

Abstract
The obligate intracellular parasite Toxoplasma gondii critically relies on host cell invasion during infection. Proteins secreted from the apical micronemes are central components for host cell recognition, invasion, egress, and virulence. Although previous work established that the sporozoite protein with an altered thrombospondin repeat (SPATR) is a micronemal protein conserved in other apicomplexan parasites, including Plasmodium, Neospora, and Eimeria, no genetic evidence of its contribution to invasion has been reported. SPATR contains a predicted epidermal growth factor domain and two thrombospondin type 1 repeats, implying a role in host cell recognition. In this study, we assess the contribution of T. gondii SPATR (TgSPATR) to T. gondii invasion by genetically ablating it and restoring its expression by genetic complementation. Δspatr parasites were ∼50% reduced in invasion compared to parental strains, a defect that was reversed in the complemented strain. In mouse virulence assays, Δspatr parasites were significantly attenuated, with ∼20% of mice surviving infection. Given the conservation of this protein among the Apicomplexa, we assessed whether the Plasmodium falciparum SPATR ortholog (PfSPATR) could complement the absence of the TgSPATR. Although PfSPATR showed correct micronemal localization, it did not reverse the invasion deficiency of Δspatr parasites, because of an apparent failure in secretion. Overall, the results suggest that TgSPATR contributes to invasion and virulence, findings that have implications for the many genera and life stages of apicomplexans that express SPATR.
INTRODUCTION
Apicomplexan parasites are obligate intracellular pathogens that cause a broad range of human and animal diseases. Included in this phylum are Eimeria spp. (coccidiosis), Cryptosporidium spp. (cryptosporidiosis), Plasmodium spp. (malaria), and Toxoplasma gondii (toxoplasmosis). Among the most promiscuous and successful of these is T. gondii, which infects virtually any nucleated cell in vitro and has an exceptional host range in the wild. Human seroprevalence rates are estimated at 25 to 30% worldwide, but the prevalence can vary widely depending on geographic region and culinary practices (1). Humans acquire Toxoplasma by ingesting cat-derived oocysts in contaminated food or water, by ingesting tissue cysts in infected meat, or through congenital transmission from mother to fetus (2). Parasites liberated from oocysts or tissue cysts subsequently penetrate the intestinal epithelium before differentiating into the rapidly dividing tachyzoite form. During acute-phase infection, tachyzoites replicate and disseminate throughout the body, including to neural and muscle tissues, where they redifferentiate to the slowly dividing bradyzoites within tissue cysts, remaining dormant through the life of its host. Through every step of this process, the parasite must actively invade host cells to propagate and avoid aspects of the host immune response.
Although members of the Apicomplexa are biologically specialized, they nonetheless share many common cellular and molecular characteristics. Principal among these features are an apical complex, invasion-related secretory organelles, and modes of motility and invasion (3–5). Invasion, consisting of attachment and penetration, involves a coordinated sequential secretion of proteins from secretory organelles termed micronemes, rhoptries, and dense granules (5, 6). Invasion is completed upon pinching off of the newly enveloped parasite inside a parasitophorous vacuole, where replication ensues. Several microneme protein (MIC) complexes are necessary for efficient cell invasion and virulence based on genetic disruption (7–13). Many of these molecules have conserved adhesive modules, such as epidermal growth factor (EGF), Apple/PAN, thrombospondin type I repeats (TSR), and microneme adhesive motif (MAR) domains. Therefore, poorly characterized or hypothetical proteins containing such domains are likely involved in invasion.
Despite the expanding repertoire of secretory proteins shown to be important for T. gondii or Plasmodium falciparum cell invasion, only a few notable orthologs are shared between these apicomplexans. Conserved secretory components, including T. gondii MIC2 (TgMIC2)/P. falciparum thrombospondin-related anonymous protein (PfTRAP), apical membrane antigen 1 (AMA1), rhoptry neck protein 2 (RON2), and subtilisin protease 1 (SUB1), likely evolved prior to divergence of the last common ancestor and are considered core components of the invasion system (14). In light of recent studies challenging the established model of active invasion and the “essential” roles of these proteins (15, 16), the possibility that additional, less-characterized molecules could contribute to residual invasion warrants further consideration.
We previously identified and endogenously tagged one such apicomplexan-conserved MIC termed the T. gondii sporozoite protein with an altered thrombospondin repeat (TgSPATR) (17). SPATR was initially identified in Plasmodium falciparum (PfSPATR) (18), but recent whole-genome sequencing revealed orthologs in most Apicomplexa. TgSPATR was also identified in a proteomic analysis of Ca2+-ionophore-dependent secretion (19), and its basic properties were subsequently characterized, but its contribution to invasion was not addressed (20). In P. falciparum, PfSPATR is expressed at several stages of the parasite life cycle, an important consideration for vaccine development (18). Furthering its vaccine potential, Plasmodium SPATR is immunogenic in naturally infected and immunized volunteers, and antibodies to recombinant SPATR block sporozoite invasion (18, 21). These findings suggest a role in invasion, which is consistent with the ability of Plasmodium SPATR to bind HepG2 hepatoma cells (18, 21). Herein, we further dissect the significance of TgSPATR in infection by genetically ablating it in T. gondii. Our findings reveal previously unrecognized structural features of TgSPATR and suggest that it contributes to T. gondii invasion and parasite virulence during infection.
MATERIALS AND METHODS
BLAST searches.
The TgSPATR (TgME49_293900) and PfSPATR (PF3D7_0212600) deduced amino acid sequences were used to BLASTp query the deduced proteomes of Babesia, Cryptosporidium, Eimeria, Neospora, Plasmodium, Theileria, and Toxoplasma at eupathdb.org. These sequences were also used to tBLASTn query toxodb.org for Gregarina and Sarcocystis. Subjects with an E value of <1e−08 were considered significant, with subsequent manual verification of conserved residues, including cysteines indicative of the thrombomodulin EGF (TmEGF) or TSR domains. A hit was identified in Sarcocystis neurona, but the relatively low homology precluded assembly of the amino acid sequence from the genomic scaffold.
Parasite culture, transfection, and selection.
T. gondii tachyzoites were maintained by growth in human foreskin fibroblast (HFF) cells cultured in Dulbecco's modified Eagles medium (DMEM) containing 10% fetal bovine serum (FBS) (GIBCO), 2 mM glutamine, 10 mM HEPES, and 50 μg/ml penicillin-streptomycin (D10 complete medium). SPATR-yellow fluorescent protein (YFP) (17) was transfected with a TgSPATR PCR knockout (KO) construct (see below) containing a chloramphenicol acetyltransferase (CAT) selectable marker. Δspatr parasite clones were identified by PCR, immunoblotting, and immunofluorescence. The hypoxanthine xanthine-guanine phosphoribosyltransferase (HXG) selectable marker in this strain was removed by 6-thioxanthine negative selection using a Ku80 construct as described previously (22). The resulting Δspatr Δhxg strain was complemented with a TgSPATRmyc construct containing an HXG selectable marker, to generate the ΔspatrComp strain. All primers used in the study are listed in Table S1 in the supplemental material. For transfections, 1 × 107 parasites were electroporated with a Bio-Rad Gene Pulsar II with 1.5-kV voltage, 25 μF capacitance, and no resistance setting. Drug selection was applied the day after transfection, and clones were isolated by limiting dilution in 96-well plates.
Generation of anti-TgSPATR antibodies.
