

Abstract
Staphylococcus aureus is a Gram-positive pathogen that causes a diverse range of bacterial infections. Invasive S. aureus strains secrete an extensive arsenal of hemolysins, immunomodulators, and exoenzymes to cause disease. Our studies have focused on the secreted enzyme hyaluronidase (HysA), which cleaves the hyaluronic acid polymer at the β-1,4 glycosidic bond. In the study described in this report, we have investigated the regulation and contribution of this enzyme to S. aureus pathogenesis. Using the Nebraska Transposon Mutant Library (NTML), we identified eight insertions that modulate extracellular levels of HysA activity. Insertions in the sigB operon, as well as in genes encoding the global regulators SarA and CodY, significantly increased HysA protein levels and activity. By altering the availability of branched-chain amino acids, we further demonstrated CodY-dependent repression of HysA activity. Additionally, through mutation of the CodY binding box upstream of hysA, the repression of HysA production was lost, suggesting that CodY is a direct repressor of hysA expression. To determine whether HysA is a virulence factor, a ΔhysA mutant of a community-associated methicillin-resistant S. aureus (CA-MRSA) USA300 strain was constructed and found to be attenuated in a neutropenic, murine model of pulmonary infection. Mice infected with this mutant strain exhibited a 4-log-unit reduction in bacterial burden in their lungs, as well as reduced lung pathology and increased levels of pulmonary hyaluronic acid, compared to mice infected with the wild-type, parent strain. Taken together, these results indicate that S. aureus hyaluronidase is a CodY-regulated virulence factor.
INTRODUCTION
Staphylococcus aureus is a leading cause of both hospital and community-associated infections in the developed world, accounting for $14.5 billion in health care costs annually in the United States alone (1). These infections range from mild skin and soft tissue infections to potentially fatal infections, such as pneumonia, endocarditis, osteomyelitis, sepsis, and toxic shock syndrome (2). Additionally, the increasing prevalence of methicillin-resistant S. aureus (MRSA) strains in both the hospital and community has exacerbated the problem (3–5). S. aureus is a successful pathogen due to its extensive arsenal of virulence factors that consist of both surface-associated proteins, such as microbial surface components recognizing adhesive matrix molecules (MSCRAMMs), and secreted proteins, including hemolysins, immunomodulators, and a number of exoenzymes (2, 3). Many emerging MRSA isolates are known for causing tissue-destructive infections (5, 6), and it has been proposed that the excessive damage is due, in part, to the large quantities of toxins and secreted enzymes produced by these new isolates (7).
Our studies have focused on the exoenzyme hyaluronidase (also called hyaluronate lyase), encoded by the gene hysA (8). Hyaluronidases are bacterial enzymes that cleave the β-1,4 glycosidic bond of hyaluronic acid (HA), a high-molecular-weight polymer composed of repeating disaccharide units of N-acetylglucosamine and d-glucuronic acid (9, 10). Hyaluronic acid is synthesized and secreted from the plasma membrane of mammalian cells, and it is abundant in the skin, skeletal tissue, umbilical cord, lungs, heart valves, brain, and a number of other tissues (9–11). Hyaluronic acid also performs a multitude of functions for the host, including providing structure to tissues and water homeostasis, assisting with cell proliferation, and acting as an immune regulator (10, 11). Many of these tissues with high HA concentrations are frequently infected with S. aureus (2).
Cleavage of HA by bacterial hyaluronidases occurs by β elimination of the β-1,4 glycosidic bond and results in unsaturated disaccharides as the product of complete digestion (9). In contrast, the eukaryotic hyaluronidases hydrolyze the β-1,4 glycosidic bond, producing mostly tetra- and hexasaccharides as end products. For a number of Gram-positive organisms, hyaluronidases have been shown to be essential virulence factors because of their ability to disseminate cells and virulence factors through tissue (9). For example, μ-toxin, a hyaluronidase of Clostridium perfringens, was found to significantly increase the ability of α-toxin to penetrate tissue (12). Similarly, a mutant with a mutation in the hyaluronidase of Streptococcus pyogenes showed reduced tissue penetration compared to the wild type in a murine skin abscess model (13). Additionally, there have been a number of structural studies on the hyaluronidases from Streptococcus pneumoniae and Streptococcus agalactiae (14–16), and the S. aureus enzyme is reported to have a structure similar to the structures of these streptococcal enzymes (9).
Reports of S. aureus hyaluronidase date back to pioneering 1933 studies performed by Duran-Reynals (17), who identified a spreading factor found in the spent medium of invasive S. aureus strains. This spreading factor increased the lesion size in a rabbit skin infection model, as measured by the spread of India ink (17), and it could increase the size of lesions caused by other bacterial pathogens. Subsequent reports concluded that the enzyme hyaluronidase was responsible for the spreading factor activity (18–21), but Hobby et al. suggested that other agents may contribute to the phenomenon (22). A number of follow-up studies investigated the enzymatic properties and expression of S. aureus hyaluronidase (23–25), but in the intervening years, progress stagnated until the hysA gene was identified and sequenced (26). More recently, the HysA enzyme was detected in proteomic studies (27) and shown to be required for increased wound size in a murine skin infection model (28). Little is known about hysA regulation, although SarA is thought to repress hysA gene expression, and there is some evidence that the agr quorum-sensing system induces expression (28, 29).
To expand our knowledge of HysA in S. aureus, we proceeded to investigate the regulation and the contribution of this enzyme to pathogenicity. We identified genes involved in the regulation of hysA utilizing a transposon mutant library, and the most prominent of these was the global regulator CodY. We characterized the role of HysA in the pathogenesis of S. aureus using a murine pulmonary infection model, and we present evidence that HysA promotes virulence through the breakdown of hyaluronic acid during infection.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The bacterial strains and plasmids used in this study are described in Table 1. All S. aureus strains were grown in tryptic soy broth (TSB) or tryptic soy agar (TSA) with the appropriate antibiotics for plasmid maintenance or selection (ampicillin, 100 μg/ml; chloramphenicol, 10 μg/ml; erythromycin, 10 μg/ml; tetracycline, 0.625 μg/ml) at 37°C with shaking at 220 rpm with a flask-to-volume ratio of approximately 5:1, unless otherwise specified. Hyaluronic acid sodium salt purified from Streptococcus equi was purchased from Sigma-Aldrich. Plasmid DNA was purified from Escherichia coli and electroporated into S. aureus RN4220 as previously described (30). Plasmids and transposon insertions were moved from S. aureus RN4220 or S. aureus JE2 with bacteriophage Φ11 to other S. aureus strains by transduction. All of the oligonucleotides used for this study can be found in Table 2.
TABLE 1.
