

Abstract
The intracellular parasite Toxoplasma gondii has multiple strategies to alter host cell function, including the injection of rhoptry proteins into the cytosol of host cells as well as bystander populations, but the consequence of these events is unclear. Here, a reporter system using fluorescent parasite strains that inject Cre recombinase with their rhoptry proteins (Toxoplasma-Cre) was combined with Ai6 Cre reporter mice to identify cells that have been productively infected, that have been rhoptry injected but lack the parasite, or that have phagocytosed T. gondii. The ability to distinguish these host-parasite interactions was then utilized to dissect the events that lead to the production of interleukin-12 p40 (IL-12p40), which is required for resistance to T. gondii. In vivo, the use of invasion-competent or invasion-inhibited (phagocytosed) parasites with IL-12p40 (YET40) reporter mice revealed that dendritic cell (DC) and macrophage populations that phagocytose the parasite or are infected can express IL-12p40 but are not the major source, as larger numbers of uninfected cells secrete this cytokine. Similarly, the use of Toxoplasma-Cre parasite strains indicated that dendritic cells and inflammatory monocytes untouched by the parasite and not cells injected by the parasite are the primary source of IL-12p40. These results imply that a soluble host or parasite factor is responsible for the bulk of IL-12p40 production in vivo, rather than cellular interactions with T. gondii that result in infection, infection and clearance, injection of rhoptry proteins, or phagocytosis of the parasite.
INTRODUCTION
Toxoplasma gondii is an obligate intracellular parasite that infects approximately 30% of the world's population (1). The development of protective immunity to this microorganism is dependent on the innate recognition of T. gondii, which leads to the production of interleukin-12 (IL-12), which is essential for NK and T cell secretion of gamma interferon (IFN-γ) (2–8). IFN-γ in turn stimulates numerous antimicrobial effector mechanisms required for the control of T. gondii (9, 10) and is involved in amplifying the production of IL-12 (11, 12). Given the key role of IL-12 in resistance to T. gondii and other pathogens, many groups have sought to define its cellular source and to understand the mechanisms that trigger its production (13). As a result, monocytes (14), neutrophils (15, 16), macrophages (3, 17), conventional dendritic cells (cDCs) (6, 18–22), and plasmacytoid DCs (23) have all been shown to produce IL-12 during toxoplasmosis. However, recent studies have highlighted the important contribution of CD8α+ DCs (21) and monocyte-derived DCs as relevant sources of IL-12 during experimental toxoplasmosis (18).
While there is an extensive literature on the multiple sources of IL-12 during toxoplasmosis, less is known about what host-microbe interactions induce the initial production of IL-12 during this infection. The in vitro incubation of macrophages/DCs with live parasites, heat-killed organisms, or soluble parasite extracts can promote IL-12, and MyD88-Toll-like receptor (TLR) signaling has been implicated in the recognition of T. gondii and synthesis of IL-12 in several studies (19, 24–27). Of relevance to understanding the events that lead to IL-12 production is the basic cell biology of how T. gondii interacts with host cells. To date, it has not been obvious how parasite-derived pathogen-associated molecular patterns (PAMPs) are detected by the host. It has been well established that as T. gondii infects cells, it forms a unique parasitophorous vacuole (PV) that is distinct from the lysosomal system (28, 29). Within this PV, at least two secreted dense granule proteins (GRA15 and GRA24) have been linked to the induction of IL-12 production (30, 31). Nod-like receptors (NLRs) have also been implicated in recognition of intracellular Toxoplasma, although the ligands involved have yet to be determined (32). In contrast, phagocytosis of this organism leads to its degradation (33) and potential recognition of known parasite PAMPs (e.g., DNA and profilin) by endosomal TLRs such as TLR9, -11, and -12. Implicit in these models is the idea that DCs and/or macrophages that phagocytose the parasite or kill intracellular parasites would be sources of IL-12. However, there is strong evidence that profilin, a parasite-derived soluble factor, can induce expression of IL-12 (27, 34), and there may be multiple pathways/mechanisms for profilin to bind to TLR11 and -12. Nevertheless, the idea that parasite lysis is required for the release of profilin (35) would suggest that endocytosis of this released, soluble profilin is a primary mechanism to drive IL-12 production in vivo.
While IL-12 is key for resistance to T. gondii, there is a literature showing that this organism can interfere with the production of this cytokine. T. gondii can inject effector proteins into the host cell cytosol that activate the host cell transcription factors STAT3 and STAT6, two transcription factors associated with inhibition of IL-12 (36–39). In addition, recent in vitro and in vivo studies that used parasites that inject Cre recombinase along with their normal cargo of rhoptry proteins (Toxoplasma-Cre parasites) combined with Ai6 mice bearing the Cre-dependent reporter (ZsGreen1 fluorescent protein) (40) revealed that the injection of rhoptry proteins into bystander cells (or multiple cells by serial injection) without invasion is one mechanism that results in a population previously designated uninfected-injected (U-I) cells (36, 41, 42). These previous studies indicated that U-I cells could also result from division of infected cells or from infected cells activated to kill and clear the intracellular parasite (41), and this study classifies all cells that are subject to these host-parasite interactions as U-I cells. The identification of U-I populations highlights the ability of these transgenic parasites to be used to better characterize the host-pathogen interactions (productive infection, phagocytosis, and U-I populations) that lead to IL-12 production. The in vitro studies presented here provide novel insights into the utility of these Toxoplasma-Cre reporter strains that express various fluorescent proteins (including mCherry, which is resistant to lysosomal degradation) to track different host-parasite interactions. In vivo experiments with these organisms indicate that direct interaction with the parasite (infection, phagocytosis, or injection) does not appear to be critical for the production of IL-12p40 at early time points but rather that innate immune cells that have likely interacted with soluble TLR ligands or proinflammatory cytokines are the primary source of IL-12p40.
MATERIALS AND METHODS
Mice.
C57BL/6 6- to 10-week-old mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and Taconic (Cranbury, NJ). Ai6 mice (a more sensitive Cre reporter strain with the insertion of a CAG promoter in a Rosa26 locus) and YET40 mice (IL-12p40 knock-in mice in which yellow fluorescent protein [YFP] reports transcription of the p40 allele) were originally obtained from The Jackson Laboratory and bred in the University of Pennsylvania animal facility. Ai6 mice treated with anti-IFN-γ blocking antibody (Ab) (clone XMG1.2) were given 1 mg of antibody intraperitoneally (i.p.) 1 day before infection and 3 days after infection and were euthanized at 5 days postinfection (dpi) for analysis. All procedures involving mice were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania (Animal Welfare Assurance reference number A3079-01) and were in accordance with the guidelines set forth in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Parasites and infection.