A construct representing the nearly fully mature TgSPATR (S94 to N530) was cloned into the pAcGP67b baculovirus transfer vector. Primary virus was generated in Spodoptera frugiperda cells and used to infect 2.4 liters of High Five insect cells. The insect cell culture was harvested 65 h postinfection, with cellular debris removed by centrifugation, and the clarified supernatant was concentrated and buffer exchanged into buffer A (20 mM HEPES [pH 8], 1 M NaCl, 30 mM imidazole) using tangential flow. TgSPATR was purified using Ni-agarose (elution buffer: 20 mM HEPES [pH 8.0], 250 mM imidazole, 1 M NaCl) and size exclusion chromatography (Sephacryl 200) (running buffer: 20 mM HEPES [pH 7.5], 150 mM NaCl) with fractions analyzed by SDS-PAGE and pooled based on purity. For polyclonal antibody production, 30 μg of recombinant His-tagged TgSPATR was emulsified in Freund's complete adjuvant (Sigma) and injected into the peritoneal cavity of BALB/c mice. Three weeks postinjection, mice were boosted with 30 μg of TgSPATR-His in Freund's incomplete adjuvant; 7 days after boost, mice were exsanguinated and sera were collected. The mice were given food and water ad libitum. Cages were changed twice weekly. Mice were checked daily during the infection period. All procedures and protocols were adhered to as approved by the University Committee on the Use and Care of Animals at the University of Michigan.
Complement plasmid construction and knockout PCR construct amplification.
The vector for complementing into the Ku80 locus was generated by modifying the TgM2AP complement plasmid (9). A Ku80 5′ flank was inserted upstream of the existing TgM2AP 5′ untranslated region (UTR), and a Ku80 3′ flank was inserted downstream of the HXG selectable marker (pM2AP.Ku80.HXG). To generate the TgSPATR complement vector, the TgM2AP coding sequence was excised with NsiI and PacI and replaced with the TgSPATR gene containing a myc tag upstream of the stop codon (TgSPATR.1.NsiI.F and TgSPATR.1605myc.PacI.R). The TgSPATR knockout fusion PCR product was generated by fusing a 5′ flank of TgSPATR (TgSPATR.629641.F and 5′Tub.TgSPATR.5′0.632256.R), TubCATSag1 (TgSPATR.5′0.632256.5′Tub.F and TgSPATR.3′0.638674.3′Sag1.R), and a 3′ flank of TgSPATR (3′Sag1.TgSPATR.3′0.638674.F and TgSPATR.640219.R) and amplifying the fusion KO cassette with TgSPATR.629641.F and TgSPATR.640219.R.
PfSPATR plasmid constructs.
PfSPATR was codon optimized according to T. gondii codon preferences provided by the Kazusa DNA Research Institute (http://www.kazusa.or.jp/codon/cgi-bin/showcodon.cgi?species=5811). A synthetic cassette encoding codon-optimized PfSPATR with flanking NsiI and PacI cloning sites and a C-terminal myc tag was generated (Life Technologies). This fragment was subcloned into the pM2AP.Ku80.HXG complement vector. Δspatr parasites were transfected with PfSPATR, and positive clones were identified after mycophenolic acid/xanthine selection.
Invasion and attachment assays.
Assays were performed as previously described (9). Briefly, 1 × 107 parasites were allowed to invade subconfluent HFF monolayers in 8-well chamber slides for 20 min before fixation. Slides were differentially stained with anti-SAG1 antibodies to differentiate attached versus invaded parasites. The attachment assays on glutaraldehyde-fixed host cells were performed as described previously (9). For the cytochalasin D attachment assay, parasites were treated with 2 μM cytochalasin D for 10 min at room temperature (RT) prior to inoculation onto host cell monolayers and allowed to attach for 20 min. Slides were fixed and stained for the SAG1 surface antigen to identify parasites.
Induced egress assay.
Parasites were inoculated into 8-well chamber slides and allowed to replicate for 28 h. Slides were washed twice in Hanks balanced salt solution supplemented with 1 mM CaCl2 and 1 mM MgCl2 (HBSSC) and placed in a 37°C water bath. HBSSC with either 1% dimethyl sulfoxide (DMSO) or 2 μM calcium ionophore A23187 was heated to 37°C prior to being added to the chamber slides. Induced egress was allowed to occur for 2 min, followed by the addition of 2× fixative (8% formaldehyde in phosphate-buffered saline [PBS]). Slides were fixed for 20 min at room temperature followed by indirect immunofluorescence with rabbit anti-SAG1 and mouse anti-GRA7 to identify parasites and vacuoles, respectively.
Indirect immunofluorescence microscopy.
All manipulations were carried out at RT. Tachyzoite-infected HFF cells on 8-well chamber slides were fixed with 4% paraformaldehyde for 20 min, followed by washes in PBS. Fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 15 min, washed, and blocked in 10% FBS in PBS for 30 min. The wells were then stained with the primary antibodies (mouse anti-green fluorescent protein [GFP] 1:250 [JL-8; Clontech], mouse anti-myc 1:250 [9E10; DSHB], rabbit anti-MIC5 1:500, rabbit anti-MIC2 1:500, mouse anti-SPATR 1:250) followed by Alexa 594- or Alexa 488-conjugated goat anti-mouse or goat anti-rabbit antibodies (Molecular Probes, Eugene, OR) and 4′,6-diamidino-2-phenylindole (DAPI). After washes in PBS-1% FBS-1% normal goat serum (NGS), slides were mounted in Mowiol, and fluorescent images were collected at ×1,000 total magnification with a Zeiss Axio inverted microscope.
ESA preparation and immunoblotting.
Lysed parasites were filter purified, chased with DMEM-20 mM HEPES, and resuspended to a concentration of 4 × 108/ml in DMEM-20 mM HEPES. One hundred microliters of DMEM-2% ethanol in Eppendorf tubes was preheated at 37°C prior to the addition of 100 μl of parasites, and excreted/secreted antigen (ESA) induction was performed for 2 min at 37°C. Tubes were placed immediately on ice for 5 min, followed by centrifugation at 1,000 × g for 10 min at 4°C. A total of 175 μl of the supernatant was removed and centrifuged again, followed by removal of 150 μl and the addition of 5× sample buffer. The pellet was washed with PBS and resuspended in hot 1× sample buffer. Cell pellets and ESA were run on 10% SDS-PAGE gels and blotted by semidry transfer (Bio-Rad) to a polyvinylidene fluoride (PVDF) membrane. Membranes were blocked with 5% nonfat milk, probed with mouse anti-SPATR 1:10,000, rabbit anti-MIC2 1:5,000, mouse anti-GRA1 1:20,000, and mouse anti-myc (9E10) 1:5,000 (DSHB) in 1.25% nonfat milk-PBS-0.1% Tween 20 (PBS-T), washed in PBS-T, and probed with horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit in 1.25% milk–PBS-T. Blots were incubated with enhanced chemiluminescent substrate (Super Signal West Pico; Pierce) and exposed to film.
Metabolic labeling and immunoprecipitation.