Strains and plasmids
| Strain or plasmid | Description | Reference(s) or source |
|---|---|---|
| Strains | ||
| E. coli | ||
| BW25141 | Cloning strain | 58 |
| ER2566 | Protein expression strain | New England Biolabs |
| DH5α | Cloning/protein expression strain | Gibco |
| AH2856 | HysA protein expression strain, ER2566 containing pCR03 | This work |
| S. aureus | ||
| RN4220 | Restriction-deficient strain 8325-4 | 59 |
| AH1263 | USA300 CA-MRSA Erms (LAC) | 39 |
| AH2525 | AH1263 ΔhysA | This work |
| AH2839 | AH2525 containing pHysA | This work |
| JE2 | CA-MRSA USA300 Erms, plasmid-cured LAC derivative | 40 |
| AH2914 | JE2 hysA::ΦNΣ Ermr | 40 |
| AH2936 | JE2 sarA::ΦNΣ Ermr | 40 |
| AH2937 | JE2 sigB::ΦNΣ Ermr | 40 |
| AH2938 | JE2 rsbW::ΦNΣ Ermr | 40 |
| AH2939 | JE2 rsbU::ΦNΣ Ermr | 40 |
| AH2942 | JE2 SAUSA300_0543::ΦNΣ Ermr | 40 |
| AH2945 | JE2 hslU::ΦNΣ Ermr | 40 |
| AH2946 | JE2 codY::ΦNΣ Ermr | 40 |
| AH3052 | AH1263 Δspa | This work |
| AH3134 | AH1263 Δspa codY::ΦNΣ Ermr | This work |
| AH3207 | AH1263 Δspa codY::ΦNΣ Tetr hysA::ΦNΣ Ermr | This work |
| AH1292 | AH1263 Δagr::tetM | 60 |
| AH2032 | AH1263 Δagr::tetM ΔsigB | 60, 61 |
| AH1483 | AH1263 ΔsigB | 62 |
| AH3453 | AH1263 Δagr::tetM codY::ΦNΣ Ermr | This work |
| AH3172 | AH1263 ΔhysA codY::ΦNΣ Ermr | This work |
| Plasmids | ||
| pET28a | E. coli protein expression vector | Novagen |
| pJB38 | S. aureus gene knockout vector, Camr | 31 |
| pTet | Antibiotic cassette replacement vector, Camr Ampr | 32 |
| pSKerm-MCS | E. coli-S. aureus shuttle vector, Ermr Camr | 63 |
| pALC2073 | E. coli-S. aureus shuttle vector, Camr | 64 |
| pALC2073-RNAIII | Tetracycline-inducible RNAIII expression vector, Camr | 60 |
| pCR02 | pJB38 hysA knockout vector, Camr | This work |
| pHysA (pCR01) | hysA complementation plasmid, Camr Ermr | This work |
| pCR03 | hysA 6× His-tagged E. coli protein expression vector, Kanr | This work |
| pCR04 | pCR01 mutated CodY box, Camr Ermr | This work |
| pCR05 | pCR01 mutated CodY box, Camr Ermr | This work |
| pCR06 | pCR01 mutated CodY box, Camr Ermr | This work |
TABLE 2.
Oligonucleotides used in this study
The ΔhysA mutant was constructed using the pJB38-hysA modified pKOR1 system as described previously (31). Briefly, 600-bp flanking regions upstream and downstream of hysA were amplified and joined by overlap PCR extension using genomic DNA from strain AH1263 as the template and oligonucleotides CBR12 and CBR13 to generate the upstream fragment and oligonucleotides CBR14 and CBR15 to generate the downstream fragment. The outermost primers (CBR12 and CBR15) were engineered to include SbfI and XmaI sites at their 5′ ends, respectively, and the inner primers (CBR13 and CBR14) were similarly engineered with XhoI and MluI sites at their 5′ ends to aid with the overlap extension. PCR products were ligated with T4 DNA ligase (Invitrogen) into pJB38 at the SbfI and XmaI sites and electroporated into E. coli strain BW25141. Plasmid DNA was purified from this strain, isolated using a high-speed plasmid minikit (IBI Scientific), and electroporated into RN4220. The plasmid construct was isolated from S. aureus RN4220 and transduced into AH1263 using bacteriophage Φ11. The hysA mutation was constructed on the chromosome as previously outlined in the pJB38 method (31). The final colonies were screened for plasmid loss by determination of antibiotic susceptibility, and loss of the plasmid was confirmed by PCR.
The complementing plasmid pHysA (pCR01) was created by amplifying the hysA promoter region (∼600 bp upstream of the putative translational start site) through the stop codon with the flanking primers CBR35 and CBR29. Amplified DNA containing the hysA coding sequence and the promoter region was restriction digested with BamHI-HF and NheI-HF (New England BioLabs) and ligated into linearized pSKerm-MCS plasmid DNA at the BamHI and NheI sites. Ligated DNA was electroporated into E. coli strain BW25141 using standard molecular techniques.
To create the HysA overexpression construct pCR03, the hysA gene without the encoded signal sequence was amplified from AH1263 genomic DNA using primers CBR41 and CBR42, which contain NheI and XhoI sites at their 5′ ends, respectively. The resulting PCR product was ligated into pET28a linearized with NheI and XhoI and electroporated into E. coli strain ER2566 to generate strain AH2856.
To mutate the CodY binding box upstream of hysA, plasmids pCR04, pCR05, and pCR06 were constructed using overlap PCR to engineer the six nucleotide changes. A 5′ region introducing the mutations into the CodY box upstream of hysA was amplified using CBR35 and either CBR83, CBR85, or CBR87. Using homologous primers to introduce the same mutations and amplify the remainder of the hysA promoter region and hysA, a corresponding 3′ region was generated using CBR29 and either CBR82, CBR84, or CBR86. The 5′ and 3′ fragments were mixed at a ratio of 1:1 in combinations, as follows: CBR35-CBR83–CBR82-CBR29, CBR35-CBR85–CBR84-CBR29, and CBR35-CBR87-CBR86-CBR29. Overlap PCR was done using the mixed fragments as a template and CBR29 and CBR35 to amplify hysA and the promoter region containing the mutated CodY boxes. pSKermMCS and the PCR product inserts were restriction digested with BamHI-HF and NheI-HF and ligated using T4 DNA ligase. The ligations were transformed into E. coli strain DH5α as previously described. The resulting plasmids were confirmed by sequencing.
The spa mutant was generated using the same protocol for deleting hysA described above. Flanking regions of spa (SAUSA300_0113) were amplified with primer pair spa_delA_EcoRI and spa_delB and primer pair spa_delC and spa_delD_SalI. The products were fused using overlap extension PCR with primers spa_delA_EcoRI and spa_delD_SalI. The resulting fragment was digested with EcoRI and SalI and ligated into pJB38. The spa deletion was confirmed by PCR with primers spa_upstream and spa_downstream. The construct with the spa and codY double mutation was generated by transducing the codY::ΦNΣ cassette into S. aureus strain LAC Δspa with bacteriophage Φ11. To generate strain AH3207, the pTet marker switching plasmid was used to replace the erythromycin resistance cassette of the codY::ΦNΣ transposon insertion in S. aureus strain AH2946 with a tetracycline resistance cassette, as described by Bose et al. (32). This construct was then transduced by bacteriophage Φ11 into the spa deletion strain that also harbored the hysA::ΦNΣ transposon insertion.
Protein purification.
AH2856 was grown overnight at 37°C in LB medium containing kanamycin (50 μg/ml) and subcultured into 1 liter of fresh LB medium at a ratio of 1:250. The culture was grown at 30°C to mid-logarithmic phase, IPTG (isopropyl-β-d-thiogalactopyranoside) was added to a 1 mM final concentration, and the culture was allowed to grow for an additional 4 h. Cells were harvested by centrifugation, washed once with water, pelleted by centrifugation, and frozen at −20°C. Cells were mechanically lysed using a Microfluidics microfluidizer (model LV1; Newton, MA) at 25,000 lb/in2 and running the samples through the machine twice. Cell lysate was clarified by centrifugation for 20 min at 15,000 × g and 4°C. Protein was purified using Fractogel His-Bind resin (Novagen) per the manufacturer's instructions, dialyzed into phosphate-buffered saline (PBS), concentrated using a Millipore centrifugal filter unit (30,000-molecular-weight cutoff), and brought to 10 mg/ml in 1× PBS. The protein suspension was frozen in 10% glycerol at −20°C until used.
Transposon library screen.
The Nebraska Transposon Mutant Library (NTML) was grown overnight in TSB in 96-well microtiter plates. Cultures were stamped onto hyaluronic acid (HA) plates (TSB, 1% agarose, 0.4 mg/ml HA, 1% [wt/vol] bovine serum albumin [BSA]), prepared as described previously (8), and incubated at 37°C for 4 h. Acetic acid (10%) was added to the plates to visualize zones of clearing. Plates were scored for their hyaluronidase activity, and mutations that increased or decreased activity were rescreened in the same manner. Mutations that affected activity in both screens were quantitatively assessed for hyaluronidase activity by growing each strain overnight in TSB, subculturing to an optical density at 600 nm (OD600) of 1.0, and dispensing the diluted culture as five aliquots (4 μl) onto HA plates. After 6 h of incubation, the plates were flooded with 10% acetic acid, and zones resulting from HA cleavage were enumerated with a caliper.
