

Abstract
Legionella pneumophila, an intracellular pathogen responsible for the severe pneumonia Legionnaires' disease, uses its dot/icm-encoded type IV secretion system (T4SS) to translocate effector proteins that promote its survival and replication into the host cell cytosol. However, by introducing bacterial products into the host cytosol, L. pneumophila also activates cytosolic immunosurveillance pathways, thereby triggering robust proinflammatory responses that mediate the control of infection. Thus, the pulmonary cell types that L. pneumophila infects not only may act as an intracellular niche that facilitates its pathogenesis but also may contribute to the immune response against L. pneumophila. The identity of these host cells remains poorly understood. Here, we developed a strain of L. pneumophila producing a fusion protein consisting of β-lactamase fused to the T4SS-translocated effector RalF, which allowed us to track cells injected by the T4SS. Our data reveal that alveolar macrophages and neutrophils both are the primary recipients of T4SS-translocated effectors and harbor viable L. pneumophila during pulmonary infection of mice. Moreover, both alveolar macrophages and neutrophils from infected mice produced tumor necrosis factor and interleukin-1α in response to T4SS-sufficient, but not T4SS-deficient, L. pneumophila. Collectively, our data suggest that alveolar macrophages and neutrophils are both an intracellular reservoir for L. pneumophila and a source of proinflammatory cytokines that contribute to the host immune response against L. pneumophila during pulmonary infection.
INTRODUCTION
Legionella pneumophila is a Gram-negative bacterium found ubiquitously in freshwater environments, where it is often found in association with its natural host, protozoan amoebae (1). L. pneumophila recently has become a human pathogen due to modern technologies, such as cooling towers and air conditioners, which can aerosolize freshwater contaminated with L. pneumophila (2–4). Humans then can inhale these contaminated droplets, allowing L. pneumophila to gain access to the pulmonary airway. L. pneumophila infection can lead to a severe bacterial pneumonia known as Legionnaires' disease (2), with mortality rates approaching 30% (5).
Once in the lung, L. pneumophila encounters a specialized subset of pulmonary phagocytes called alveolar macrophages (6). Following phagocytosis, the Legionella-containing phagosome avoids endocytic maturation and bacterial degradation and is converted into an endoplasmic reticulum (ER)-derived vacuole that supports bacterial replication (7). To establish infection, L. pneumophila utilizes its type IV secretion system (T4SS), encoded by the dot/icm genes, to translocate approximately 300 effector proteins into the host cell cytosol (8–15). Many of these effector proteins are thought to be involved in recruiting ER-derived vacuoles to the Legionella-containing vacuole or prevent endocytic maturation (15). Other effector proteins modulate host cell processes such as autophagy or host protein synthesis (16–20). These virulence activities ultimately prevent destruction of L. pneumophila and allow for its replication within host cells. The T4SS is essential for the ability of L. pneumophila to survive and replicate within host cells, as L. pneumophila mutants lacking a functional T4SS do not replicate and reside in phagosomes that mature along a canonical endocytic pathway (10, 11).
While the Dot/Icm T4SS is essential for L. pneumophila to survive intracellularly and to cause disease, cytosolic immune surveillance systems activate host defense responses to T4SS activity that are critical for the control of L. pneumophila infection (21). For example, the NAIP5/NLRC4 inflammasome detects T4SS-dependent delivery of flagellin, leading to the caspase-1-dependent secretion of interleukin-1 (IL-1) family cytokines and pyroptotic cell death (22–24). Cytosolic detection of T4SS activity also is required for the robust secretion of inflammasome-independent cytokines, such as tumor necrosis factor (TNF) (25–27). The IL-1 family cytokines and TNF are critical for host defense against L. pneumophila (20, 28–30). Thus, the cells that interact with L. pneumophila in the lung and receive T4SS-translocated effectors may have a dual role during in vivo infection, in that they can enable intracellular survival of the pathogen and also contribute directly to the immune response by detecting T4SS-translocated products. However, the identities of the pulmonary cell types that interact with L. pneumophila and receive T4SS-translocated effectors are poorly understood.
Alveolar macrophages are thought to be the primary cell type infected by L. pneumophila and to support bacterial replication in vivo (31). However, it is unknown whether other immune phagocytes in the lung, such as neutrophils, inflammatory monocytes, or dendritic cells, also receive T4SS-translocated effectors and contribute to the immune response or support L. pneumophila survival. Previous studies have demonstrated that in addition to alveolar macrophages, L. pneumophila can be detected in neutrophils during pulmonary infection (30). Neutrophils are thought to be highly bactericidal, and their presence in the lung and airway space during pulmonary L. pneumophila infection correlates with lower bacterial burden (20, 28, 32–34). Whether L. pneumophila can survive within neutrophils and translocate T4SS effectors into these cells during pulmonary infection is unknown. L. pneumophila can be taken up by a wide variety of cell types in vitro, such as neutrophils, bone marrow-derived dendritic cells, type I and type II alveolar epithelial cells, endothelial cells, and plasmacytoid dendritic cells (35–39). However, the efficiency of L. pneumophila replication within these cell types varies greatly, and whether these cell types are injected by the T4SS or productively infected in vivo is unknown. Thus, we decided to investigate which cell types receive T4SS-translocated effectors and, as a result, may support L. pneumophila survival and contribute to cytosolic immunosurveillance during pulmonary infection.
Using a fluorescence resonance energy transfer (FRET)-based reporter of T4SS translocation, we were able to detect effector translocation into macrophages, dendritic cells, and airway epithelial cells in vitro. We also demonstrate that only T4SS-injected cells contain viable L. pneumophila, whereas infected cells that have not received T4SS effectors do not contain viable bacteria. In vivo, alveolar macrophages and neutrophils in the airway space and lung tissue were the primary recipients of T4SS-translocated effectors and harbored viable bacteria. Consistent with the critical role of immune sensing of T4SS activity in triggering host cytokine production, alveolar macrophages and neutrophils from mice infected with T4SS-competent L. pneumophila, but not T4SS-deficient bacteria, secreted the cytokines TNF and IL-1α, which are known to be important for immune-mediated clearance of infection (28–30). We did not observe T4SS-mediated injection into other lung cell populations, including airway epithelial cells and dendritic cells, suggesting that these cells are not a primary intracellular niche for L. pneumophila and do not directly participate in cytosolic immunosurveillance of T4SS activity during lung infection. Collectively, our data indicate that alveolar macrophages and neutrophils play a dual role as both an intracellular niche and immune mediator during pulmonary L. pneumophila infection.
MATERIALS AND METHODS
Ethics statement.
All experiments performed in this study were done so in accordance with the Animal Welfare Act (AWA) and the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (40). The Institutional Animal Care and Use Committee of the University of Pennsylvania approved all procedures (protocols 803465, 803459, and 804928).
Bacterial strains and plasmids.
All experiments used Legionella pneumophila serogroup 1 strains. For in vitro studies, macrophages, dendritic cells, and epithelial cells were infected with Lp02 (rpsL, hsdR, thyA), a thymidine auxotroph derived from strain Lp01, or a ΔdotA or ΔflaA isogenic mutant strain (10). For in vivo studies, mice were infected with the JR32-derived (rpsL, hsdR) ΔdotA or ΔflaA isogenic mutant strain (22, 41). For in vivo experiments requiring cell sorting, the aforementioned Lp02 strains were used. For in vitro and in vivo studies, L. pneumophila was cultured on charcoal yeast extract agar containing 6.25 μg/ml chloramphenicol for 48 h at 37°C prior to infection (42, 43). For studies requiring motile L. pneumophila, 48-h cultures grown on CYE agar were grown overnight in AYE broth containing chloramphenicol with shaking at 37°C until >50% of the bacteria were observed to be motile by light microscopy. Plasmids encoding M45-tagged β-lactamase-RalF fusion protein or M45-tagged β-lactamase were generated as follows. Briefly, the pJB1806 plasmid (RSF1010 ori, tdΔI, Ampr, Cmr) first was modified by cloning the icmR promoter and M45 epitope tag into the EcoRI and BamHI sites (44). The mature TEM-1 β-lactamase gene (BlaM) then was amplified from a Yersinia pseudotuberculosis YopE-BlaM-encoding plasmid using primers that introduced a 5′ BglII site (5′-AATAAGATCTTGCACCCAGAAACGCTGGTG-3′) and 3′ BamHI site (5′-GCCTCACTGATTAAGCATTGGGGGATCCAATA-3′) (45). The resulting PCR product was digested with BglII and BamHI and cloned into the BamHI site of the pJB1806 PicmR:M45 plasmid to create a plasmid encoding M45-tagged β-lactamase. To generate the plasmid encoding a translational fusion of M45-β-lactamase-RalF, RalF was amplified from Lp01 genomic DNA using primers that introduced BamHI sites at the 5′ and 3′ ends (5′-AATAGGATCCGGCATCCAGAAATTGAAAAAGCCC-3′) and (5′-GAAAAAGGTAGACAATTAAAATTTTAAGGATCCAATA-3′). The resulting PCR product was digested with BamHI and cloned into the BamHI site downstream of the gene encoding M45-BlaM. The resulting plasmids then were electroporated into L. pneumophila, and transformed colonies were selected for with chloramphenicol (8).
Mice.