Pulse-chase labeling was performed as previously described (23) with the following modifications. A total of 2 × 108/ml extracellular RH strain parasites were purified in methionine- and cysteine-free DMEM supplemented with 1% fetal bovine serum and labeled with 37 kBq/ml (final concentration) of 35S-methionine and cysteine (Amersham) for 10 min at 37°C. Labeled parasites were then “chased” in DMEM growth medium containing 1% FBS and 5 mM methionine and 5 mM cysteine for 0, 15, 30, 45, and 60 min (2 × 107 tachyzoites per time point). Parasites collected at each time point were then spun down and lysed in RIPA buffer (50 mM Tris-HCl [pH 7.5], 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS, 150 mM NaCl, supplemented with DNase and RNase) plus protease inhibitors (Roche), followed by immunoprecipitation with mouse anti-SPATR or rabbit anti-M2AP antibodies. Following immunoprecipitation, samples were run on SDS-PAGE gels, which were then incubated in Amplify (Amersham) and dried in cellophane. Dried gels were exposed to a phosphoimaging screen and imaged on a Typhoon Imagine system.
In vivo virulence assay.
Groups of 5 (experiment 1) or 14 (experiment 2) 6-week-old female Swiss Webster mice were infected intraperitoneally with 20 tachyzoites (in 200 μl PBS) of Δku80::HXG, SPATR-YFP, Δspatr, or ΔspatrComp strains or 200 tachyzoites of the Δspatr strain. At the same time, 200 μl of each diluted parasite strain containing 20 tachyzoites was inoculated into 6-well plates and incubated for 7 days before being stained with 2% crystal violet (Sigma) to visualize plaques. Seroconversion of all surviving mice was confirmed by enzyme-linked immunosorbent assay (ELISA) analysis 3 weeks postinfection.
Statistical analyses.
Prism GraphPad software was used for statistical analysis. Two-tailed Student's t tests were used for analysis of invasion, attachment, gliding, and egress assays. A P value of <0.05 was considered significant. An unpaired t test with Welch's correction, and without an assumption of equal standard deviation, was also performed on the data points.
Protein binding assay.
Tachyzoite sonicate was prepared by resuspending 2 × 108 tachyzoites in 1 ml PBS, 1 mM CaCl2, and 1 mM MgCl2 (PBS-CM) and sonication on ice with a Micronix microtip sonicator 6 times for 30 s with 20-s rests. Tachyzoite ESA fractions were generated by induced secretion of parasites at 1 × 109/ml with 1% ethanol for 30 min and collection of the supernatant. The sonicates were centrifuged at 13,000 × g for 10 min at 4°C. Confluent host cells (HFF, CHO, HeLa) in 6-well plates were washed with PBS-CM, followed by blocking with 10% FBS in PBS-CM for 30 min at 12°C. Tachyzoite sonicate, ESA, or TgSPATR-His recombinant protein (1 μg/ml) was added to the host cells and incubated for 1 h at 12°C. Cells were washed 4 times with PBS-CM, and cell-bound fractions (CBF) were collected by lysing the monolayer in 250 μl RIPA buffer. Ten microliters of unbound fraction (1% of total) and 100% of CBF (chloroform/methanol precipitation followed by resuspension in hot 1× sample buffer) were loaded per lane, and membranes were probed with mouse anti-SPATR to detect the presence of TgSPATR. Blots were also probed with mouse anti-MIC4 as a positive control.
RESULTS
Identification of novel SPATR structural features and orthologs within the Apicomplexa.
The recent emergence of whole-genome sequences for several apicomplexan parasites prompted us to perform bioinformatic searches to identify new SPATR orthologs. Basic local alignment search tool protein (BLASTp) searches of apicomplexan sequences (eupathdb.org) with TgSPATR identified orthologs in Hammondia hammondi, Neospora caninum, and Sarcocystis neurona (Table 1), suggesting conservation among coccidian parasites. A putative ortholog was also identified in Eimeria tenella, although conservation was seen only within the C-terminal TSR region. Orthologs were also identified in hemosporinid parasites (Plasmodium spp.) but not in piroplasms (Babesia, Theileria), cryptosporidia (Cryptosporidium spp.), or gregarines (Gregarina niphandrodes) despite querying with SPATR sequences from several genera. Multiple sequence alignments (see Fig. S1 in the supplemental material) permitted assignment of the domain structure (Fig. 1A). Although it was previously noted that TgSPATR and PfSPATR harbor an EGF-like domain, our analysis suggests that this domain belongs to the thrombomodulin EGF (TmEGF) subfamily based on conservation of a signature CXCPXGY motif (Fig. 1B and C). This domain is conserved in all of the SPATR orthologs except E. tenella SPATR. Multiple sequence analysis (see Fig. S1 in the supplemental material) and structural modeling (see Fig. S2) of the C-terminal region revealed a second TSR domain (termed TSR1) adjacent to previously recognized TSR (termed TSR2) at the end of the protein. Although TSR1 contains an N-terminal extension relative to TSR2, it is predicted to adopt a classic TSR fold (see Fig. S2). TgSPATR and its coccidian orthologs (H. hammondi and N. caninum SPATR) also possess two unique domains (UD1 and UD2) flanking the TmEGF domain, which accounts for an increase in size relative to the Plasmodium orthologs.
TABLE 1.
Identification of SPATR orthologs in apicomplexan parasites
| SPATR orthologg | Gene identifiera | Length (aa)b | MWc | Stage expressiond | Reference or source |
|---|---|---|---|---|---|
| TgSPATR | TGME49_293900 | 535 | 47.5 | TZ, BZ, SZ | 20; toxodb.org |
| HhSPATR | AHJH01000553e | 535 | 47.5 | ND | toxodb.org |
| NcSPATR | NCLIV_000860 | 546 | 45.7 | ND | toxodb.org |
| SnSPATR | Sneu_scaffold00013e | NDf | ND | ND | toxodb.org |
| EtSPATR | ETH_00041180 | 339 | 38.8 | ND | toxodb.org |
| PfSPATR | PF3D7_0212600 | 251 | 26.7 | S, LS, MZ, GA, OO, SZ | 18; eupathdb.org |
| PvSPATR | PVX_002900 | 249 | 26.3 | ND | eupathdb.org |
| PcSPATR | PCHAS_031160 | 275 | 28.4 | ND | eupathdb.org |
| PySPATR | PY17X_031000 | 249 | 26.5 | ND | eupathdb.org |
From eupathdb.org (v20) or toxodb.org (v10.0).
Predicted length in amino acids, including the signal peptide.
Predicted molecular weight (MW) after removal of the signal peptide.
Obtained from the indicated reference or source or from transcriptional or proteomic data in eupathdb.org. Abbreviations are as follows: TZ, tachyzoite; BZ, bradyzoite; SZ, sporozoites; S, schizont; LS, late schizont; MZ, merozoite; GA, gametocyte; OO, ookinete.
Scaffold identifier since the genome is not annotated currently.
ND, not determined because the relatively low homology did not allow assembly of the protein sequence from the genomic scaffold.
Tg, T. gondii; Hh, H. hammondi; Nc, N. caninum; Sn, S. neurona; Et, E. tenella; Pf, P. falciparum; Pv, Plasmodium vivax; Pc, Plasmodium chabaudi; Py, Plasmodium yoelii.
FIG 1.

