

Abstract
Toll-like receptors (TLRs) play a key role in the innate immune responses to periodontal pathogens in periodontal disease. The present study was performed to determine the roles of TLR2 and TLR4 signaling in alveolar bone resorption, using a Porphyromonas gingivalis-associated ligature-induced periodontitis model in mice. Wild-type (WT), Tlr2−/−, and Tlr4−/− mice (8 to 10 weeks old) in the C57/BL6 background were used. Silk ligatures were applied to the maxillary second molars in the presence or absence of live P. gingivalis infection. Ligatures were removed from the second molars on day 14, and mice were kept for another 2 weeks before sacrifice for final analysis (day 28). On day 14, there were no differences in alveolar bone resorption and gingival RANKL expression between mice treated with ligation plus P. gingivalis infection and mice treated with ligation alone. Gingival interleukin-1β (IL-1β) and tumor necrosis factor alpha (TNF-α) expression was increased, whereas IL-10 expression was decreased in WT and Tlr2−/− mice but not in Tlr4−/− mice. On day 28, WT and Tlr4−/− mice treated with ligation plus P. gingivalis infection showed significantly increased bone loss and gingival RANKL expression compared to those treated with ligation alone, whereas such an increase was diminished in Tlr2−/− mice. Gingival TNF-α upregulation and IL-10 downregulation were observed only in WT and Tlr4−/− mice, not in Tlr2−/− mice. In all mice, bone resorption induced by ligation plus P. gingivalis infection was antagonized by local anti-RANKL antibody administration. This study suggests that P. gingivalis exacerbates ligature-induced, RANKL-dependent periodontal bone resorption via differential regulation of TLR2 and TLR4 signaling.
INTRODUCTION
Periodontal disease is one of the most common chronic diseases in humans (1) and is associated with microbial pathogens that reside in the oral cavity. Most compelling evidence now indicates that periodontal disease is not a conventional infectious disease but is an inflammatory disease (2, 3) triggered by the host immune response to the dysbiotic microbiota (4, 5).
Innate immunity is considered to act as a sentinel for the immune system and is promptly activated after recognition of the diverse repertoire of microbial pathogens. Innate immune cells express various pattern recognition receptors (PRRs), which recognize signature molecules of microorganisms (6). Toll-like receptors (TLRs) are a well-characterized class of PRRs in mammalian species (7, 8). In addition to their regulation of innate immunity, a subset of TLR-induced signals is dedicated to the control of adaptive immunity (9). Bacteria express various pathogen-associated molecular patterns (PAMPs) that can be detected by TLRs (10). Two members of the TLR family, TLR2 and TLR4, have been identified as the principal signaling receptors for bacterial cell wall components. TLR2 recognizes a wide variety of PAMPs, such as lipoproteins and peptidoglycans from both Gram-positive and Gram-negative bacteria, as well as lipoteichoic acid from Gram-positive bacteria (11). TLR4 recognizes lipopolysaccharide (LPS) from Gram-negative bacteria (12).
Porphyromonas gingivalis is a Gram-negative anaerobic bacterium implicated as a major periodontal pathogen (13, 14). Recent studies demonstrated that P. gingivalis orchestrates inflammation through manipulation of host immunity and community-wide effects on the periodontal microbiota and that periodontal disease is initiated by polymicrobial synergy and dysbiosis (15, 16). Surface components of P. gingivalis, such as LPS, lipoproteins, and fimbriae, interact with TLR2 and TLR4 expressed by host cells and stimulate production of proinflammatory cytokines (17), such as interleukin-1β (IL-1β) and tumor necrosis factor alpha (TNF-α), and anti-inflammatory cytokines (18, 19), such as IL-10, as well as production of receptor activator of NF-κB ligand (RANKL), an osteoclast differentiation factor that promotes osteoclastogenesis (20). The elevated osteoclastogenic activity shifts the balance of bone metabolism toward catabolism, and ultimately leads to the induction of alveolar bone resorption. Previous studies have shown that TLR2 is required for the innate response to P. gingivalis both in vitro (21) and in vivo (22, 23). Most recently, a TLR2- and TNF-dependent, macrophage-specific mechanism was identified as underlying P. gingivalis-induced inflammatory bone loss in vivo (24). The roles of TLR4 signaling in P. gingivalis-induced immune responses have also been implicated in innate immunity, dendritic cell activation, and atherosclerosis progression (25–28).
The purpose of the present study was to determine the roles of TLR2 and TLR4 signaling in P. gingivalis-associated ligature-induced experimental periodontal bone resorption in mice in vivo. Our results indicate that P. gingivalis exacerbates ligature-induced periodontal bone resorption via differential regulation of both TLR2 and TLR4 signaling and that this is RANKL dependent.
MATERIALS AND METHODS
Mice.
Wild-type (WT) C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Tlr2−/− and Tlr4−/− mice derived from the C57BL/6 background were a kind gift from Toshihisa Kawai (Forsyth Institute, Cambridge, MA). All mice used in the study (8 to 10 weeks old) were maintained in specific-pathogen-free (SPF) units of the Forsyth Institute Animal Facility. The mice were kept on a 12-h light-dark cycle. The experimental protocols were approved by the Institutional Animal Care and Use Committee of the Forsyth Institute.
Bacterial culture.
P. gingivalis (strain ATCC 33277) was grown on anaerobic blood agar plates (NHK agar; Northeast Laboratory Services, Waterville, ME) in an anaerobic chamber with 85% N2, 5% H2, and 10% CO2. A single colony of P. gingivalis was isolated from the plate and grown in Trypticase soy broth (BD Biosciences, San Diego, CA) containing 1% yeast extract, 5 μg/ml hemin, and 2.5 μg/ml menadione as previously described (29). Bacterial numbers in culture broth were determined by reading absorbance values, using a spectrophotometer, and comparing them to a curve derived from a standard plate count. After incubation at 37°C for 5 days, the bacteria were harvested and washed three times with sterile phosphate-buffered saline (PBS). P. gingivalis was resuspended at 1 × 1010 cells/ml in PBS and mixed thoroughly with an equal volume of sterile 2% (wt/vol) low-viscosity carboxymethyl cellulose (CMC) as the vehicle.