C57BL/6 mice were purchased from Jackson Laboratories. Mice were maintained in accordance with the guidelines of the University of Pennsylvania Institutional Animal Use and Care Committee. For infections, 8- to 12-week-old female mice were anesthetized by intraperitoneal injection of a ketamine-xylazine-phosphate-buffered saline (PBS) solution at a dose of 100 mg ketamine/kg of body weight and 10 mg/kg xylazine. Mice then were infected intranasally with 40 μl of a bacterial suspension containing 5 × 106 CFU L. pneumophila or PBS vehicle control. At the indicated time points after infection, mice were sacrificed. To isolate lung airway cells, bronchoalveolar lavage was performed 3 to 5 times with 1 ml of cold PBS each time. Lungs then were excised and digested for 30 min at 37°C with occasional shaking in 5 ml of PBS containing 5% fetal bovine serum (FBS), 250 U/ml of collagenase IV (Worthington Biochem), and 20 U/ml DNase I (Roche). Lungs then were mechanically homogenized, and a single-cell suspension was obtained. To determine bacterial burden, lungs were mechanically homogenized in sterile, distilled H2O, and a portion of the lysate was spread onto CYE plates containing either chloramphenicol or streptomycin.
Cell culture.
For macrophages, C57BL/6 mouse bone marrow cells were differentiated in RPMI containing 30% L929 cell supernatant and 20% FBS at 37°C, 5% CO2 in a humidified incubator. The macrophages were replated in RPMI containing 15% L929 cell supernatant and 10% FBS (28). For dendritic cells, bone marrow cells were differentiated in RPMI containing 10% FBS, 50 μM β-mercaptoethanol, 2 mM l-glutamine, and 20 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF; Peprotech) (46). Semiadherent dendritic cells then were isolated and replated in medium lacking GM-CSF. A549 cells (ATCC) were cultured in Dulbecco's modified Eagle medium (DMEM) containing 10% FBS (47). For infections, cells were treated with 10 ng/ml LPS from E. coli strain 055:B5 (Sigma), 10 μl of bacterial suspension, or 10 μl of PBS vehicle control.
Flow cytometry, fluorescence-based imaging flow cytometry, and cell sorting.
For in vitro experiments, infected cells were lifted and loaded with CCF4-AM (Invitrogen) per the manufacturer's instructions. Cells then were washed and treated with Live/Dead fixable dead cell stain (Invitrogen). Bone marrow-derived dendritic cells were stained with antibodies specific for CD11c and major histocompatibility complex class II (MHC-II; eBioscience). To stain for intracellular L. pneumophila, cells were fixed with BD Cytofix, permeabilized with BD Phosflow perm buffer III (BD Biosciences), and then stained with a rabbit polyclonal antibody against L. pneumophila followed by a rabbit-specific secondary antibody tagged to a fluorophore (Invitrogen). For in vivo studies, lung and airway cells were loaded with CCF4-AM and treated with the Live/Dead stain. Cells then were stained with antibodies specific for the cell surface antigens CD45, CD11c, Ly6G, Ly6C, NK1.1 (BioLegend), MHCII, CD19, CD3ε, CD31, CD326 (eBioscience), Siglec F, CD11b, and Ter119 (BD Biosciences). Data were collected on an LSR II flow cytometer (BD Biosciences), and postcollection data were analyzed using FlowJo (TreeStar). For fluorescent imaging experiments, data and images were collected on an Amnis ImageStreamX Mark II, and data were analyzed using IDEAS software (EMD Millipore). Cells were gated on live singlets that had retained the CCF4-AM dye. Cell sorting experiments were performed on a FACSAria II flow cytometer (BD Biosciences).
Enzyme-linked immunosorbent assay (ELISA).
Harvested supernatants from cultured cells or bronchoalveolar lavage specimens were assayed using capture and detection antibodies specific for IL-1α and TNF (BioLegend).
Immunoblot analysis.
Legionella pneumophila cells expressing the appropriate reporter plasmids were harvested from a 2-day heavy patch and lysed. Lysates then were subjected to SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membrane, and probed with an anti-M45 epitope monoclonal antibody (48).
Statistical analysis.
The plotting of data and statistical analysis were performed using GraphPad Prism software. Statistical significance was determined using the unpaired, two-tailed Student's t test or one-way analysis of variance (ANOVA) with Tukey's posttest. Differences were considered significant if the P value was <0.05.
RESULTS
A reporter system tracks translocation of type IV secretion system effectors by Legionella pneumophila into mammalian cells.
L. pneumophila uses its T4SS to translocate effector proteins into the cytosol of host cells. To track this translocation, we constructed a plasmid in which the well-characterized L. pneumophila icmR promoter drives transcription of a gene encoding a translational fusion of the mature TEM-1 BlaM and the well-characterized T4SS effector protein RalF and introduced this plasmid into L. pneumophila (see Fig. S1A and B in the supplemental material) (49–51). We chose RalF because it is translocated into the cytosol of infected cells immediately following the intimate interaction of L. pneumophila with host cells (52, 53). Following infection of host cells by bacterial strains expressing the BlaM-RalF fusion protein, the enzymatic activity of translocated BlaM-RalF was detected in host cells by means of the membrane-permeable BlaM substrate CCF4-AM (51). CCF4-AM consists of coumarin joined to fluorescein by a β-lactam ring. When excited at 409 nm, fluorescence resonance energy transfer (FRET) between coumarin and fluorescein results in green fluorescence emission at 518 nm. T4SS-injected BlaM will cleave the CCF4-AM substrate in the host cytosol and eliminate FRET, resulting in blue fluorescence emission at 447 nm.
We generated L. pneumophila strains expressing either BlaM or BlaM-RalF and infected C57BL/6 bone marrow-derived macrophages (BMDM) with these strains for 8 h. Following infection, the cells were loaded with CCF4-AM and analyzed by flow cytometry to determine whether blue fluorescence emitted by cleaved CCF4-AM was detected (see Fig. S1C in the supplemental material). Approximately 20 to 25% of macrophages infected with BlaM-RalF-expressing wild-type (WT) L. pneumophila and L. pneumophila lacking flagellin (ΔflaA mutant), which evade NAIP5 inflammasome responses, were positive for blue fluorescence resulting from cleaved CCF4-AM, but this was not the case following infection with strains lacking a functional T4SS (ΔdotA mutants). This indicates that CCF4-AM is efficiently cleaved only by BlaM-RalF translocated by T4SS-sufficient bacteria and that BlaM-RalF remaining within bacteria does not generate a detectable signal in this assay. The frequencies of injected cells in WT and ΔflaA mutant L. pneumophila infections were comparable, although the frequency of injection was consistently lower in WT infections (Fig. 1A and 2A; also see Fig. S1C). The robust detection of injection by WT L. pneumophila is surprising considering that a higher percentage of vacuoles containing WT L. pneumophila fail to avoid rapid endocytic maturation and that flagellin induces NAIP5-dependent cell death in C57BL/6 macrophages (22, 23, 54–56). Following infection with L. pneumophila strains expressing BlaM alone, we found that a much lower percentage of macrophages became positive for blue CCF4-AM fluorescence compared to macrophages infected with L. pneumophila expressing BlaM-RalF (see Fig. S1C). Importantly, this small percentage of CCF4-AM positive cells still was dependent on infection with T4SS-sufficient bacteria, suggesting that BlaM lacking a canonical T4SS signal sequence is inefficiently delivered into the host cytosol by the T4SS.
FIG 1.

Viable Legionella pneumophila organisms are associated predominantly with macrophages positive for T4SS-dependent translocation. (A) BMDMs were left untreated (Unfx) or were infected with the WT, ΔdotA, or ΔflaA strain for 8 h at an MOI of 5. Cells then were loaded with CCF4-AM, fixed, permeabilized, and stained with a polyclonal antibody against L. pneumophila. Cells were analyzed by flow cytometry. The percentages of cells positive for L. pneumophila as determined by staining and cells positive for T4SS injection are denoted within the gates and quantified in the bar graph shown. (B and C) Cells were treated as described for panel A but were infected at an MOI of 10 for 4 h and analyzed using an Amnis ImageStream imaging flow cytometer. (D) BMDMs were left untreated or infected with the ΔdotA or ΔflaA mutant strain for 8 h at an MOI of 5 in the presence of exogenous thymidine (100 μg/ml) and loaded with CCF4-AM. Samples infected with the ΔdotA mutant were sorted in bulk for loaded cells. Cells infected with the ΔflaA mutant then were sorted based on uncleaved or cleaved CCF4-AM signal, lysed, and plated on CYE plates. CFU then were enumerated. Bar graphs show means only or means ± standard errors of the means (SEM) from triplicate samples. Results are representative of 2 independent experiments. ns, not significant.
FIG 2.

Legionella pneumophila T4SS-dependent translocation is detected in dendritic cells and alveolar epithelial cells during in vitro infection. BMDMs (A), bone marrow-derived dendritic cells (BMDCs) (B), and A549 cells (C) were left untreated or were infected with the WT, ΔdotA, or ΔflaA strain for 4, 8, or 12 h at an MOI of 5. Cells then were loaded with CCF4-AM and analyzed for injection by flow cytometry. Representative plots show injection 8 h postinfection (PI). Graphs show means ± SEM from triplicate wells. Results are representative of 2 independent experiments (n = 3).
T4SS-injected host cells contain viable Legionella pneumophila.
