populations as indicated. ***, P < 0.001 for P. intermedia versus all other species by 1-way ANOVA. (D) The phagocytic abilities of cell populations isolated from chambers at the indicated time points were determined as for Fig. 5. Total phagocytosis is the sum of the specific phagocytosis values at all three time points. ***, P < 0.001. (E) Production of reactive oxygen species was determined as for Fig. 5. *, P < 0.05 for total F. nucleatum versus all other species by 1-way ANOVA. Pi, P. intermedia; Fn, F. nucleatum; Si, S. intermedius; Pm, P. micra.
DISCUSSION
Our results demonstrate that while the innate immune response to a nonpathogenic species functions efficiently to kill bacteria inoculated into the subcutaneous chamber model, this response is completely inhibited in infections containing P. intermedia. This inhibition results from a cytotoxic effect on infiltrating neutrophils resulting in cell death through an apparently necrotic pathway and a strong suppression of antibacterial functions. The physical structure of the root canal and associated bone likely restricts access of innate and other immune cells to the site of the infection, contributing to the development of necrosis and the failure of innate immunity in these infections (27). Our results for the first time demonstrate that cytotoxic activity of bacterial species typically associated with these infections, especially P. intermedia, may also contribute to the persistence of these infections. Previous experiments have demonstrated that endodontic pathogens can persist and form transmissible abscesses in animal models (28), but ours is the first demonstration of the mechanism of this persistence.
The effect of the endodontic pathogens on neutrophils is complex. An obvious marker of this effect is the generation of the R2 population, with PI staining at a level intermediate between those for live and dead cells. Two possible mechanisms can account for this intermediate staining: either the ability of PI to enter the cells is limited, or the amount of DNA available to bind PI is reduced. The first mechanism has been proposed in the case of neutrophils treated in vitro with the cytolysin streptolysin O (SLO), where the PI-intermediate cells have taken up PI through pores formed by SLO but these pores have subsequently been resealed, resulting in a limited amount of PI uptake (29). This mechanism is probably not applicable in our case, because the PI staining is performed after cell isolation and FACS staining and is performed on ice, so the neutrophils should be metabolically inactive, and ongoing pore formation and resealing would be limited. In addition, staining with LIVE/DEAD stain (Fig. 6) confirms that the R2 cells have permeable or disrupted membranes. The TEM results (Fig. 7) suggest that the R2 neutrophils have an extremely distorted structure, with fragmented or even absent nuclei, supporting the alternative hypothesis that there is a reduced amount of cellular DNA in the R2 population to bind PI. Neutrophil extracellular trap formation (30) involves release of chromatin fibers and could result in a reduced amount of intracellular DNA. However, peritoneal neutrophils treated with phorbol myristate acetate (PMA) to induce NET formation did not result in generation of the R2 population (data not shown), and apparent NET formation in neutrophils from S. mitis-infected chambers is not associated with generation of the R2 population (Fig. 7). Thus, it is unlikely that production of NETs generates the R2 population; rather, it appears that the EP and P. intermedia induce the R2 population through direct damage resulting in nuclear fragmentation and loss.
Clearly the death of neutrophils caused by EP/P. intermedia is distinct from the neutrophil death that occurs in the S. mitis-infected chambers. In the latter case, the annexin V/strong PI positivity and the membrane blebbing seen in TEM support the idea that these cells are apoptotic, although their nuclei are not condensed, and we have not seen evidence of early apoptotic cells (data not shown). On the other hand, the R2 population is annexin V negative, and the disrupted appearance of these cells by TEM resembles primary necrosis (31), which can be associated with cell lysis. Since cells undergoing pyroptosis also appear necrotic (32), we cannot rule this out as a potential mechanism of cell death, especially as pyroptosis is caused by several bacterial species (33). However, clearly the determination of the mechanism of cell death caused by P. intermedia is an area of great interest for future work.
