

Abstract
The bacterial pathogen Pseudomonas aeruginosa causes acute infections associated with significant morbidity and mortality. P. aeruginosa elicits strong innate immune responses in immunocompetent hosts, and the resulting recruitment of neutrophils to the site of infection is necessary for bacterial clearance. P. aeruginosa lipopolysaccharide and flagellin are recognized by extracellular Toll-like receptors, but the most rapid responses to infection occur when cytosolic receptors sense flagellin or type 3 secretion system (T3SS) structural proteins. The subsequent activation of the NLRC4 inflammasome and caspase-1 generates an interleukin-1β (IL-1β) signal that is required for the rapid neutrophilic response. A T3SS effector, exotoxin U (ExoU), can inhibit activation of the NLRC4 inflammasome and caspase-1. Thus, our observation that IL-1 receptor (IL-1R)-mediated signals were still required to initiate a response to ExoU-producing bacteria was unexpected. As both IL-1α and IL-1β signal via the IL-1R, we examined immune responses in mice lacking either of these cytokines. IL-1β-deficient mice responded to ExoU-producing P. aeruginosa bacteria similarly to wild-type animals; however, IL-1α-deficient mice had an attenuated immune response. The situation was reversed following infections by ExoU-negative bacteria: here, IL-1α was dispensable for neutrophil recruitment, while IL-1β was required. IL-1α secretion by macrophages infected with ExoU-producing P. aeruginosa isolates was independent of both caspase-1 and caspase-11. This study documents distinct roles for IL-1α and IL-1β in the response to P. aeruginosa infection as a function of the T3SS effectors produced by the infecting strain. The redundancy of these two cytokines nonetheless allows the infected host to mount a response to ExoU-positive and -negative bacterial isolates.
INTRODUCTION
Pseudomonas aeruginosa is an opportunistic human pathogen capable of causing acute infections, such as ventilator-associated pneumonia, as well as chronic colonization of the respiratory tract in patients with cystic fibrosis or bronchiectasis (1). Infections with P. aeruginosa can progress rapidly both in human patients and in mouse models; thus, it is not surprising that innate immune responses are important for control of this pathogen (2). Neutrophils are the primary cell type implicated in P. aeruginosa clearance, and in their absence, very small bacterial inocula (<100 CFU) are capable of causing murine death (3). Multiple innate immune receptors can respond to P. aeruginosa infection, but the quickest responses triggered by low bacterial inocula appear to involve interleukin-1 (IL-1) receptor (IL-1R)-mediated signals (4). Two forms of IL-1, designated IL-1α and IL-1β, bind to this receptor and elicit similar biological responses (5). These two cytokines are encoded by distinct genes and exhibit only ca. 25% sequence identity.
Il1b gene expression is positively regulated by NF-κB-mediated signaling, but the protein is produced as an inactive precursor. Processing and secretion of pro-IL-1β by macrophages require inflammasome activation, which can be triggered by NLRC4-mediated recognition of P. aeruginosa type 3 secretion system (T3SS) structural proteins or by the intracellular delivery of P. aeruginosa flagellin (6–8). Assembly of the inflammasome is followed by proteolytic activation of caspase-1 and subsequent cleavage and secretion of biologically active IL-1β. Secreted neutrophil proteases can also generate bioactive IL-1β by cleaving pro-IL-1β at a site different from the caspase-1 cleavage site.
IL-1α is more widely and constitutively expressed than IL-1β and has intracellular functions, but it also acts locally in a membrane-bound form by activating IL-1R1 (5, 9). Additionally, the passive release of IL-1α upon cell death can trigger a sterile inflammatory response to dying cells (10). Although IL-1α can be cleaved, cleavage is not mediated by caspase-1 and is not required for binding to IL-1R1 (11–13).
Recent studies have shown that myeloid cells secrete IL-1α in response to many classical inflammasome activators (9, 14). These responses are dependent on canonical inflammasome components and result in the cosecretion of IL-1α and cleaved IL-1β; however, particulate NLRP3 activators also induced inflammasome-independent IL-1α secretion in one study (14). Even in the absence of IL-1α secretion, however, bioactive, surface-bound IL-1α is produced by monocytes solely in response to NF-κB-activating signals and does not require inflammasome components, caspase-1, or IL-1β (9).