The T4SS is essential for the survival of L. pneumophila within host cells. To determine whether cells injected with BlaM-RalF contain L. pneumophila, we infected BMDMs with these reporter strains and loaded the cells with CCF4-AM. After loading, the macrophages were fixed, permeabilized, and stained with an antibody specific for L. pneumophila (Fig. 1A). Infection with all three strains (the WT and ΔdotA and ΔflaA mutants) of L. pneumophila resulted in macrophages staining positive for the presence of bacteria. Ninety to 100% of cells that were positive for BlaM-RalF injection also were positive for L. pneumophila staining in both the WT and ΔflaA strains. With both strains, we detected a subset of cells that was positive for L. pneumophila, but translocation of BlaM-RalF was not within a detectible range, revealing heterogeneity in BlaM-RalF translocation at the single-cell level. The percentage of cells positive for L. pneumophila but negative for BlaM-RalF translocation could result from bacteria that failed to successfully translocate T4SS effectors into the host cell, either because they were nonviable, were not in the transmissive phase, or failed to efficiently evade rapid endocytic maturation.
To determine whether the L. pneumophila organisms associated with injected or uninjected macrophages were intact or degraded, we analyzed these macrophages with fluorescence-based imaging flow cytometry (Fig. 1B; also see Fig. S2A in the supplemental material). The majority of macrophages infected with the ΔdotA strain showed dim L. pneumophila staining, with multiple small puncta present per cell (Fig. 1C). Because the ΔdotA mutants are unable to evade endocytic maturation due to their lack of a functional T4SS, punctate staining could result from bacteria that were degraded. Alternatively, punctate staining could represent uninfected cells that had phagocytosed bacterial debris. When we infected macrophages with the L. pneumophila ΔflaA mutant encoding a functional T4SS, again we could identify T4SS-injected and uninjected cells. Many of the uninjected cells stained positive for intracellular L. pneumophila (see Fig. S2B), but the majority of these cells exhibited dim, punctate staining similar to the staining seen for ΔdotA mutant-infected macrophages (Fig. 1B and C). This may represent cells containing bacteria that had not successfully evaded endocytic maturation or uninfected cells that had phagocytosed bacterial debris. In contrast, the majority of injected cells showed a single bright punctum of L. pneumophila staining, indicating the presence of an intact bacterium that had not been transported to a hydrolytic compartment.
To test whether injected macrophages contain viable L. pneumophila, we sorted infected macrophages that were either positive or negative for the cleaved CCF4-AM signal, lysed the macrophages, and enumerated bacterial CFU in these distinct cell populations (Fig. 1D). T4SS-injected cells recovered from an L. pneumophila ΔflaA mutant infection contained the vast majority (nearly 6 bacteria for every injected BMDM) of viable L. pneumophila as determined by CFU count. Uninjected cells from the same infection contained a minimal number of viable L. pneumophila (less than 1 bacterium for each uninjected BMDM), comparable to the number of viable bacteria recovered from a ΔdotA mutant infection. Similar results were obtained with the JR32 strains of L. pneumophila (see Fig. S1D and E in the supplemental material). To exclude the possibility that the uninjected cells contained viable L. pneumophila that lost the BlaM-RalF reporter plasmid encoding chloramphenicol resistance, we also plated cell lysates in the presence or absence of chloramphenicol (see Fig. S3A). The CFU obtained on plates with and without chloramphenicol were indistinguishable, suggesting that the plasmid is stably maintained during in vitro infection in the absence of antibiotics. Collectively, our data indicate that viable bacteria are associated primarily with cells that have received translocated BlaM-Ralf, whereas uninjected cells either are not infected or contain nonviable bacteria.
Translocation by the type IV secretion system can be detected in dendritic cells and alveolar epithelial cells in vitro.
L. pneumophila can infect a variety of cell types in vitro, including dendritic cells and airway epithelial cells (35–37). Thus, we examined whether T4SS-mediated translocation into these cell types could be detected using the β-lactamase reporter system. At a given MOI, compared to infected BMDMs, we detected a much lower frequency of T4SS-mediated injection into bone marrow-derived dendritic cells (BMDCs) infected with WT or ΔflaA mutant L. pneumophila (Fig. 2A and B). We also infected A549 cells, an alveolar epithelial cell line, and detected a low frequency of injection into these cells (Fig. 2C).
Other researchers have noted an increase in bacterial uptake by host cells when L. pneumophila is grown under conditions that promote bacterial motility (22). Indeed, infection of macrophages with motile L. pneumophila resulted in a large increase in the frequency of injected macrophages, as the percentage of injected cells increased from 10% to more than 80% (Fig. S4B). In contrast, in A549 cells, we did not observe an increase in injection regardless of bacterial motility, in that 1.3% of cells infected with nonmotile or motile L. pneumophila were injected (see Fig. S4A). For all cell types, the percentage of cells injected by the T4SS of L. pneumophila increased over time (Fig. 2). In all instances, cleaved CCF4-AM signal required the expression of a functional T4SS, suggesting that the β-lactamase reporter operates in a T4SS-dependent manner in a variety of cell types. As we observed more robust injection into macrophages and dendritic cells than into nonphagocytic alveolar epithelial cells, these data suggest that both increased cell contact and efficient uptake by professional phagocytes contribute to the ability of L. pneumophila to efficiently translocate effector proteins.
Legionella pneumophila translocates bacterial effectors into alveolar macrophages and neutrophils during pulmonary infection.
Our data suggest that the L. pneumophila T4SS can translocate effectors into alveolar epithelial cells, dendritic cells, and macrophages during in vitro infection. During pulmonary infection, replicating L. pneumophila can be detected in alveolar macrophages (31), indicating that alveolar macrophages receive T4SS-translocated effectors. However, whether alveolar epithelial cells, dendritic cells, and other cell types receive T4SS-translocated effectors in vivo has not been investigated. To identify the cells that receive translocated effectors during a permissive model of in vivo infection, we intranasally infected C57BL/6 mice with the L. pneumophila ΔflaA mutant expressing BlaM-RalF, as WT L. pneumophila does not establish a productive infection in mice that encode a functional NAIP5 allele (see Fig. S3D in the supplemental material) (22, 23). In this model, similar to WT L. pneumophila infection of A/J mice expressing a hypomorphic NAIP5 allele, the lungs of C57BL/6 mice exhibit an approximately 1-log increase in ΔflaA mutant CFU by 24 to 48 h postinfection (hpi) (see Fig. S3C and D) (57). The mice subsequently are able to control infection, with minimal bacterial CFU detected in the lungs by 5 days postinfection (see Fig. S3C and D) (23). Expression of the plasmid containing BlaM-RalF did not affect the replication of the ΔflaA mutant in vivo (see Fig. S3C). After intranasal inoculation with L. pneumophila, we performed bronchoalveolar lavage to isolate cells from the airway space at various time points and loaded them with CCF4-AM to detect T4SS-mediated injection of BlaM-RalF. At 4 h postinoculation, we detected T4SS-mediated translocation of β-lactamase activity in nearly 50% of cells recovered from the airway of mice infected with the ΔflaA mutant (Fig. 3A). Greater than 95% of the T4SS-injected cells were alveolar macrophages, as indicated by their expression of CD11c and Siglec F (58, 59). Similar results were obtained with WT L. pneumophila at this time point (data not shown). Consistent with our in vitro data, we did not observe injection of BlaM-RalF in mice infected with the ΔdotA strain, which is unable to translocate effectors into host cells and cannot establish a productive infection in vivo.
FIG 3.

Alveolar macrophages and neutrophils in the airway space are injected by the Legionella pneumophila T4SS. C57BL/6J mice were infected intranasally with PBS vehicle control or the ΔdotA or ΔflaA mutant. At 4 (A) or 24 (B) h postinfection, cells in the airway space were isolated, enumerated, loaded with CCF4-AM, and stained for cell surface markers. Cells then were analyzed for injection by flow cytometry and cell surface marker expression. Results are representative of 2 independent experiments, with n = 4 mice per group. (C and D) Alveolar macrophages (MΦ) and neutrophils from the airway space of mice infected with the ΔflaA strain were isolated at 4, 24, 48, or 72 h postinfection and loaded with CCF4-AM. The percentage (C) and total number (D) of cells injected by L. pneumophila in each population were quantified. Graphs show means ± SEM (n = 3 to 4 mice per group).
At later times postinfection, we detected recruitment of a large population of neutrophils to the airway space of ΔflaA mutant-infected mice that did not occur in mice infected with the ΔdotA mutant (see Fig. S5A and B in the supplemental material), consistent with previous studies indicating that neutrophil recruitment is T4SS dependent (20, 28, 30, 60). When we identified cells injected by L. pneumophila in the airway space at 24 hpi, we again identified alveolar macrophages as being positive for T4SS-mediated injection, but we also could identify injected cells that expressed high levels of Ly6G and were negative for MHC-II (Fig. 3B). We determined that these injected Ly6G+ cells were neutrophils, as they expressed low levels of Ly6C, a cell surface marker highly expressed on inflammatory monocytes (see Fig. S2C) (61, 62). The frequency of injected neutrophils was much lower than that of injected alveolar macrophages (Fig. 3C). However, due to the large influx of neutrophils, the total number of injected neutrophils was comparable to or greater than the total number of injected alveolar macrophages at 24, 48, and 72 hpi (Fig. 3D).