Hyaluronidase activity assay.
The hyaluronidase activity assay was performed as previously described (28), with some modifications. Briefly, S. aureus strains were grown overnight in TSB at 37°C with shaking, subcultured 1:1,000, and grown at 37°C for the time specified below. Spent culture medium was isolated with Spin-X filters (pore size, 0.22 μm) and frozen at −20°C until assayed. Hyaluronic acid (100 μl at 1 mg/ml) was mixed with 50 μl of spent medium and allowed to react at 37°C for 15 min. The reaction was stopped by adding 25 μl of potassium tetraborate solution (0.8 M, pH 9.1), and the reaction mixture was vortexed and boiled for 3 min. In parallel, 12.5 μl of spent medium was added to 31.25 μl of stop solution (0.8 mg/ml hyaluronic acid, 0.8 M potassium tetraborate, pH 9.1) at time zero, vortexed, and boiled for 3 min. The samples were dispensed into a 96-well microtiter plate in quadruplicate, and freshly prepared DMAB solution (10% [wt/vol] p-dimethylaminobenzaldehyde, 12.5% [vol/vol] 10 M HCl [Sigma-Aldrich], and 87.5% [vol/vol] glacial acetic acid [Fisher Scientific]) was added to each well. The plate was incubated at 37°C for 20 min to allow the color reaction to take place. A Tecan Infinite M200 plate reader was used to measure the absorbance at 590 nm. Hyaluronidase specific activity is expressed as 103 × the number of μmol N-acetylglucosamine (NAG; Sigma-Aldrich) released ml−1 min−1 per OD600 unit and is calculated by the equation 103 × 3 × (1/15) × (1/m) × ΔA590 × (1/OD600), where ΔA590 represents the difference in absorbance between the readings at 15 min and time zero for each sample, m represents the slope of the standard curve of NAG, OD600 is the optical density of the culture when the sample was taken, 3 is the dilution factor of the sample tested in substrate, and 1/15 is the reciprocal of the reaction time allowed at 37°C. The assay was performed in triplicate for each condition tested.
To determine the impact of RNAIII on hyaluronidase activity, strains with pALC2073 or pALC2073-RNAIII were grown overnight in TSB at 37°C and were subcultured 1:1,000 into fresh TSB with anhydrotetracycline (aTc) at various concentrations. Cultures were grown for 8 h, cells were pelleted, and spent medium was filtered and assayed for activity.
Assays with limiting to rich medium.
Chemically defined medium (CDM) was made as described by Pohl et al. (33), except that the group 6 vitamin solution was replaced with nicotinamide (500 μg/liter), thiamine (500 μg/liter), pantothenate (500 μg/liter), and biotin (500 μg/liter) (final concentrations). Single amino acids were omitted or added in excess, as indicated in the figure legends. The medium was buffered to pH 7.0. S. aureus strains were grown overnight in TSB and subcultured 1:1,000 into CDM or CDM lacking isoleucine. Bacteria were grown for 5 h, at which point cells were pelleted, washed once with 1× PBS, and suspended into fresh CDM. For those cells grown in CDM lacking isoleucine, they were then suspended in fresh CDM containing 5× branched-chain amino acids (BCAAs; isoleucine, valine, leucine). Cells were grown in the new medium for an additional 2 h. Growth was monitored spectrophotometrically at 600 nm throughout by measuring the optical density of 100 μl of culture in a 96-well microtiter plate and a Tecan infinite M200 plate reader. Additionally, 200 μl of each culture was removed at the designated time points indicated in the figures, and spent medium was isolated using 0.22-μm-pore-size Spin-X centrifuge tube filters (Costar). Spent media were used to determine hyaluronidase activity and the presence or absence of Panton-Valentine leukocidin (PVL) (see below).
Protein immunoblot for PVL.
Spent culture medium isolated from cultures of cells grown in CDM, CDM lacking isoleucine, or CDM with 5× BCAAs as described above (5 μl for 4 h or 1 μl for the other time points) was brought to a total volume of 10 μl in SDS loading buffer containing 1% β-mercaptoethanol. Proteins were resolved by PAGE using Tricine-SDS gels containing 12% polyacrylamide and 170 kV. Proteins were transferred (160 mA for 1 h) onto nitrocellulose membranes and blocked overnight at 4°C in 0.1% Tris-buffered saline containing 0.1% Tween 20 (TBST) and 5% skim milk. The membrane was probed for PVL using Luk-S polyclonal antibody (IBT Bioservices, Gaithersburg, MD) at 1:10,000 in TBST containing 5% skim milk for 2 h at room temperature and then washed three times for 10 min each time with TBST. Peroxidase-conjugated AffiniPure goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) was applied at 1:20,000 in blocking buffer at room temperature for 1 h. Membranes were washed three times for 10 min each time with 0.1% TBST, and SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) was added to detect proteins, followed by exposure to Classic X-ray film (Research Products International Corporation). Bands were quantified using ImageJ software.
Protein immunoblot for HysA.
S. aureus strains AH3052, AH3134, and AH3207 were grown overnight in TSB at 37°C with shaking at 220 rpm. The strains were subcultured at a ratio of 1:100 into fresh TSB and grown for 8 h at 37°C with shaking at 220 rpm. Cultures were normalized to an OD600 of 3.0 with TSB, spent media were isolated by filtration using a 0.22-μm-pore-size Millex-GS syringe filter (Millipore), concentrated using Amicon Ultra 10,000-molecular-weight-cutoff centrifugal filters (Millipore), and frozen at −20°C. The 10-μl samples were brought to a total volume of 15 μl in SDS loading buffer containing 1% β-mercaptoethanol, and proteins were resolved by PAGE using Tricine-SDS gels containing 9% polyacrylamide and 170 kV. Proteins were transferred (160 mA for 1 h) to nitrocellulose membranes and blocked overnight at 4°C in blocking buffer (0.1% TBST containing 5% skim milk). A rabbit polyclonal antibody to HysA was generated by Pacific Immunology, and the antibody was purified as described above. HysA was probed using a dilution of 1:40,000 with the anti-HysA rabbit serum for 1 h at room temperature in blocking buffer. The membrane was then washed three times for 10 min each time in 0.1% TBST. Peroxidase-conjugated AffiniPure goat anti-rabbit IgG was applied at a 1:20,000 dilution in blocking buffer at room temperature for 1 h. Membranes were washed three times for 10 min each time with 0.1% TBST, and SuperSignal West Pico chemiluminescent substrate was added to detect proteins, followed by exposure to Classic X-ray film.
Neutropenic murine pneumonia model.
Female BALB/c mice were purchased from the National Cancer Institute (Frederick, MD). All animals were housed under specific-pathogen-free conditions in filter-top cages at the Walter Reed Army Institute of Research (WRAIR) Animal Facility and provided sterile food and water ad libitum. Six- to 8-week-old mice were injected intraperitoneally with 150 mg/kg of body weight of cyclophosphamide (Baxter) 4 days prior to infection and 100 mg/kg of cyclophosphamide 1 day prior to infection, as previously described (34). Mice were anesthetized with isoflurane and infected intranasally with 1.0 × 107 CFU of S. aureus suspended in 25 μl of PBS. At 48 h postinfection, the mice were euthanized and lungs were extracted, homogenized, and serially diluted before suspensions of lung tissue were plated onto TSA. The CFU were enumerated to assess the bacterial burden. For survival studies, infected mice were monitored twice a day for signs of morbidity. Following sacrifice, paraffin-embedded lung tissue samples from infected and uninfected mice were sectioned and stained with hematoxylin-eosin (H&E). All procedures were performed in accordance with a protocol approved by the WRAIR Institutional Animal Care and Use Committee (protocol number IB02-10). All animal research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to the principles stated in the Guide for the Care and Use of Laboratory Animals (35).