Structural features of apicomplexan SPATR orthologs. (A) Schematic illustrations of SPATR from T. gondii, N. caninum, E. tenella, and P. falciparum showing domain structures and approximate relative sizes. Illustrations are based on representatives from the multiple sequence analysis shown in Fig. S1 in the supplemental material. (B) Schematic representation of TgSPATR along with multiple sequence alignments of the conserved TmEGF and TSR domains. Residues conserved among all four sequences are colored red, whereas those conserved among two or three sequences are blue. Conserved cysteine residues are denoted with an asterisk. Gaps are indicated by a hyphen. An arrow indicates the beginning of the mature TgSPATR sequence. The boxed region highlights a conserved motif that is a hallmark of the thrombomodulin EGF subfamily. Secondary structure elements predicted by structural modeling (see Fig. S2) are indicated above the TSR multiple sequence alignments. (C) Pfam hidden Markov model representative of the thrombomodulin EGF signature sequence, including the conserved motif corresponding to the boxed region in panel B. (D) Pulse-chase analysis of mouse anti-SPATR immunoprecipitations show a single band for TgSPATR through various chase times of 15, 30, 45, and 60 min. Parallel analysis of TgM2AP processing was included as a positive control. (E) Alignment of the coccidian SPATR sequences adjacent to the established cleavage site for TgSPATR (arrow). Conserved P1 and P3 residues are indicated.
MICs typically contain a short (15- to 20-amino-acid [aa]) signal peptide that is removed cotranslationally by signal peptidase in the endoplasmic reticulum (ER). MICs can also contain an N-terminal signal anchor sequence or a propeptide that is cleaved during vesicular transport to the micronemes. Immunoblotting such proteins in parasite lysates usually detects both the signal anchor/propeptide and mature forms of the protein. Although TgSPATR was detected as a single band in parasite lysates (20), the N-terminal sequence of the secreted form of TgSPATR begins 93 aa downstream from the initiator methionine (19, 20). To distinguish whether TgSPATR contains a signal anchor/propeptide or an unusually long signal peptide, we performed a metabolic labeling pulse-chase immunoprecipitation experiment. After a 15-min pulse with [35S]methionine/cysteine, a single band of ∼70 kDa was immunoprecipitated with mouse anti-SPATR antibodies (Fig. 1D, top). No change in the size of this species was observed during the subsequent chase periods of 15, 30, 45, and 60 min. A parallel analysis of TgM2AP revealed the expected processing of the pro to mature forms (Fig. 1D, bottom) (24). The findings imply that TgSPATR has a 93-aa signal peptide, a notion that is further supported by conservation of small residues (G, A, or V) in the P1 and P3 positions (Fig. 1E), making it a suitable cleavage site for signal peptidase (25). We conclude that TgSPATR is not synthesized as an immature precursor but instead has an unusually long signal peptide.
Genetic ablation and complementation of TgSPATR.
Next we sought to create a strain deficient in expression of TgSPATR to assess its role in T. gondii infection. The SPATR-YFP strain (Table 2), generated in a previous study from Δku80::HXG parasites amenable to genetic manipulation (17), was transfected with a knockout construct containing a chloramphenicol acetyltransferase (CAT) selectable marker (Fig. 2A). The absence of TgSPATR as observed by immunofluorescence (Fig. 2B) and immunoblotting (Fig. 2C) suggested successful deletion of TgSPATR, which was confirmed by PCR analysis showing the integration of the selectable marker in the spatr locus and the lack of the spatr gene in the Δspatr strain (see Fig. S3 in the supplemental material). This strain possesses the HXG selectable marker in the Ku80 locus. To allow reuse of HXG for genetic complementation, we removed it by targeted deletion and negative selection with 6-thioxanthine to create the Δspatr Δhxg strain. This knockout strain was then genetically complemented to restore expression of TgSPATR with a C-terminal myc tag by integration with HXG into the “empty” Ku80 locus, creating the ΔspatrComp strain. Thus, the grandparent (Δku80::HXG), parent (SPATR-YFP), knockout (Δspatr), and complement (ΔspatrComp) strains all express HXG, eliminating it as a potentially confounding factor.
TABLE 2.
T. gondii strains used in this study
| Strain | Alternative name | Genotype | Reference or source |
|---|---|---|---|
| Δku80 strain | Grandparent | RHΔku80::HXG | 17 |
| SPATR-YFP strain | Parent | RHΔku80::HXG/SPATR-YFP | 17 |
| Δspatr strain | Knockout | RHΔku80::HXGΔspatr-yfp::CAT | This study |
| Δspatr Δhxg strain | Cleanup | RHΔku80ΔhxgΔspatr-yfp::CAT | This study |
| ΔspatrComp strain | Complement | RHΔku80::SPATRmycHXG/Δspatr-yfp::CAT | This study |
| PfSPATRmyc strain | P. falciparum complement | RHΔku80::PfSPATRmycHXG/Δspatr-yfp::CAT | This study |
FIG 2.

Genetic disruption and complementation of TgSPATR. (A) The YFP-tagged TgSPATR gene was knocked out by homologous recombination with the CAT selectable marker. The HXG selectable marker in the Ku80 locus was removed by negative selection with 6-thioxanthine, followed by transfection with a construct containing a myc-tagged TgSPATR gene and the HXG marker. (B) Indirect immunofluorescence of intracellular (first four rows) and extracellular (EC; fifth row) parasites with mouse anti-TgSPATR and rabbit anti-TgMIC5 antibodies. (C) Parasite lysate (pellet) and ESA fractions from the indicated strains. ESA fractions were collected from culture supernatants after parasites were treated for 2 min at 37°C with 1% ethanol to stimulate microneme secretion. Treated parasites were pelleted to create the parasite lysates. GRA1 was immunoblotted as a loading control for each strain.
It was shown previously that TgSPATR localizes to the micronemes (20). We confirmed this localization with mouse polyclonal antibodies generated against full-length TgSPATR (Fig. 2B, top row). The apical staining in the SPATR-YFP parasites verified that the protein was targeted properly to the micronemes, although some nonmicronemal staining was observed (Fig. 2B, second row), as is sometimes the case with a large tag. Immunoblotting of excreted/secreted antigens (ESA) showed that this mistargeting of SPATR-YFP also led to reduced secretion despite normal levels of expression. SPATR-YFP was also proteolytically processed within the parasite, creating a tagless species that was preferentially secreted. The specificity of the anti-SPATR antibody as well as the loss of the TgSPATR protein was confirmed by the absence of fluorescence in Δspatr parasites (Fig. 2B, third row). Micronemal localization of the myc-tagged TgSPATR was also confirmed, in both intracellular and extracellular parasites (Fig. 2B, fourth and fifth rows). Although the majority of the TgSPATRmyc in the ΔspatrComp parasites localized to the micronemes, additional staining was observed in other parts of the parasite. This may be due to the overexpression of this construct, as is evident in both the pellet and the ESA fractions (Fig. 2C).
Δspatr parasites are invasion deficient.
The presence of putative adhesive domains and their localization in the micronemes alluded to a potential role for TgSPATR in invasion. Because the spatr knockout parasites were generated from the SPATR-YFP strain, which was derived from the Δku80::HXG “grandparent,” both of these strains were used as controls in the assays. The parental strains in addition to the knockout and complemented strains were subjected to a red-green invasion assay in which extracellular (attached) parasites are immunostained red and intracellular (invaded) parasites are stained green. In this assay, parasites defective in attachment typically show a decrease in both intracellular parasites and extracellular parasites, but the latter is sometimes difficult to detect due to fewer parasites identified at this stage. Parasites defective in penetration exhibit a decrease in intracellular parasites, and they sometimes show a concomitant increase in extracellular parasites due to arrest at the attachment stage. Δspatr parasites showed ∼50% fewer intracellular parasites than Δku80::HXG or SPATR-YFP parasites (Fig. 3A). This defect was fully reversed in the complemented strain, indicating that the reduced invasion was due to the loss of TgSPATR. Although TgSPATR-deficient parasites also showed a trend toward fewer extracellular parasites than the parental and complement strains, the difference was not statistically significant. A second independent knockout clone and its respective complement showed a similar invasion deficiency (data not shown).