As we could detect robust T4SS-mediated injection of BlaM-RalF into cells of the airway space, we wanted to determine whether cells within the lung interstitium were injected by L. pneumophila as well. Notably, we again observed T4SS-mediated injection into alveolar macrophages and neutrophils within lung homogenates (Fig. 4A). As in the airway space, we detected a large influx of neutrophils into the lung tissue of ΔflaA mutant-infected mice but not in mice infected with the ΔdotA strain of L. pneumophila (see Fig. S5C and D in the supplemental material). Although in vitro we observed T4SS-mediated injection into bone marrow-derived dendritic cells as well as A549 alveolar epithelial cells (Fig. 2B and C), we did not detect injection into lung dendritic cells or CD326+ airway epithelial cells, suggesting that L. pneumophila does not efficiently infect or translocate effectors into these cell types during a permissive mouse model of infection (Fig. 4A). We also did not observe injection into inflammatory monocytes, plasmacytoid dendritic cells, eosinophils, B cells, T cells, NK cells, or endothelial cells within the lung tissue at any time assayed postinfection (see Fig. S2C and D in the supplemental material). The frequency of T4SS-injected neutrophils in the lung tissue was much lower than that seen in alveolar macrophages, similar to what we observed in the airway space (Fig. 4B). At 4 hpi, the majority of cells receiving T4SS-translocated effectors in the lung tissue were alveolar macrophages, but at later times, many of the T4SS-injected cells were neutrophils (Fig. 4C). Importantly, L. pneumophila recovered at 48 and 72 hpi retained the reporter plasmid, indicating that the plasmid is stably maintained during in vivo infection even in the absence of antibiotic selection (see Fig. S3B). A previous study examining a nonpermissive model of C57BL/6 mice infected with WT L. pneumophila also found that CD45-negative cells or lung epithelial cells did not appear to have taken up L. pneumophila, with alveolar macrophages appearing to be the primary cells infected at early time points postinfection, followed by infection of recruited neutrophils at 1 day postinfection (30), suggesting that in both permissive and nonpermissive mouse models, similar lung cell types are infected.
FIG 4.
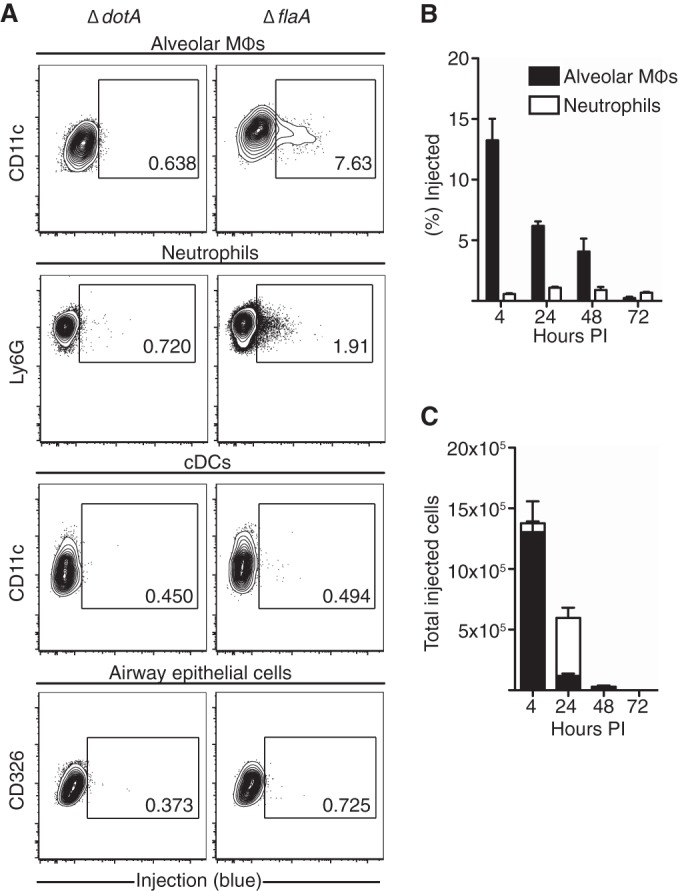
Legionella pneumophila T4SS injects alveolar macrophages and neutrophils in the lung. (A) Mice were infected intranasally with the L. pneumophila ΔdotA or ΔflaA mutant for 24 h. Lung cells then were isolated, loaded with CCF4-AM, and stained. Flow plots are pregated on the denoted cell populations. Results are representative of 3 independent experiments, with n = 4 mice per group. (B and C) Mice were infected with the ΔflaA mutant, and lung cells were isolated at various times postinfection. The percentage (B) and total number (C) of cells injected by L. pneumophila in each population were quantified. Graphs show means ± SEM (n = 3 to 4 mice per group). cDC, conventional dendritic cell.
Neutrophils and alveolar macrophages in the lungs of infected mice harbor viable Legionella pneumophila bacteria and produce cytokines.
Alveolar macrophages are thought to be the primary cell type that is infected by L. pneumophila and supports bacterial replication (31). A previous study using a nonpermissive model of C57BL/6 mice infected with WT L. pneumophila also found that recruited neutrophils take up L. pneumophila in the lung, but whether L. pneumophila could translocate effectors into neutrophils or survive within these cells was not examined (30). As we observed that both alveolar macrophages and neutrophils in L. pneumophila-infected lungs were injected by the T4SS, we sought to determine whether in addition to alveolar macrophages, neutrophils also contained viable bacteria. Therefore, we sorted total alveolar macrophages and neutrophils from the lungs of mice infected intranasally with L. pneumophila and enumerated bacteria from lysed cells. As a comparison, we also sorted total inflammatory monocytes, a population of cells negative for T4SS injection, from these infected mice as well. As expected, alveolar macrophages isolated 24 h after infection contained viable bacteria (Fig. 5A). In contrast, inflammatory monocytes contained very few viable L. pneumophila organisms, consistent with the lack of observed T4SS-dependent translocation into these cells (Fig. 5A). Interestingly, neutrophils from ΔflaA mutant-infected mice contained viable bacteria at a frequency consistent with the extent of injection, suggesting that injected neutrophils harbor viable bacteria in the airway and lung tissue. Although the absolute frequency of viable L. pneumophila in alveolar macrophages was greater than that within neutrophils (nearly 10 bacteria per 100 alveolar macrophages versus 1 bacterium per 100 neutrophils), the higher absolute numbers of neutrophils present during infection results in the unexpected finding that neutrophils actually contain nearly twice as many viable bacteria as alveolar macrophages (Fig. 5B). To examine whether this also was the case at the peak of pulmonary bacterial load, we sorted alveolar macrophages and neutrophils from mice 48 hpi (Fig. 5C and D). As with samples from 24 hpi, although a higher frequency of alveolar macrophages contained viable L. pneumophila, in total there were more L. pneumophila organisms in neutrophils than alveolar macrophages.
FIG 5.
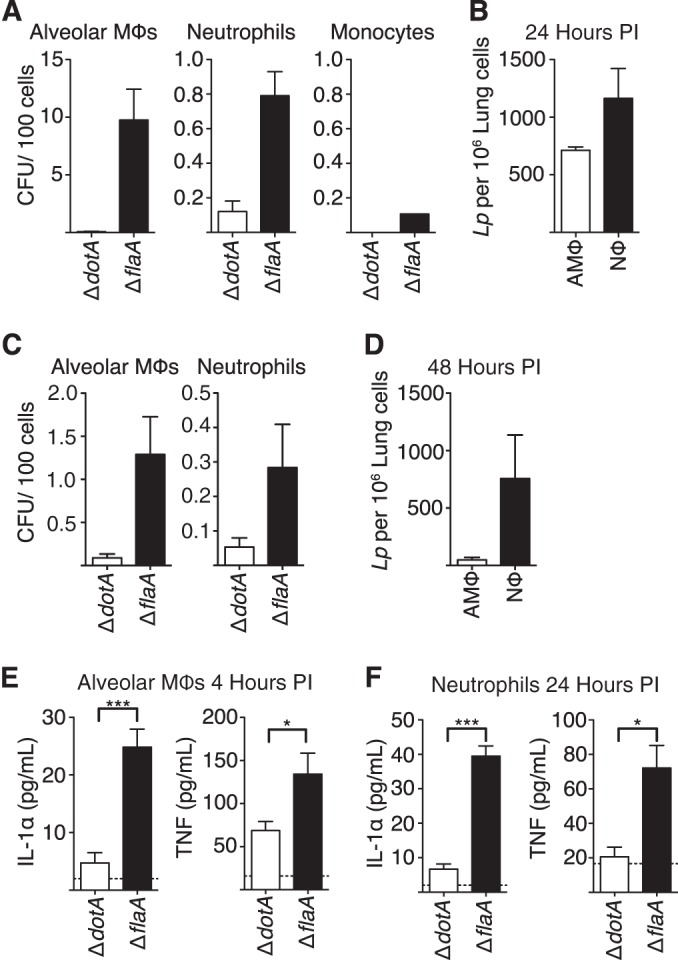
Alveolar macrophages and neutrophils from infected mice contain viable Legionella pneumophila and secrete cytokines. (A and C) Mice were intranasally infected with the ΔdotA or ΔflaA mutant. At 24 (A) or 48 (C) hours postinfection, the lungs and airway space were homogenized, and then single-cell suspensions were stained for cell surface markers to identify alveolar macrophages, neutrophils, and inflammatory monocytes. These cell populations were sorted using a flow cytometer, lysed, and plated on CYE agar to enumerate L. pneumophila CFU. (B) The frequency of viable bacteria per cell type shown in panel A then was multiplied by the frequency of the appropriate cell type found in a total of 106 lung cells. (D) Similarly, the frequency of viable bacteria per cell type shown in panel C was multiplied by the frequency of the appropriate cell type found in a total of 106 lung cells. (E) Alveolar macrophages were isolated by bronchoalveolar lavage 4 h after intranasal infection with the ΔdotA or ΔflaA mutant and cultured overnight. Supernatants were collected, and TNF and IL-1α concentrations were determined by ELISA. (F) Neutrophils were isolated and sorted at 24 hpi from mice treated as described for panel A and then cultured overnight. Graphs show means ± SEM. *, P < 0.05; ***, P < 0.0005.