Immunohistochemistry.
The lungs of PBS-treated and infected mice were harvested at 48 h postinfection, fixed in 4.0% paraformaldehyde, and cut into 5-μm sections. As a control, uninfected lung tissue samples were pretreated with 100 turbidity reducing units (TRU) of Streptomyces hyalurolyticus hyaluronidase (Sigma) for 2 h at 60°C (36). Hyaluronic acid was labeled as previously described (37). Briefly, samples were incubated in 0.2% BSA for 2 h at 4°C to block nonspecific binding. Following incubation, samples were treated with 2 μg/ml of biotinylated hyaluronic acid binding protein (EMD Biosciences) for 16 h at 4°C. Fluorescein isothiocyanate-conjugated streptavidin (Invitrogen) was used to visualize hyaluronic acid. The presence of hyaluronic acid in lung sections was quantified by measuring the mean fluorescent intensity of 40 fields per section. Images were acquired with an Olympus AX80 microscope and analyzed with Image Pro (version 7.0) software.
Statistical analysis.
The statistical significance of all data was analyzed using the Student t test with GraphPad Prism (version 6) software (GraphPad, La Jolla, CA), except for data from survival studies. Data from the survival studies were analyzed by the Mantel-Cox test (P = 0.0022).
RESULTS
The gene encoding hyaluronidase (hysA) is conserved across all S. aureus clonal lineages (8), and only one hyaluronidase is carried by the genome of a community-associated MRSA (CA-MRSA) USA300 strain (38). The hysA gene (SAUSA300_2161) is located near a number of genes of unknown function and is transcribed by itself in the opposite direction of neighboring genes (Fig. 1A). To begin our studies on hysA, we created a markerless deletion construct in strain AH1263 (39), an erythromycin-sensitive version of the USA300 isolate LAC (here called LAC wild type [LAC-WT]). The ΔhysA knockout mutant was confirmed by PCR and a lack of enzymatic cleavage of HA by an agar plate assay (Fig. 1B). We also verified the absence of hyaluronidase activity in the knockout mutant using a quantitative, spectrophotometric assay (Fig. 1C). The HysA activity was complemented to LAC-WT levels by providing hysA on a plasmid (Fig. 1), demonstrating that the phenotype was due to the mutant construction and not secondary or polar effects.
FIG 1.
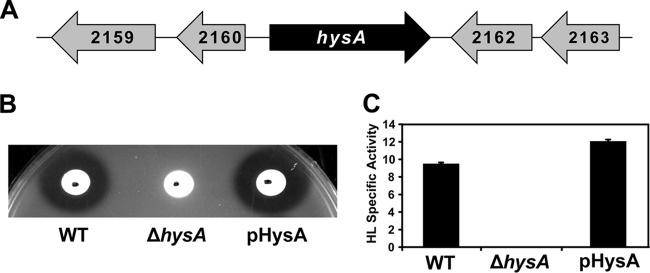
Generation and initial characterization of the CA-MRSA USA300 ΔhysA mutant and complemented strains. (A) Location of hysA on the CA-MRSA USA300 LAC chromosome. Numbers indicate SAUSA300 locus numbers. The hysA gene is surrounded by genes with unknown function and is transcribed by itself on the chromosome. (B and C) HA plate and hyaluronidase (HL) specific activity assays of LAC-WT, LAC ΔhysA, and complemented (pHysA) strains. As shown, HysA activity is abolished for the ΔhysA mutant and restored to WT levels when it is complemented in both assays.
The regulation of hysA has received only limited attention, although there are reports for the potential involvement of global regulators sarA and agr (28, 29). To identify genetic loci that contribute to hysA regulation, we screened 1,952 mutants from the Nebraska Transposon Mutant Library (NTML) (40) for hyaluronidase activity using an HA plate assay. Screening of the NTML resulted in the identification of eight bursa aurealis transposon insertions that significantly altered hyaluronidase activity. To determine the relative extent to which each mutation affected activity, five replicates of each mutant were spotted on HA plates and the zone size was enumerated with a caliper (Fig. 2A). NTML WT strain JE2 had an average zone size of 1.01 ± 0.02 cm, while a mutation in hysA resulted in no detectable zone of clearing. Multiple strains with insertions in the sigma factor B operon (sigB, rsbU, and rsbW) and strains with insertions in sarA, hslU, and codY had elevated hyaluronidase activity. Interestingly, insertion into the putative deaminase, SAUSA300_0543, gave a reduced zone of clearing of HA (0.76 ± 0.03 cm) and was the only mutation in the library besides hysA to cause the strain to have a zone size smaller than the WT zone size.
FIG 2.

Transposon mutagenesis library screen for altered HysA activity. (A) Summary of results of screen for HysA activity on HA agarose plates. Values of HysA activity are reported as diameters (cm) of zones of clearing, measured with a caliper, and represent the means ± standard deviations of five technical replicates for each strain plated on HA agarose. Pictures are representative images of each strain. (B) Hyaluronidase specific activity assay of strains containing mutations that significantly altered HysA activity on HA agarose plates. The data are presented as the fold changes compared to the activity of USA300 WT strain JE2. Values represent the means and standard deviations of four technical replications for three independent biological determinations. Statistical significance (****, P < 0.0001) was determined by Student's t test.
To quantitatively measure the effect of the identified transposon insertions on hyaluronidase activity, we performed a spectrophotometric specific activity assay on spent medium of 8-h cultures (Fig. 2B). In support of the results of the HA plate assay, the codY transposon mutant was found to have activity that was increased 12-fold compared to that of the WT. Additionally, multiple insertions in the sigB operon, as well as one in hslU, increased the activity approximately 3-fold, and an insertion in sarA resulted in a 10-fold increase. The mutant with the transposon insertion in SAUSA300_0543 had one-third of the activity of the WT (Fig. 2B), similar to the reduction in activity observed in the HA plate assay. As expected, no activity by the hysA transposon mutant was observed in this assay.
Despite previous reports of agr quorum-sensing involvement in hysA regulation (28), we did not find any insertions in the agr locus that significantly altered hyaluronidase activity in our screen. We hypothesized that this was perhaps due to the fact that S. aureus strain 8325-4, which has a known mutation in rsbU that results in a defect in SigB activity, was used in the previous study. As shown in Fig. 2, we found multiple mutations in the sigB operon, including a mutation in rsbU, that increase hyaluronidase activity. Thus, a role for agr could be masked in the USA300 background that has an intact rsbU gene. To investigate this question, we shifted our studies to the USA300 LAC (LAC-WT) background, and we quantified HysA activity in LAC-WT as well as the agr and sigB single mutant and agr sigB double mutant strains. We found that HysA activity was unchanged in an agr mutant and increased significantly in a sigB mutant. Comparing the sigB agr double mutant to the sigB mutant (Fig. 3A), there was a significant decrease in activity when agr was deactivated. This observation is in agreement with published reports (28), and we also made similar observations in the USA400 MW2 background (data not shown), indicating that this effect occurs across multiple S. aureus strain types.
FIG 3.

Role of the agr system in hysA gene regulation. Cultures of various LAC strains were grown for 8 h, spent medium was prepared, and the hyaluronidase specific activity in each sample was measured. (A) To assess the agr and SigB interaction, activity was measured in the WT, agr and sigB single mutant, and agr sigB double mutant strains and plotted. (B) To assess the agr and CodY interaction, activity was measured in the WT, agr and codY single mutant, and codY agr double mutant strains. For panels A and B, statistical significance (****, P < 0.0001) was determined by Student's t test. (C) Hyaluronidase specific activity for the WT containing the empty vector (black bars) or RNAIII under the control of a tetracycline-inducible promoter (gray bars). Student's t test was used to determine statistical significance (**, P < 0.01; ****, P < 0.0001). (D) Model for SigB and CodY regulation of HysA through the agr system.