Animal model.
Mice of each strain (wild type, Tlr2−/−, and Tlr4−/−) were randomly divided into 3 groups: (i) control untreated group (n = 10), (ii) group receiving ligation alone (n = 10), and (iii) group receiving ligation plus P. gingivalis infection (n = 18). Control groups were treated with vehicle alone. For the ligation-only group, vehicle-soaked ligatures (USP size 5-0) were placed subgingivally around the maxillary second molars on day 0 and were retained for 2 weeks. For the ligation-P. gingivalis infection group, P. gingivalis-soaked ligatures were placed subgingivally around the maxillary second molars on day 0 and were retained for 2 weeks. In addition, 10 μl of P. gingivalis (1 × 1010/ml) in PBS mixed with CMC was applied to the sulci around the maxillary second molars on days 1 and 2 to enhance the P. gingivalis colonization. Two weeks after treatment, five mice (n = 5) from each group were sacrificed, and tissues were collected for analysis of cytokine production and bone loss. Ligatures were removed from the remaining animals in group 2 (n = 5) and group 3 (n = 13), and mice were kept for another 2 weeks before termination. During this 2-week period, some mice from group 3 were also injected with either rabbit anti-mouse RANKL IgG (n = 8) or control normal rabbit IgG (n = 8) in interdental papilla (1 μg/site) on both the buccal and lingual sides, on days 15, 17, and 21. Both anti-RANKL IgG and normal rabbit IgG were obtained from Peprotech (Rocky Hill, NJ). All the remaining animals were sacrificed and tissues collected for analysis on day 28.
Tissue collection and sample preparation.
Animals were euthanized by CO2 inhalation, and the maxillae were removed from each group (n = 5) after 2 or 4 weeks. First, gingival tissues were isolated under a surgical microscope for homogenate preparation. The maxilla were then defleshed by a dermestid beetle colony, bleached, and stained with a 1% toluidine blue solution for bone resorption measurement as previously described (30). Additional samples from group 3 were analyzed for cytokine production and bone resorption after injection with either anti-RANKL IgG (n = 5) or normal IgG (n = 5). Formation of osteoclasts along alveolar bone surfaces was determined in separate samples from group 3 after injection with either anti-RANKL IgG (n = 3) or normal IgG (n = 3). Briefly, maxillary samples were fixed in 4% paraformaldehyde for 3 days at 4°C and decalcified in 10% EDTA in 0.1 M Tris buffer at 4°C for 12 days. Sagittal paraffin sections (20 sections per animal) were prepared with a 5-μm thickness, and the interproximal areas between the maxillary molars were first examined by hematoxylin and eosin (HE) staining. The sections were then subjected to specific staining for the identification of osteoclasts.
Measurements of bone resorption.
Images were captured under a digital stereomicroscope system on a custom-made stage holder to facilitate visualization of the cementoenamel junction (CEJ) and alveolar bone level. The polygonal area was measured using ImageJ (NIH) on buccal and palatal surfaces for each segment, and a standard calibrator was used for calibration at the same magnification. The bone resorption area was enclosed coronally by the CEJ of the molars, laterally by the exposed distal root of the first molar and the exposed mesial root of the third molar, and apically by the alveolar crest. For indicated experiments, the bone resorption outcomes were further verified by scanning with a Scanco μCT 40 machine (Scanco USA, Inc., Wayne, PA) and by quantitative three-dimensional (3D) analysis using Mimics software (Materialise, Plymouth, MI). The alveolar bone region of interest (ROI) was selected by setting the furcation of the second molar as the center of sphere, including the distal root of the first molar and the mesial root of the third molar. The designated ROI was analyzed for differences in cancellous bone parameters. Four criteria were used to analyze the level of bone resorption: remaining bone volume (mm3), bone volume density (%), trabecular thickness (mm), and trabecular number (1/mm). All the bone resorption measurements were performed without prior knowledge of the group designation of the mice, and the recordings were verified by a second examiner.
Reverse transcription and quantitative real-time PCR.
Total RNA was extracted from gingival tissues by use of TRIzol reagent (Sigma) and was reverse transcribed using a SuperScript II reverse transcriptase kit (Invitrogen). Predesigned primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), RANKL, osteoprotegerin (OPG), IL-1β, IL-10, and TNF-α were purchased from Sigma. Quantitative PCR was conducted using LightCycler SYBR green I master solution and a LightCycler 480 system (Roche), and the cycling conditions were set up according to the manufacturer's instructions. Oral swabs from each animal were resolved in 200 μl PBS containing a protease inhibitor cocktail (Roche), and P. gingivalis bacterial colonization in the mouse oral cavity at the termination of the experiment (day 28) was confirmed by real-time PCR using P. gingivalis 16S rRNA gene-specific primers. A universal 16S rRNA gene primer was used to quantitate the total number of bacteria recovered from each animal. The quantity of P. gingivalis DNA or total bacterial DNA from each sample was extrapolated from the DNA standard curve derived from serial dilutions of bacteria with known concentrations. P. gingivalis numbers were normalized to the total bacterial count in each sample.
ELISA.
Collected gingival tissues were homogenized with a Dounce glass homogenizer in RIPA buffer and proteinase inhibitor cocktail (Sigma). The RANKL levels in gingival homogenates were detected using a murine RANKL enzyme-linked immunosorbent assay (ELISA) development kit (Peprotech).
Statistical analysis.
All quantitative data are expressed as means ± standard errors (SE). The effects of treatments on periodontal bone resorption, cytokine gene expression assessed by PCR, RANKL production assessed by ELISA, and multinucleated TRAP+ cell formation were evaluated by the Student-Newman-Keuls (SNK) multiple-comparison test following one-way analysis of variance (ANOVA). P values of <0.05 were considered statistically significant.
RESULTS
Alveolar bone resorption and gingival RANKL production on day 14.