The EP/P. intermedia could be directly killing neutrophils through pore formation caused by a cytolysin. By TEM, the R2 neutrophils seem to be already lysed or in the process of lysis (Fig. 7), although these cells appear intact by phase-contrast microscopy (data not shown) and are seen as intact cells in flow cytometry. Cells with membranes perforated by pores could have these characteristics. Furthermore, the observation that myeloperoxidase levels are higher in fluid from chambers infected with EP than in that from chambers infected with S. mitis (Fig. 4D) supports a cell lytic mechanism. Myeloperoxidase is present in preformed neutrophil granules, which are released upon activation (34). Perhaps granule release during the process of pore formation/cell lysis contributes to the observed myeloperoxidase activity.
Therefore, our results support the idea that P. intermedia produces a cytolysin in the chamber environment that damages or kills neutrophils. While cytolysin activity has not previously been described for P. intermedia, this species is beta-hemolytic, and several genes encoding putative hemolysins have been identified but are not well characterized (35, 36). Whether the described hemolysins are also cytotoxic to neutrophils is not known, and we have not determined if the EP/P. intermedia are cytotoxic to cell types other than neutrophils, such as red blood cells. P. intermedia also produces a protease, interpain A, that can inactivate complement (37), but this activity is unlikely to cause the extreme damage to neutrophils that occurred in the chambers. Alternatively, both P. intermedia and F. nucleatum can generate succinate during metabolic activity (38, 39), and succinate has the ability to suppress neutrophil phagocytosis (40). While this activity may contribute to the toxic activity of the EP, it is unlikely to act alone, since succinate suppresses phagocytosis in neutrophils but does not directly kill them. In addition, its activity requires an acidic medium, and the pH of the chamber fluid is near neutral (data not shown). Thus, it is likely that the cytotoxic activity we described for P. intermedia has not been previously characterized.
In this work, we compared infection with a single Gram-negative species to that with a mixture of Gram-negative and Gram-positive species. The increased efficiency of the innate immune response in eradicating S. mitis in the chamber may to some extent be expected, since increased virulence of polymicrobial infections over those with individual species has been demonstrated previously (41–43). However, the results of infection with individual species confirm that there is a significant difference among the responses to all the species. The comparison between F. nucleatum and P. intermedia is informative, since these are both Gram-negative species with very different effects on the innate immune response. None of the endodontic pathogens are cleared as efficiently as S. mitis (Fig. 8), so it is likely that they all can inhibit neutrophil function to some extent. F. nucleatum expresses a cell surface-associated protein that induces apoptosis in human lymphocytes (44), and S. intermedius produces a human-specific cholesterol-dependent cytolysin (intermedilysin) (45). It is likely, therefore, that the four pathogens generate a combination of virulence factors that together contribute to the neutrophil damage we observed. Nevertheless, the role of P. intermedia appears to be critical.
Formation of biofilms by bacterial species contributes to resistance of the bacteria to antibiotic and innate immune killing (46). It is widely thought that biofilm formation contributes to the persistence of endodontic infections, and indeed biofilms have been directly stained in infected pulp chambers (47, 48). It is likely that biofilms form inside the titanium chambers in our infection model, and our SEM results are consistent with the appearance of biofilm in chambers infected with EP (Fig. 2B to D). Material adhering to the chamber material during S. mitis infection at 3 days, however, appears to be comprised primarily of neutrophils, and individual bacteria were present but not common. This strain has been shown to form biofilm under appropriate conditions in vitro (49), suggesting that biofilm forms in the chamber model as well. Biofilm formation may contribute to the persistence of the bacteria in the EP-infected chambers and may account to some extent for the difference in clearance between EP and S. mitis. On the other hand, the biofilms may accumulate in the EP-infected chambers because the bacteria are not being killed by the host, while ongoing bacterial destruction in the S. mitis-infected chambers limits biofilm formation. Further work is required to distinguish these possibilities. Biofilms have been shown to be susceptible in vitro to neutrophil killing, (50, 51), so it will be of interest in future experiments to define the role of biofilm formation in this model.