Several papers have observed a role for caspase-11 in flagellin-independent IL-1α release from murine macrophages infected with T3SS- or T4SS-expressing pathogens, such as Yersinia pseudotuberculosis or Legionella pneumophila ΔflaA (15, 16). IL-1α plays an important role in initiating innate immune responses to L. pneumophila, as genetic ablation or antibody blockade of IL-1α inhibits neutrophil recruitment to infected airways (15, 17).
Given the importance of IL-1-dependent signals for neutrophil recruitment following bacterial infection, it is not surprising that many pathogens possess mechanisms that attenuate or interfere with IL-1 signaling. As an example, two of the four known P. aeruginosa T3SS effectors, exotoxin U (ExoU) and ExoS, have inhibitory effects on inflammasome activation and IL-1β production through mechanisms that have not yet been delineated (7, 18). Although it is very rare for P. aeruginosa strains to express and translocate both ExoU and ExoS, almost all type 3 secretion system-positive strains express one or the other effector. We had previously reported that caspase-1-dependent immune responses to P. aeruginosa were robustly triggered by nonflagellated strains lacking all of the known T3SS effectors but still expressing a complete, translocation-competent T3SS apparatus (4), consistent with the ability of inflammasome adaptors, such as Naip1 and Naip2, to recognize components of the T3SS apparatus (19–21). However, we also observed that IL-1R-dependent signaling events were still important in innate immune responses to bacteria expressing the caspase-1 inhibitor ExoU in a murine acute pneumonia model (22). The basis for these seemingly contradictory observations was explored using transgenic mouse models deficient in defined aspects of IL-1 signaling.
MATERIALS AND METHODS
Mice.
All animal work was conducted according to relevant national and international guidelines. Protocols for all animal studies were approved by the Yale Institutional Animal Care and Use Committee. Interleukin-1β-deficient (Il1b−/−) and interleukin-1α-deficient (Il1a−/−) mice with a C57BL/6 background (23) were generously provided by Yoichiro Iwakura (Tokyo University of Science). casp1−/− (16) and casp1 casp11−/− (24) mice backcrossed to C57BL/6 (N10) mice were generously provided by Richard Flavell (Yale University). Interleukin-1R1-deficient (Il1r−/−) mice with a C57BL/6 background were purchased from the Jackson Laboratory. Wild-type C57BL/6 mice were purchased from NCI (NIH). All mice were housed in a specific-pathogen-free facility in microisolator cages. Age-matched (8 to 10 weeks) and sex-matched littermates were used for all experiments. In some experiments, Il1r−/− mice were injected intraperitoneally with etanercept (Enbrel; anti-tumor necrosis factor receptor [anti-TNFR]; 250 μg) twice at 24-h intervals and then infected intranasally with bacterial strains.
Bacterial strains and growth conditions.
All bacterial strains were maintained as frozen stocks at −80°C and freshly streaked to Vogel-Bonner minimal medium (VBM) or Luria-Bertani (LB) agar prior to each experiment. P. aeruginosa strain PA103 is a nonflagellated clinical isolate that produces two T3SS effectors, ExoU and ExoT (25). PA103 ΔexoU carries an unmarked deletion of the exoU gene (26).
Mouse infections.
Bacteria grown overnight in LB medium in a 37°C shaker were diluted 1:50 and grown for 2 to 4 h in LB at 37°C (250 rpm). The log-phase culture was again diluted to an optical density at 600 nm (OD600) of 0.06 and grown for an additional 1 to 2 h (37°C, 250 rpm). Bacteria were washed, resuspended, and diluted in phosphate-buffered saline (PBS) to achieve an inoculum of ca. 5 × 104 CFU in 40 μl. Animals were lightly anesthetized with 30% (vol/vol) isoflurane in propylene glycol and infected intranasally. The actual inoculum for each experiment was determined by plating serial dilutions of the inoculum to VBM agar. Mice were euthanized at 4 h postinfection. Bronchoalveolar lavage (BAL) was performed 3 times with 1 ml PBS prior to dissection of lungs. Single-cell lung suspensions were prepared as described previously and plated to VBM agar to determine the bacterial load in tissue (22). The data shown represent those from 2 to 4 independent experiments each involving 3 to 8 mice per group.
Cell counts and differentials.