FIG 3.

TgSPATR knockout parasites are defective in host cell invasion. (A) Red-green invasion assays of parasites after 20 min of incubation with HFF host cells. Parasites were stained as described in Materials and Methods. *, P < 0.05. (B) Red-green invasion assays of parasites after 0.5, 1, 2, 4, and 8 h of incubation with HFF host cells. *, P < 0.05. (C) Attachment of parasites onto glutaraldehyde-fixed host cells. HFF host cells were fixed with glutaraldehyde, blocked with ethanolamine, and exposed to parasites for 20 min before parasites were stained with anti-SAG1. BAPTA-AM-treated Δku80::HXG parasites are a negative control for attachment. ns, not significant. (D) Attachment of cytochalasin D-treated parasites to HFF host cells. Parasites were treated with cytochalasin D and allowed to attach to HFF host cells for 15 min in the continued presence of cytochalasin D before being stained with anti-SAG1. ns, not significant. (E) Enumeration of nonmotile parasites or parasites performing twirling, helical, or circular gliding by video microscopy. ns, not significant. Data in all graphs are means + standard errors of the means (SEM) from three independent experiments, each with triplicate samples, with the exception of data in panel E, which are means − standard deviations (SD) from 18 individual videos of each parasite strain in 3 independent experiments. (F) Induced egress. Parasites grown for 28 h in chamber slides were induced with A23187, and occupied versus unoccupied vacuoles were enumerated.
The invasion assay allows parasites to invade for 20 min. It is possible that the invasion deficiency can be overcome with additional time if parasites are given a longer incubation period. To assess this, parasites were allowed to invade host cells for 0.5, 1, 2, 4, and 8 h prior to differential immunostaining. Through all time points tested, the Δspatr parasites showed a consistently lower (∼50%) invasion rate than Δku80::HXG parental parasites (Fig. 3B); this gap, however, is narrowed by the 8-h time point. Together, these findings suggest that TgSPATR contributes to cell invasion by T. gondii.
We attempted to distinguish the extent that the Δspatr invasion defect is due to decreased parasite attachment by performing adhesion assays on glutaraldehyde-fixed host cells. The average number of attached Δspatr parasites showed a trend toward being lower than that of the parental or complemented strains, but the difference was not statistically significant (Fig. 3C). As a positive control, the Δku80::HXG strain treated with 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid-acetoxymethyl ester (BAPTA-AM) to chelate intracellular calcium and preclude microneme secretion showed virtually no attachment. For a secondary assessment of attachment, the strains were treated with cytochalasin D to immobilize parasites and preclude penetration into host cells but still allow attachment. Results were obtained that were nearly identical to those for the glutaraldehyde-fixed host cells (Fig. 3D). While the findings are not definitive, Δspatr parasites show features that are more consistent with a defect in attachment than penetration.
In addition to the loss of the TgSPATR gene, the invasion defect observed in the Δspatr strain could be the result of mislocalization of one of the other MIC proteins. Of the MICs tested (MIC1, MIC2, MIC3, MIC4, MIC5, MIC6, MIC8, MIC10, MIC11, SUB1, PLP1, AMA1, and TLN4), all were correctly targeted to the micronemes (data not shown). Since MIC protein complexes often rely on each member of the complex for correct trafficking, the proper targeting of these MICs reduces the likelihood that they are partners of TgSPATR.
Gliding motility and egress are unaltered in Δspatr parasites.
MIC proteins involved in invasion often also play a role in gliding motility. We performed static assays to initially assess whether Δspatr parasites are defective in gliding motility. Δku80::HXG and Δspatr parasites were allowed to glide on glass slides, and the trails deposited by gliding parasites were visualized by staining for the SAG1 surface antigen. No differences were noted in the abundance or types of trails (helical or circular) left by the parasites (data not shown). To examine gliding more extensively, we visualized parasite motility by video microscopy. Consistent with the static assay, no differences in parasite helical gliding, circular gliding, or twirling were seen between parental and knockout parasites (Fig. 3E).
The parasite lytic cycle culminates in egress from the host cell. Since micronemes are required for efficient egress (26), we tested the ability of Δspatr parasites to exit host cells in response to induction with the calcium ionophore A23187. No difference was seen in the percentage of occupied vacuoles after induced egress (Fig. 3F), indicating that TgSPATR does not play a role in parasite exit under the conditions tested. We also quantified in these experiments the number of vacuoles containing 4 or 8 parasites and observed no difference between wild-type (WT) and Δspatr strains (data not shown). These findings imply that TgSPATR does not contribute to parasite replication, which is consistent with a previous study reporting that micronemes are not necessary for intracellular growth (27).
Δspatr parasites are attenuated in virulence.
To assess the role of TgSPATR in Toxoplasma infection, mice were infected with Δku80::HXG, SPATR-YFP, Δspatr, and ΔspatrComp strains. Since the type I strain used in this study is highly virulent, with a lethal dose of a single viable parasite (28), a low dose of 10 tachyzoites was used for infection by intraperitoneal injection. By day 10 postinfection, all mice infected with parental or complemented strains became moribund and were humanely euthanized with the exception of one mouse infected with the SPATR-YFP strain, which survived until day 17 postinfection (Fig. 4). In contrast, mice infected with 10 Δspatr tachyzoites showed a statistically significant delayed time until moribund state, with mice surviving an average of 4.5 days longer than those infected with parental strains. Furthermore, ∼20% of the mice (4 of 19 mice) survived until the endpoint of the experiment (3 months postinfection). All surviving mice were seropositive. Increasing the dose to 200 Δspatr tachyzoites resulted in the loss of most infected mice, indicating that the knockout is not profoundly avirulent. The numbers of parasites injected were corroborated with concomitant plaque assays. Overall, these findings suggest that expression of TgSPATR is required for full virulence and that its role in invasion contributes to the outcome of infection.
FIG 4.
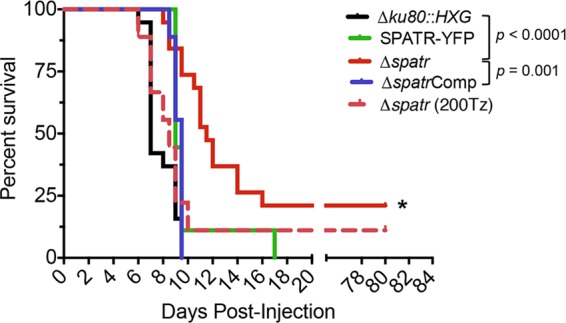
Δspatr parasites are attenuated in in vivo virulence. Mice were infected intraperitoneally with 10 tachyzoites (or the indicated number for Δspatr parasites) in 2 independent experiments, which were combined for the data. An asterisk denotes a statistically significant time until moribundity of the Δspatr strain compared to the Δku80::HXG and ΔspatrComp strains as determined using the Kaplan-Meier estimator (P < 0.05). Twenty percent of Δspatr strain-infected mice survived to the end point.
TgSPATR is sensitive to structural perturbations.