Transgenic Pru-tdTomato was generated as previously described (43). Toxoplasma-Cre strains of T. gondii (RH-Cre-mCherry, Pru-Cre-mCherry, Pru-mCherry, and Pru-Cre-tdTomato) were generated as previously described (41, 44). Briefly, parental parasites (Pru-tdTomatoΔhpt) which lack the endogenous gene for hypoxanthine xanthine guanine phosphoribosyl transferase (HPT) were transfected with the previously described vector which expresses the selectable HPT marker and the epitope-tagged rhoptry protein fused to Cre recombinase (ptoxofilin-Cre) (44). Parasites were then subjected to several rounds of selection for the expression of HPT using medium containing 25 μg/ml of mycophenolic acid and 50 μg/ml of xanthine before being cloned by limiting dilution. Single-cell clones that were HPT positive (HPT+) were then confirmed to efficiently cause Cre-mediated recombination in a Cre reporter cell line as previously described (44). Stable transgenic parasite lines were maintained in vitro by serial passage through human foreskin fibroblasts in parasite culture medium (Dulbecco modified Eagle medium [DMEM] [Invitrogen, Carlsbad, CA], 20% medium M199 [Invitrogen], 10% fetal bovine serum [FBS] [Serum Source International, Charlotte, NC], 1% penicillin-streptomycin [Invitrogen], 25 μg/ml gentamicin [Gibco]). Tachyzoites of each strain were prepared for infection by serial needle passage and filtered through a 5-μm-pore-size filter. Mice were infected i.p. with 104 or 106 live, invasion-competent parasites. To prevent parasite invasion and rhoptry secretion (which promotes phagocytic uptake of live parasites), RH-Cre-mCherry parasites were resuspended in fetal calf serum (FCS) and treated with 100 μM 4-bromophenacyl bromide (4-BPB) dissolved in dimethyl sulfoxide (DMSO) (Sigma) for 10 min at 37°C. Parasites were then rinsed with phosphate-buffered saline (PBS) prior to injection or addition to in vitro cell culture assay mixtures.
In vitro cell culture and imaging.
Wild-type (WT) and Ai6 bone marrow macrophages (BMM) and bone marrow DCs (BMDCs) were grown by isolating bone marrow from the long bones of Ai6 mice, and red blood cells (RBCs) were lysed with 0.86% ammonium chloride. Bone marrow cells were then plated at 2 × 106 per 100-mm non-tissue culture-treated plate in 10 ml medium. For BMM, macrophage medium (complete DMEM with 25 mM HEPES, 1% penicillin-streptomycin, 1 mM sodium pyruvate [Gibco], and 0.1% β-mercaptoethanol) was supplemented with 30% L929 supernatant. BMM were then fed an additional 10 ml of macrophage medium with 30% L929 on days 3 and 6. Between days 7 and 9, cells were harvested using ice-cold PBS with 5 μM EDTA, and 106 cells were plated in 35-mm MatTek imaging dishes in macrophage medium. BMDCs were cultured in 10 ml of complete RPMI (RPMI with 10% FCS, 1% penicillin-streptomycin, 1 mM sodium pyruvate, 1% nonessential amino acids [Gibco], and 0.1% β-mercaptoethanol) supplemented with 20 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) (Peprotech) and fed an additional 10 ml of complete RPMI with GM-CSF on days 3 and 6. On day 7, cells were harvested using ice-cold PBS and cultured in polypropylene tubes at 106 cells/ml. Splenic DCs were generated by subcutaneous injection of 3 × 106 B16 melanoma cells expressing FLT3 ligand in Ai6 mice. CD11c+ DCs were then purified by magnetic-activated cell sorting (MACS) from single-cell suspensions of spleens from these tumor-bearing mice.
For the measurement of the kinetics of ZsGreen1 expression by flow cytometry, Pru-Cre-tdTomato parasites were added at a multiplicity of infection (MOI) of 1 to the BMM. Cells were then harvested at different time points and analyzed. For the measurement of ZsGreen1 kinetics by fluorescence microscopy, RH-Cre-mCherry parasites were added to Ai6 BMM at an MOI of 1. BMM were subsequently imaged for 20 h using a Leica DMI4000 microscope equipped with a Yokogawa CSU10 spinning-disk confocal unit, a Hamamatsu ImagEM EMCCD camera, and an environmental chamber maintaining 37°C, 5% CO2, and high humidity. For phagocytosis assays, RH-Cre-mCherry parasites were treated with 4-BPB and fed to WT BMM. At 4 h after infection, medium containing LysoTracker Green at 70 nM was added to the cells and left for 30 min at 37°C. Cells were subsequently rinsed with macrophage medium and imaged with the spinning-disk confocal microscope described above. For IL-12p40 assays, BMDCs or splenic DCs were infected for 18 to 20 h with Pru-Cre-tdTomato at an MOI of 1.
Flow cytometry and Amnis ImageStream.
Single-cell suspensions were prepared from peritoneal exudate cells (PECs) by a peritoneal wash with 8 ml ice-cold PBS. PECs were then resuspended in complete RPMI 1640. Single-cell suspensions were prepared from spleens and mediastinal and parathymic lymph nodes (LN). RBCs were lysed using 0.86% ammonium chloride. Cells for staining (2 × 106) were washed with flow cytometry buffer (PBS, 1% bovine serum albumin [BSA] [Sigma-Aldrich], 2 mM EDTA [Life Technologies]) and blocked in 10 μl Fc block solution (flow cytometry buffer, 1 μg/ml anti-CD16/32 2.4G2, 1 μg/ml normal rat IgG [Life Technologies]) containing Live/Dead Fixable Aqua dead cell stain (Life Technologies) at 4°C for 10 min. Cells were then stained in 50 μl at 4°C for 15 to 20 min and washed in flow cytometry buffer for acquisition by LSR Fortessa or Amnis ImageStream. For intracellular cytokine staining, 106 cells from the peritoneal exudate, spleen, or LN were plated in a 96-well plate and incubated with 1× brefeldin A at 37°C for 6 h. Harvested cells were surface stained as described above, fixed using 2% paraformaldehyde at room temperature for 10 min, and then washed with flow cytometry buffer. Cells were then stained for intracellular cytokines in 50 μl of 0.5% saponin (Sigma-Aldrich) in flow cytometry buffer at 4°C for 1 h. Cells were washed and resuspended in flow cytometry buffer for acquisition. Abs used were anti-Ly6C–peridinin chlorophyll protein (PerCP)-Cy5.5 (eBioscience, clone HK1.4), anti-major histocompatibility complex II (MHCII)–Alexa Fluor 700 (eBioscience, clone M5/114.15.2), anti-Ly6G–allophycocyanin (APC) (BioLegend, clone 1A8), anti-CD3–PacBlue (BioLegend, clone 17A2), anti-NK1.1–PacBlue (BioLegend, clone PK136), anti-CD19–eFluor 450 (eBioscience, clone 1D3), anti-CD11b–APC-eFluor780 (eBioscience, clone M1/70), anti-CD11c–phycoerythrin (PE)-Cy7 (eBioscience, clone N418), anti-CD8a–eFluor650NC (eBioscience, clone 53-6.7), and anti-IL-12/23p40–Alexa Fluor 647 (eBioscience, clone C17.8). Results were analyzed using FlowJo 9.2 (Tree Star, Ashland, OR) and Amnis Ideas software.
Statistical analyses.
To determine statistical significance, Student t tests were performed.
RESULTS
Interaction of Toxoplasma-Cre strains with Ai6 Cre reporter macrophages.