Cells from BAL fluid were collected by centrifugation. Red blood cells (RBCs) were lysed (RBC lysing buffer; Sigma-Aldrich) before total cell counts were determined using a hemacytometer. Cytospin samples were prepared using a Shandon cytocentrifuge (Shadon EZ Single Cytofunnel; Thermo Scientific) and dried prior to staining with a Diff-Quik stain kit (IMEB, Inc., San Marcos, CA). At least 200 cells were counted to determine macrophage and neutrophil percentages in BAL fluid.
Bone marrow-derived macrophages (BMDMs).
Bone marrow was harvested from mouse femurs, and cells were differentiated into macrophages by culture in RPMI supplemented with 30% L929 cell supernatant and 10% fetal bovine serum (FBS) in a humidified incubator at 37°C. Macrophages were replated 1 day prior to infection in RPMI plus 10% FBS in 24-well plates at 2 × 105 cells/well. Cells were primed with lipopolysaccharide (LPS; 0.5 μg/ml) for 2.5 h prior to infection with P. aeruginosa strains (multiplicity of infection [MOI], 20) grown as described above for animal infections. Mock-infected samples were inoculated with PBS.
Cytotoxicity assay.
Lactate dehydrogenase (LDH) release was measured using an LDH cytotoxicity assay kit (Clontech) according to the manufacturer's instructions. Total LDH release was determined by lysing uninfected cells with Triton X-100 (0.1%) and measuring the amount of LDH released.
Cytokine measurements.
Supernatants prepared from the first milliliter of BAL fluid and from BMDMs were stored at −20°C and assayed for cytokine levels by enzyme-linked immunosorbent assay (ELISA; DuoSet; R&D Systems) according to the manufacturer's protocol. The IL-1β DuoSet ELISA can recognize both processed and unprocessed IL-1β (data not shown).
Statistical analysis.
Statistical analysis was carried out using Prism (version 5) software (GraphPad).
RESULTS
IL-1R-dependent and -independent pathways contribute to P. aeruginosa recognition in vivo.
P. aeruginosa PA103 is an ExoU/ExoT-producing T3SS-positive strain originally isolated from a patient with acute pneumonia (25). This aflagellate strain has been extensively characterized in vitro and in mouse models of acute pneumonia. ExoU, a phospholipase A2, causes rapid necrotic cell death (27, 28); interestingly, mouse BMDMs infected with ExoU-positive P. aeruginosa strains fail to activate caspase-1, though other signaling events that accompany bacterial infection, such as activation of mitogen-activated protein kinase and NF-κB pathways, are observed (7). Mice infected with PA103 rapidly recruited neutrophils to the airways; this host response was not significantly altered by the absence of caspase-1 or caspase-1/11 signaling pathways (Fig. 1). Surprisingly, mice lacking IL-1R signaling showed attenuated neutrophil recruitment following PA103 infection, suggesting that a cytokine sensed by IL-1R but produced in a caspase-1- and caspase-11-independent manner was important for this response. Myeloid differentiation factor 88 (MyD88) is an adaptor for IL-1 family receptors and Toll-like receptors (TLRs), with the exception of TLR-3 (29). Myd88−/− mice showed an even greater attenuation of neutrophil recruitment, indicating the presence of IL-1R-independent signaling following bacterial infection, most likely due to activation of TLR4 by these aflagellate extracellular bacteria. Slight increases in bacterial burden were noted in Il1r−/− and Myd88−/− animals at this early postinfection time point (4 h), likely reflecting decreased numbers of phagocytic cells in the airways.
FIG 1.