While endodontic infections are invariably polymicrobial, Prevotella species are strongly associated with both primary endodontic and periapical infections (4, 52). In one study of endodontic infections of deciduous teeth, P. intermedia was the most frequently identified species out of 83 taxa assessed: overall, 96.5% of the 40 samples tested contained P. intermedia (53). Another recent sequencing-based study identified Prevotella species in 91 to 94% of patients (52). In many studies, Prevotella species are the first or second most frequently identified species (4, 52). These observations can be explained by our discovery of a cytotoxic activity associated with P. intermedia. Since this activity suppresses neutrophil function, it can protect all the species in the infected teeth in addition to P. intermedia. Our results suggest that as long as P. intermedia is present, all species can survive and the infections can persist. P. intermedia is also associated with the severe oral infections necrotizing gingivitis and noma (54) and with several other polymicrobial infections, including diabetic foot ulcers (55) and bacterial vaginosis (56), and is found in lungs of cystic fibrosis patients (57). It is possible that the cytotoxic activity we have described may be important in these other infections as well.
Our studies used strains of endodontic pathogens obtained from ATCC. These strains have been used for many years in studies of the host response to endodontic infection in a mouse model (6, 7, 11) and have the advantage that they are readily available for others to reproduce the results. It will be of great interest to assess the association of the cytotoxic activity we identified with clinical isolates of P. intermedia and with different Prevotella species that are also strongly associated with endodontic infections, such as P. denticola and P. nigrescens (4). It must be noted, however, that the four species used in this study represent just a fraction of the species found associated with endodontic infections, where recent sequencing studies have identified more than 900 individual species (58).
Several aspects of P. intermedia have been studied in the past, so it is of interest why the cytotoxic activity has not been identified previously. We suggest that the unique attributes of the chamber model allow the observation of this phenotype. Importantly, the chamber is likely to support a relatively anaerobic environment with its small size and restricted tissue exposure. In addition, the solid surface provides an excellent substrate for biofilm formation. It is likely that the chamber model provides appropriate conditions allowing the EP to survive and express the neutrophil-damaging activity. However, the effectiveness of the neutrophils in chambers infected with S. mitis and several of the individual species in reducing the number of live bacteria demonstrates that the innate immune system is fully capable of clearing infections in these chambers. In addition, the high density of bacteria in these chambers raises the possibility that the neutrophil-damaging activity may be upregulated at high bacterial concentrations or in the specific environment of the subcutaneous chamber. Such a phenomenon would explain why in previous experiments using different model systems with lower bacterial inocula, P. intermedia was able to be cleared by the host (59). Conditions similar to those in the chambers may prevail in the tooth root canal, including limited volume, a solid surface conducive to biofilm formation, the presence of neutrophils associated with biofilms (48), and limited access by infiltrating cells. The total inoculum used in our experiments may be higher than that found in the pulp chamber, even taking into account the larger volume of the titanium chambers compared to the pulp chamber. However, locally high bacterial concentrations may exist in the pulp chamber, especially in biofilms, and our model aims to reproduce these high concentrations.
In summary, we describe here for the first time a novel virulence activity associated with P. intermedia that induces neutrophil dysfunction and death, allowing infections with this organism to evade neutrophil control. This activity can explain in part why endodontic infections including this species are so difficult to control, since they directly impair the innate immune defense of the host. These observations may also explain the persistence of other polymicrobial infections where Prevotella species have also been identified. Future work to characterize this activity and its mechanism of action will be important and may lead to new therapeutic approaches for control of endodontic and other infections.
ACKNOWLEDGMENTS
We thank Rui Nakata for technical assistance, Rani Singh for providing bone marrow neutrophils, and Tommy Hui for help with experiments.
This work was supported by National Institute of Dental and Craniofacial Research of NIH under award R01DE22380.