Previous studies have described the use of Cre-mediated reporters (enhanced green fluorescent protein [eGFP] and luciferase) to detect the ability of T. gondii to inject Cre (41, 44), but the Ai6 reporter mouse strain has emerged as a more sensitive reporter, especially in adult cells (40, 41). To better understand the kinetics and fidelity of this reporter in macrophages, a cell type critical for immunity to T. gondii, BMM were derived from the Ai6 mice. These cells were infected with the avirulent Prugniaud (Pru) strain expressing tdTomato (Pru-tdTomato) or with a virulent RH or avirulent Pru strain engineered to express the Cre fusion protein along with mCherry or tdTomato (RH-Cre-mCherry and Pru-Cre-tdTomato). At 20 h postinfection (hpi), the ZsGreen1 reporter protein was expressed by cells infected with the Pru-Cre-tdTomato strain but not by those infected with the Pru-tdTomato strain. Thus, reporter expression was exclusive to Toxoplasma-Cre parasites and was not induced by infection alone (Fig. 1A), which is consistent with previous work (41). Flow cytometric analysis of these cultures reveals four populations of interest: cells containing a parasite but not expressing the ZsGreen1 reporter (quadrant a), infected cells expressing the reporter (quadrant b), uninfected-injected (U-I) cells (quadrant c), and untouched cells which had not been infected or injected or phagocytosed T. gondii and thus did not express the reporter or contain a detectable parasite-derived material (quadrant d). Images of populations a to c were captured using an Amnis ImageStream, and representative images that illustrate the presence or absence of the parasites expressing tdTomato are presented in Fig. 1B. As in previous studies (41), expression of ZsGreen1 was not dependent on the strain of Toxoplasma-Cre parasite used. Similarly, the relative frequency of the four populations described above was essentially identical for the Cre-expressing RH and Pru strains, suggesting that there are no intrinsic strain-specific effects in these in vitro assays. In both RH-Cre-mCherry (Fig. 1C) and Pru-Cre-tdTomato (Fig. 1D) infections, ZsGreen1 expression can be detected as early as 7 hpi by live cell fluorescence microscopy or flow cytometry, reaching peak intensity by approximately 18 hpi. However, the slow kinetics of ZsGreen1 expression makes it difficult to distinguish cells that had phagocytosed T. gondii from those that were newly infected but had yet to express detectable levels of ZsGreen1.
FIG 1.

Characterization of Ai6 reporter cells after Toxoplasma-Cre infection. (A) Primary Ai6 BMM were analyzed by flow cytometry for ZsGreen1 expression at 20 h after infection with the control Pru-tdTomato or Pru-Cre-tdTomato strain. Populations were further divided into Toxo+ ZsGreen1− (a), Toxo+ ZsGreen1+ (b), Toxo− ZsGreen1+ (uninfected-injected [U-I]) (c), and Toxo− ZsGreen1− (untouched) (d) subsets. (B) ImageStream analysis (magnification, ×40) of Ai6 BMM subsets (a to c) at 20 h after infection, showing bright-field (BF), Toxo plus BF overlay, ZsGreen1 alone (ZsG), and overlay of the Toxo and ZsGreen1 channels. (C) Ai6 BMM were infected with RH-Cre-mCherry, and the kinetics of ZsGreen1 expression was measured by live-cell spinning-disk microscopy for 20 h. (D) Flow cytometric analysis of ZsGreen1 expression in Ai6 BMM infected with Pru-Cre-tdTomato for 25 h after infection.
When these kinetic studies were performed using Toxoplasma-Cre parasites expressing mCherry, a population of cells that contained a diffuse red staining suggestive of parasite debris was revealed by fluorescence microscopy (data not shown). Images obtained by ImageStream revealed that Ai6 BMM infected with mCherry-expressing parasite strains contained either diffuse or punctate red fluorescence (Fig. 2A). To determine whether cells containing diffuse mCherry fluorescence would appear as Toxo+ ZsGreen1− cells by flow cytometry, the fluorescence intensities of cells containing punctate versus diffuse mCherry fluorescence were compared (Fig. 2B). To delineate these two populations, the area of mCherry fluorescence was plotted against the mean mCherry fluorescence intensity. This plot distinguishes cells containing small, intact, highly fluorescent parasites from cells containing dim, diffuse, fluorescent parasite debris. At 20 h after infection, when plotted as mCherry versus ZsGreen1, the total mCherry fluorescence intensity of these two distinct populations of diffuse and punctate mCherry+ cells overlaps in mCherry fluorescence intensity (Fig. 2C). This analysis also revealed that the vast majority of cells that contained diffuse mCherry fluorescence were ZsGreen1−. This observation suggests that this mCherry+ parasite debris results from phagocytosis of the tachyzoites rather than the clearance of the parasite from an infected cell that would have been exposed to rhoptry-injected Cre-recombinase and then become ZsGreen1+.
FIG 2.

Distinguishing infected cells from parasite phagocytosis. (A) ImageStream images of Ai6 primary macrophages containing intact RH-Cre-mCherry parasites that have punctate fluorescence or diffuse mCherry staining consistent with parasite debris. (B) ImageStream analysis of mCherry fluorescence intensity versus fluorescence area reveals two distinct populations containing either intact, punctate mCherry+ parasites or diffuse mCherry debris. (C) Back gating of the punctate and diffuse mCherry populations plotted as Toxo-mCherry versus ZsGreen1 to show the equivalent total mCherry fluorescence intensity of each population. (D) RH-Cre-mCherry parasites were treated with the invasion inhibitor 4-BPB, added to cultures of primary BMM, and left for 4 h. Cells were subsequently stained with LysoTracker Green for 30 min and imaged at a magnification of ×160 by spinning-disk confocal microscopy. Colocalization of the LysoTracker Green and Toxo-mCherry channels shows that 4-BPB-inhibited parasites are phagocytosed by macrophages. (E) Flow cytometric analysis of Ai6 BMM incubated with 4-BPB-treated RH-Cre-mCherry for 20 h allows detection of phagocytosed parasites and confirms that phagocytosis of the parasite does not induce expression of the ZsGreen1 reporter. (F) ImageStream analysis of the data from panel E confirms that mCherry+ cells contain diffuse mCherry debris, while the one cell that has punctate mCherry fluorescence (arrow) is a result of an extracellular parasite attached to the cell surface.
To directly determine whether these diffuse red cells result from phagocytosis of the parasite, Ai6 BMM were cultured in the presence of RH-Cre-mCherry treated with 4-BPB, an inhibitor of rhoptry injection which prevents parasite invasion (45). In these experiments, this treatment results in phagocytosis of the parasite, as shown by the presence of the mCherry+ parasite in the lysosomal system at 4 h after addition of the parasites (Fig. 2D). Flow cytometric analysis after 18 h showed a significant enrichment of untouched or Toxo+ ZsGreen1− cells (Fig. 2E), and the vast majority of mCherry+ cells contained characteristic diffuse red staining (Fig. 2F). The sensitivity of this system is illustrated by the detection of a single cell (circled in Fig. 2F) with punctate mCherry fluorescence. The image of this cell clearly shows a macrophage with an intact parasite attached to its surface. Thus, ImageStream analysis of this reporter system can reliably distinguish whether a Toxo+ ZsGreen1− cell is recently infected or has phagocytosed the parasite and contains fluorescent parasite debris.
Tracking host-parasite interactions in vivo.
To examine host-parasite interactions in vivo, Ai6 Cre reporter mice were infected i.p. with 104 Pru-mCherry or Pru-Cre-mCherry tachyzoites, and the peritoneal exudate cells (PECs) were examined by flow cytometry at day 5 postinfection (Fig. 3A). This analysis combined with images from the ImageStream (Fig. 3B) showed that the same host-parasite interactions detected during in vitro infections with Ai6 BMM could be detected in vivo at the site of infection. In quadrants a to c, Toxo+ ZsGreen1− cells containing punctate or diffuse red fluorescence as well as Toxo+ ZsGreen1+ and U-I populations were observed. In contrast to the in vitro infections (Fig. 1A), the ratio of U-I to Toxo+ ZsGreen1+ cells in vivo is increased 18-fold (from 1:6.6 in vitro to 1:0.4 in vivo), indicating that while all the same host-parasite interactions are detected, their relative frequencies may be different.
FIG 3.