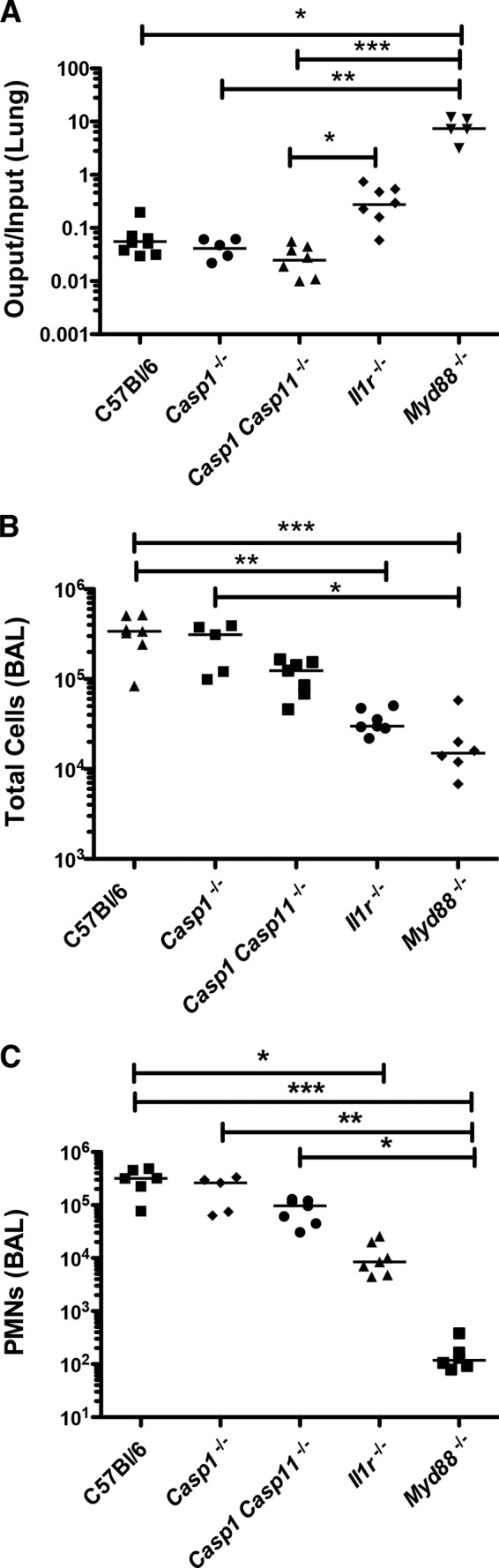
Neutrophil recruitment following PA103 infection requires IL-1R- and MyD88-dependent signaling but is independent of caspase-1 or caspase-11. Mice of the indicated genotypes were intranasally infected with ca. 5 × 104 CFU of PA103 and then euthanized at 4 hpi. (A) Ratio of the number of CFU recovered from lung/number of CFU in the inoculum; (B) total cell count in BAL fluid; (C) number of neutrophils (polymorphonuclear leukocytes [PMN]) in BAL fluid. Each symbol represents one mouse; bars indicate the geometric mean (A) or median (B, C) for each group. Statistical significance was tested by analysis of variance (Kruskal-Wallis test), followed by Dunn's multiple-comparison test. *, P < 0.5; **, P < 0.01; ***, P < 0.001.
IL-1β signaling is not required for early recognition of ExoU-positive P. aeruginosa bacteria.
The preserved responses of Casp1−/− and Casp1 Casp11−/− mice to PA103 infection, as reflected by neutrophil recruitment, suggested that cytokines generated by caspase-1 activity, such as IL-1β, were unlikely to play an important signaling role during early phases of infection. However, active IL-1β can be produced by the activity of other proteases on pro-IL-1β (30–32). We measured the IL-1β levels in BAL fluid collected from PA103-infected Casp1−/− Casp11−/− mice at 4 h postinfection (hpi) and found significantly elevated IL-1β levels in these samples, suggesting that caspase-1-independent production of this cytokine was occurring, as has been reported in other models of P. aeruginosa infection (33) (see Fig. S1 in the supplemental material). We therefore tested whether this cytokine was itself required for innate immune responses after infection by using knockout mice carrying a disrupted Il1b gene. Il1b−/− mice infected with PA103 responded with a rapid recruitment of neutrophils indistinguishable from that observed in infected wild-type (C57BL/6) mice (Fig. 2).
FIG 2.

IL-1β signaling plays a significant role in the host response to P. aeruginosa lacking ExoU. Mice of the indicated genotypes were infected with 5 × 104 PA103 or isogenic ΔexoU bacteria and then sacrificed at 4 hpi. (A) Lung bacteria, expressed as the ratio of the number of CFU recovered/number of CFU in the inoculum; lines indicate the geometric mean for each group. (B) Total number of cells in BAL fluid; lines indicate the median. (C) Number of neutrophils in BAL fluid; lines indicate the median. Each symbol represents an individual animal. (D) Proportion of macrophages and neutrophils in BAL fluid; bars indicate means ± SEMs (n = 9 to 12). diff, differential cell count. The statistical significance of pairwise comparisons was determined using analysis of variance (Kruskal-Wallis test), followed by Dunn's multiple-comparison test. n. s., not significant; *, P < 0.5; **, P < 0.01; ***, P < 0.001.