The levels of alveolar bone resorption and gingival RANKL production in mice were first analyzed at an early stage, on day 14 after initial treatments. Compared to that of the control group, ligation alone induced significant bone resorption in WT, Tlr2−/−, and Tlr4−/− mice (P < 0.001), suggesting that ligation-induced bone loss is independent of TLR2 and TLR4 (Fig. 1A). No significant difference was observed in the level of bone resorption between the ligation-only group and the ligation-P. gingivalis infection group for all three types of mice (Fig. 1A). Compared to those of the control group, the gingival RANKL concentrations were significantly increased after ligation (P < 0.01), but no difference was observed between mice treated with ligation alone and those treated with ligation plus P. gingivalis infection (Fig. 1B). These results indicated that compared to ligation alone, P. gingivalis infection did not result in additional bone resorption or RANKL production at an early stage of the inflammatory response (2 weeks).
FIG 1.
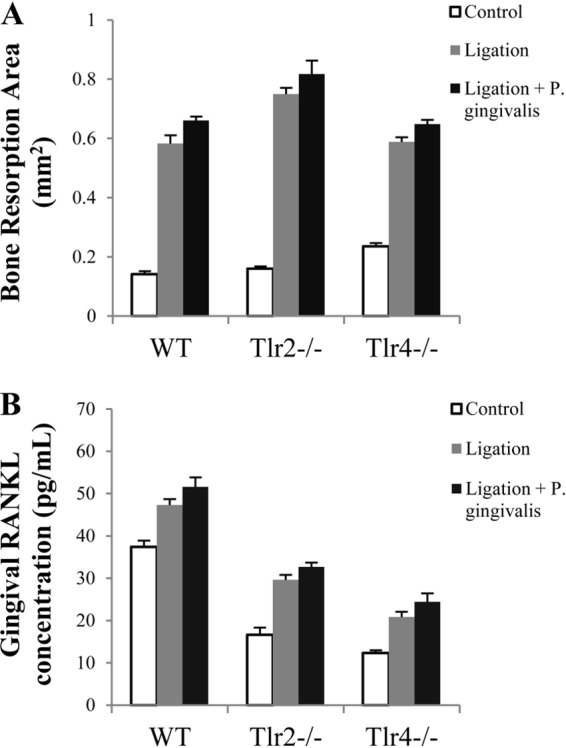
Alveolar bone resorption and gingival RANKL concentrations following 2 weeks of treatment in WT, Tlr2−/−, and Tlr4−/− mice. (A) Alveolar bone resorption around maxillary second molars (left and right), induced by ligation or ligation plus P. gingivalis infection, was analyzed after 2 weeks of treatment of WT, Tlr2−/−, and Tlr4−/− mice and is presented as the area of bone loss (mm2). (B) Total protein was isolated from gingival tissues, and the RANKL concentration was analyzed by ELISA using a specific antibody. The nontreatment groups were used as controls. Data are means and SE (n = 10).
Gingival cytokine expression on day 14.
We analyzed the cytokine expression (IL-1β, TNF-α, and IL-10) in gingival tissues 2 weeks following treatment for WT, Tlr2−/−, and Tlr4−/− mice. Compared to that in controls, ligation significantly increased gingival IL-1β and TNF-α expression but greatly reduced gingival IL-10 expression in all three types of mice, in a TLR-independent manner (Fig. 2A to C). Compared to treatment with ligation alone, P. gingivalis infection in addition to ligation induced higher levels of IL-1β and TNF-α mRNAs (Fig. 2A and B) and lower levels of IL-10 mRNA (Fig. 2C) in the gingival tissues from WT and Tlr2−/− mice but not in those from Tlr4−/− mice. These results suggest that P. gingivalis enhances ligation-induced upregulation of IL-1β and TNF-α and downregulation of IL-10 via TLR4, but not TLR2, at an early stage of the inflammatory response (2 weeks).
FIG 2.
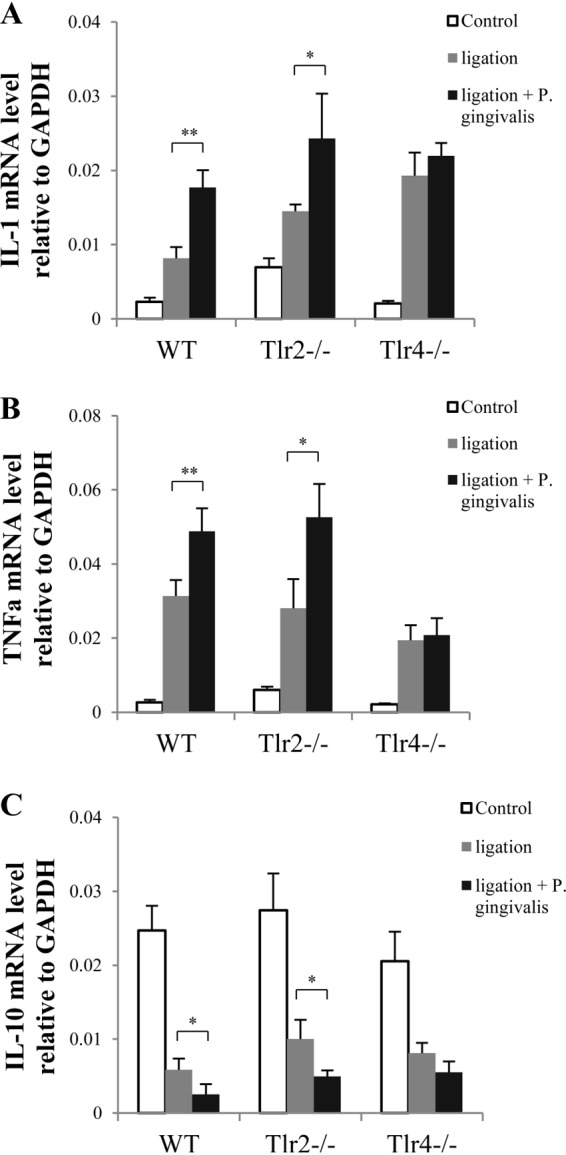
Inflammatory cytokine production from gingival tissues of WT, Tlr2−/−, and Tlr4−/− mice following 2 weeks of treatment. Two weeks after mice were treated with ligation alone or ligation plus P. gingivalis infection, total RNA was isolated from gingival tissues around maxillary second molars (left and right) and was analyzed by real-time PCR using gene-specific primers to detect IL-1β mRNA expression (A), TNF-α mRNA expression (B), and IL-10 mRNA expression (C). Amplification of the GAPDH gene was used as a control. Data are means and SE (n = 10). *, P < 0.05; **, P < 0.01.