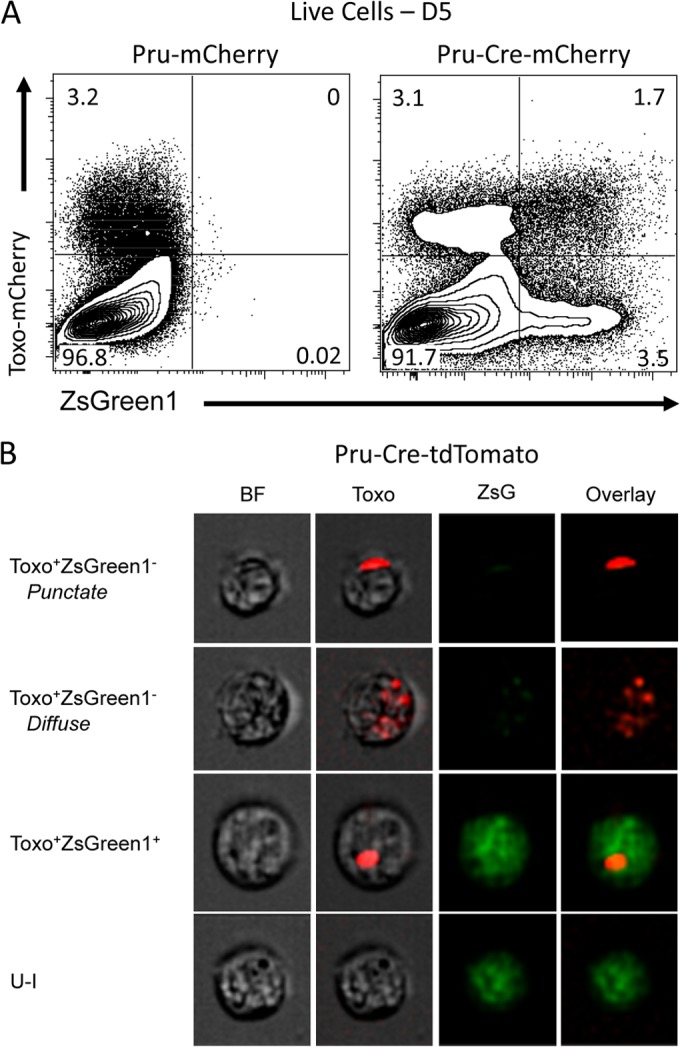
Examining host-parasite interactions in vivo. (A) Ai6 mice were infected i.p. with 104 control Pru-mCherry or Pru-Cre-mCherry parasites, and PECs were analyzed by flow cytometry at 5 dpi. The expression of ZsGreen1 was limited to the Pru-Cre-mCherry strain. (B) PECs from Ai6 mice infected i.p. with 104 Pru-Cre-tdTomato parasites were imaged at a magnification of ×40 using the ImageStream. Toxo+ ZsGreen1− populations containing either punctate or diffuse tdTomato fluorescence were detected along with Toxo+ ZsGreen1+ and U-I populations.
To better understand how these interactions between host immune cells and the parasite change with time in different tissues, Ai6 mice were infected with 104 Pru-Cre-tdTomato i.p., and the phenotypes of infected and ZsGreen1+ cells in the peritoneal exudate, spleen, and draining lymph nodes were assessed at 3, 5, and 10 days postinfection (dpi). At 3 dpi, most ZsGreen1+ cells at the site of infection are DCs, monocytes, and resident macrophages (Fig. 4A), and no detectable infected or ZsGreen1+ populations were observed in the spleen and draining LN (data not shown). As the infection progresses to day 5, the cellular composition of the peritoneum changes as monocytes, DCs, and neutrophils continue to be recruited to the site of infection. These cells are increasingly infected (Toxo+) compared to those at day 3 and are accompanied by an increase in the U-I population (Fig. 4B). At day 10, the infection has started to resolve in the peritoneum as indicated by the reduced frequency of Toxo+ cells, but the fraction and number of U-I cells remain high. When the spleen and draining LN were examined, few infected and/or ZsGreen1+ cells were detected at days 3 and 5, but these cells were present by day 10 in the spleen and draining lymph nodes (see Fig. S1 in the supplemental material). The lack of DCs or macrophages that had interacted with T. gondii in these secondary lymphoid organs at days 3 and 5 was surprising, as these sites have been shown to be critical for cytokine production as well as priming of the adaptive immune response.
FIG 4.

Kinetics of host-parasite interactions at the site of infection. (A) Ai6 mice were infected with 104 Pru-Cre-tdTomato parasites i.p., and the PECs were subsequently analyzed by flow cytometry at 3, 5, and 10 dpi. Innate cell populations were first gated through singlets, live cells, and dump− (CD3−, CD19−, NK1.1−). Further division into subsets allowed for differentiation of DCs (CD11c+, MHCII+), monocytes (non-DCs, CD11b+, Ly6G−, Ly6C+), resident macrophages (non-DCs, CD11bHI, Ly6G−, Ly6C−), and neutrophils (non-DCs, CD11b+, Ly6G+, Ly6Cint). The interactions of each of these subsets with the parasite were analyzed by plotting Toxo-tdTomato versus ZsGreen1 fluorescence. The average percentage of each quadrant is shown. (B) Quantitation of the percentage and number of cells of each fluorescent quadrant, i.e., Toxo+ ZsGreen1− (Toxo+), Toxo+ ZsGreen1+ (DP), and Toxo− ZsGreen1+ (U-I), versus days postinfection shows the kinetics of each type of host-parasite interaction at the site of infection. (n = 3 for each time point; averages ± standard errors of the means [SEM] are shown).
The experiments with the avirulent Toxoplasma-Cre parasites revealed a high frequency of U-I monocytes, macrophages, and DCs. Although the injection of rhoptry proteins without productive invasion by the parasite (kiss-and-run model) could account for these populations, it was also possible that in vivo these cells were activated by IFN-γ to destroy intracellular parasites (46, 47), giving rise to a subpopulation of cells with a U-I phenotype due to injection of the Cre during invasion and subsequent death of the parasite. To address this possibility, Ai6 mice were treated with control rat IgG antibody or anti-IFN-γ monoclonal antibody and infected with Pru-Cre-mCherry. Analysis of the PECs at 5 dpi showed that mice treated with control rat IgG had significant populations of U-I dendritic cells, resident macrophages, and inflammatory monocytes (Fig. 5A). When mice were treated with anti-IFN-γ, the populations of Toxo+ ZsGreen1+ cells were markedly increased, while the percentage and number of U-I cells in these cell populations were significantly reduced (Fig. 5B and C). These data suggest that a subset of the U-I population may reflect parasite clearance by infected cells, as previously proposed (41, 44).
FIG 5.

Role of IFN-γ in the development of U-I cells. (A) Ai6 mice were treated with either rat IgG or anti-IFN-γ at 1 day prior to infection and 3 dpi. These mice were infected with 104 Pru-Cre-mCherry parasites i.p., and at 5 dpi the PECs were harvested. The presence of the fluorescent parasite and expression of ZsGreen1 in innate cell populations in the PECs (DCs, monocytes, resident macrophages, and neutrophils) were analyzed by flow cytometric analysis. (B) Quantitation of the percentage of each subset of innate cells (Toxo+, Toxo+ ZsGreen1+, or U-I) was compared between rat IgG-treated or anti-IFN-γ-treated mice. (C) The number of cells of each innate cell population in each Toxo/ZsGreen1 subset for both rat IgG- and anti-IFN-γ-treated mice was determined (n = 4; results are representative of 3 separate experiments [average ± SEM]; *, P < 0.05; **, P < 0.01; ***, P < 0.001).