We had previously reported an important role for caspase-1-dependent innate immune responses to P. aeruginosa bacteria that did not express ExoU. These experiments were carried out using mice that were subsequently determined to be deficient in both caspase-1 and caspase-11 activity (34). We therefore repeated these infections using singly deficient Casp1−/− mice (16) and confirmed that strains lacking exoU required caspase-1 signaling to initiate the characteristic neutrophil recruitment seen in wild-type mice after infection (see Fig. S2 in the supplemental material). In order to test whether this requirement for caspase-1 reflected a requirement for IL-1β signaling, we carried out infections in Il1b−/− mice using the isogenic ExoU-deficient strain PA103 ΔexoU. IL-1β signaling did contribute to the recognition of PA103 ΔexoU bacteria in the lung, as knockout animals showed a significant attenuation in neutrophil recruitment following infection (Fig. 2). Induction of the neutrophil chemokine keratinocyte chemoattractant (KC) was dependent on IL-1β signaling in PA103 ΔexoU-infected mice but was apparently independent of IL-1β in PA103-infected animals (Fig. 3A). Thus, the relative importance of IL-1β during infection was a function of the effectors secreted by the infecting bacteria.
FIG 3.
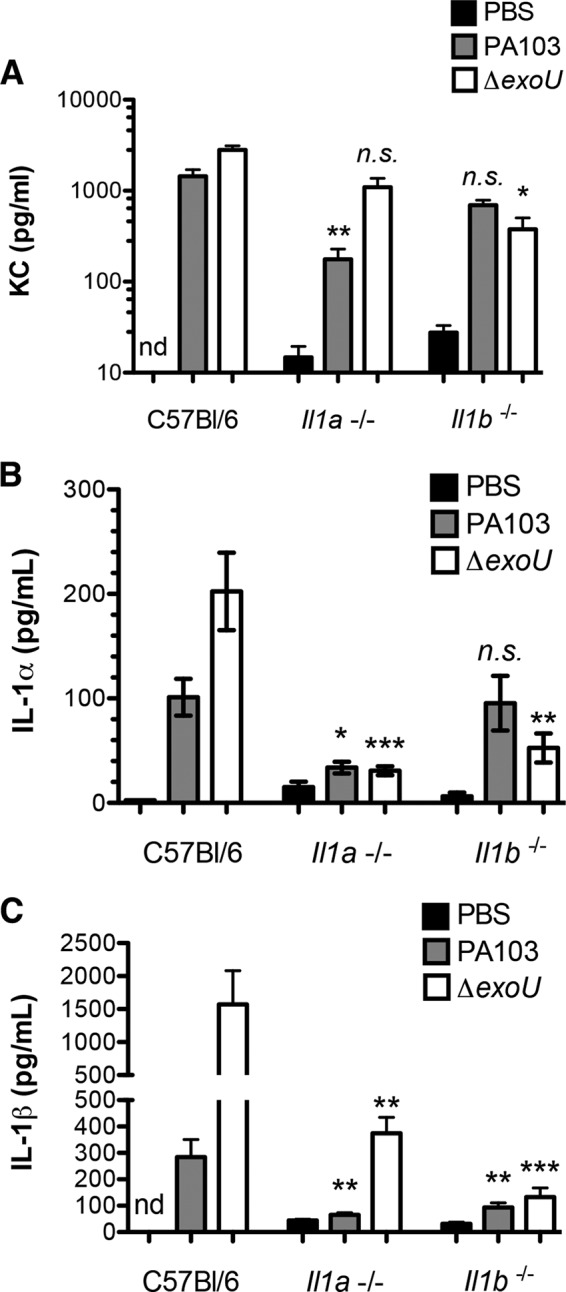
Dependence of cytokine production in vivo on IL-1α and IL-1β. KC (A), IL-1α (B), and IL-1β (C) levels in mouse BAL fluid were measured at 4 h postinfection with either PA103 or PA103 ΔexoU bacteria. Each bar shows the mean ± SEM for 5 (PBS controls) or 8 to 15 (infected) animals. Analysis of variance followed by Dunn's multiple-comparison posttest was used to determine whether cytokine levels differed significantly in pairwise comparisons between C57BL/6 and each knockout mouse infected with the same bacterial strain. nd, not detected; n. s., not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
IL-1α is required for early recognition of ExoU-positive P. aeruginosa bacteria.