Alveolar bone resorption and gingival RANKL production on day 28.
Two weeks after the initial treatments, ligatures were removed from the maxillary second molars, and animals were kept for another 2 weeks to determine the effect of continued P. gingivalis infection on bone resorption independent of ligation. At the 4-week time point (day 28), WT and Tlr4−/− mice treated with ligation plus P. gingivalis infection showed significantly larger bone resorption areas than those treated with ligation alone (Fig. 3A). However, there was no such difference observed between the two groups of Tlr2−/− mice (Fig. 3A). To further verify the observed difference in bone resorption between mice treated with ligation plus P. gingivalis and those treated with ligation alone, we performed 3D quantitative bone morphometric analysis using micro-computed tomography (micro-CT). The results consistently showed the difference in bone resorption between mice treated with ligation plus P. gingivalis and those treated with ligation alone (Fig. 3B to E). These data demonstrated that the presence of P. gingivalis infection significantly aggravated alveolar bone loss after ligature removal in WT and Tlr4−/− mice but not in Tlr2−/− mice. On day 28, WT and Tlr4−/− mice, but not Tlr2−/− mice, showed significantly increased gingival RANKL production for treatment with ligation plus P. gingivalis infection compared to treatment with ligation alone (Fig. 3F). These results suggested that the level of RANKL production was positively correlated with the level of bone resorption in all three types of mice treated with ligation plus P. gingivalis infection.
FIG 3.

Bone resorption and gingival RANKL concentrations after ligature removal. Two weeks after treatment by ligation or ligation plus P. gingivalis infection, ligatures were removed from mice to allow wound healing for another 2 weeks. (A) The alveolar bone resorption around maxillary second molars (left and right) in WT, Tlr2−/−, and Tlr4−/− mice was measured and is presented as the area of bone loss (mm2). To further verify the observed difference in bone resorption between mice treated with ligation plus P. gingivalis and those treated with ligation alone, 3D bone assessment was performed using micro-CT to determine the remaining bone volume (mm3) (B), the bone volume density (% of bone surface/bone volume) (C), the trabecular thickness of remaining bone (mm) (D), and the trabecular number of remaining bone (1/mm) (E). (F) Total protein was isolated from gingival tissues, and RANKL production in gingival tissues was analyzed by ELISA using a specific antibody. Data are means and SE (n = 10). *, P < 0.05; **, P < 0.01.
Gingival cytokine expression on day 28.
To determine the effects of P. gingivalis infection on pro- and anti-inflammatory cytokine expression after removal of ligatures, we assessed the mRNA levels of IL-1β, TNF-α, and IL-10 in the gingival tissues of WT, Tlr2−/−, and Tlr4−/− mice 2 weeks after ligature removal (day 28). At this time point, all three types of mice showed increased expression of IL-1β when treated with ligation plus P. gingivalis infection compared to ligation alone (Fig. 4A). Increased gingival TNF-α expression (Fig. 4B) and reduced gingival IL-10 expression (Fig. 4C) could be detected only in WT and Tlr4−/− mice, not in Tlr2−/− mice. These results suggest that in the absence of ligation, P. gingivalis infection upregulates gingival TNF-α expression and downregulates gingival IL-10 expression, in a TLR2-dependent manner.
FIG 4.
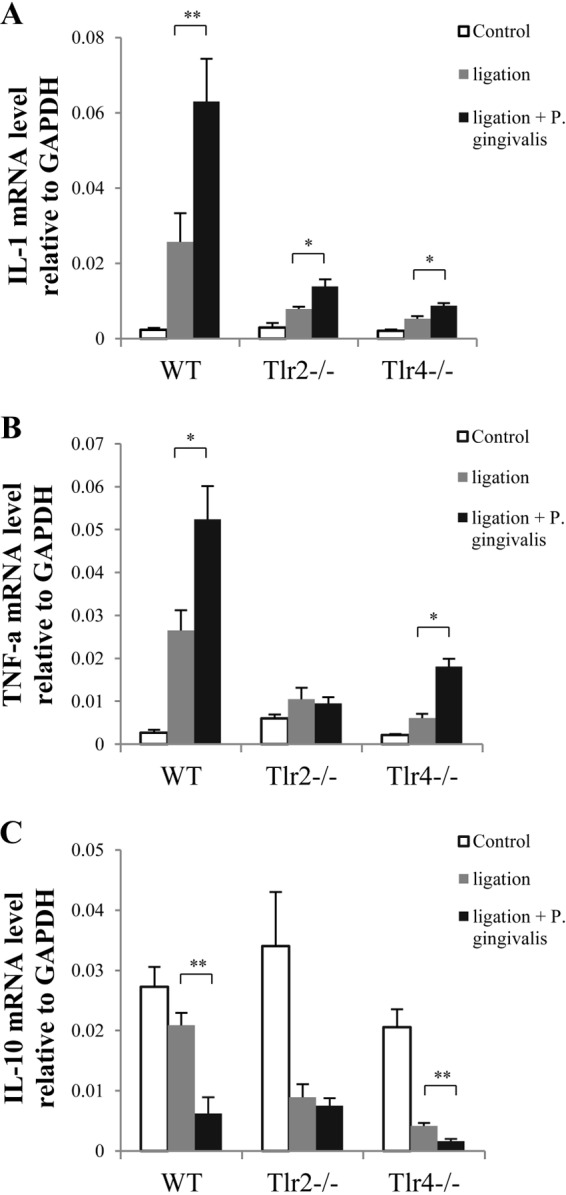
Inflammatory cytokine production from gingival tissues of WT, Tlr2−/−, and Tlr4−/− mice after ligature removal. Two weeks after treatment by ligation or ligation plus P. gingivalis infection, ligatures were removed from mice to allow wound healing for another 2 weeks. Total RNA was isolated from gingival tissues around maxillary second molars (left and right) and was analyzed by real-time PCR using gene-specific primers to detect IL-1β mRNA (A), TNF-α mRNA (B), and IL-10 mRNA (C) expression. Amplification of the GAPDH gene was used as a control. Data are means and SE (n = 10). *, P < 0.05; **, P < 0.01.