Tracking host-parasite interactions that lead to IL-12 production.
These newly developed techniques to track host-parasite interactions were then applied to understand the mechanisms that lead to IL-12p40 expression. As DCs have been implicated as the required source of IL-12p40 in vivo, bone marrow DCs (BMDCs) and CD8α+ splenic DCs derived from Ai6 mice were infected with Pru-Cre-tdTomato. At 20 hpi, DCs were incubated for 6 h in brefeldin A and analyzed for IL-12p40 production by intracellular cytokine staining as a function of infection and ZsGreen1 expression. Infection of BMDCs showed that infection with the parasite (Toxo+ and Toxo+ ZsGreen1+ [DP]) drove IL-12p40 production more than injection of rhoptry proteins alone (U-I) or the presence of the parasite alone (untouched) (Fig. 6A). In contrast, the addition of parasites to cultures of CD8α+ splenic DCs resulted in marked IL-12p40 production, but this was not preferentially associated with any host-parasite interaction (Toxo+, DP, or U-I) (Fig. 6B). Indeed, the finding that “untouched” populations produced similar levels of IL-12p40 suggests that a soluble factor may be responsible for activating all the cells in these cultures, regardless of whether they have been infected or injected.
FIG 6.

Analysis of DC-parasite interactions that induce IL-12p40 expression in vitro. (A) Ai6 BMDCs were infected with Pru-Cre-tdTomato at an MOI of 1 for 20 h and stained for intracellular IL-12p40 after incubation for 6 h in brefeldin A. BMDCs were then divided into subsets by Toxo-tdTomato and ZsGreen1 expression, and the percentage of p40+ cells was quantified by flow cytometry. (B) Splenic DCs were expanded in vivo by FLT3L-expressing tumors, isolated by magnetic-activated cell sorting (MACS) column purification, and infected at an MOI of 1 for 20 h. CD8α+ CD11b− splenic DCs were incubated in brefeldin A for 6 h and then analyzed by flow cytometry for IL-12p40 production in cells divided into subsets for Toxo-tdTomato and ZsGreen1 expression. The percentage of CD8α+ DCs producing p40 was then quantified.
To examine IL-12p40 production in vivo, Ai6 mice were infected with Pru-Cre-tdTomato i.p. and the cellular sources of IL-12p40 were analyzed at 3 dpi. In contrast to the in vitro studies with BMDC, flow cytometric analysis of the innate cells in the PECs showed that while dendritic cells (Fig. 7A) and inflammatory monocytes (Fig. 7B) were the primary sources of IL-12p40+ cells, less than 10% of the IL-12p40+ populations had interacted with T. gondii (Fig. 7C). One limitation to this approach was that the fixation necessary to detect intracellular IL-12p40 resulted in the loss of the diffuse mCherry or tdTomato signal that results from the phagocytosis of the parasite (but not the punctate staining of the parasites themselves [data not shown]), making it difficult to assess whether the cells that were making IL-12p40 had phagocytosed T. gondii. To directly address this issue, IL-12p40 reporter knock-in mice (YET40) that express the p40 gene linked with eYFP via a viral internal ribosome entry site element (48) were infected with 106 Pru-tdTomato parasites i.p. or injected with 2 × 106 RH parasites that expressed mCherry and had been treated with 4-BPB to prevent invasion and induce phagocytosis. RH-mCherry parasites were used in place of Pru-tdTomato because the fluorescence of mCherry is maintained after phagocytosis of parasites in vivo (49). Although the RH and Pru strains of T. gondii can have different effects on host cells, treatment with the 4-BPB inhibitor prevents the injection of the parasite rhoptry proteins and results in phagocytosis of the parasite that also prevents the intracellular injection of dense granule proteins. Because rhoptry and dense granule proteins are primarily responsible for eliciting the phenotypic differences between parasite strains, the use of different parasite strains should not have an impact on the ability of cells to respond to phagocytosed parasites. Similar to the IL-12p40 staining following challenge, YFP was expressed primarily in uninfected DCs, while few infected innate cells were YFP+ (Fig. 8A) as determined by cell number (Fig. 8B). The percentage of infected cells that are YFP+ is comparable to that in uninfected populations (Fig. 8C), indicating that while infected cells are capable of making IL-12p40, infection alone does not drive production of the cytokine. In mice dosed with live but invasion-inhibited parasites, the major sources of YFP+ cells were untouched DCs and resident macrophages (Fig. 8D and E). Phagocytosis of T. gondii (mCherry+) did result in an increased frequency of YFP+ cells compared to mCherry− cells (Fig. 8D and F); however, this increase was less than what has been previously reported in other models of innate activation (48). In addition, the quantitation of the percentage of YFP+ cells for each cell type revealed that cells that have been infected by or have phagocytosed the parasite are not deficient in making IL-12p40 compared to uninfected cells (Fig. 8E and F). To conclude, infection by or phagocytosis of T. gondii is not associated with the preferential expression of IL-12p40.
FIG 7.

Assessing the host-parasite interactions that induce IL-12p40 in Ai6 mice infected with Toxoplasma-Cre. (A) DCs in the peritoneums of Ai6 mice infected with Pru-Cre-tdTomato were analyzed at 3 dpi for IL-12p40 production. DCs that were IL-12p40+ were then divided into subsets and quantified based on Toxo-tdTomato and ZsGreen1 expression. The percentage of each subset was then quantified. (B) Monocytes were also analyzed for IL-12p40 production, and IL-12p40+ cells were analyzed for Toxo-tdTomato and ZsGreen1. (C) The numbers of untouched, Toxo+ ZsGreen1−, Toxo+ ZsGreen1+, and U-I DCs and monocytes expressing IL-12p40 in the peritoneum were quantified.
FIG 8.

Infection of host cells and phagocytosis of the parasite do not initiate IL-12p40 production at the site of infection. (A) YET40 mice were infected i.p. with 106 Pru-tdTomato parasites for 20 h, and the expression of YFP was assessed in Toxo− and Toxo+ DCs, monocytes, and resident macrophages in the peritoneum. (B and C) Numbers and percentages of YFP+ DCs, monocytes, and resident macrophages that were either from mice injected with PBS alone or from infected mice and gated on Toxo− or Toxo+ cells. (D) RH-mCherry parasites were treated with 4-BPB to inhibit rhoptry injection and invasion of host cells, and then 2 × 106 parasites were injected i.p. into YET40 mice. The expression of YFP by DCs, monocytes, and resident macrophages that were untouched (Toxo−) or had phagocytosed the parasite (Toxo+) was analyzed and quantified by flow cytometry. (E and F) Numbers and percentages of YFP+ DCs, monocytes, and resident macrophages that were either from mice injected with PBS alone or from mice injected with 4-BPB inhibited RH-mCherry and gated on Toxo− or Toxo+ (phagocytosis) cells.