To investigate the contributions of TgSPATR domains to subcellular trafficking and secretion of TgSPATR, we generated a series of domain deletions, as illustrated in Fig. 5A. Expression of these constructs in Δspatr parasites resulted in either accumulation within the ER (ΔEGF mutant) or mistrafficking (ΔTSR, ΔTSR1, ΔTSR2 mutants) (Fig. 5B). Moreover, although these TgSPATR mutants were secreted by parasites, they were released in a calcium-independent manner (Fig. 5C), consistent with a lack of targeting to the micronemes. TgSPATR does not contain transmembrane or cytosolic domains, implying that mistargeting is not due to effects on membrane association. Consistent with the defects in localization and secretion, expression of the domain deletion constructs failed to restore normal invasion (Fig. 5D). These findings suggest that TgSPATR is sensitive to structural perturbations that affect its localization.
FIG 5.

TgSPATR domain deletion mutants are mislocalized. (A) Schematic illustrations of individual domain deletions of EGF, TSR, TSR1, and TSR2. (B) Indirect immunofluorescence localization of intracellular parasites using mouse anti-myc and rabbit anti-M2AP. (C) Induced (1% ethanol for 2 min) and constitutive (20 min) ESA fractions of all domain deletions. BAPTA-AM treatment for 10 min blocked the majority of secretion of a control MIC protein, TgMIC2, in both induced and constitutive secretion but not of the domain deletion mutants. Blots probed with mouse anti-myc or mouse anti-MIC2. (D) Red-green invasion assays of parasites after 20 min of incubation with HFF host cells. Parasites were stained as described in Materials and Methods. Data are means + SEM from two independent experiments, each with triplicate samples. The ΔEGF strain was not included in the secretion or invasion assays because it showed protein arrest in the ER.
PfSPATR cannot functionally complement the absence of TgSPATR because of a defect in secretion.
Next, we sought to determine whether PfSPATR is a functional ortholog capable of restoring the invasion deficiency of Δspatr parasites. To accomplish this, we replaced TgSPATRmyc in the complementation vector with a synthetic myc-tagged PfSPATR cassette codon optimized for T. gondii. This construct was transfected into the Δspatr strain, and individual clones were isolated, including a representative clone termed PfSPATRmyc. Using antibodies against the myc tag, these clones were shown to express PfSPATR by immunoblotting and that it was targeted properly to the micronemes by immunofluorescence (Fig. 6A and B). However, a product was not detected in the ESA fraction, despite concentrating the ESA and running the entire sample (equivalent to secretions from 5 × 108 parasites). Secretions from ΔspatrComp parasites were used as a positive control for induced secretion and inhibition by BAPTA-AM, and probing the immunoblots with anti-MIC2 demonstrated the presence of secreted products in the PfSPATRmyc samples. The lack of secretion is likely responsible for the inability of PfSPATRmyc to restore normal invasion (Fig. 6C).
FIG 6.
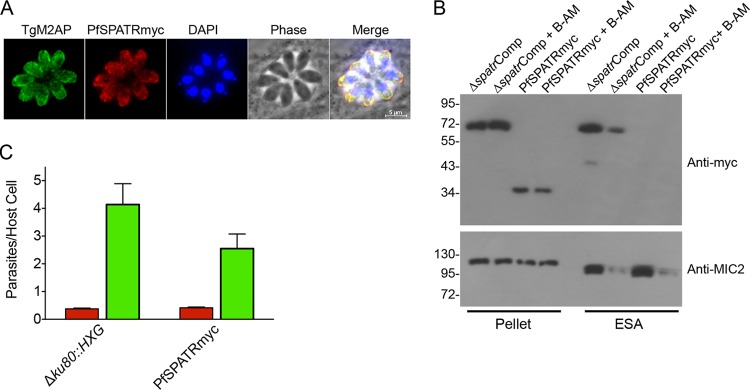
Secretion of PfSPATR is not detectable, and PfSPATR cannot functionally complement TgSPATR. (A) Myc-tagged PfSPATR is trafficked to the micronemes based on colocalization with TgM2AP. (B) Expression of PfSPATR in the Δspatr strain is detected in pellets but not ESA in immunoblot analysis with anti-myc antibody. ΔspatrComp lysate and ESA are included as controls. The top blot was stripped and reprobed with anti-MIC2 to control for the presence of ESA products in the PfSPATR lanes. B-AM, BAPTA-AM. (C) Red-green invasion assay of PfSPATRmyc relative to Δku80::HXG. Data are means + SEM from three independent experiments, each with triplicate samples.
DISCUSSION
T. gondii and Plasmodium spp. utilize a repertoire of molecules during their mission to invade host cells for propagation and survival. These invasion-related proteins are principally secreted from two types of organelles, the micronemes and rhoptries. While increasing numbers of microneme and rhoptry proteins continue to be characterized, only a few are known to have a significant role in invasion based on genetic studies. For genes that could not be directly knocked out, technological advances, such as the use of Cre-lox, have allowed the genetic disruption of these previously considered “essential” genes (15, 16). Residual invasion by such mutants could be conferred by (i) the upregulation of related proteins, e.g., as for T. gondii AMA1 (15), (ii) gliding independent mechanisms, as proposed recently (29), (iii) the presence of alternative invasion pathways involving a greater role for host components, as suggested in references 30 and 31), or (iv) the utilization of additional invasion proteins capable of conferring residual invasion in the absence of key proteins. Potentially relevant to the latter possibility, we herein present data on the genetic disruption of one such conserved protein, TgSPATR.
Previous studies on TgSPATR established its micronemal localization and secretion from the micronemes in a calcium-dependent manner. To assess its role in invasion, we generated a direct TgSPATR knockout strain and detected an ∼50% reduction in invasion efficiency, which was restored upon reintroducing TgSPATR. Two independent types of attachment assays suggested a potential contribution of TgSPATR to parasite attachment, but confirmation of this awaits the development of better assays with lower interexperiment variation. That T. gondii secretes a variety of other adhesive proteins likely also reduces the impact of the knockout on attachment. The invasion deficiency could also be the result of mistrafficking and mislocalization of another MIC, potentially a partner of TgSPATR. The localization of MICs for which antibodies are available was tested and all were targeted properly to the micronemes, reducing the likelihood that any of these are partner proteins of TgSPATR. Since TgSPATR appears to lack features for anchoring, it may conspire with a membrane-anchored partner protein to fulfill a putative role in attachment. Future analysis of prospective associated proteins may yield insight into the mechanism of TgSPATR function.
Several parasite proteins containing EGF or TSR-like domains have adhesive properties (8, 32, 33). The presence of EGF and TSR-like domains indicated that TgSPATR could also have host cell binding properties. However, TgSPATR cell binding activity was not detected despite testing TgSPATR from parasite lysate, ESA, or recombinant protein on several types of host cells (HFF, CHO, and HeLa) (data not shown). This could be attributed to multiple factors, including a low affinity of monomeric TgSPATR for a host cell receptor in the absence of high-avidity presentation on the parasite surface, or, despite being produced in insect cells, the recombinant protein could be folded incorrectly or the affinity tag needed for purification could interfere with function.
The decrease in invasion by Δspatr parasites likely contributes to the delayed-time-until-moribund-state kinetics and survival of Δspatr strain-infected mice. Similar effects on invasion due to deletion of other microneme proteins such as M2AP (9) or MIC3 (7) also cause delayed lethality, presumably due to a slower progression of the infection leading to more effective immune clearance. A recent study showed that EGF-containing MICs, including MIC3, activate Akt signaling via EGF receptors to diminish autophagic killing of the parasite in host cells (34). However, recombinant TgSPATR did not promote epidermal growth factor receptor (EGFR)-Akt activation (L. Muniz-Feliciano, J. Van Grol, J. A. Portillo, L. Liew, B. Liu, C. R. Carlin, V. B. Carruthers, S. Matthews, and C. S. Subauste, unpublished data). While we cannot rule out that SPATR affects immune pathways via other mechanisms, the available evidence supports virulence attenuation due to deficient cell invasion.