The presence of neutrophils in the airway space during infection correlates with lower bacterial burden thought to be due in part to their potent bactericidal activity (20, 28, 30). However, as our data suggest that L. pneumophila cells inject and survive within neutrophils, potentially activating cytosolic immunosurveillance pathways within these cells, we next examined whether or not infected neutrophils also contribute to the T4SS-dependent production of proinflammatory cytokines important for bacterial clearance. To test this, following intranasal infection with either the L. pneumophila ΔdotA or ΔflaA mutant, we measured cytokines secreted by alveolar macrophages isolated at 4 h postinfection or neutrophils isolated at 24 h postinfection. Alveolar macrophages from ΔflaA mutant-infected mice secreted TNF and IL-1α, whereas macrophages from ΔdotA mutant-infected mice did not. Interestingly, neutrophils from mice infected with the ΔflaA mutant also secreted substantial amounts of TNF and IL-1α, whereas neutrophils from mice infected with the ΔdotA mutant did not secrete detectable levels of IL-1α and secreted significantly less TNF, which correlates with the lack of detectable cytokine production observed during pulmonary infection with the ΔdotA mutant (Fig. 5E and F). Intriguingly, these data demonstrate that in addition to alveolar macrophages, neutrophils also produce proinflammatory cytokines in the context of T4SS-competent L. pneumophila infection. This indicates that in addition to their potent bactericidal activity, neutrophils contribute to the control of infection by other immune effector mechanisms, such as cytokine production.
DISCUSSION
Legionella pneumophila uses its T4SS to inject a large number of effector proteins into the cytosol of host phagocytes (63). The T4SS is necessary for intracellular replication and pathogenesis, as L. pneumophila mutants lacking a functional T4SS fail to establish a replicative niche and do not cause pathology in mice (8, 11, 20, 30). In addition to being required for L. pneumophila pathogenesis, T4SS activity potently activates multiple cytosolic immunosurveillance pathways (25, 64–66). Thus, cells that interact with L. pneumophila and receive T4SS-translocated effectors serve as a potential replicative niche but also may contribute to the immune response against L. pneumophila. However, the precise identity of such cells is unknown. Therefore, we set out to identify host cells that receive T4SS-translocated effectors during infection with L. pneumophila. BlaM reporter systems have been used during in vivo infection with Yersinia pseudotuberculosis (67, 68), Yersinia pestis (69, 70), Yersinia enterocolitica (71), Salmonella enterica serovar Typhimurium (72, 73), and Pseudomonas aeruginosa (74, 75) to detect the translocation of effectors into host cells by the type III and type IV secretion systems. We demonstrate in this study that by using β-lactamase (BlaM) translationally fused to the T4SS-translocated effector protein RalF, we can successfully track injection by the T4SS into host cells during both in vitro and in vivo infection, and we describe the first use of this BlaM reporter during in vivo pulmonary infection with L. pneumophila.
We observed robust T4SS-mediated injection into alveolar macrophages at 4 h postinfection, consistent with previous observations that these cells are the primary cell type infected by L. pneumophila during pulmonary infection in human patients (31). At later time points postinfection, we find that in addition to alveolar macrophages, a large number of the cells injected by L. pneumophila in vivo are neutrophils. Most likely this is due to the large influx of neutrophils into the lungs and airway space during infection (60, 76–80). Other researchers have shown that neutrophils contain intracellular L. pneumophila in a nonpermissive mouse model of pulmonary infection, but they did not examine whether L. pneumophila could survive within neutrophils or whether neutrophils are capable of receiving T4SS-translocated effectors (30). In vitro studies have suggested that Legionella species are resistant to the highly bactericidal activity of neutrophils but cannot replicate within these cells (39, 81). Thus, we initially presumed that although neutrophils might be injected during in vivo infection, the majority of bacteria eventually would be cleared due to a failure to replicate in these cells, and we would not be able to detect large numbers of viable bacteria within these cells. To our surprise, we obtained viable L. pneumophila cells in numbers that roughly corresponded to the frequency of injection seen with our reporter system, suggesting that L. pneumophila can survive within neutrophils during in vivo infection. Unexpectedly, given the large number of neutrophils that enter the lung, the total number of L. pneumophila CFU harbored by neutrophils is greater than the total number of L. pneumophila CFU found within the alveolar macrophage population 24 and 48 hpi. Given the large numbers of infected neutrophils that we observed, it would be of interest to determine whether L. pneumophila could establish an ER-derived vacuole and successfully replicate within neutrophils, as this could represent another intracellular niche for L. pneumophila. Most bacteria are thought not to survive or replicate within neutrophils, but there are a few exceptions, including Neisseria gonorrhoeae (82), Anaplasma phagocytophilum (83, 84), and pathogenic Escherichia coli (85).
As we were able to detect robust T4SS-dependent injection only into alveolar macrophages and neutrophils, we conclude that phagocytic cells in the airway space are the primary recipients of T4SS-translocated effectors during pulmonary L. pneumophila infection. Whether or not T4SS-injected cells survive infection and traffic to other organs, including lymph nodes, also is unknown. However, previous studies have reported that alveolar macrophages do traffic to lymph nodes when given allergic stimuli (86). Thus, it would be of interest to investigate whether T4SS-injected alveolar macrophages either induce adaptive immunity or participate in the dissemination of infection to other organs (86). Surprisingly, we were unable to detect T4SS-injected conventional dendritic cells during in vivo infection. Dendritic cells undergo rapid apoptosis in response to L. pneumophila T4SS activity (87), which could account for why we do not detect injection in dendritic cells that is as robust as that of macrophages in vitro and in vivo. It would be interesting to determine whether more robust injection could be detected in DCs lacking apoptotic regulators, such as BAX and BAK, that are resistant to L. pneumophila-induced apoptosis. However, C57BL/6-derived macrophages undergo rapid pyroptosis in response to WT L. pneumophila infection, yet we still detect robust levels of injection in this cell type, suggesting that L. pneumophila-induced cell death is an insufficient explanation to account for the lack of detectable injection in DCs (57). Given that phagocytosis is required for T4SS-mediated translocation, another possibility is that dendritic cells in the lung do not efficiently phagocytose L. pneumophila (88). Alternatively, pulmonary dendritic cells and L. pneumophila may be spatially separated during in vivo infection.
Using the A549 alveolar epithelial cell line during in vitro infection, we detected a low percentage of T4SS-injected cells under conditions using both nonmotile and motile bacteria. Many researchers utilize A549 cells as a model for L. pneumophila infection and can detect productive bacterial replication within these cells (37). However, these studies either use higher MOIs than those used in this study or opsonize the bacteria prior to infection. These discrepancies in technique may explain why we are unable to detect higher percentages of injected alveolar epithelial cells during in vitro infection. We also were unable to detect robust T4SS-dependent translocation into airway epithelial cells or other nonphagocytic cells during in vivo infection. Utilizing a nonpermissive model of C57BL/6 mice infected with WT L. pneumophila, other researchers also found that lung epithelial cells did not appear to contain L. pneumophila (30). Our data argue against a direct role for airway epithelial cells in the cytosolic sensing of L. pneumophila T4SS activity during pulmonary infection. Airway epithelial cells have been shown to indirectly respond to L. pneumophila infection by producing the chemokine CXCL1 in response to IL-1 produced by macrophages (30).
We found that alveolar macrophages secreted TNF and IL-1α 4 h postinfection in vivo. Both TNF and IL-1α are important for controlling L. pneumophila infection. The inflammasome-regulated cytokines IL-1α and IL-1β are critical for neutrophil recruitment to the lung airway during L. pneumophila infection through a mechanism involving the IL-1R-dependent induction of CXCL1 from alveolar epithelial cells (20, 28, 30). It is unclear whether other cells in the lung also produce cytokines so early during infection. However, as IL-1α production in vivo is T4SS dependent and we could detect T4SS injection only into alveolar macrophages at 4 h postinfection, our data suggest that during the first few hours of infection, alveolar macrophages are the primary source of IL-1α, consistent with another study indicating that hematopoietic cells are an early source of IL-1α (20).