DISCUSSION
Over the last 20 years, many advances have allowed the engineering of transgenic T. gondii to better understand how these organisms interact with host cells (43, 50–52). The development of parasite strains that express various fluorescent proteins has been useful to identify infected cells but also has limitations. For example, studies in mice challenged with T. gondii that expresses YFP or red fluorescent protein (RFP) were important for the recognition that infected cells were not major sources of IL-12 (18, 34). However, RFP and YFP quickly lose their fluorescent signal with decreases in pH, which makes it difficult to follow the fate of phagocytosed parasites. In contrast, the mCherry protein is stable at low pH (53), which may explain the ability to detect diffuse staining in cells that have phagocytosed parasites that express this fluorescent marker. However, because mCherry is dimmer and is susceptible to fixation, it is technically challenging to combine this marker with intracellular staining for cytokines, whereas tdTomato can be used for this purpose. Thus, the opportunity to use Toxoplasma-Cre strains that express different fluorescent proteins combined with Ai6 reporter mice allows the differentiation of viable intracellular organisms from phagocytosed parasites and offers new insights into the events that contribute to the uninfected but injected (U-I) population. There do remain caveats to use of this reporter system, which include the slow kinetics of ZsGreen1 expression, but this does provide the opportunity to differentiate recently infected populations from those infected for longer periods of time. Nevertheless, these studies illustrate the utility of access to a range of genetically encoded markers and reporters that can be used to help identify different host-parasite interactions, principles that are also relevant to other intracellular pathogens.
While in vitro analysis revealed a small population of U-I cells, the in vivo experiments with the avirulent Pru strain showed that these cells became the most frequent population at later time points after infection. Because IFN-γ-meditated mechanisms mediate control of this strain of T. gondii, it was possible that clearance of intracellular tachyzoites might contribute to the U-I population in vivo. Indeed, experiments blocking IFN-γ led to a marked reduction in the frequency of these U-I cells, which would support this hypothesis. An alternative explanation is that the increased parasite burden in the absence of IFN-γ results in the infection of cell populations that would have normally been subject to the kiss-and-run interaction. Nevertheless, because the U-I population was not entirely ablated when IFN-γ was neutralized, it remains likely that a population of U-I leukocytes results from kiss-and-run interactions with T. gondii, as appears to be the case for neurons in an infected brain (41). Additional studies are needed to determine whether these populations are phenotypically distinct from infected cells and if they influence the outcome of infection.
As noted in the introduction, there are data that imply that direct interactions between T. gondii and DCs and/or macrophages are required for IL-12 production (31, 54), while other reports have shown that the main source of IL-12 is uninfected cells (14, 18, 34). These previous studies would not have been able to distinguish uninfected cells from populations that (i) had been injected but never infected, (ii) had been infected but then had cleared the parasites, or (iii) had phagocytosed dead T. gondii. The in vitro studies that compare the abilities of BMDCs and CD8α+ splenic DCs to produce IL-12p40 when incubated with live T. gondii reflect the variation in the literature. For BMDCs, direct interactions with T. gondii through infection or phagocytosis are associated with the production of IL-12, whereas the CD8α+ splenic DCs appear to be most responsive to soluble factors produced in these cultures. The in vivo analysis concurs with current models that the most prominent source of IL-12 during toxoplasmosis is DCs, especially CD8α+ splenic DCs, but also suggests that these populations have not interacted with T. gondii in any way (infected, injected, or phagocytosed). These observations contribute to our understanding of the cellular interactions between T. gondii and immune populations in several ways and suggest that it is the interaction between a soluble factor and these immune populations that drives IL-12p40 production. There is a well-developed literature showing that profilin, a soluble cytoplasmic molecule involved in parasite motility, is primarily responsible for the induction of IL-12p40, which is dependent on the MyD88, UNC93B1, and TLR11/12 components of the TLR sensing pathways (24, 25, 27, 34, 54). Recent evidence has shown that UNC93B1, TLR11, and TLR12 are localized to the endolysosome upon treatment with soluble profilin (34, 54). Because T. gondii actively invades host cells and resides in the nonfusogenic PV, it has been hypothesized that endocytosis of soluble profilin (released as a consequence of parasite lysis) or phagocytosis of the intact parasite allows TLR11/12 access to profilin. While these studies have demonstrated that phagocytosis of the intact parasite does not drive IL-12p40 production and suggests that the mechanism is instead endocytosis of a soluble parasite factor such as profilin (34, 35), further work is needed to rule out other host-derived factors induced by infection.
One of the surprising questions raised by this work is why macrophages and DCs at the site of infection, which are infected or had phagocytosed T. gondii, are not more prominent sources of IL-12. Prior work, along with this study, has shown that following i.p. challenge with T. gondii, the resident cell populations in the peritoneum (i.e., not those recruited) do not produce IL-12p40 (18). In both studies, infected cells were not induced to make IL-12p40, while these studies also demonstrate that phagocytosis of the parasite does not drive IL-12p40 production. This lack of responsiveness is reminiscent of the TLR desensitization observed in resident macrophages in other tissues (55). Another possibility is that the lack of IFN-γ at the site of infection at early time points after infection mutes the response to the TLR stimulus. It has long been appreciated that IFN-γ priming can promote the IL-12 production by macrophages and DCs, and IFN-γ can promote the remodeling of the chromatin structures that allows increased access to the IL-12 gene (11).
Finally, in the context of studies that show that infected DCs are required for the development of CD4+ and CD8+ T cell responses to T. gondii (43, 56, 57), the finding that IL-12p40 is produced by cells that are not infected (or have not been injected or phagocytosed T. gondii) suggests that cells that are presenting parasite antigen are not making IL-12. It is unclear whether this result indicates that the presentation of antigen and production of IL-12 necessary for adaptive T cell responses are performed by distinct DC subsets or is simply a reflection of temporal interactions of accessory cells with parasites and parasite-derived materials that cannot be distinguished using this reporter system. The ability to further refine the reporter systems utilized here combined with the ability to image these host populations in vivo should provide an opportunity to dissect the events that are required for the initiation of a protective immune response during toxoplasmosis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grants AI104998, AI42334, AI21423, AI098374, and NS065116 and by the Commonwealth of Pennsylvania.
We thank the members of the Hunter Laboratory for critical reading of the manuscript.
Footnotes
Published ahead of print 14 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01643-14.