Effects of local anti-RANKL antibody administration on alveolar bone resorption and gingival RANKL production.
We determined the effect of gingival administration of anti-RANKL antibody on alveolar bone resorption on day 28. After treatment with ligation plus P. gingivalis infection, mice of all three types injected with anti-RANKL antibody demonstrated significantly reduced alveolar bone resorption compared to the control group. Such an effect was not observed in mice injected locally with normal IgG (Fig. 5A) after treatment with ligation plus P. gingivalis infection. Gingival RANKL production was significantly decreased in the anti-RANKL antibody-treated group compared to the control group. No difference was observed in the level of gingival RANKL production between the control group and the normal IgG-treated group (Fig. 5B). To evaluate the ratio of RANKL to OPG expression, we simultaneously detected the mRNA levels of RANKL and OPG in gingival tissues by real-time PCR. The results demonstrated that regardless of the type of mice tested, the gingival RANKL/OPG expression ratio was significantly reduced in mice treated with anti-RANKL antibody compared to those in the control group (Fig. 5C).
FIG 5.
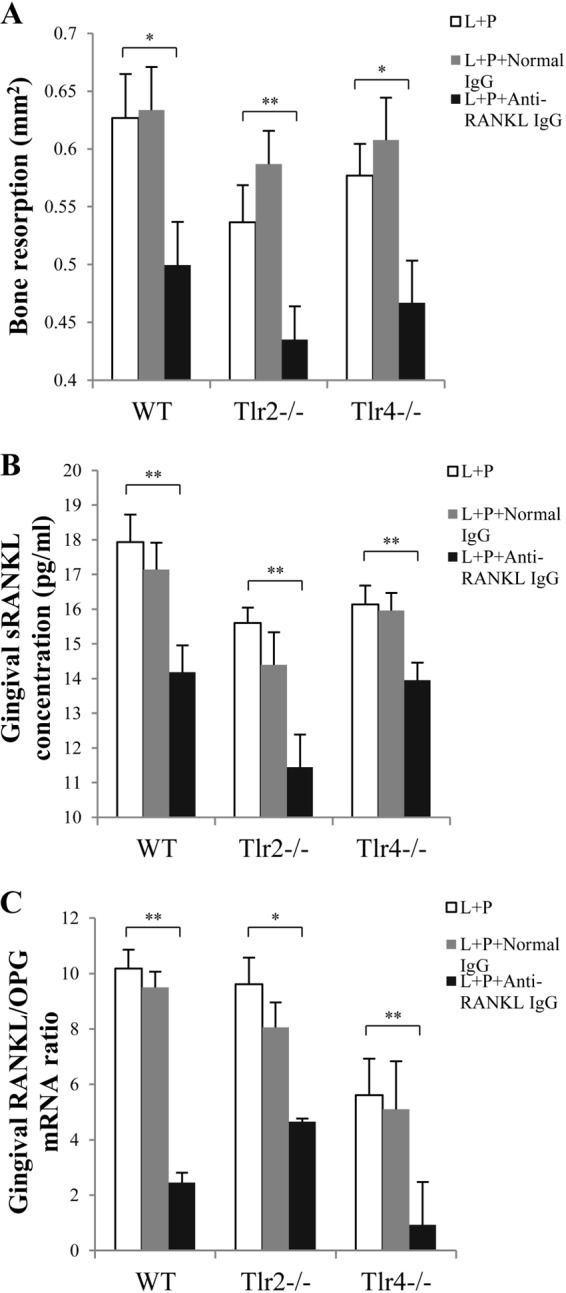
Bone resorption and gingival RANKL and OPG expression after RANKL blockade in WT, Tlr2−/−, and Tlr4−/− mice. Two weeks after treatment by ligation or ligation plus P. gingivalis infection, ligatures were removed from mice to allow wound healing for another 2 weeks. During this 2-week period, mice were injected with either rabbit anti-mouse RANKL IgG or normal rabbit IgG in the interdental papilla (1 μg/site) on both the buccal and lingual sides of the maxillary second molars (left and right), on days 15, 17, and 21. (A) At the termination of the experiment (day 28), the alveolar bone resorption around maxillary second molars (left and right) in WT, Tlr2−/−, and Tlr4−/− mice was measured and is presented as the area of bone loss (mm2). (B) Total protein was isolated from gingival tissues, and gingival RANKL concentrations were measured by ELISA. (C) Total RNA was isolated from gingival tissues, and RANKL mRNA expression and OPG mRNA expression in gingival tissues were detected by real-time PCR. The RANKL/OPG ratio was then calculated. Amplification of the GAPDH gene was used as a control. L, ligation; P, P. gingivalis infection. Data are means and SE (n = 10). *, P < 0.05; **, P < 0.01.
P. gingivalis colonization and animal weight monitoring.
Four weeks after initial treatments, P. gingivalis colonization in gingival tissues was verified by real-time PCR (see Fig. S1 in the supplemental material). Similar bacterial counts were detected in WT, Tlr2−/−, and Tlr4−/− mice treated with ligation plus P. gingivalis infection, indicating that the subsequent colonization of P. gingivalis bacteria in the gingival sulcus after initial infection (with P. gingivalis-soaked ligatures) was TLR independent. To ensure that animals were not chronically stressed due to the treatments, we also monitored animal weight throughout the 4-week period, and the results demonstrated similar patterns of stress recovery for all three types of mice used. The weights of the mice decreased initially after treatments, reached the maximum loss on day 3 (less than 15%), gradually returned to the original level by day 21, and were slightly increased at the termination of the experiment, on day 28 (see Fig. S2). This indicated that the treatments (ligation and P. gingivalis infection) did not generate any irreversible stress and metabolic variations among WT, Tlr2−/−, and Tlr4−/− mice.
DISCUSSION
Periodontal disease is an inflammatory disease that leads to alveolar bone loss and may exert an adverse impact on systemic health. It is widely accepted that host responses to periodontal pathogens play key roles in the onset and progression of periodontal diseases. Although host immune responses facilitate a protective mechanism against pathogenic colonization and infection, both hypo- and hyperimmune responses can result in pathogenesis. The host innate immune response to pathogens is largely mediated via TLR signaling (31).