At 24 h postinfection, we found that neutrophils recruited to the lungs of mice infected with the ΔflaA mutant also secrete the proinflammatory cytokines TNF and IL-1α, but not mice infected with the ΔdotA mutant. Therefore, these data indicate that, like alveolar macrophages, neutrophils also secrete cytokines in response to cytosolic sensing of T4SS-translocated bacterial products. It has been reported previously that neutrophils can secrete cytokines, but the signaling pathways that control cytokine production and secretion in neutrophils are poorly understood. Neutrophils are known to release TNF-containing granules in response to a variety of stimuli, including various bacterial infections (89, 90). Previous research has demonstrated that neutrophils can release IL-1α in a model of sterile inflammation or IL-1β independently of caspase-1 and caspase-11 in a mouse model of arthritis and during bacterial infection (91–93). In an intravenous infection model of L. pneumophila infection, splenic neutrophils were shown to produce IL-18, an IL-1 family cytokine, which induces IFN-γ production from NK cells (94). Previous studies demonstrated that IL-1α secretion is regulated by both inflammasome-dependent and -independent pathways during in vivo WT L. pneumophila infection (20, 28), but it is unknown which of these pathways are used by macrophages and neutrophils to secrete IL-1α in vivo. It would be of interest to determine the host and bacterial components required for release of IL-1 and other cytokines from macrophages and neutrophils in response to in vivo infection with L. pneumophila.
Overall, our study is the first to define the cell types that receive T4SS-translocated effectors during pulmonary L. pneumophila infection. We reveal that both alveolar macrophages and neutrophils receive translocated effector proteins, harbor viable bacteria, and respond to infection by producing inflammatory cytokines. Collectively, our data indicate that alveolar macrophages and neutrophils provide not only an intracellular reservoir for L. pneumophila but also an important source of proinflammatory cytokines that contribute to a successful host immune response during pulmonary L. pneumophila infection.
Supplementary Material
ACKNOWLEDGMENTS
We thank Andree Hubber for generating the pJB908 plasmid containing the icmR promoter and M45 epitope tag, the laboratory of Michael Betts, specifically Morgan Reuter, for the use of their imaging flow cytometer and the help required to operate it, Sara Cherry and members of the Brodsky laboratory for helpful discussions, and Igor Brodsky and Liam Bradley for critical review of the manuscript.
This work was supported in part by National Institutes of Health grants K99/R00AI087963 (to S.S.), R01AI0487700 (to C.R.R.), and T32GM007229 (to A.M.C.), as well as funds from the American Lung Association (grant RG-268528-N to S.S.), the University of Pennsylvania University Research Foundation (to S.S.), the American Heart Association (grant 13BGIA14780070 to S.S.), and the Roy and Diana Vagelos Scholars Program in Molecular Life Sciences (to M.M.D.).
This material is based upon work supported by the National Science Foundation under grant no. DGE-0822 (C.N.C., graduate research fellowship).
Footnotes
Published ahead of print 4 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01891-14.
REFERENCES
- 1. Fields BS. 1996. The molecular ecology of legionellae. Trends Microbiol. 4:286–290. 10.1016/0966-842X(96)10041-X [DOI] [PubMed] [Google Scholar]
- 2. Fraser DW, Tsai TR, Orenstein W, Parkin WE, Beecham HJ, Sharrar RG, Harris J, Mallison GF, Martin SM, McDade JE, Shepard CC, Brachman PS. 1977. Legionnaires' disease: description of an epidemic of pneumonia. N. Engl. J. Med. 297:1189–1197. 10.1056/NEJM197712012972201 [DOI] [PubMed] [Google Scholar]
- 3. McDade JE, Shepard CC, Fraser DW, Tsai TR, Redus MA, Dowdle WR. 1977. Legionnaires' disease: isolation of a bacterium and demonstration of its role in other respiratory disease. N. Engl. J. Med. 297:1197–1203. 10.1056/NEJM197712012972202 [DOI] [PubMed] [Google Scholar]
- 4. Phin N, Parry-Ford F, Harrison T, Stagg HR, Zhang N, Kumar K, Lortholary O, Zumla A, Abubakar I. 23 June 2014. Epidemiology and clinical management of Legionnaires' disease. Lancet Infect. Dis. 10.1016/S1473-3099(14)70713-3 [DOI] [PubMed] [Google Scholar]
- 5. Domínguez A, Alvarez J, Sabrià M, Carmona G, Torner N, Oviedo M, Cayla J, Minguell S, Barrabeig I, Sala M, Godoy P, Camps N. 2009. Factors influencing the case-fatality rate of Legionnaires' disease. Int. J. Tuberc. Lung Dis. 13:407–412 [PubMed] [Google Scholar]
- 6. Chandler FW, Hicklin MD, Blackmon JA. 1977. Demonstration of the agent of Legionnaires' disease in tissue. N. Engl. J. Med. 297:1218–1220. 10.1056/NEJM197712012972206 [DOI] [PubMed] [Google Scholar]
- 7. Shin S, Roy CR. 2008. Host cell processes that influence the intracellular survival of Legionella pneumophila. Cell Microbiol. 10:1209–1220. 10.1111/j.1462-5822.2008.01145.x [DOI] [PubMed] [Google Scholar]
- 8. Marra A, Blander SJ, Horwitz MA, Shuman HA. 1992. Identification of a Legionella pneumophila locus required for intracellular multiplication in human macrophages. Proc. Natl. Acad. Sci. U. S. A. 89:9607–9611. 10.1073/pnas.89.20.9607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ensminger AW, Isberg RR. 2009. Legionella pneumophila Dot/Icm translocated substrates: a sum of parts. Curr. Opin. Microbiol. 12:67–73. 10.1016/j.mib.2008.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berger KH, Isberg RR. 1993. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol. Microbiol. 7:7–19. 10.1111/j.1365-2958.1993.tb01092.x [DOI] [PubMed] [Google Scholar]
- 11. Roy CR, Berger KH, Isberg RR. 1998. Legionella pneumophila DotA protein is required for early phagosome trafficking decisions that occur within minutes of bacterial uptake. Mol. Microbiol. 28:663–674. 10.1046/j.1365-2958.1998.00841.x [DOI] [PubMed] [Google Scholar]
- 12. Segal G, Purcell M, Shuman HA. 1998. Host cell killing and bacterial conjugation require overlapping sets of genes within a 22-kb region of the Legionella pneumophila genome. Proc. Natl. Acad. Sci. U. S. A. 95:1669–1674. 10.1073/pnas.95.4.1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vogel JP, Andrews HL, Wong SK, Isberg RR. 1998. Conjugative transfer by the virulence system of Legionella pneumophila. Science 279:873–876. 10.1126/science.279.5352.873 [DOI] [PubMed] [Google Scholar]
- 14. Nagai H. 2002. A Bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 295:679–682. 10.1126/science.1067025 [DOI] [PubMed] [Google Scholar]
- 15. Hubber A, Roy CR. 2010. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 26:261–283. 10.1146/annurev-cellbio-100109-104034 [DOI] [PubMed] [Google Scholar]
- 16. Belyi Y, Tabakova I, Stahl M, Aktories K. 2008. Lgt: a family of cytotoxic glucosyltransferases produced by Legionella pneumophila. J. Bacteriol. 190:3026–3035. 10.1128/JB.01798-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shen X, Banga S, Liu Y, Xu L, Gao P, Shamovsky I, Nudler E, Luo Z-Q. 2009. Targeting eEF1A by a Legionella pneumophila effector leads to inhibition of protein synthesis and induction of host stress response. Cell Microbiol. 11:911–926. 10.1111/j.1462-5822.2009.01301.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Choy A, Dancourt J, Mugo B, O'Connor TJ, Isberg RR, Melia TJ, Roy CR. 2012. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 338:1072–1076. 10.1126/science.1227026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Belyi Y, Niggeweg R, Opitz B, Vogelsgesang M, Hippenstiel S, Wilm M, Aktories K. 2006. Legionella pneumophila glucosyltransferase inhibits host elongation factor 1A. Proc. Natl. Acad. Sci. U. S. A. 103:16953–16958. 10.1073/pnas.0601562103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barry KC, Fontana MF, Portman JL, Dugan AS, Vance RE. 2013. IL-1α signaling initiates the inflammatory response to virulent Legionella pneumophila in vivo. J. Immunol. 190:6329–6339. 10.4049/jimmunol.1300100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shin S. 2012. Innate immunity to intracellular pathogens: lessons learned from Legionella pneumophila. Adv. Appl. Microbiol. 79:43–71. 10.1016/B978-0-12-394318-7.00003-6 [DOI] [PubMed] [Google Scholar]