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Published ahead of print 14 July 2014
REFERENCES
- 1. Marton IJ, Kiss C. 2000. Protective and destructive immune reactions in apical periodontitis. Oral Microbiol. Immunol. 15:139–150. 10.1034/j.1399-302x.2000.150301.x [DOI] [PubMed] [Google Scholar]
- 2. American Dental Association. 2007. 2005-2006 survey of dental services rendered. American Dental Association, Chicago, IL [Google Scholar]
- 3. de Chevigny C, Dao TT, Basrani BR, Marquis V, Farzaneh M, Abitbol S, Friedman S. 2008. Treatment outcome in endodontics: the Toronto study—phase 4: initial treatment. J. Endod. 34:258–263. 10.1016/j.joen.2007.10.017 [DOI] [PubMed] [Google Scholar]
- 4. Siqueira JF, Jr, Rocas IN. 2009. Diversity of endodontic microbiota revisited. J. Dent. Res. 88:969–981. 10.1177/0022034509346549 [DOI] [PubMed] [Google Scholar]
- 5. Chae P, Im M, Gibson F, Jiang Y, Graves DT. 2002. Mice lacking monocyte chemoattractant protein 1 have enhanced susceptibility to an interstitial polymicrobial infection due to impaired monocyte recruitment. Infect. Immun. 70:3164–3169. 10.1128/IAI.70.6.3164-3169.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hou L, Sasaki H, Stashenko P. 2000. Toll-like receptor 4-deficient mice have reduced bone destruction following mixed anaerobic infection. Infect. Immun. 68:4681–4687. 10.1128/IAI.68.8.4681-4687.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sasaki H, Hou L, Belani A, Wang CY, Uchiyama T, Muller R, Stashenko P. 2000. IL-10, but not IL-4, suppresses infection-stimulated bone resorption in vivo. J. Immunol. 165:3626–3630. 10.4049/jimmunol.165.7.3626 [DOI] [PubMed] [Google Scholar]
- 8. Rittling SR, Zetterberg C, Yagiz K, Skinner S, Suzuki N, Fujimura A, Sasaki H. 2010. Protective role of osteopontin in endodontic infection. Immunology 129:105–114. 10.1111/j.1365-2567.2009.03159.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stashenko P, Teles R, D'Souza R. 1998. Periapical inflammatory responses and their modulation. Crit. Rev. Oral Biol. Med. 9:498–521. 10.1177/10454411980090040701 [DOI] [PubMed] [Google Scholar]
- 10. Sasaki H, Balto K, Kawashima N, Eastcott J, Hoshino K, Akira S, Stashenko P. 2004. Gamma interferon (IFN-gamma) and IFN-gamma-inducing cytokines interleukin-12 (IL-12) and IL-18 do not augment infection-stimulated bone resorption in vivo. Clin. Diagn. Lab. Immunol. 11:106–110. 10.1128/CDLI.11.1.106-110.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Teles R, Wang CY, Stashenko P. 1997. Increased susceptibility of RAG-2 SCID mice to dissemination of endodontic infections. Infect. Immun. 65:3781–3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Balto K, Sasaki H, Stashenko P. 2001. Interleukin-6 deficiency increases inflammatory bone destruction. Infect. Immun. 69:744–750. 10.1128/IAI.69.2.744-750.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sadik CD, Kim ND, Luster AD. 2011. Neutrophils cascading their way to inflammation. Trends Immunol. 32:452–460. 10.1016/j.it.2011.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Soehnlein O, Lindbom L, Weber C. 2009. Mechanisms underlying neutrophil-mediated monocyte recruitment. Blood 114:4613–4623. 10.1182/blood-2009-06-221630 [DOI] [PubMed] [Google Scholar]
- 15. Kolaczkowska E, Kubes P. 2013. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13:159–175. 10.1038/nri3399 [DOI] [PubMed] [Google Scholar]
- 16. Genco CA, Arko RJ. 1994. Animal chamber models for study of host-parasite interactions. Methods Enzymol. 235:120–140. 10.1016/0076-6879(94)35136-8 [DOI] [PubMed] [Google Scholar]