A protein is generally accepted as functionally conserved if an ortholog from another species can preclude or reverse the phenotype(s) observed from a gene disruption in another species. For example, previous work showed that precomplementation of T. gondii with the Eimeria tenella MIC1 cDNA encoding an ortholog of TgMIC2 allowed the targeted deletion of TgMIC2 (35). Along these lines, we attempted to complement the Δspatr parasites with PfSPATR. While PfSPATR was expressed and targeted properly to the micronemes, it was not detected in the ESA fraction. This suggests a failure of PfSPATR to exit the micronemes upon fusion with the plasma membrane. A similar pattern of correct microneme localization but reduced secretion was seen with a particular mutant of TgM2AP (36). Although the basis for this is unclear, it is possible that one or both of the unique domains (UD1 and UD2) present in TgSPATR but not in PfSPATR are needed for secretion. If PfSPATR is defective in secretion, this is likely the basis for a failure to restore normal invasion in Δspatr parasites. We cannot, however, rule out other possible explanations, such as the proteolytic removal of the myc tag upon discharge, although we have not noted this with other MICs.
Our findings are consistent with previous studies suggesting a role for SPATR in hepatocyte invasion by malaria sporozoites. Chattopadhyay and colleagues showed that antibodies to PfSPATR reduced P. falciparum invasion of HepG2 cells by ∼80% (18). The same study showed that recombinant PfSPATR binds to HepG2 cells. More recent work identified two PfSPATR peptides with cell binding activity, including one peptide (141CKDGSNKDYADFEIIGASGY160) with binding activity for both HepG2 cells and human erythrocytes (37). Although the corresponding peptide in TgSPATR (416CKDEGTKDAPAVDFIYELNTY436) encompasses strand A of the newly recognized TSR1 domain (Fig. 1), the functional significance of this region remains unknown since our attempts to delete TSR1 resulted in mistrafficking of the protein. Sera from sporozoite-immunized volunteers and malaria patients recognize recombinant SPATR (18, 38), confirming its immunogenicity and antigenicity. These findings, together with evidence that Plasmodium SPATR is expressed throughout the malaria life cycle, including intraerythrocytic, sexual, and transmissive stages, provide support for future efforts to evaluate SPATR as a vaccine candidate along with other functionally important antigens.
Supplementary Material
ACKNOWLEDGMENTS
We thank John Boothroyd for providing the anti-SAG1 antibody, all members of the Carruthers lab for helpful discussions, and Tracey Schultz for technical support.
This work was supported by operating grants from the Canadian Institutes of Health Research (MOP82915 to M.J.B.) and the United States National Institutes of Health Grant (R01AI046675 to V.B.C.).
Footnotes
Published ahead of print 4 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01877-14.
REFERENCES
- 1. Montoya JG, Liesenfeld O. 2004. Toxoplasmosis. Lancet 363:1965–1976. 10.1016/S0140-6736(04)16412-X [DOI] [PubMed] [Google Scholar]
- 2. Robert-Gangneux F, Darde ML. 2012. Epidemiology of and diagnostic strategies for toxoplasmosis. Clin. Microbiol. Rev. 25:264–296. 10.1128/CMR.05013-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Besteiro S, Dubremetz JF, Lebrun M. 2011. The moving junction of apicomplexan parasites: a key structure for invasion. Cell. Microbiol. 13:797–805. 10.1111/j.1462-5822.2011.01597.x [DOI] [PubMed] [Google Scholar]
- 4. Santos JM, Soldati-Favre D. 2011. Invasion factors are coupled to key signaling events leading to the establishment of infection in apicomplexan parasites. Cell. Microbiol. 13:787–796. 10.1111/j.1462-5822.2011.01585.x [DOI] [PubMed] [Google Scholar]
- 5. Carruthers VB, Boothroyd JC. 2007. Pulling together: an integrated model of Toxoplasma cell invasion. Curr. Opin. Microbiol. 10:83–89. 10.1016/j.mib.2006.06.017 [DOI] [PubMed] [Google Scholar]
- 6. Carruthers VB, Sibley LD. 1997. Sequential protein secretion from three distinct organelles of Toxoplasma gondii accompanies invasion of human fibroblasts. Eur. J. Cell Biol. 73:114–123 [PubMed] [Google Scholar]
- 7. Cerede O, Dubremetz JF, Soete M, Deslee D, Vial H, Bout D, Lebrun M. 2005. Synergistic role of micronemal proteins in Toxoplasma gondii virulence. J. Exp. Med. 201:453–463. 10.1084/jem.20041672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huynh MH, Carruthers VB. 2006. Toxoplasma MIC2 is a major determinant of invasion and virulence. PLoS Pathog. 2:e84. 10.1371/journal.ppat.0020084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huynh MH, Rabenau KE, Harper JM, Beatty WL, Sibley LD, Carruthers VB. 2003. Rapid invasion of host cells by Toxoplasma requires secretion of the MIC2-M2AP adhesive protein complex. EMBO J. 22:2082–2090. 10.1093/emboj/cdg217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mital J, Meissner M, Soldati D, Ward GE. 2005. Conditional expression of Toxoplasma gondii apical membrane antigen-1 (TgAMA1) demonstrates that TgAMA1 plays a critical role in host cell invasion. Mol. Biol. Cell 16:4341–4349. 10.1091/mbc.E05-04-0281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lagal V, Binder EM, Huynh MH, Kafsack BF, Harris PK, Diez R, Chen D, Cole RN, Carruthers VB, Kim K. 2010. Toxoplasma gondii protease TgSUB1 is required for cell surface processing of micronemal adhesive complexes and efficient adhesion of tachyzoites. Cell. Microbiol. 12:1792–1808. 10.1111/j.1462-5822.2010.01509.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kessler H, Herm-Gotz A, Hegge S, Rauch M, Soldati-Favre D, Frischknecht F, Meissner M. 2008. Microneme protein 8—a new essential invasion factor in Toxoplasma gondii. J. Cell Sci. 121:947–956. 10.1242/jcs.022350 [DOI] [PubMed] [Google Scholar]
- 13. Cerede O, Dubremetz JF, Bout D, Lebrun M. 2002. The Toxoplasma gondii protein MIC3 requires pro-peptide cleavage and dimerization to function as adhesin. EMBO J. 21:2526–2536. 10.1093/emboj/21.11.2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sharma P, Chitnis CE. 2013. Key molecular events during host cell invasion by apicomplexan pathogens. Curr. Opin. Microbiol. 16:432–437. 10.1016/j.mib.2013.07.004 [DOI] [PubMed] [Google Scholar]