- 22. Ren T, Zamboni DS, Roy CR, Dietrich WF, Vance RE. 2006. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2:e18. 10.1371/journal.ppat.0020018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Molofsky AB, Byrne BG, Whitfield NN, Madigan CA, Fuse ET, Tateda K, Swanson MS. 2006. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J. Exp. Med. 203:1093–1104. 10.1084/jem.20051659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lightfield KL, Persson J, Brubaker SW, Witte CE, Moltke von J, Dunipace EA, Henry T, Sun Y-H, Cado D, Dietrich WF, Monack DM, Tsolis RM, Vance RE. 2008. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 9:1171–1178. 10.1038/ni.1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shin S, Case CL, Archer KA, Nogueira CV, Kobayashi KS, Flavell RA, Roy CR, Zamboni DS. 2008. Type IV secretion-dependent activation of host MAP kinases induces an increased proinflammatory cytokine response to Legionella pneumophila. PLoS Pathog. 4:e1000220. 10.1371/journal.ppat.1000220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Spörri R, Joller N, Albers U, Hilbi H, Oxenius A. 2006. MyD88-dependent IFN-gamma production by NK cells is key for control of Legionella pneumophila infection. J. Immunol. 176:6162–6171. 10.4049/jimmunol.176.10.6162 [DOI] [PubMed] [Google Scholar]
- 27. Fontana MF, Banga S, Barry KC, Shen X, Tan Y, Luo Z-Q, Vance RE. 2011. Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila. PLoS Pathog. 7:e1001289. 10.1371/journal.ppat.1001289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Casson CN, Copenhaver AM, Zwack EE, Nguyen HT, Strowig T, Javdan B, Bradley WP, Fung TC, Flavell RA, Brodsky IE, Shin S. 2013. Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog. 9:e1003400. 10.1371/journal.ppat.1003400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brieland JK, Remick DG, Freeman PT, Hurley MC, Fantone JC, Engleberg NC. 1995. In vivo regulation of replicative Legionella pneumophila lung infection by endogenous tumor necrosis factor alpha and nitric oxide. Infect. Immun. 63:3253–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. LeibundGut-Landmann S, Weidner K, Hilbi H, Oxenius A. 2011. Nonhematopoietic cells are key players in innate control of bacterial airway infection. J. Immunol. 186:3130–3137. 10.4049/jimmunol.1003565 [DOI] [PubMed] [Google Scholar]
- 31. Nash TW, Libby DM, Horwitz MA. 1984. Interaction between the Legionnaires' disease bacterium (Legionella pneumophila) and human alveolar macrophages. Influence of antibody, lymphokines, and hydrocortisone. J. Clin. Investig. 74:771–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nauseef WM. 2007. How human neutrophils kill and degrade microbes: an integrated view. Immunol. Rev. 219:88–102. 10.1111/j.1600-065X.2007.00550.x [DOI] [PubMed] [Google Scholar]
- 33. Tateda K, Moore TA, Deng JC, Newstead MW, Zeng X, Matsukawa A, Swanson MS, Yamaguchi K, Standiford TJ. 2001. Early recruitment of neutrophils determines subsequent T1/T2 host responses in a murine model of Legionella pneumophila pneumonia. J. Immunol. 166:3355–3361. 10.4049/jimmunol.166.5.3355 [DOI] [PubMed] [Google Scholar]
- 34. Tateda K, Moore TA, Newstead MW, Tsai WC, Zeng X, Deng JC, Chen G, Reddy R, Yamaguchi K, Standiford TJ. 2001. Chemokine-dependent neutrophil recruitment in a murine model of Legionella pneumonia: potential role of neutrophils as immunoregulatory cells. Infect. Immun. 69:2017–2024. 10.1128/IAI.69.4.2017-2024.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neild AL, Roy CR. 2003. Legionella reveal dendritic cell functions that facilitate selection of antigens for MHC class II presentation. Immunity 18:813–823. 10.1016/S1074-7613(03)00140-7 [DOI] [PubMed] [Google Scholar]
- 36. Mody CH, Paine R, Shahrabadi MS, Simon RH, Pearlman E, Eisenstein BI, Toews GB. 1993. Legionella pneumophila replicates within rat alveolar epithelial cells. J. Infect. Dis. 167:1138–1145. 10.1093/infdis/167.5.1138 [DOI] [PubMed] [Google Scholar]
- 37. Maruta K, Miyamoto H, Hamada T, Ogawa M, Taniguchi H, Yoshida S. 1998. Entry and intracellular growth of Legionella dumoffii in alveolar epithelial cells. Am. J. Respir. Crit. Care Med. 157:1967–1974 [DOI] [PubMed] [Google Scholar]
- 38. Ang DKY, Oates CVL, Schuelein R, Kelly M, Sansom FM, Bourges D, Boon L, Hertzog PJ, Hartland EL, van Driel IR. 2010. Cutting edge: pulmonary Legionella pneumophila is controlled by plasmacytoid dendritic cells but not type I IFN. J. Immunol. 184:5429–5433. 10.4049/jimmunol.1000128 [DOI] [PubMed] [Google Scholar]
- 39. Horwitz MA, Silverstein SC. 1981. Interaction of the Legionnaires' disease bacterium (Legionella pneumophila) with human phagocytes. I. L. pneumophila resists killing by polymorphonuclear leukocytes, antibody, and complement. J. Exp. Med. 153:386–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC [Google Scholar]
- 41. Marra A, Shuman HA. 1989. Isolation of a Legionella pneumophila restriction mutant with increased ability to act as a recipient in heterospecific matings. J. Bacteriol. 171:2238–2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Feeley JC, Gibson RJ, Gorman GW, Langford NC, Rasheed JK, Mackel DC, Baine WB. 1979. Charcoal-yeast extract agar: primary isolation medium for Legionella pneumophila. J. Clin. Microbiol. 10:437–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Neild A, Murata T, Roy CR. 2005. Processing and major histocompatibility complex class II presentation of Legionella pneumophila antigens by infected macrophages. Infect. Immun. 73:2336–2343. 10.1128/IAI.73.4.2336-2343.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bardill JP, Miller JL, Vogel JP. 2005. IcmS-dependent translocation of SdeA into macrophages by the Legionella pneumophila type IV secretion system. Mol. Microbiol. 56:90–103. 10.1111/j.1365-2958.2005.04539.x [DOI] [PubMed] [Google Scholar]
- 45. Brodsky IE, Medzhitov R. 2008. Reduced secretion of YopJ by Yersinia limits in vivo cell death but enhances bacterial virulence. PLoS Pathog. 4:e1000067. 10.1371/journal.ppat.1000067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. 1992. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 176:1693–1702. 10.1084/jem.176.6.1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wickremasinghe MI, Thomas LH, Friedland JS. 1999. Pulmonary epithelial cells are a source of IL-8 in the response to Mycobacterium tuberculosis: essential role of IL-1 from infected monocytes in a NF-kappa B-dependent network. J. Immunol. 163:3936–3947 [PubMed] [Google Scholar]
- 48. Obert S, O'Connor RJ, Schmid S, Hearing P. 1994. The adenovirus E4–6/7 protein transactivates the E2 promoter by inducing dimerization of a heteromeric E2F complex. Mol. Cell. Biol. 14:1333–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Knapp T, Hare E, Feng L, Zlokarnik G, Negulescu P. 2003. Detection of beta-lactamase reporter gene expression by flow cytometry. Cytometry A 51:68–78. 10.1002/cyto.a.10018 [DOI] [PubMed] [Google Scholar]
- 50. Gal-Mor O, Zusman T, Segal G. 2002. Analysis of DNA regulatory elements required for expression of the Legionella pneumophila icm and dot virulence genes. J. Bacteriol. 184:3823–3833. 10.1128/JB.184.14.3823-3833.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zlokarnik G, Negulescu PA, Knapp TE, Mere L, Burres N, Feng L, Whitney M, Roemer K, Tsien RY. 1998. Quantitation of transcription and clonal selection of single living cells with beta-lactamase as reporter. Science 279:84–88. 10.1126/science.279.5347.84 [DOI] [PubMed] [Google Scholar]
- 52. Nagai H, Cambronne ED, Kagan JC, Amor JC, Kahn RA, Roy CR. 2005. A C-terminal translocation signal required for Dot/Icm-dependent delivery of the Legionella RalF protein to host cells. Proc. Natl. Acad. Sci. U. S. A. 102:826–831. 10.1073/pnas.0406239101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ninio S, Roy CR. 2007. Effector proteins translocated by Legionella pneumophila: strength in numbers. Trends Microbiol. 15:372–380. 10.1016/j.tim.2007.06.006 [DOI] [PubMed] [Google Scholar]
- 54. Amer A, Franchi L, Kanneganti T-D, Body-Malapel M, Ozören N, Brady G, Meshinchi S, Jagirdar R, Gewirtz A, Akira S, Nuñez G. 2006. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 281:35217–35223. 10.1074/jbc.M604933200 [DOI] [PubMed] [Google Scholar]
- 55. Amer AO, Swanson MS. 2005. Autophagy is an immediate macrophage response to Legionella pneumophila. Cell Microbiol. 7:765–778. 10.1111/j.1462-5822.2005.00509.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Byrne BG, Dubuisson J-F, Joshi AD, Persson JJ, Swanson MS. 2013. Inflammasome components coordinate autophagy and pyroptosis as macrophage responses to infection. mBio 4:e00620–12. 10.1128/mBio.00620-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Case CL, Shin S, Roy CR. 2009. Asc and Ipaf inflammasomes direct distinct pathways for caspase-1 activation in response to Legionella pneumophila. Infect. Immun. 77:1981–1991. 10.1128/IAI.01382-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sung S-SJ, Fu SM, Rose CE, Gaskin F, Ju S-T, Beaty SR. 2006. A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J. Immunol. 176:2161–2172. 10.4049/jimmunol.176.4.2161 [DOI] [PubMed] [Google Scholar]