- 17. Niederman R, Kelderman H, Socransky S, Ostroff G, Genco C, Kent R, Jr, Stashenko P. 2002. Enhanced neutrophil emigration and Porphyromonas gingivalis reduction following PGG-glucan treatment of mice. Arch. Oral Biol. 47:613–618. 10.1016/S0003-9969(02)00042-0 [DOI] [PubMed] [Google Scholar]
- 18. Mizrahi B, Shapira L, Domb AJ, Houri-Haddad Y. 2006. Citrus oil and MgCl2 as antibacterial and anti-inflammatory agents. J. Periodontol. 77:963–968. 10.1902/jop.2006.050278 [DOI] [PubMed] [Google Scholar]
- 19. Burns E, Eliyahu T, Uematsu S, Akira S, Nussbaum G. 2010. TLR2-dependent inflammatory response to Porphyromonas gingivalis is MyD88 independent, whereas MyD88 is required to clear infection. J. Immunol. 184:1455–1462. 10.4049/jimmunol.0900378 [DOI] [PubMed] [Google Scholar]
- 20. Houri-Haddad Y, Soskolne WA, Halabi A, Barak V, Shapira L. 2000. Repeat bacterial challenge in a subcutaneous chamber model results in augmented tumour necrosis factor-alpha and interferon-gamma response, and suppression of interleukin-10. Immunology 99:215–220. 10.1046/j.1365-2567.2000.00965.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Swamydas M, Lionakis MS. 2013. Isolation, purification and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. J. Vis. Exp. 2013:e50586. 10.3791/50586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Herigstad B, Hamilton M, Heersink J. 2001. How to optimize the drop plate method for enumerating bacteria. J. Microbiol. Methods 44:121–129. 10.1016/S0167-7012(00)00241-4 [DOI] [PubMed] [Google Scholar]
- 23. Harper-Owen R, Dymock D, Booth V, Weightman AJ, Wade WG. 1999. Detection of unculturable bacteria in periodontal health and disease by PCR. J. Clin. Microbiol. 37:1469–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Geissmann F, Jung S, Littman DR. 2003. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19:71–82. 10.1016/S1074-7613(03)00174-2 [DOI] [PubMed] [Google Scholar]
- 25. Kennedy AD, DeLeo FR. 2009. Neutrophil apoptosis and the resolution of infection. Immunol. Res. 43:25–61. 10.1007/s12026-008-8049-6 [DOI] [PubMed] [Google Scholar]
- 26. Turina M, Miller FN, McHugh PP, Cheadle WG, Polk HC., Jr 2005. Endotoxin inhibits apoptosis but induces primary necrosis in neutrophils. Inflammation 29:55–63. 10.1007/s10753-006-8970-6 [DOI] [PubMed] [Google Scholar]
- 27. Siqueira JF, Jr, Rocas IN. 2013. Microbiology and treatment of acute apical abscesses. Clin. Microbiol. Rev. 26:255–273. 10.1128/CMR.00082-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sundqvist GK, Eckerbom MI, Larsson AP, Sjogren UT. 1979. Capacity of anaerobic bacteria from necrotic dental pulps to induce purulent infections. Infect. Immun. 25:685–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Keyel PA, Loultcheva L, Roth R, Salter RD, Watkins SC, Yokoyama WM, Heuser JE. 2011. Streptolysin O clearance through sequestration into blebs that bud passively from the plasma membrane. J. Cell Sci. 124:2414–2423. 10.1242/jcs.076182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. 2007. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 176:231–241. 10.1083/jcb.200606027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li HN, Barlow PG, Bylund J, Mackellar A, Bjorstad A, Conlon J, Hiemstra PS, Haslett C, Gray M, Simpson AJ, Rossi AG, Davidson DJ. 2009. Secondary necrosis of apoptotic neutrophils induced by the human cathelicidin LL-37 is not proinflammatory to phagocytosing macrophages. J. Leukoc. Biol. 86:891–902. 10.1189/jlb.0209050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kepp O, Galluzzi L, Zitvogel L, Kroemer G. 2010. Pyroptosis—a cell death modality of its kind? Eur. J. Immunol. 40:627–630. 10.1002/eji.200940160 [DOI] [PubMed] [Google Scholar]
- 33. Fink SL, Cookson BT. 2006. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 8:1812–1825. 10.1111/j.1462-5822.2006.00751.x [DOI] [PubMed] [Google Scholar]