REFERENCES
- 1. Robert-Gangneux F, Darde ML. 2012. Epidemiology of and diagnostic strategies for toxoplasmosis. Clin. Microbiol. Rev. 25:264–296. 10.1128/CMR.05013-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gazzinelli RT, Hieny S, Wynn TA, Wolf S, Sher A. 1993. Interleukin 12 is required for the T-lymphocyte-independent induction of interferon gamma by an intracellular parasite and induces resistance in T-cell-deficient hosts. Proc. Natl. Acad. Sci. U. S. A. 90:6115–6119. 10.1073/pnas.90.13.6115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gazzinelli RT, Wysocka M, Hayashi S, Denkers EY, Hieny S, Caspar P, Trinchieri G, Sher A. 1994. Parasite-induced IL-12 stimulates early IFN-gamma synthesis and resistance during acute infection with Toxoplasma gondii. J. Immunol. 153:2533–2543 [PubMed] [Google Scholar]
- 4. Hunter CA, Candolfi E, Subauste C, Van Cleave V, Remington JS. 1995. Studies on the role of interleukin-12 in acute murine toxoplasmosis. Immunology 84:16–20 [PMC free article] [PubMed] [Google Scholar]
- 5. Lieberman LA, Cardillo F, Owyang AM, Rennick DM, Cua DJ, Kastelein RA, Hunter CA. 2004. IL-23 provides a limited mechanism of resistance to acute toxoplasmosis in the absence of IL-12. J. Immunol. 173:1887–1893. 10.4049/jimmunol.173.3.1887 [DOI] [PubMed] [Google Scholar]
- 6. Scharton-Kersten T, Contursi C, Masumi A, Sher A, Ozato K. 1997. Interferon consensus sequence binding protein-deficient mice display impaired resistance to intracellular infection due to a primary defect in interleukin 12 p40 induction. J. Exp. Med. 186:1523–1534. 10.1084/jem.186.9.1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scharton-Kersten TM, Wynn TA, Denkers EY, Bala S, Grunvald E, Hieny S, Gazzinelli RT, Sher A. 1996. In the absence of endogenous IFN-gamma, mice develop unimpaired IL-12 responses to Toxoplasma gondii while failing to control acute infection. J. Immunol. 157:4045–4054 [PubMed] [Google Scholar]
- 8. Yap G, Pesin M, Sher A. 2000. Cutting edge: IL-12 is required for the maintenance of IFN-gamma production in T cells mediating chronic resistance to the intracellular pathogen, Toxoplasma gondii. J. Immunol. 165:628–631. 10.4049/jimmunol.165.2.628 [DOI] [PubMed] [Google Scholar]
- 9. Hunter CA, Sibley LD. 2012. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat. Rev. Microbiol. 10:766–778. 10.1038/nrmicro2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. MacMicking JD. 2012. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol. 12:367–382. 10.1038/nri3210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qiao Y, Giannopoulou EG, Chan CH, Park SH, Gong S, Chen J, Hu X, Elemento O, Ivashkiv LB. 2013. Synergistic activation of inflammatory cytokine genes by interferon-gamma-induced chromatin remodeling and Toll-like receptor signaling. Immunity 39:454–469. 10.1016/j.immuni.2013.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ma X, Chow JM, Gri G, Carra G, Gerosa F, Wolf SF, Dzialo R, Trinchieri G. 1996. The interleukin 12 p40 gene promoter is primed by interferon gamma in monocytic cells. J. Exp. Med. 183:147–157. 10.1084/jem.183.1.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yarovinsky F. 2014. Innate immunity to Toxoplasma gondii infection. Nat. Rev. Immunol. 14:109–121. 10.1038/nri3598 [DOI] [PubMed] [Google Scholar]
- 14. Mordue DG, Sibley LD. 2003. A novel population of Gr-1+-activated macrophages induced during acute toxoplasmosis. J. Leukoc. Biol. 74:1015–1025. 10.1189/jlb.0403164 [DOI] [PubMed] [Google Scholar]
- 15. Bliss SK, Butcher BA, Denkers EY. 2000. Rapid recruitment of neutrophils containing prestored IL-12 during microbial infection. J. Immunol. 165:4515–4521. 10.4049/jimmunol.165.8.4515 [DOI] [PubMed] [Google Scholar]
- 16. Bliss SK, Zhang Y, Denkers EY. 1999. Murine neutrophil stimulation by Toxoplasma gondii antigen drives high level production of IFN-gamma-independent IL-12. J. Immunol. 163:2081–2088 [PubMed] [Google Scholar]
- 17. Robben PM, Mordue DG, Truscott SM, Takeda K, Akira S, Sibley LD. 2004. Production of IL-12 by macrophages infected with Toxoplasma gondii depends on the parasite genotype. J. Immunol. 172:3686–3694. 10.4049/jimmunol.172.6.3686 [DOI] [PubMed] [Google Scholar]
- 18. Goldszmid RS, Caspar P, Rivollier A, White S, Dzutsev A, Hieny S, Kelsall B, Trinchieri G, Sher A. 2012. NK cell-derived interferon-gamma orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity 36:1047–1059. 10.1016/j.immuni.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hou B, Benson A, Kuzmich L, DeFranco AL, Yarovinsky F. 2011. Critical coordination of innate immune defense against Toxoplasma gondii by dendritic cells responding via their Toll-like receptors. Proc. Natl. Acad. Sci. U. S. A. 108:278–283. 10.1073/pnas.1011549108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu CH, Fan YT, Dias A, Esper L, Corn RA, Bafica A, Machado FS, Aliberti J. 2006. Cutting edge: dendritic cells are essential for in vivo IL-12 production and development of resistance against Toxoplasma gondii infection in mice. J. Immunol. 177:31–35. 10.4049/jimmunol.177.1.31 [DOI] [PubMed] [Google Scholar]
- 21. Mashayekhi M, Sandau MM, Dunay IR, Frickel EM, Khan A, Goldszmid RS, Sher A, Ploegh HL, Murphy TL, Sibley LD, Murphy KM. 2011. CD8alpha(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity 35:249–259. 10.1016/j.immuni.2011.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reis e Sousa C, Hieny S, Scharton-Kersten T, Jankovic D, Charest H, Germain RN, Sher A. 1997. In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J. Exp. Med. 186:1819–1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pepper M, Dzierszinski F, Wilson E, Tait E, Fang Q, Yarovinsky F, Laufer TM, Roos D, Hunter CA. 2008. Plasmacytoid dendritic cells are activated by Toxoplasma gondii to present antigen and produce cytokines. J. Immunol. 180:6229–6236. 10.4049/jimmunol.180.9.6229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koblansky AA, Jankovic D, Oh H, Hieny S, Sungnak W, Mathur R, Hayden MS, Akira S, Sher A, Ghosh S. 2013. Recognition of profilin by Toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity 38:119–130. 10.1016/j.immuni.2012.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Melo MB, Kasperkovitz P, Cerny A, Konen-Waisman S, Kurt-Jones EA, Lien E, Beutler B, Howard JC, Golenbock DT, Gazzinelli RT. 2010. UNC93B1 mediates host resistance to infection with Toxoplasma gondii. PLoS Pathog. 6:e1001071. 10.1371/journal.ppat.1001071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Scanga CA, Aliberti J, Jankovic D, Tilloy F, Bennouna S, Denkers EY, Medzhitov R, Sher A. 2002. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J. Immunol. 168:5997–6001. 10.4049/jimmunol.168.12.5997 [DOI] [PubMed] [Google Scholar]
- 27. Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, Sher A. 2005. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308:1626–1629. 10.1126/science.1109893 [DOI] [PubMed] [Google Scholar]
- 28. Mordue DG, Hakansson S, Niesman I, Sibley LD. 1999. Toxoplasma gondii resides in a vacuole that avoids fusion with host cell endocytic and exocytic vesicular trafficking pathways. Exp. Parasitol. 92:87–99. 10.1006/expr.1999.4412 [DOI] [PubMed] [Google Scholar]
- 29. Mordue DG, Sibley LD. 1997. Intracellular fate of vacuoles containing Toxoplasma gondii is determined at the time of formation and depends on the mechanism of entry. J. Immunol. 159:4452–4459 [PubMed] [Google Scholar]