In this study, we investigated which TLR exerts the effects of P. gingivalis-associated osteoclastogenesis by using a mouse model of ligature-induced periodontal bone resorption. We note that in this animal model, P. gingivalis functions in accordance with the rest of the microbiota. The ligature facilitates biofilm accumulation (including that of P. gingivalis and other indigenous bacteria) and thereby disrupts tissue homeostasis and promotes inflammatory bone loss. Thus, P. gingivalis acts as a keystone pathogen (15) in this mouse model, creating a dysbiosis between the host and dental biofilm, an altered oral commensal microbiota, which is responsible for the initiation of pathological bone loss.
An important advantage of using the ligature-induced periodontitis model in mice is that disease can be initiated at a set time, with a predictable sequence of alveolar bone resorption in a relatively short time (32). Our data showed that P. gingivalis infection did not enhance the bone resorption induced by ligation on day 14 for all three types of mice (Fig. 1A). However, significant increases in bone loss were observed in WT and Tlr4−/− mice, but not Tlr2−/− mice, in the presence of P. gingivalis infection on day 28 (Fig. 3A). Since we observed that ligation induced a greater level of bone resorption (3- to 4-fold higher) than that induced by P. gingivalis infection in a relatively short period, it is reasonable to postulate that P. gingivalis-induced, TLR2-mediated alveolar bone resorption was masked at the early stage of the inflammatory response. Experimental animal models are critical tools for investigating mechanisms of periodontal pathogenesis and testing new therapeutic approaches. Our findings suggest that when bacterial infection is used in combination with ligation, the contribution of bacteria to bone resorption at an early stage should be interpreted with caution. In our model, the removal of ligatures after 2 weeks (day 14) could be advantageous to study the independent effect of sustained P. gingivalis infection when ligation is no longer present.
Studies have shown that the TLR2 signaling pathway is indispensable for P. gingivalis infection-associated bone resorption in mice and that TLR2 deficiency attenuates induced alveolar bone resorption (22). The results of this study are consistent with these prior studies demonstrating that TLR2 signaling mediates P. gingivalis-induced alveolar bone loss. Studies have shown that P. gingivalis can produce unique sphingolipids that promote RANKL expression from osteoblast cells via ligation to TLR2 (33). In contrast to the LPS produced by a majority of Gram-negative bacteria, which binds to TLR4 (34), P. gingivalis LPS is known to activate TLR2 (35, 36). This may be derived partly from the chemical structure of P. gingivalis lipid A, the bioactive moiety of LPS, which differs remarkably from lipid A produced by enterobacteria, such as Escherichia coli (37).
However, P. gingivalis microbial products may have stimulatory effects on different immune cells through different TLRs. Recent studies demonstrated that the tetra- and penta-acylated lipid A structures of P. gingivalis LPS differentially activate TLR4-mediated signaling pathways (38). Studies have also shown that TLR4 signaling is required for the regulation of dendritic cell and CD4+ T cell responses to the P. gingivalis antigen hemagglutinin B (27, 39). Our results demonstrate that TLR4 but not TLR2 signaling is predominately involved in P. gingivalis infection-associated upregulation of IL-1β and TNF-α, and downregulation of IL-10, at an early stage of the inflammatory response (Fig. 2). These results suggest that TLR2/4 signaling is differentially regulated by P. gingivalis infection during different stages of periodontal bone loss. A further understanding of the underlying mechanism may have profound implications for the development of therapeutic options for periodontal disease.
Our previous rat models of periodontal disease showed similar results, i.e., the levels of RANKL mRNA and protein expression positively correlated with the level of bone resorption, and RANKL blockage by anti-RANKL antibody led to significantly decreased bone loss (40, 41). In this study, the decreased RANKL/OPG ratio in mouse gingival tissue injected with anti-RANKL antibody further substantiated the effect of RANKL blockage in the bone resorption process (Fig. 5C). Taken together, these data indicate that P. gingivalis-associated ligature-induced bone loss is RANKL dependent.
The extensive release of TLR-triggered inflammatory mediators may harm the host by accelerating inflammation and activating bone and tissue destruction, as seen in periodontal disease (7, 8, 42). Our study demonstrated that 2 weeks after removal of ligatures (day 28), TNF-α was significantly higher only in WT and Tlr4−/− mice, not Tlr2−/− mice, treated with ligation plus P. gingivalis infection compared to those treated with ligation alone (Fig. 4B). Interestingly, our results showed that anti-inflammatory IL-10 production was unexpectedly decreased in WT and Tlr4−/− mice treated with ligation plus P. gingivalis infection compared to those treated with ligation alone, while such a difference was not observed between these two groups of Tlr2−/− mice (Fig. 4C). Studies have shown that IL-10 production can be induced by TLR activation in various immune cells, such as dendritic cells, monocytes, and macrophages (43–46). Further studies are warranted to determine the temporal and spatial IL-10 regulation in P. gingivalis-associated ligature-induced periodontal bone loss. Nonetheless, these findings suggest that P. gingivalis-associated bone resorption may be associated with TLR2-mediated activation of TNF-α and inhibition of IL-10. Together with the data shown in Fig. 2, these results indicate that TLR2/4 signaling is differentially regulated for the control of cytokine production at different stages of periodontal pathogenesis.
In summary, the present study demonstrates that P. gingivalis exacerbates ligature-induced alveolar bone resorption through differential regulation of TLR2 and TLR4 signaling in a RANKL-dependent manner. Insights from the current work using this novel animal model will facilitate the design of targeted therapeutic strategies against immune-mediated periodontal bone resorption.
Supplementary Material
ACKNOWLEDGMENTS
We thank Xiaoqing Song, from the Department of Applied Oral Sciences, Forsyth Institute, for her technical assistance. We are also grateful to Jun-O Jin, from the Department of Immunology and Infectious Diseases, Forsyth Institute, for sharing his research experience by carrying out some experiments.
This work was supported by NIH grants DE-003420 and DE-021837 from the National Institute of Dental and Craniofacial Research.
We have no conflicts of interest to report.
Footnotes
Published ahead of print 21 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02084-14.