CodY is reported to regulate the agr system (41, 42), and we hypothesized that some hysA regulation might be occurring indirectly through the known CodY-agr interactions. To address this question, we initially confirmed that the effect of CodY was consistent in the LAC background by crossing the transposon insertion into LAC-WT, and we observed that HysA activity increased 16-fold in the codY mutant over that in LAC-WT (data not shown). Next, we quantified HysA activity in LAC-WT, as well as in the agr and codY single mutant and agr codY double mutant strains (Fig. 3B). As anticipated, HysA activity was unchanged in an agr mutant and increased in a codY mutant. However, HysA activity was significantly decreased in the agr codY double mutant compared to that in the codY mutant (Fig. 3B). We speculated that the unnaturally high levels of RNAIII in the codY mutant could be distorting HysA production. To test this question, we transformed USA300 WT with a plasmid that expresses RNAIII under the control of a tetracycline-inducible promoter. In the absence of the inducer, there was essentially no difference in HysA activity between this strain and an empty vector control (Fig. 3C). However, HysA activity increased with the induction of RNAIII expression compared to the activity of the control (Fig. 3C). Collectively, these data lead us to suggest the model depicted in Fig. 3D for CodY and agr-mediated regulation of HysA.
In considering the regulatory model (Fig. 3D), it still remained possible that CodY directly regulated hysA gene expression. The CodY regulon has been investigated previously (41–43), and hysA was not identified as a target in any of these studies. We inspected the sequence upstream of hysA for the presence of the previously published CodY consensus binding box (41), and we found a well-conserved CodY binding box 202 bp upstream of the putative translational start site (Fig. 4A), with one nucleic acid change from a cytosine to a thymine being found in the region between the inverted repeats. The presence of this box led us to predict that CodY may interact with the hysA putative promoter region. To confirm that we were not being misled by the results of the hyaluronidase activity assays, we tested to see if the HysA protein levels increased accordingly in a codY mutant. We used an antibody to HysA that was developed and found by immunoblotting that enzyme levels were increased in a codY mutant compared to those in the WT strain (Fig. 4B). To eliminate background binding in the immunoblot, protein A was inactivated in each of these strains. The band corresponding to HysA was not detected in a codY hysA double mutant, demonstrating that the antibody was specific and suggesting that the increase in hyaluronidase activity is due to elevated HysA levels. Also of note, HysA was not stable in spent medium, and we frequently observed lower-molecular-weight breakdown products by immunoblotting (Fig. 4B).
FIG 4.

The CodY binding box is required for hysA regulation. (A) Location of the putative CodY binding box in the hysA promoter region. An alignment of the consensus CodY binding box and the proposed hysA box is shown. (B) Immunoblotting for HysA performed with a LAC-WT strain. Spent medium was concentrated 20- or 50-fold from the WT and the codY and codY hysA mutant strains, as indicated, and immunoblotted for HysA. In each strain, a Δspa deletion was engineered to remove background antibody binding. (C) Mutations generated in the CodY box in plasmids pCR04, pCR05, and pCR06. (D) Hyaluronidase specific activity assay at 8 h for the WT (LAC ΔhysA with one of the indicated plasmid constructs) or a codY mutant (the LAC ΔhysA codY double mutant with one of the indicated plasmid constructs) containing the pHysA, pCR04, pCR05, or pCR06 construct. Statistical analysis was performed using a Student's t test (****, P < 0.0001).
To assess whether CodY was acting to directly regulate hysA, we constructed plasmids with CodY binding box mutations in the hysA promoter region (Fig. 4C). As our testing system, we used the LAC ΔhysA mutant with plasmid pHysA, which complements hyaluronidase activity to nearly WT levels (Fig. 1C). Using pHysA as a backbone, we introduced five to six nucleotide substitutions from the consensus sequence into the CodY box, and these were based on previous studies in Bacillus subtilis, where this number of changes was sufficient to disrupt CodY-based repression (44). These constructs were tested by transforming them into the LAC ΔhysA mutant or ΔhysA codY double mutant and measuring HysA activity from plasmid expression. We first introduced A-to-C and T-to-C changes into the first half site of the box in constructs pCR04 and pCR05 (Fig. 4C). In testing these constructs, the CodY-based repression of HysA activity was reduced markedly to approximately 2-fold (Fig. 4D). We made additional, independent nucleotide changes in the second box half site in construct pCR06 (Fig. 4C), and CodY-based repression of HysA activity was completely eliminated (Fig. 4D). These results provide strong support for the suggestion that CodY directly regulates hysA expression through binding to the box identified in the promoter region.
Finally, we took advantage of the CodY response to the availability of branched-chain amino acids (BCAAs) to test the possibility that CodY represses HysA production in response to environmental changes. CodY gains a high affinity for target promoters when BCAAs are available, leading to repression of its targets. When BCAAs are limiting, such as when cells are in stationary phase or under starvation, CodY loses this affinity, resulting in relieved repression of CodY targets (45, 46). We grew LAC-WT in chemically defined medium (CDM) lacking isoleucine or in CDM with standard levels of BCAAs until the cells began to enter stationary phase (Fig. 5A). If hysA is under CodY control, we would anticipate increased HysA activity in the cells grown without isoleucine compared to that in cells grown in CDM with standard levels of BCAAs. In support of this hypothesis, there was a more than 2.5-fold increase in hyaluronidase activity when BCAAs were limiting compared to that when bacteria were grown in CDM at both 4 and 5 h (Fig. 5B).
FIG 5.

Effect of nutrient availability on hysA regulation. (A) Growth curve of LAC-WT grown in CDM or in CDM lacking isoleucine for the first 5 h. Cells were harvested by centrifugation, washed 1 time in PBS, and suspended in either fresh CDM or CDM containing 5× BCAAs. Arrows, times when samples were taken or where medium was switched, as indicated. (B) Hyaluronidase specific activity of samples taken at the time points indicated in panel A. Statistical analysis was performed using Student's t test (****, P < 0.0001). (C) Immunoblot for the LukS subunit of PVL at the time points indicated in panel A. (D) Quantification of LukS immunoblot using ImageJ software. post, samples taken at time points after the medium switch.
We also tested the impact of high levels of BCAAs on CodY repression. The cells grown in medium lacking isoleucine were switched to medium that contained excess BCAAs. If hysA is under CodY control, we would anticipate that the excess BCAAs would bind to CodY and repress expression. As predicted, when BCAAs were supplied in excess, HysA production dropped over 5-fold compared to that in cells grown in CDM (Fig. 5B). These activity changes were not due to differences in growth (Fig. 5A). As a control, samples were taken from each time point and immunoblot assays for Panton-Valentine leukocidin (PVL) (Fig. 5C and D), which is known to be regulated by CodY in USA300 lineages, were performed (47). The trends in PVL production mirrored those of HysA production under all the conditions tested, suggesting that they are regulated in a similar manner.
To determine whether HysA is required for virulence, neutropenic BALB/c mice were infected intranasally with 1 × 107 CFU of either LAC-WT or the ΔhysA mutant. After 4 days of infection, ∼80% of mice infected with LAC-WT succumbed to infection, which is in striking contrast to the death of only ∼20% of mice infected with the ΔhysA mutant (Fig. 6A). At 48 h postinfection, a 4-log-unit reduction in the bacterial burden was observed in the lungs of mice infected with the ΔhysA mutant compared to the bacterial burden in the lungs of mice infected with LAC-WT (Fig. 6B), suggesting that the virulence defect is likely caused by an inability of the mutant to replicate in the lungs of infected mice.
FIG 6.