- 34. Lacy P, Eitzen G. 2008. Control of granule exocytosis in neutrophils. Front. Biosci. 13:5559–5570. 10.2741/3099 [DOI] [PubMed] [Google Scholar]
- 35. Suzuki N, Fukamachi H, Arimoto T, Yamamoto M, Igarashi T. 2012. Contribution of hly homologs to the hemolytic activity of Prevotella intermedia. Anaerobe 18:350–356. 10.1016/j.anaerobe.2012.04.005 [DOI] [PubMed] [Google Scholar]
- 36. Beem JE, Nesbitt WE, Leung KP. 1998. Identification of hemolytic activity in Prevotella intermedia. Oral Microbiol. Immunol. 13:97–105. 10.1111/j.1399-302X.1998.tb00719.x [DOI] [PubMed] [Google Scholar]
- 37. Potempa M, Potempa J, Kantyka T, Nguyen KA, Wawrzonek K, Manandhar SP, Popadiak K, Riesbeck K, Eick S, Blom AM. 2009. Interpain A, a cysteine proteinase from Prevotella intermedia, inhibits complement by degrading complement factor C3. PLoS Pathog. 5:e1000316. 10.1371/journal.ppat.1000316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saito K, Takahashi N, Horiuchi H, Yamada T. 2001. Effects of glucose on formation of cytotoxic end-products and proteolytic activity of Prevotella intermedia, Prevotella nigrescens and Porphyromonas gingivalis. J. Periodont. Res. 36:355–360. 10.1034/j.1600-0765.2001.360602.x [DOI] [PubMed] [Google Scholar]
- 39. Takahashi N, Saito K, Schachtele CF, Yamada T. 1997. Acid tolerance and acid-neutralizing activity of Porphyromonas gingivalis, Prevotella intermedia and Fusobacterium nucleatum. Oral Microbiol. Immunol. 12:323–328. 10.1111/j.1399-302X.1997.tb00733.x [DOI] [PubMed] [Google Scholar]
- 40. Rotstein OD, Vittorini T, Kao J, McBurney MI, Nasmith PE, Grinstein S. 1989. A soluble Bacteroides by-product impairs phagocytic killing of Escherichia coli by neutrophils. Infect. Immun. 57:745–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Polak D, Wilensky A, Shapira L, Halabi A, Goldstein D, Weiss EI, Houri-Haddad Y. 2009. Mouse model of experimental periodontitis induced by Porphyromonas gingivalis/Fusobacterium nucleatum infection: bone loss and host response. J. Clin. Periodontol. 36:406–410. 10.1111/j.1600-051X.2009.01393.x [DOI] [PubMed] [Google Scholar]
- 42. Polak D, Shapira L, Weiss EI, Houri-Haddad Y. 2013. Virulence mechanism of bacteria in mixed infection: attenuation of cytokine levels and evasion of polymorphonuclear leukocyte phagocytosis. J. Periodontol. 84:1463–1468. 10.1902/jop.2012.120528 [DOI] [PubMed] [Google Scholar]
- 43. Byrne DP, Potempa J, Olczak T, Smalley JW. 2013. Evidence of mutualism between two periodontal pathogens: co-operative haem acquisition by the HmuY haemophore of Porphyromonas gingivalis and the cysteine protease interpain A (InpA) of Prevotella intermedia. Mol. Oral Microbiol. 28:219–229. 10.1111/omi.12018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kaplan CW, Ma X, Paranjpe A, Jewett A, Lux R, Kinder-Haake S, Shi W. 2010. Fusobacterium nucleatum outer membrane proteins Fap2 and RadD induce cell death in human lymphocytes. Infect. Immun. 78:4773–4778. 10.1128/IAI.00567-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Macey MG, Whiley RA, Miller L, Nagamune H. 2001. Effect on polymorphonuclear cell function of a human-specific cytotoxin, intermedilysin, expressed by Streptococcus intermedius. Infect. Immun. 69:6102–6109. 10.1128/IAI.69.10.6102-6109.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Scherr TD, Heim CE, Morrison JM, Kielian T. 2014. Hiding in plain sight: interplay between staphylococcal biofilms and host immunity. Front. Immunol. 5:37. 10.3389/fimmu.2014.00037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ricucci D, Siqueira JF, Jr, Bate AL, Pitt Ford TR. 2009. Histologic investigation of root canal-treated teeth with apical periodontitis: a retrospective study from twenty-four patients. J. Endod. 35:493–502. 10.1016/j.joen.2008.12.014 [DOI] [PubMed] [Google Scholar]