REFERENCES
- 1. Oliver RC, Brown LJ, Loe H. 1998. Periodontal diseases in the United States population. J. Periodontol. 69:269–278. 10.1902/jop.1998.69.2.269 [DOI] [PubMed] [Google Scholar]
- 2. Michalek SM, Katz J, Childers NK, Martin M, Balkovetz DF. 2002. Microbial/host interactions: mechanisms involved in host responses to microbial antigens. Immunol. Res. 26:223–234. 10.1385/IR:26:1-3:223 [DOI] [PubMed] [Google Scholar]
- 3. Taubman MA, Valverde P, Han X, Kawai T. 2005. Immune response: the key to bone resorption in periodontal disease. J. Periodontol. 76:2033–2041. 10.1902/jop.2005.76.11-S.2033 [DOI] [PubMed] [Google Scholar]
- 4. Hajishengallis G, Lamont RJ. 2014. Breaking bad: manipulation of the host response by Porphyromonas gingivalis. Eur. J. Immunol. 44:328–338. 10.1002/eji.201344202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hajishengallis G. 2014. Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends Immunol. 35:3–11. 10.1016/j.it.2013.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goutagny N, Fitzgerald KA. 2006. Pattern recognition receptors: an update. Expert Rev. Clin. Immunol. 2:569–583. 10.1586/1744666X.2.4.569 [DOI] [PubMed] [Google Scholar]
- 7. Beutler BA. 2009. TLRs and innate immunity. Blood 113:1399–1407. 10.1182/blood-2008-07-019307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kawai T, Akira S. 2011. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34:637–650. 10.1016/j.immuni.2011.05.006 [DOI] [PubMed] [Google Scholar]
- 9. Iwasaki A, Medzhitov R. 2004. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5:987–995. 10.1038/ni1112 [DOI] [PubMed] [Google Scholar]
- 10. Akira S, Hemmi H. 2003. Recognition of pathogen-associated molecular patterns by TLR family. Immunol. Lett. 85:85–95. 10.1016/S0165-2478(02)00228-6 [DOI] [PubMed] [Google Scholar]
- 11. Oliveira-Nascimento L, Massari P, Wetzler LM. 2012. The role of TLR2 in infection and immunity. Front. Immunol. 3:79. 10.3389/fimmu.2012.00079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beutler B. 2000. Tlr4: central component of the sole mammalian LPS sensor. Curr. Opin. Immunol. 12:20–26. 10.1016/S0952-7915(99)00046-1 [DOI] [PubMed] [Google Scholar]
- 13. Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr 1998. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25:134–144. 10.1111/j.1600-051X.1998.tb02419.x [DOI] [PubMed] [Google Scholar]
- 14. Socransky SS, Haffajee AD, Ximenez-Fyvie LA, Feres M, Mager D. 1999. Ecological considerations in the treatment of Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis periodontal infections. Periodontol. 2000 20:341–362. 10.1111/j.1600-0757.1999.tb00165.x [DOI] [PubMed] [Google Scholar]
- 15. Darveau RP, Hajishengallis G, Curtis MA. 2012. Porphyromonas gingivalis as a potential community activist for disease. J. Dent. Res. 91:816–820. 10.1177/0022034512453589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hajishengallis G, Lamont RJ. 2012. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol. Oral Microbiol. 27:409–419. 10.1111/j.2041-1014.2012.00663.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jung YO, Cho ML, Lee SY, Oh HJ, Park JS, Park MK, Park MJ, Ju JH, Kim SI, Park SH, Kim HY, Min JK. 2009. Synergism of Toll-like receptor 2 (TLR2), TLR4, and TLR6 ligation on the production of tumor necrosis factor (TNF)-alpha in a spontaneous arthritis animal model of interleukin (IL)-1 receptor antagonist-deficient mice. Immunol. Lett. 123:138–143. 10.1016/j.imlet.2009.03.004 [DOI] [PubMed] [Google Scholar]
- 18. Liu BS, Cao Y, Huizinga TW, Hafler DA, Toes RE. 19 March 2014. TLR-mediated STAT3 and ERK activation controls IL-10 secretion by human B cells. Eur. J. Immunol. 10.1002/eji.201344341 [DOI] [PubMed] [Google Scholar]
- 19. Qian C, Jiang X, An H, Yu Y, Guo Z, Liu S, Xu H, Cao X. 2006. TLR agonists promote ERK-mediated preferential IL-10 production of regulatory dendritic cells (diffDCs), leading to NK-cell activation. Blood 108:2307–2315. 10.1182/blood-2006-03-005595 [DOI] [PubMed] [Google Scholar]
- 20. Takahashi N, Udagawa N, Suda T. 1999. A new member of tumor necrosis factor ligand family, ODF/OPGL/TRANCE/RANKL, regulates osteoclast differentiation and function. Biochem. Biophys. Res. Commun. 256:449–455. 10.1006/bbrc.1999.0252 [DOI] [PubMed] [Google Scholar]
- 21. Zhang P, Liu J, Xu Q, Harber G, Feng X, Michalek SM, Katz J. 2011. TLR2-dependent modulation of osteoclastogenesis by Porphyromonas gingivalis through differential induction of NFATc1 and NF-kappaB. J. Biol. Chem. 286:24159–24169. 10.1074/jbc.M110.198085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burns E, Bachrach G, Shapira L, Nussbaum G. 2006. Cutting edge: TLR2 is required for the innate response to Porphyromonas gingivalis: activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J. Immunol. 177:8296–8300. 10.4049/jimmunol.177.12.8296 [DOI] [PubMed] [Google Scholar]
- 23. Ukai T, Yumoto H, Gibson FC, 3rd, Genco CA. 2008. Macrophage-elicited osteoclastogenesis in response to bacterial stimulation requires Toll-like receptor 2-dependent tumor necrosis factor-alpha production. Infect. Immun. 76:812–819. 10.1128/IAI.01241-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Papadopoulos G, Weinberg EO, Massari P, Gibson FC, 3rd, Wetzler LM, Morgan EF, Genco CA. 2013. Macrophage-specific TLR2 signaling mediates pathogen-induced TNF-dependent inflammatory oral bone loss. J. Immunol. 190:1148–1157. 10.4049/jimmunol.1202511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bainbridge BW, Coats SR, Darveau RP. 2002. Porphyromonas gingivalis lipopolysaccharide displays functionally diverse interactions with the innate host defense system. Ann. Periodontol. 7:29–37. 10.1902/annals.2002.7.1.29 [DOI] [PubMed] [Google Scholar]