HysA is required for USA300 virulence. Mice were intranasally inoculated with 1.0 × 107 CFU of either LAC-WT, its ΔhysA mutant strain, or PBS. (A) Mice (n = 27) were monitored daily for signs of morbidity and mortality. **, P = 0.0022, Mantel-Cox test. (B) Groups of mice (n = 21) were euthanized after 48 h, and the bacterial burden in the lungs was quantified by CFU enumeration. **, P = 0.0095. (C) Histopathology of the lungs from mice treated with PBS or infected with either LAC-WT or the ΔhysA mutant. Black arrowheads, areas shown in the enlarged images in the insets. Data are representative of those from three independent experiments.
The hyaluronidase activity of multiple Gram-positive pathogens has been shown to cause severe tissue damage in vivo (12, 13, 28). Therefore, we investigated whether the loss of virulence of the ΔhysA mutant strain was associated with reduced tissue damage in the lung. Histological analysis showed that the lungs of mice infected with the ΔhysA mutant exhibited less pathology and influx of alveolar macrophages than the lungs of LAC-WT-infected mice (Fig. 6C). To determine whether hyaluronidase activity contributed to the virulence of S. aureus in vivo, HA was fluorescently labeled and quantified in the lungs of infected mice. There was a significant increase in the amount of HA (P < 0.0001) present in the lungs of ΔhysA mutant-infected mice compared with the amount present in the lungs of LAC-WT-infected mice (Fig. 7A and B). In fact, the level of HA present in the lungs of wild-type-infected mice was similar to that found in the lungs of mice treated with purified hyaluronidase from Streptomyces hyalurolyticus (Fig. 7C). Taken together, these findings show that hysA is an important determinant as a cause of S. aureus infection.
FIG 7.

HysA is required for the breakdown of hyaluronic acid in vivo. (A and B) Micrographs of immunofluorescently labeled HA in the lungs of mice inoculated with 1.0 × 107 CFU of either strain LAC-WT or its ΔhysA mutant strain. (C) The mean fluorescence intensity was quantified for each sample (40 fields per section). A sample treated with hyaluronidase from Streptomyces hyalurolyticus (Strep Hys) was also included as a control. NS, not significant; ****, P < 0.0001.
DISCUSSION
Hyaluronidases are thought to act as spreading factors, allowing dissemination of pathogens throughout mammalian tissue during infection (9). In fact, the earliest studies on spreading factors described them as agents that increase lesion size when combined with various bacterial and viral pathogens (17, 21). In this report, we investigated the regulation and in vivo function of HysA during S. aureus infection using a CA-MRSA USA300 strain, and we found that HysA is indeed an important factor for causing an infection.
Through our screen of the NTML, we identified eight mutations that significantly altered HysA activity, including a mutation in sarA, multiple mutations in the sigB operon, and a mutation in an uncharacterized nucleoside deaminase encoded by the SAUSA300_0543 gene. The deaminase was the only target identified that is required for the full expression of HysA. Surprisingly, we did not identify mutations in the agr locus during the screen, although it was reported that agr is required for HysA production (28). Considering that the previous studies were done with a strain with a natural mutation in rsbU, which is known to mimic a sigB mutation and elevate agr function (48), we investigated this question. When an agr deletion was introduced into a sigB mutant, we observed a drop in HysA activity (Fig. 3A), supporting the findings of the previous studies. Taken together, when the agr system is hyperactivated, HysA levels increase, and by removing the agr locus, these levels drop, which supports the previously identified agr-dependent regulation. We speculated that this agr role occurred only at unnaturally high levels of RNAIII, and we confirmed this by expressing RNAIII from a plasmid and boosting HysA output. Thus, agr has a role in HysA production, but it seems to be important only in situations where RNAIII is elevated beyond normal WT levels.
Interestingly, our screen of the NTML revealed that a mutation in the global regulator CodY resulted in increased HysA activity. CodY responds to intracellular concentrations of branched-chain amino acids in S. aureus to control target gene expression (33, 41, 42, 45). We initially speculated that the CodY regulation might be indirect through the agr system, and indeed, the introduction of an agr deletion reduced the level of HysA production in a codY mutant (Fig. 3B). However, we observed that the hysA promoter region had a nearly perfect CodY binding box (Fig. 4A), and importantly, nucleotide changes to the conserved positions of the box eliminated CodY-based repression (Fig. 4C and D). Thus, there are indirect and direct mechanisms of CodY-dependent regulation occurring to coordinate HysA production in S. aureus.
CodY-dependent regulation suggests that HysA has a potential nutrient-scavenging function. HA is a major component of the viscous substance produced by connective tissues, and it acts as a host defense mechanism by creating a protective barrier that impedes the penetration of bacteria and bacterial products into deeper tissues (49). Sequestration of essential nutrients, such as iron (50) and tryptophan (51), is another mechanism of bacterial infection prevention that works through the generation of nutrient-limited environments. Bacterial pathogens can rapidly adapt to these hostile situations by regulating the expression of genes important for virulence and metabolism (52), and our findings suggest that S. aureus uses CodY to regulate HysA in this manner. HA can be broken down by hyaluronidases produced by a variety of bacterial pathogens, such as Streptococcus pyogenes, Streptococcus pneumoniae, and Mycobacterium tuberculosis (9), and metabolized as a source of carbon (53–55). At this time, it has not been clearly demonstrated that S. aureus can use HA as a carbon source in vivo or whether HysA is involved in this growth mechanism. Both questions are in need of further examination. However, it is possible that the reduction in bacterial burden in a ΔhysA mutant during pulmonary (Fig. 6B) and soft tissue (28) infections can be explained by an inability of S. aureus to utilize an alternative carbon source under nutrition-limited conditions in vivo.
This finding is consistent with the findings of previous work showing that CodY regulates other virulence factors to act as a link between metabolism and virulence (33). For example, the hemolysins (α and β), as well as the secreted proteases SspA and SspB, have been shown to be regulated directly by CodY. HysA was required for the breakdown of HA in vivo, and this correlated with a loss of structural integrity and enhanced necrosis of pulmonary tissues, allowing the pathogen access to deeper tissues. In addition to HysA, S. aureus possesses other mechanisms for penetrating the host. One example is a secreted cysteine protease, staphopain A, which is able to induce vascular leakage during a guinea pig infection (56), and this effect can be augmented by the addition of another secreted cysteine protease, staphopain B. Additionally, lipases have been observed to enhance S. aureus tissue penetration (57). Taken together these results indicate that HysA is part of a class of S. aureus secreted proteins that act in a coordinated fashion as spreading factors by breaking down host tissues to facilitate bacterial dissemination (22). Interestingly, the target for each enzyme differs, despite the functional redundancy of these proteins. Considering the dynamic range of infections caused by S. aureus, these findings lead us to speculate that these spreading factors may be a part of a fine-tuned virulence mechanism utilized by this pathogen to nonspecifically break down host barriers and promote dissemination from any site of infection.
Our studies give new insights into the regulation and role of the S. aureus virulence factor HysA. This work identified an important link between metabolism and virulence by illustrating a relationship between the metabolic regulator CodY and the virulence factor HysA. By therapeutic targeting of HysA and other spreading factors, it might be possible to prevent localized infections from evolving into more complicated, systemic disease.
ACKNOWLEDGMENTS
We thank Heidi Crosby for assistance with strain construction.
We acknowledge National Institutes of Health training grant 5T32GM008365-22 and NIH/NIDCR award T90 DE023520 for supporting C.B.I. during this work. The D. V. Zurawski lab is supported by multiple grants from the Military Infectious Diseases Research Program (MIDRP) and the Defense Medical Research and Development Program (DMRDP).
The findings and opinions expressed herein belong to the authors and do not necessarily reflect the official views of the WRAIR, the U.S. Army, the U.S. Department of Defense, or the U.S Food and Drug Administration.