- 48. Ricucci D, Siqueira JF., Jr 2010. Biofilms and apical periodontitis: study of prevalence and association with clinical and histopathologic findings. J. Endod. 36:1277–1288. 10.1016/j.joen.2010.04.007 [DOI] [PubMed] [Google Scholar]
- 49. Duran-Pinedo AE, Baker VD, Frias-Lopez J. 2014. The periodontal pathogen Porphyromonas gingivalis induces expression of transposases and cell-death of Streptococcus mitis in a biofilm model. Infect. Immun. 82:3374–3382. 10.1128/IAI.01976-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gunther F, Wabnitz GH, Stroh P, Prior B, Obst U, Samstag Y, Wagner C, Hansch GM. 2009. Host defence against Staphylococcus aureus biofilms infection: phagocytosis of biofilms by polymorphonuclear neutrophils (PMN). Mol. Immunol. 46:1805–1813. 10.1016/j.molimm.2009.01.020 [DOI] [PubMed] [Google Scholar]
- 51. Daw K, Baghdayan AS, Awasthi S, Shankar N. 2012. Biofilm and planktonic Enterococcus faecalis elicit different responses from host phagocytes in vitro. FEMS Immunol. Med. Microbiol. 65:270–282. 10.1111/j.1574-695X.2012.00944.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Anderson AC, Al-Ahmad A, Elamin F, Jonas D, Mirghani Y, Schilhabel M, Karygianni L, Hellwig E, Rehman A. 2013. Comparison of the bacterial composition and structure in symptomatic and asymptomatic endodontic infections associated with root-filled teeth using pyrosequencing. PLoS One 8:e84960. 10.1371/journal.pone.0084960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tavares WL, Neves de Brito LC, Teles RP, Massara ML, Ribeiro Sobrinho AP, Haffajee AD, Socransky SS, Teles FR. 2011. Microbiota of deciduous endodontic infections analysed by MDA and Checkerboard DNA-DNA hybridization. Int. Endod. J. 44:225–235. 10.1111/j.1365-2591.2010.01805.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bolivar I, Whiteson K, Stadelmann B, Baratti-Mayer D, Gizard Y, Mombelli A, Pittet D, Schrenzel J, Geneva Study Group on Noma 2012. Bacterial diversity in oral samples of children in Niger with acute noma, acute necrotizing gingivitis, and healthy controls. PLoS Negl. Trop. Dis. 6:e1556. 10.1371/journal.pntd.0001556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dowd SE, Wolcott RD, Sun Y, McKeehan T, Smith E, Rhoads D. 2008. Polymicrobial nature of chronic diabetic foot ulcer biofilm infections determined using bacterial tag encoded FLX amplicon pyrosequencing (bTEFAP). PLoS One 3:e3326. 10.1371/journal.pone.0003326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shipitsyna E, Roos A, Datcu R, Hallen A, Fredlund H, Jensen JS, Engstrand L, Unemo M. 2013. Composition of the vaginal microbiota in women of reproductive age—sensitive and specific molecular diagnosis of bacterial vaginosis is possible? PLoS One 8:e60670. 10.1371/journal.pone.0060670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Surette MG. 2014. The cystic fibrosis lung microbiome. Ann. Am. Thorac. Soc. 11(Suppl 1):S61–S65. 10.1513/AnnalsATS.201306-159MG [DOI] [PubMed] [Google Scholar]
- 58. Santos AL, Siqueira JF, Jr, Rocas IN, Jesus EC, Rosado AS, Tiedje JM. 2011. Comparing the bacterial diversity of acute and chronic dental root canal infections. PLoS One 6:e28088. 10.1371/journal.pone.0028088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hafstrom C, Dahlen G. 1997. Pathogenicity of Prevotella intermedia and Prevotella nigrescens isolates in a wound chamber model in rabbits. Oral Microbiol. Immunol. 12:148–154. 10.1111/j.1399-302X.1997.tb00371.x [DOI] [PubMed] [Google Scholar]