- 59. Stevens WW, Kim TS, Pujanauski LM, Hao X, Braciale TJ. 2007. Detection and quantitation of eosinophils in the murine respiratory tract by flow cytometry. J. Immunol. Methods 327:63–74. 10.1016/j.jim.2007.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Frutuoso MS, Hori JI, Pereira MSF, Junior DSL, Sônego F, Kobayashi KS, Flavell RA, Cunha FQ, Zamboni DS. 2010. The pattern recognition receptors Nod1 and Nod2 account for neutrophil recruitment to the lungs of mice infected with Legionella pneumophila. Microbes Infect. 12:819–827. 10.1016/j.micinf.2010.05.006 [DOI] [PubMed] [Google Scholar]
- 61. Fleming TJ, Fleming ML, Malek TR. 1993. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J. Immunol. 151:2399–2408 [PubMed] [Google Scholar]
- 62. Sunderkötter C, Nikolic T, Dillon MJ, van Rooijen N, Stehling M, Drevets DA, Leenen PJM. 2004. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J. Immunol. 172:4410–4417. 10.4049/jimmunol.172.7.4410 [DOI] [PubMed] [Google Scholar]
- 63. Chen C, Banga S, Mertens K, Weber MM, Gorbaslieva I, Tan Y, Luo Z-Q, Samuel JE. 2010. Large-scale identification and translocation of type IV secretion substrates by Coxiella burnetii. Proc. Natl. Acad. Sci. U. S. A. 107:21755–21760. 10.1073/pnas.1010485107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Losick VP, Isberg RR. 2006. NF-kappaB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J. Exp. Med. 203:2177–2189. 10.1084/jem.20060766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Casson CN, Shin S. 2013. Inflammasome-mediated cell death in response to bacterial pathogens that access the host cell cytosol: lessons from legionella pneumophila. Front. Cell Infect. Microbiol. 3:111. 10.3389/fcimb.2013.00111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. von Moltke J, Ayres JS, Kofoed EM, Chavarría-Smith J, Vance RE. 2013. Recognition of bacteria by inflammasomes. Annu. Rev. Immunol. 31:73–106. 10.1146/annurev-immunol-032712-095944 [DOI] [PubMed] [Google Scholar]
- 67. Maldonado-Arocho FJ, Green C, Fisher ML, Paczosa MK, Mecsas J. 2013. Adhesins and host serum factors drive Yop translocation by Yersinia into professional phagocytes during animal infection. PLoS Pathog. 9:e1003415. 10.1371/journal.ppat.1003415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Harmon DE, Davis AJ, Castillo C, Mecsas J. 2010. Identification and characterization of small-molecule inhibitors of Yop translocation in Yersinia pseudotuberculosis. Antimicrob. Agents Chemother. 54:3241–3254. 10.1128/AAC.00364-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pan NJ, Brady MJ, Leong JM, Goguen JD. 2009. Targeting type III secretion in Yersinia pestis. Antimicrob. Agents Chemother. 53:385–392. 10.1128/AAC.00670-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pechous RD, Sivaraman V, Price PA, Stasulli NM, Goldman WE. 2013. Early host cell targets of Yersinia pestis during primary pneumonic plague. PLoS Pathog. 9:e1003679. 10.1371/journal.ppat.1003679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Köberle M, Klein-Günther A, Schütz M, Fritz M, Berchtold S, Tolosa E, Autenrieth IB, Bohn E. 2009. Yersinia enterocolitica targets cells of the innate and adaptive immune system by injection of Yops in a mouse infection model. PLoS Pathog. 5:e1000551. 10.1371/journal.ppat.1000551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yoon H, Ansong C, Adkins JN, Heffron F. 2011. Discovery of Salmonella virulence factors translocated via outer membrane vesicles to murine macrophages. Infect. Immun. 79:2182–2192. 10.1128/IAI.01277-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Geddes K, Cruz F, Heffron F. 2007. Analysis of cells targeted by Salmonella type III secretion in vivo. PLoS Pathog. 3:e196. 10.1371/journal.ppat.0030196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Diaz MH, Hauser AR. 2010. Pseudomonas aeruginosa cytotoxin ExoU is injected into phagocytic cells during acute pneumonia. Infect. Immun. 78:1447–1456. 10.1128/IAI.01134-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kung VL, Khare S, Stehlik C, Bacon EM, Hughes AJ, Hauser AR. 2012. An rhs gene of Pseudomonas aeruginosa encodes a virulence protein that activates the inflammasome. Proc. Natl. Acad. Sci. U. S. A. 109:1275–1280. 10.1073/pnas.1109285109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Brieland J, Freeman P, Kunkel R, Chrisp C, Hurley M, Fantone J, Engleberg C. 1994. Replicative Legionella pneumophila lung infection in intratracheally inoculated A/J mice. A murine model of human Legionnaires' disease. Am. J. Pathol. 145:1537–1546 [PMC free article] [PubMed] [Google Scholar]
- 77. Winn WC, Glavin FL, Perl DP, Keller JL, Andres TL, Brown TM, Coffin CM, Sensecqua JE, Roman LN, Craighead JE. 1978. The pathology of Legionnaires' disease. Fourteen fatal cases from the 1977 outbreak in Vermont. Arch. Pathol. Lab. Med. 102:344–350 [PubMed] [Google Scholar]
- 78. Winn WC, Myerowitz RL. 1981. The pathology of the Legionella pneumonias. A review of 74 cases and the literature. Hum. Pathol. 12:401–422 [DOI] [PubMed] [Google Scholar]
- 79. Ang DKY, Ong SY, Brown AS, Hartland EL, van Driel IR. 2012. A method for quantifying pulmonary Legionella pneumophila infection in mouse lungs by flow cytometry. BMC Res. Notes 5:448. 10.1186/1756-0500-5-448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Berrington WR, Iyer R, Wells RD, Smith KD, Skerrett SJ, Hawn TR. 2010. NOD1 and NOD2 regulation of pulmonary innate immunity to Legionella pneumophila. Eur. J. Immunol. 40:3519–3527. 10.1002/eji.201040518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Weinbaum DL, Bailey J, Benner RR, Pasculle AW, Dowling JN. 1983. The contribution of human neutrophils and serum to host defense against Legionella micdadei. J. Infect. Dis. 148:510–517. 10.1093/infdis/148.3.510 [DOI] [PubMed] [Google Scholar]
- 82. Simons MP, Nauseef WM, Apicella MA. 2005. Interactions of Neisseria gonorrhoeae with adherent polymorphonuclear leukocytes. Infect. Immun. 73:1971–1977. 10.1128/IAI.73.4.1971-1977.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chen SM, Dumler JS, Bakken JS, Walker DH. 1994. Identification of a granulocytotropic Ehrlichia species as the etiologic agent of human disease. J. Clin. Microbiol. 32:589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ohashi N, Zhi N, Lin Q, Rikihisa Y. 2002. Characterization and transcriptional analysis of gene clusters for a type IV secretion machinery in human granulocytic and monocytic ehrlichiosis agents. Infect. Immun. 70:2128–2138. 10.1128/IAI.70.4.2128-2138.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nazareth H, Genagon SA, Russo TA. 2007. Extraintestinal pathogenic Escherichia coli survives within neutrophils. Infect. Immun. 75:2776–2785. 10.1128/IAI.01095-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kirby AC, Coles MC, Kaye PM. 2009. Alveolar macrophages transport pathogens to lung draining lymph nodes. J. Immunol. 183:1983–1989. 10.4049/jimmunol.0901089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nogueira CV, Lindsten T, Jamieson AM, Case CL, Shin S, Thompson CB, Roy CR. 2009. Rapid pathogen-induced apoptosis: a mechanism used by dendritic cells to limit intracellular replication of Legionella pneumophila. PLoS Pathog. 5:e1000478. 10.1371/journal.ppat.1000478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Charpentier X, Gabay JE, Reyes M, Zhu JW, Weiss A, Shuman HA. 2009. Chemical genetics reveals bacterial and host cell functions critical for type IV effector translocation by Legionella pneumophila. PLoS Pathog. 5:e1000501. 10.1371/journal.ppat.1000501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bennouna S, Bliss SK, Curiel TJ, Denkers EY. 2003. Cross-talk in the innate immune system: neutrophils instruct recruitment and activation of dendritic cells during microbial infection. J. Immunol. 171:6052–6058. 10.4049/jimmunol.171.11.6052 [DOI] [PubMed] [Google Scholar]
- 90. Tsuda Y, Takahashi H, Kobayashi M, Hanafusa T, Herndon DN, Suzuki F. 2004. Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant Staphylococcus aureus. Immunity 21:215–226. 10.1016/j.immuni.2004.07.006 [DOI] [PubMed] [Google Scholar]
- 91. Guma M, Ronacher L, Liu-Bryan R, Takai S, Karin M, Corr M. 2009. Caspase 1-independent activation of interleukin-1beta in neutrophil-predominant inflammation. Arthritis Rheum. 60:3642–3650. 10.1002/art.24959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Karmakar M, Sun Y, Hise AG, Rietsch A, Pearlman E. 2012. Cutting edge: IL-1β processing during Pseudomonas aeruginosa infection is mediated by neutrophil serine proteases and is independent of NLRC4 and caspase-1. J. Immunol. 189:4231–4235. 10.4049/jimmunol.1201447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, White MR, Dinarello CA, Apte RN. 2011. IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J. Immunol. 187:4835–4843. 10.4049/jimmunol.1102048 [DOI] [PubMed] [Google Scholar]
- 94. Spörri R, Joller N, Hilbi H, Oxenius A. 2008. A novel role for neutrophils as critical activators of NK cells. J. Immunol. 181:7121–7130. 10.4049/jimmunol.181.10.7121 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.