Footnotes
Published ahead of print 28 July 2014
REFERENCES
- 1. Noskin GA, Rubin RJ, Schentag JJ, Kluytmans J, Hedblom EC, Jacobson C, Smulders M, Gemmen E, Bharmal M. 2007. National trends in Staphylococcus aureus infection rates: impact on economic burden and mortality over a 6-year period (1998-2003). Clin. Infect. Dis. 45:1132–1140. 10.1086/522186 [DOI] [PubMed] [Google Scholar]
- 2. Lowy FD. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339:520–532. 10.1056/NEJM199808203390806 [DOI] [PubMed] [Google Scholar]
- 3. Gordon RJ, Lowy FD. 2008. Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clin. Infect. Dis. 46(Suppl. 5):S350–S359. 10.1086/533591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. David MZ, Daum RS. 2010. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 23:616–687. 10.1128/CMR.00081-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DeLeo FR, Chambers HF. 2009. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J. Clin. Invest. 119:2464–2474. 10.1172/JCI38226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chambers HF, Deleo FR. 2009. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 7:629–641. 10.1038/nrmicro2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li M, Diep BA, Villaruz AE, Braughton KR, Jiang X, DeLeo FR, Chambers HF, Lu Y, Otto M. 2009. Evolution of virulence in epidemic community-associated methicillin-resistant Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 106:5883–5888. 10.1073/pnas.0900743106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hart ME, Hart MJ, Roop AJ. 2009. Genotypic and phenotypic assessment of hyaluronidase among type strains of a select group of Staphylococcal species. Int. J. Microbiol. 2009:614371. 10.1155/2009/614371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hynes WL, Walton SL. 2000. Hyaluronidases of Gram-positive bacteria. FEMS Microbiol. Lett. 183:201–207. 10.1111/j.1574-6968.2000.tb08958.x [DOI] [PubMed] [Google Scholar]
- 10. Laurent TC, Fraser JR. 1992. Hyaluronan. FASEB J. 6:2397–2404 [PubMed] [Google Scholar]
- 11. Jiang DL, Liang J, Noble PW. 2011. Hyaluronan as an immune regulator in human diseases. Physiol. Rev. 91:221–264. 10.1152/physrev.00052.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith LD. 1979. Virulence factors of Clostridium perfringens. Rev. Infect. Dis. 1:254–262. 10.1093/clinids/1.2.254 [DOI] [PubMed] [Google Scholar]
- 13. Starr CR, Engleberg NC. 2006. Role of hyaluronidase in subcutaneous spread and growth of group A Streptococcus. Infect. Immun. 74:40–48. 10.1128/IAI.74.1.40-48.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li S, Jedrzejas MJ. 2001. Hyaluronan binding and degradation by Streptococcus agalactiae hyaluronate lyase. J. Biol. Chem. 276:41407–41416. 10.1074/jbc.M106634200 [DOI] [PubMed] [Google Scholar]
- 15. Li S, Kelly SJ, Lamani E, Ferraroni M, Jedrzejas MJ. 2000. Structural basis of hyaluronan degradation by Streptococcus pneumoniae hyaluronate lyase. EMBO J. 19:1228–1240. 10.1093/emboj/19.6.1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zheng M, Zhang H, Xu D. 2012. Initial events in the degradation of hyaluronan catalyzed by hyaluronate lyase from Streptococcus pneumoniae: QM/MM simulation. J. Phys. Chem. B 116:11166–11172. 10.1021/jp306754a [DOI] [PubMed] [Google Scholar]
- 17. Duran-Reynals F. 1933. Studies on a certain spreading factor existing in bacteria and its significance for bacterial invasiveness. J. Exp. Med. 58:161–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chain E, Duthie ES. 1940. Identity of hyaluronidase and spreading factor. Br. J. Exp. Pathol. 21:324–338 [Google Scholar]
- 19. Favilli G. 1940. Mucolytic effect of natural and artificial spreading factors: mucolytic effect of several diffusing agents and of a dlazotized compound. Nature 145:866–867. 10.1038/145866a0 [DOI] [Google Scholar]
- 20. McClean D, Hale CW. 1940. Mucolytic effect of natural and artificial spreading factors: mucinase and tissue permeability. Nature 145:867–868. 10.1038/145867a0 [DOI] [Google Scholar]
- 21. Duran-Reynals F. 1942. Tissue permeability and the spreading factors in infection: a contribution to the host:parasite problem. Bacteriol. Rev. 6:197–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hobby GL, Dawson MH, Meyer K, Chaffee E. 1941. The relationship between spreading factor and hyaluronidase. J. Exp. Med. 73:109–123. 10.1084/jem.73.1.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rogers H. 1953. Variant populations within a hyaluronidase-producing culture of Staphylococcus aureus. J. Pathol. Bacteriol. 66:545–551. 10.1002/path.1700660226 [DOI] [PubMed] [Google Scholar]
- 24. Rogers H. 1954. The rate of formation of hyaluronidase, coagulase and total extracellular protein by strains of Staphylococcus aureus. J. Gen. Microbiol. 10:209–220. 10.1099/00221287-10-2-209 [DOI] [PubMed] [Google Scholar]
- 25. Rogers H. 1957. The preferential suppression of hyaluronidase formation in cultures of Staphylococcus aureus. J. Gen. Microbiol. 16:22–37. 10.1099/00221287-16-1-22 [DOI] [PubMed] [Google Scholar]
- 26. Farrell AM, Taylor D, Holland KT. 1995. Cloning, nucleotide sequence determination and expression of the Staphylococcus aureus hyaluronate lyase gene. FEMS Microbiol. Lett. 130:81–85. 10.1111/j.1574-6968.1995.tb07702.x [DOI] [PubMed] [Google Scholar]
- 27. Jones RC, Deck J, Edmondson RD, Hart ME. 2008. Relative quantitative comparisons of the extracellular protein profiles of Staphylococcus aureus UAMS-1 and its sarA, agr, and sarA agr regulatory mutants using one-dimensional polyacrylamide gel electrophoresis and nanocapillary liquid chromatography coupled with tandem mass spectrometry. J. Bacteriol. 190:5265–5278. 10.1128/JB.00383-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Makris G, Wright JD, Ingham E, Holland KT. 2004. The hyaluronate lyase of Staphylococcus aureus—a virulence factor? Microbiology 150:2005–2013. 10.1099/mic.0.26942-0 [DOI] [PubMed] [Google Scholar]
- 29. Hart ME, Tsang LH, Deck J, Daily ST, Jones RC, Liu H, Hu H, Hart MJ, Smeltzer MS. 2013. Hyaluronidase expression and biofilm involvement in Staphylococcus aureus UAMS-1 and its sarA, agr and sarA agr regulatory mutants. Microbiology 159:782–791. 10.1099/mic.0.065367-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schenk S, Laddaga RA. 1992. Improved method for electroporation of Staphylococcus aureus. FEMS Microbiol. Lett. 94:133–138. 10.1111/j.1574-6968.1992.tb05302.x [DOI] [PubMed] [Google Scholar]
- 31. Wormann ME, Reichmann NT, Malone CL, Horswill AR, Grundling A. 2011. Proteolytic cleavage inactivates the Staphylococcus aureus lipoteichoic acid synthase. J. Bacteriol. 193:5279–5291. 10.1128/JB.00369-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bose JL, Fey PD, Bayles KW. 2013. Genetic tools to enhance the study of gene function and regulation in Staphylococcus aureus. Appl. Environ. Microbiol. 79:2218–2224. 10.1128/AEM.00136-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pohl K, Francois P, Stenz L, Schlink F, Geiger T, Herbert S, Goerke C, Schrenzel J, Wolz C. 2009. CodY in Staphylococcus aureus: a regulatory link between metabolism and virulence gene expression. J. Bacteriol. 191:2953–2963. 10.1128/JB.01492-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thompson MG, Black CC, Pavlicek RL, Honnold CL, Wise MC, Alamneh YA, Moon JK, Kessler JL, Si Y, Williams R, Yildirim S, Kirkup BC, Jr, Green RK, Hall ER, Palys TJ, Zurawski DV. 2014. Validation of a novel murine wound model of Acinetobacter baumannii infection. Antimicrob. Agents Chemother. 58:1332–1342. 10.1128/AAC.01944-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC [Google Scholar]