- 26. Darveau RP, Pham TT, Lemley K, Reife RA, Bainbridge BW, Coats SR, Howald WN, Way SS, Hajjar AM. 2004. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both Toll-like receptors 2 and 4. Infect. Immun. 72:5041–5051. 10.1128/IAI.72.9.5041-5051.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gaddis DE, Michalek SM, Katz J. 2009. Requirement of TLR4 and CD14 in dendritic cell activation by hemagglutinin B from Porphyromonas gingivalis. Mol. Immunol. 46:2493–2504. 10.1016/j.molimm.2009.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hayashi C, Papadopoulos G, Gudino CV, Weinberg EO, Barth KR, Madrigal AG, Chen Y, Ning H, LaValley M, Gibson FC, 3rd, Hamilton JA, Genco CA. 2012. Protective role for TLR4 signaling in atherosclerosis progression as revealed by infection with a common oral pathogen. J. Immunol. 189:3681–3688. 10.4049/jimmunol.1201541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Taubman MA, Han X, Larosa KB, Socransky SS, Smith DJ. 2007. Periodontal bacterial DNA suppresses the immune response to mutans streptococcal glucosyltransferase. Infect. Immun. 75:4088–4096. 10.1128/IAI.00623-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sasaki H, White SH. 2008. Aggregation behavior of an ultra-pure lipopolysaccharide that stimulates TLR-4 receptors. Biophys. J. 95:986–993. 10.1529/biophysj.108.129197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Akira S, Takeda K. 2004. Toll-like receptor signalling. Nat. Rev. Immunol. 4:499–511. 10.1038/nri1391 [DOI] [PubMed] [Google Scholar]
- 32. Abe T, Hajishengallis G. 2013. Optimization of the ligature-induced periodontitis model in mice. J. Immunol. Methods 394:49–54. 10.1016/j.jim.2013.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang YH, Jiang J, Zhu Q, AlAnezi AZ, Clark RB, Jiang X, Rowe DW, Nichols FC. 2010. Porphyromonas gingivalis lipids inhibit osteoblastic differentiation and function. Infect. Immun. 78:3726–3735. 10.1128/IAI.00225-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Akira S. 2003. Toll-like receptor signaling. J. Biol. Chem. 278:38105–38108. 10.1074/jbc.R300028200 [DOI] [PubMed] [Google Scholar]
- 35. Bainbridge BW, Darveau RP. 2001. Porphyromonas gingivalis lipopolysaccharide: an unusual pattern recognition receptor ligand for the innate host defense system. Acta Odontol. Scand. 59:131–138. 10.1080/000163501750266710 [DOI] [PubMed] [Google Scholar]
- 36. Kikkert R, de Groot ER, Aarden LA. 2008. Cytokine induction by pyrogens: comparison of whole blood, mononuclear cells, and TLR-transfectants. J. Immunol. Methods 336:45–55. 10.1016/j.jim.2008.03.010 [DOI] [PubMed] [Google Scholar]
- 37. Ogawa T, Asai Y, Makimura Y, Tamai R. 2007. Chemical structure and immunobiological activity of Porphyromonas gingivalis lipid A. Front. Biosci. 12:3795–3812. 10.2741/2353 [DOI] [PubMed] [Google Scholar]
- 38. Herath TD, Darveau RP, Seneviratne CJ, Wang CY, Wang Y, Jin L. 2013. Tetra- and penta-acylated lipid A structures of Porphyromonas gingivalis LPS differentially activate TLR4-mediated NF-kappaB signal transduction cascade and immuno-inflammatory response in human gingival fibroblasts. PLoS One 8:e58496. 10.1371/journal.pone.0058496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gaddis DE, Michalek SM, Katz J. 2011. TLR4 signaling via MyD88 and TRIF differentially shape the CD4+ T cell response to Porphyromonas gingivalis hemagglutinin B. J. Immunol. 186:5772–5783. 10.4049/jimmunol.1003192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Han X, Lin X, Yu X, Lin J, Kawai T, LaRosa KB, Taubman MA. 2013. Porphyromonas gingivalis infection-associated periodontal bone resorption is dependent on receptor activator of NF-kappaB ligand. Infect. Immun. 81:1502–1509. 10.1128/IAI.00043-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin X, Han X, Kawai T, Taubman MA. 2011. Antibody to receptor activator of NF-kappaB ligand ameliorates T cell-mediated periodontal bone resorption. Infect. Immun. 79:911–917. 10.1128/IAI.00944-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Akira S, Uematsu S, Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124:783–801. 10.1016/j.cell.2006.02.015 [DOI] [PubMed] [Google Scholar]
- 43. Bzowska M, Nogiec A, Skrzeczynska-Moncznik J, Mickowska B, Guzik K, Pryjma J. 2012. Oxidized LDLs inhibit TLR-induced IL-10 production by monocytes: a new aspect of pathogen-accelerated atherosclerosis. Inflammation 35:1567–1584. 10.1007/s10753-012-9472-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Okazaki N, Hazeki K, Izumi T, Nigorikawa K, Hazeki O. 2011. C5a controls TLR-induced IL-10 and IL-12 production independent of phosphoinositide 3-kinase. J. Biochem. 149:265–274. 10.1093/jb/mvq136 [DOI] [PubMed] [Google Scholar]
- 45. Ma F, Liu X, Li D, Wang P, Li N, Lu L, Cao X. 2010. MicroRNA-466l upregulates IL-10 expression in TLR-triggered macrophages by antagonizing RNA-binding protein tristetraprolin-mediated IL-10 mRNA degradation. J. Immunol. 184:6053–6059. 10.4049/jimmunol.0902308 [DOI] [PubMed] [Google Scholar]
- 46. Yanagawa Y, Onoe K. 2007. Enhanced IL-10 production by TLR4- and TLR2-primed dendritic cells upon TLR restimulation. J. Immunol. 178:6173–6180. 10.4049/jimmunol.178.10.6173 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.