- 30. Braun L, Brenier-Pinchart MP, Yogavel M, Curt-Varesano A, Curt-Bertini RL, Hussain T, Kieffer-Jaquinod S, Coute Y, Pelloux H, Tardieux I, Sharma A, Belrhali H, Bougdour A, Hakimi MA. 2013. A Toxoplasma dense granule protein, GRA24, modulates the early immune response to infection by promoting a direct and sustained host p38 MAPK activation. J. Exp. Med. 210:2071–2086. 10.1084/jem.20130103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rosowski EE, Lu D, Julien L, Rodda L, Gaiser RA, Jensen KD, Saeij JP. 2011. Strain-specific activation of the NF-kappaB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J. Exp. Med. 208:195–212. 10.1084/jem.20100717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ewald SE, Chavarria-Smith J, Boothroyd JC. 2014. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect. Immun. 82:460–468. 10.1128/IAI.01170-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sibley LD, Weidner E, Krahenbuhl JL. 1985. Phagosome acidification blocked by intracellular Toxoplasma gondii. Nature 315:416–419. 10.1038/315416a0 [DOI] [PubMed] [Google Scholar]
- 34. Pifer R, Benson A, Sturge CR, Yarovinsky F. 2011. UNC93B1 is essential for TLR11 activation and IL-12-dependent host resistance to Toxoplasma gondii. J. Biol. Chem. 286:3307–3314. 10.1074/jbc.M110.171025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Plattner F, Yarovinsky F, Romero S, Didry D, Carlier MF, Sher A, Soldati-Favre D. 2008. Toxoplasma profilin is essential for host cell invasion and TLR11-dependent induction of an interleukin-12 response. Cell Host Microbe 3:77–87. 10.1016/j.chom.2008.01.001 [DOI] [PubMed] [Google Scholar]
- 36. Whitmarsh RJ, Gray CM, Gregg B, Christian DA, May MJ, Murray PJ, Hunter CA. 2011. A critical role for SOCS3 in innate resistance to Toxoplasma gondii. Cell Host Microbe 10:224–236. 10.1016/j.chom.2011.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Butcher BA, Fox BA, Rommereim LM, Kim SG, Maurer KJ, Yarovinsky F, Herbert DR, Bzik DJ, Denkers EY. 2011. Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog. 7:e1002236. 10.1371/journal.ppat.1002236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Levings MK, Schrader JW. 1999. IL-4 inhibits the production of TNF-alpha and IL-12 by STAT6-dependent and -independent mechanisms. J. Immunol. 162:5224–5229 [PubMed] [Google Scholar]
- 39. El Kasmi KC, Holst J, Coffre M, Mielke L, de Pauw A, Lhocine N, Smith AM, Rutschman R, Kaushal D, Shen Y, Suda T, Donnelly RP, Myers MG, Jr, Alexander W, Vignali DA, Watowich SS, Ernst M, Hilton DJ, Murray PJ. 2006. General nature of the STAT3-activated anti-inflammatory response. J. Immunol. 177:7880–7888. 10.4049/jimmunol.177.11.7880 [DOI] [PubMed] [Google Scholar]
- 40. Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. 2010. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13:133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koshy AA, Dietrich HK, Christian DA, Melehani JH, Shastri AJ, Hunter CA, Boothroyd JC. 2012. Toxoplasma co-opts host cells it does not invade. PLoS Pathog. 8:e1002825. 10.1371/journal.ppat.1002825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boothroyd JC, Dubremetz JF. 2008. Kiss and spit: the dual roles of Toxoplasma rhoptries. Nat. Rev. Microbiol. 6:79–88. 10.1038/nrmicro1800 [DOI] [PubMed] [Google Scholar]
- 43. John B, Harris TH, Tait ED, Wilson EH, Gregg B, Ng LG, Mrass P, Roos DS, Dzierszinski F, Weninger W, Hunter CA. 2009. Dynamic Imaging of CD8(+) T cells and dendritic cells during infection with Toxoplasma gondii. PLoS Pathog. 5:e1000505. 10.1371/journal.ppat.1000505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Koshy AA, Fouts AE, Lodoen MB, Alkan O, Blau HM, Boothroyd JC. 2010. Toxoplasma secreting Cre recombinase for analysis of host-parasite interactions. Nat. Methods 7:307–309. 10.1038/nmeth.1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ravindran S, Lodoen MB, Verhelst SH, Bogyo M, Boothroyd JC. 2009. 4-Bromophenacyl bromide specifically inhibits rhoptry secretion during Toxoplasma invasion. PLoS One 4:e8143. 10.1371/journal.pone.0008143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang L, Pietrek M, Brinkmann MM, Havemeier A, Fischer I, Hillenbrand B, Dittrich-Breiholz O, Kracht M, Chanas S, Blackbourn DJ, Schulz TF. 2009. Identification and functional characterization of a spliced rhesus rhadinovirus gene with homology to the K15 gene of Kaposi's sarcoma-associated herpesvirus. J. Gen. Virol. 90:1190–1201. 10.1099/vir.0.007971-0 [DOI] [PubMed] [Google Scholar]
- 47. Yamamoto M, Okuyama M, Ma JS, Kimura T, Kamiyama N, Saiga H, Ohshima J, Sasai M, Kayama H, Okamoto T, Huang DC, Soldati-Favre D, Horie K, Takeda J, Takeda K. 2012. A cluster of interferon-gamma-inducible p65 GTPases plays a critical role in host defense against Toxoplasma gondii. Immunity 37:302–313. 10.1016/j.immuni.2012.06.009 [DOI] [PubMed] [Google Scholar]
- 48. Reinhardt RL, Hong S, Kang SJ, Wang ZE, Locksley RM. 2006. Visualization of IL-12/23p40 in vivo reveals immunostimulatory dendritic cell migrants that promote Th1 differentiation. J. Immunol. 177:1618–1627. 10.4049/jimmunol.177.3.1618 [DOI] [PubMed] [Google Scholar]
- 49. Dupont CD, Christian DA, Selleck EM, Pepper M, Leney-Greene M, Harms Pritchard G, Koshy AA, Wagage S, Reuter MA, Sibley LD, Betts MR, Hunter CA. 2014. Parasite fate and involvement of infected cells in the induction of CD4+ and CD8+ T cell responses to Toxoplasma gondii. PLoS Pathog. 10:e1004047. 10.1371/journal.ppat.1004047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Striepen B, He CY, Matrajt M, Soldati D, Roos DS. 1998. Expression, selection, and organellar targeting of the green fluorescent protein in Toxoplasma gondii. Mol. Biochem. Parasitol. 92:325–338. 10.1016/S0166-6851(98)00011-5 [DOI] [PubMed] [Google Scholar]
- 51. Gubbels MJ, Li C, Striepen B. 2003. High-throughput growth assay for Toxoplasma gondii using yellow fluorescent protein. Antimicrob. Agents Chemother. 47:309–316. 10.1128/AAC.47.1.309-316.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Saeij JP, Boyle JP, Grigg ME, Arrizabalaga G, Boothroyd JC. 2005. Bioluminescence imaging of Toxoplasma gondii infection in living mice reveals dramatic differences between strains. Infect. Immun. 73:695–702. 10.1128/IAI.73.2.695-702.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. 2004. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22:1567–1572. 10.1038/nbt1037 [DOI] [PubMed] [Google Scholar]
- 54. Andrade WA, Souza Mdo C, Ramos-Martinez E, Nagpal K, Dutra MS, Melo MB, Bartholomeu DC, Ghosh S, Golenbock DT, Gazzinelli RT. 2013. Combined action of nucleic acid-sensing Toll-like receptors and TLR11/TLR12 heterodimers imparts resistance to Toxoplasma gondii in mice. Cell Host Microbe 13:42–53. 10.1016/j.chom.2012.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Didierlaurent A, Goulding J, Patel S, Snelgrove R, Low L, Bebien M, Lawrence T, van Rijt LS, Lambrecht BN, Sirard JC, Hussell T. 2008. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J. Exp. Med. 205:323–329. 10.1084/jem.20070891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chtanova T, Han SJ, Schaeffer M, van Dooren GG, Herzmark P, Striepen B, Robey EA. 2009. Dynamics of T cell, antigen-presenting cell, and pathogen interactions during recall responses in the lymph node. Immunity 31:342–355. 10.1016/j.immuni.2009.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gigley JP, Fox BA, Bzik DJ. 2009. Cell-mediated immunity to Toxoplasma gondii develops primarily by local Th1 host immune responses in the absence of parasite replication. J. Immunol. 182:1069–1078. 10.4049/jimmunol.182.2.1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.