- 36. El-Chemaly S, Malide D, Zudaire E, Ikeda Y, Weinberg BA, Pacheco-Rodriguez G, Rosas IO, Aparicio M, Ren P, MacDonald SD, Wu HP, Nathan SD, Cuttitta F, McCoy JP, Gochuico BR, Moss J. 2009. Abnormal lymphangiogenesis in idiopathic pulmonary fibrosis with insights into cellular and molecular mechanisms. Proc. Natl. Acad. Sci. U. S. A. 106:3958–3963. 10.1073/pnas.0813368106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Monzon ME, Casalino-Matsuda SM, Forteza RM. 2006. Identification of glycosaminoglycans in human airway secretions. Am. J. Respir. Cell Mol. Biol. 34:135–141. 10.1165/rcmb.2005-0256OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF, Sensabaugh GF, Perdreau-Remington F. 2006. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367:731–739. 10.1016/S0140-6736(06)68231-7 [DOI] [PubMed] [Google Scholar]
- 39. Boles BR, Thoendel M, Roth AJ, Horswill AR. 2010. Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One 5:e10146. 10.1371/journal.pone.0010146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4(1):e00537–12. 10.1128/mBio.00537-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Majerczyk CD, Dunman PM, Luong TT, Lee CY, Sadykov MR, Somerville GA, Bodi K, Sonenshein AL. 2010. Direct targets of CodY in Staphylococcus aureus. J. Bacteriol. 192:2861–2877. 10.1128/JB.00220-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Majerczyk CD, Sadykov MR, Luong TT, Lee C, Somerville GA, Sonenshein AL. 2008. Staphylococcus aureus CodY negatively regulates virulence gene expression. J. Bacteriol. 190:2257–2265. 10.1128/JB.01545-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rivera FE, Miller HK, Kolar SL, Stevens SM, Shaw LN. 2012. The impact of CodY on virulence determinant production in community-associated methicillin-resistant Staphylococcus aureus. Proteomics 12:263–268. 10.1002/pmic.201100298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Belitsky BR, Sonenshein AL. 2008. Genetic and biochemical analysis of CodY-binding sites in Bacillus subtilis. J. Bacteriol. 190:1224–1236. 10.1128/JB.01780-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brinsmade SR, Kleijn RJ, Sauer U, Sonenshein AL. 2010. Regulation of CodY activity through modulation of intracellular branched-chain amino acid pools. J. Bacteriol. 192:6357–6368. 10.1128/JB.00937-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stenz L, Francois P, Whiteson K, Wolz C, Linder P, Schrenzel J. 2011. The CodY pleiotropic repressor controls virulence in gram-positive pathogens. FEMS Immunol. Med. Microbiol. 62:123–139. 10.1111/j.1574-695X.2011.00812.x [DOI] [PubMed] [Google Scholar]
- 47. Montgomery CP, Boyle-Vavra S, Roux A, Ebine K, Sonenshein AL, Daum RS. 2012. CodY deletion enhances in vivo virulence of community-associated methicillin-resistant Staphylococcus aureus clone USA300. Infect. Immun. 80:2382–2389. 10.1128/IAI.06172-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lauderdale KJ, Boles BR, Cheung AL, Horswill AR. 2009. Interconnections between sigma B, agr, and proteolytic activity in Staphylococcus aureus biofilm maturation. Infect. Immun. 77:1623–1635. 10.1128/IAI.01036-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fraser JR, Laurent TC, Laurent UB. 1997. Hyaluronan: its nature, distribution, functions and turnover. J. Intern. Med. 242:27–33. 10.1046/j.1365-2796.1997.00170.x [DOI] [PubMed] [Google Scholar]
- 50. Skaar EP. 2010. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 6:e1000949. 10.1371/journal.ppat.1000949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jones CL, Napier BA, Sampson TR, Llewellyn AC, Schroeder MR, Weiss DS. 2012. Subversion of host recognition and defense systems by Francisella spp. Microbiol. Mol. Biol. Rev. 76:383–404. 10.1128/MMBR.05027-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Somerville GA, Proctor RA. 2009. At the crossroads of bacterial metabolism and virulence factor synthesis in staphylococci. Microbiol. Mol. Biol. Rev. 73:233–248. 10.1128/MMBR.00005-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Costagliola C, Del Prete A, Winkler NR, Carpineto P, Ciancaglini M, Piccolomini R, Mastropasqua L. 1996. The ability of bacteria to use Na-hyaluronate as a nutrient. Acta Ophthalmol. Scand. 74:566–568 [DOI] [PubMed] [Google Scholar]
- 54. Marion C, Stewart JM, Tazi MF, Burnaugh AM, Linke CM, Woodiga SA, King SJ. 2012. Streptococcus pneumoniae can utilize multiple sources of hyaluronic acid for growth. Infect. Immun. 80:1390–1398. 10.1128/IAI.05756-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hirayama Y, Yoshimura M, Ozeki Y, Sugawara I, Udagawa T, Mizuno S, Itano N, Kimata K, Tamaru A, Ogura H, Kobayashi K, Matsumoto S. 2009. Mycobacteria exploit host hyaluronan for efficient extracellular replication. PLoS Pathog. 5:e1000643. 10.1371/journal.ppat.1000643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Imamura T, Tanase S, Szmyd G, Kozik A, Travis J, Potempa J. 2005. Induction of vascular leakage through release of bradykinin and a novel kinin by cysteine proteinases from Staphylococcus aureus. J. Exp. Med. 201:1669–1676. 10.1084/jem.20042041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stehr F, Kretschmar M, Kröger C, Hube B, Schäfer W. 2003. Microbial lipases as virulence factors. J. Mol. Catalysis B Enzym. 22:347–355. 10.1016/S1381-1177(03)00049-3 [DOI] [Google Scholar]
- 58. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nair D, Memmi G, Hernandez D, Bard J, Beaume M, Gill S, Francois P, Cheung AL. 2011. Whole-genome sequencing of Staphylococcus aureus strain RN4220, a key laboratory strain used in virulence research, identifies mutations that affect not only virulence factors but also the fitness of the strain. J. Bacteriol. 193:2332–2335. 10.1128/JB.00027-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Benson MA, Lilo S, Wasserman GA, Thoendel M, Smith A, Horswill AR, Fraser J, Novick RP, Shopsin B, Torres VJ. 2011. Staphylococcus aureus regulates the expression and production of the staphylococcal superantigen-like secreted proteins in a Rot-dependent manner. Mol. Microbiol. 81:659–675. 10.1111/j.1365-2958.2011.07720.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mootz JM, Malone CL, Shaw LN, Horswill AR. 2013. Staphopains modulate Staphylococcus aureus biofilm integrity. Infect. Immun. 81:3227–3238. 10.1128/IAI.00377-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kiedrowski MR, Kavanaugh JS, Malone CL, Mootz JM, Voyich JM, Smeltzer MS, Bayles KW, Horswill AR. 2011. Nuclease modulates biofilm formation in community-associated methicillin-resistant Staphylococcus aureus. PLoS One 6:e26714. 10.1371/journal.pone.0026714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Olson ME, King JM, Yahr TL, Horswill AR. 2013. Sialic acid catabolism in Staphylococcus aureus. J. Bacteriol. 195:1779–1788. 10.1128/JB.02294-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bateman BT, Donegan NP, Jarry TM, Palma M, Cheung AL. 2001. Evaluation of a tetracycline-inducible promoter in Staphylococcus aureus in vitro and in vivo and its application in demonstrating the role of sigB in microcolony formation. Infect. Immun. 69:7851–7857. 10.1128/IAI.69.12.7851-7857.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]