- 15. Bargieri DY, Andenmatten N, Lagal V, Thiberge S, Whitelaw JA, Tardieux I, Meissner M, Menard R. 2013. Apical membrane antigen 1 mediates apicomplexan parasite attachment but is dispensable for host cell invasion. Nat. Commun. 4:2552. 10.1038/ncomms3552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Andenmatten N, Egarter S, Jackson AJ, Jullien N, Herman JP, Meissner M. 2013. Conditional genome engineering in Toxoplasma gondii uncovers alternative invasion mechanisms. Nat. Methods 10:125–127. 10.1038/nmeth.2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huynh MH, Carruthers VB. 2009. Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot. Cell 8:530–539. 10.1128/EC.00358-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chattopadhyay R, Rathore D, Fujioka H, Kumar S, de la Vega P, Haynes D, Moch K, Fryauff D, Wang R, Carucci DJ, Hoffman SL. 2003. PfSPATR, a Plasmodium falciparum protein containing an altered thrombospondin type I repeat domain is expressed at several stages of the parasite life cycle and is the target of inhibitory antibodies. J. Biol. Chem. 278:25977–25981. 10.1074/jbc.M300865200 [DOI] [PubMed] [Google Scholar]
- 19. Kawase O, Nishikawa Y, Bannai H, Zhang H, Zhang G, Jin S, Lee EG, Xuan X. 2007. Proteomic analysis of calcium-dependent secretion in Toxoplasma gondii. Proteomics 7:3718–3725. 10.1002/pmic.200700362 [DOI] [PubMed] [Google Scholar]
- 20. Kawase O, Nishikawa Y, Bannai H, Igarashi M, Matsuo T, Xuan X. 2010. Characterization of a novel thrombospondin-related protein in Toxoplasma gondii. Parasitol. Int. 59:211–216. 10.1016/j.parint.2010.02.001 [DOI] [PubMed] [Google Scholar]
- 21. Mahajan B, Jani D, Chattopadhyay R, Nagarkatti R, Zheng H, Majam V, Weiss W, Kumar S, Rathore D. 2005. Identification, cloning, expression, and characterization of the gene for Plasmodium knowlesi surface protein containing an altered thrombospondin repeat domain. Infect. Immun. 73:5402–5409. 10.1128/IAI.73.9.5402-5409.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Beck JR, Rodriguez-Fernandez IA, Cruz de Leon J, Huynh MH, Carruthers VB, Morrissette NS, Bradley PJ. 2010. A novel family of Toxoplasma IMC proteins displays a hierarchical organization and functions in coordinating parasite division. PLoS Pathog. 6:e1001094. 10.1371/journal.ppat.1001094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brydges SD, Sherman GD, Nockemann S, Loyens A, Daubener W, Dubremetz JF, Carruthers VB. 2000. Molecular characterization of TgMIC5, a proteolytically processed antigen secreted from the micronemes of Toxoplasma gondii. Mol. Biochem. Parasitol. 111:51–66. 10.1016/S0166-6851(00)00296-6 [DOI] [PubMed] [Google Scholar]
- 24. Rabenau KE, Sohrabi A, Tripathy A, Reitter C, Ajioka JW, Tomley FM, Carruthers VB. 2001. TgM2AP participates in Toxoplasma gondii invasion of host cells and is tightly associated with the adhesive protein TgMIC2. Mol. Microbiol. 41:537–547. 10.1046/j.1365-2958.2001.02513.x [DOI] [PubMed] [Google Scholar]
- 25. Zhang Z, Henzel WJ. 2004. Signal peptide prediction based on analysis of experimentally verified cleavage sites. Protein Sci. 13:2819–2824. 10.1110/ps.04682504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kafsack BF, Pena JD, Coppens I, Ravindran S, Boothroyd JC, Carruthers VB. 2009. Rapid membrane disruption by a perforin-like protein facilitates parasite exit from host cells. Science 323(5913):530–533. 10.1126/science.1165740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Breinich MS, Ferguson DJ, Foth BJ, van Dooren GG, Lebrun M, Quon DV, Striepen B, Bradley PJ, Frischknecht F, Carruthers VB, Meissner M. 2009. A dynamin is required for the biogenesis of secretory organelles in Toxoplasma gondii. Curr. Biol. 19(4):277–286. 10.1016/j.cub.2009.01.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pfefferkorn ER, Pfefferkorn LC. 1976. Toxoplasma gondii: isolation and preliminary characterization of temperature-sensitive mutants. Exp. Parasitol. 39:365–376. 10.1016/0014-4894(76)90040-0 [DOI] [PubMed] [Google Scholar]
- 29. Egarter S, Andenmatten N, Jackson AJ, Whitelaw JA, Pall G, Black JA, Ferguson DJ, Tardieux I, Mogilner A, Meissner M. 2014. The Toxoplasma Acto-MyoA motor complex is important but not essential for gliding motility and host cell invasion. PLoS One 9:e91819. 10.1371/journal.pone.0091819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gonzalez V, Combe A, David V, Malmquist NA, Delorme V, Leroy C, Blazquez S, Menard R, Tardieux I. 2009. Host cell entry by Apicomplexa parasites requires actin polymerization in the host cell. Cell Host Microbe 5:259–272. 10.1016/j.chom.2009.01.011 [DOI] [PubMed] [Google Scholar]
- 31. Gaji RY, Huynh MH, Carruthers VB. 2013. A novel high throughput invasion screen identifies host actin regulators required for efficient cell entry by Toxoplasma gondii. PLoS One 8:e64693. 10.1371/journal.pone.0064693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chappel JA, Holder AA. 1993. Monoclonal antibodies that inhibit Plasmodium falciparum invasion in vitro recognise the first growth factor-like domain of merozoite surface protein-1. Mol. Biochem. Parasitol. 60:303–311. 10.1016/0166-6851(93)90141-J [DOI] [PubMed] [Google Scholar]
- 33. Sultan AA, Frevert TVU, Robson KJ, Crisanti ANV, Nussenzweig RS, Menard R. 1997. TRAP is necessary for gliding motility and infectivity of Plasmodium sporozoites. Cell 90:511–522. 10.1016/S0092-8674(00)80511-5 [DOI] [PubMed] [Google Scholar]
- 34. Muniz-Feliciano L, Van Grol J, Portillo J-AC, Liew L, Liu B, Carlin CR, Carruthers VB, Matthews S, Subauste CS. 2013. Toxoplasma gondii-induced activation of EGFR prevents autophagy protein-mediated killing of the parasites. PLoS Pathog. 9(12):e1003809. 10.1371/journal.ppat.1003809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huynh MH, Opitz C, Kwok LY, Tomley FM, Carruthers VB, Soldati D. 2004. Trans-genera reconstitution and complementation of an adhesion complex in Toxoplasma gondii. Cell. Microbiol. 6:771–782. 10.1111/j.1462-5822.2004.00403.x [DOI] [PubMed] [Google Scholar]
- 36. Harper JM, Huynh MH, Coppens I, Parussini F, Moreno S, Carruthers VB. 2006. A cleavable propeptide influences Toxoplasma infection by facilitating the trafficking and secretion of the TgMIC2-M2AP invasion complex. Mol. Biol. Cell 17:4551–4563. 10.1091/mbc.E06-01-0064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Curtidor H, Garcia J, Vanegas M, Puentes F, Forero M, Patarroyo ME. 2008. Identification of peptides with high red blood cell and hepatocyte binding activity in the Plasmodium falciparum multistage invasion proteins: PfSPATR and MCP-1. Biochimie 90:1750–1759. 10.1016/j.biochi.2008.08.003 [DOI] [PubMed] [Google Scholar]
- 38. Palaeya V, Lau YL, Mahmud R, Chen Y, Fong MY. 2013. Cloning, expression, and immunocharacterization of surface protein containing an altered thrombospondin repeat domain (SPATR) from Plasmodium knowlesi. Malar. J. 12:182. 10.1186/1475-2875-12-182 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.