Many cells produce pro-IL-1α in response to proinflammatory stimuli. Pro-IL-1α is bioactive in its unprocessed form and can be released by damaged or dying cells (10). We hypothesized that IL-1α might be released from cells undergoing ExoU-mediated necrosis and could provide the IL-1R-dependent signal that we had observed. This was tested by infecting Il1a−/− mice with PA103 and then analyzing immune cell recruitment to the airways at 4 h postinfection. As seen in Fig. 4, neutrophil recruitment was significantly attenuated in Il1a−/− mice infected with the ExoU-positive P. aeruginosa strain, and KC levels were significantly lower in BAL fluid collected from knockout mice (Fig. 3A). Wild-type and Il1a−/− mice were also infected with the isogenic PA103 ΔexoU mutant. Neutrophil recruitment was indistinguishable between these two groups of mice, demonstrating that IL-1α signals were either not generated or irrelevant during infection by a P. aeruginosa strain lacking ExoU.
FIG 4.

IL-1α signaling is specifically required for innate immune recognition of ExoU-positive P. aeruginosa bacteria. Mice of the indicated genotypes were infected with 5 × 104 PA103 or isogenic ΔexoU bacteria and then sacrificed at 4 hpi. (A) Lung bacteria, expressed as the ratio of the number of CFU recovered/number of CFU in the inoculum; lines indicate the geometric mean for each group. (B) Total number of cells in BAL fluid; lines indicate the median. (C) Number of neutrophils in BAL fluid; lines indicate the median. Each symbol represents an individual animal. (D) Proportion of macrophages and neutrophils in BAL fluid; bars indicate means ± SEMs (n = 9 to 12). The statistical significance of pairwise comparisons was determined using analysis of variance (Kruskal-Wallis test), followed by Dunn's multiple-comparison test. n.s., not significant; ***, P < 0.001.
We also measured IL-1α and IL-1β levels in mouse BAL fluid at the 4-hpi time point (Fig. 3B and C). IL-1α was induced following PA103 infection, as we had observed previously (22), but the absolute amount of measured cytokine was low and similar to that seen in ΔexoU mutant-infected animals. In contrast, IL-1β secretion was strongly induced in ΔexoU mutant-infected mice. Genetic ablation of IL-1α was associated with the absence of IL-1β in PA103-infected mice and significantly decreased IL-1β levels in ΔexoU mutant-infected animals. This suggests that IL-1α signaling is required for IL-1β secretion in the setting of PA103 infection, while IL-1β is generated by both IL-1α-dependent and -independent signals during ΔexoU mutant infection.
Rapid IL-1α release from BMDMs following infection with ExoU-positive P. aeruginosa bacteria is independent of caspase-1 and caspase-11.
Our data suggest that IL-1α signals make a significant contribution to the innate immune recognition of PA103, but the levels of this cytokine in BAL fluid are unimpressive. We reasoned, however, that a requirement for the early generation of this cytokine upon infection of resident airway cells, such as macrophages or epithelial cells, could account for our findings. Bacteria expressing type 3 or type 4 secretion systems can activate a caspase-11-dependent, caspase-1-independent pathway in murine BMDMs that leads to the release of IL-1α (15). We tested whether P. aeruginosa also stimulated IL-1α release from BMDMs and if cytokine secretion was affected by ExoU expression. LPS-primed BMDMs infected with PA103 (MOI, 20) released small amounts of IL-1β at early time points postinfection, and the amounts were much less than those observed for PA103 ΔexoU-infected BMDMs (Fig. 5). PA103-infected BMDMs also secreted IL-1α in amounts equal to or larger than the amount secreted by PA103 ΔexoU-infected cells. Secretion of both IL-1α and IL-1β was abrogated in BMDMs derived from Casp1−/− Casp11−/− mice infected with PA103 ΔexoU, but macrophages from these same mice showed no change in IL-1α release following PA103 infection. Thus, ExoU-expressing bacteria elicit IL-1α release independently of the caspase-11 pathway described for other T3SS- or T4SS-positive pathogens, most likely as the result of ExoU-mediated necrosis.
FIG 5.
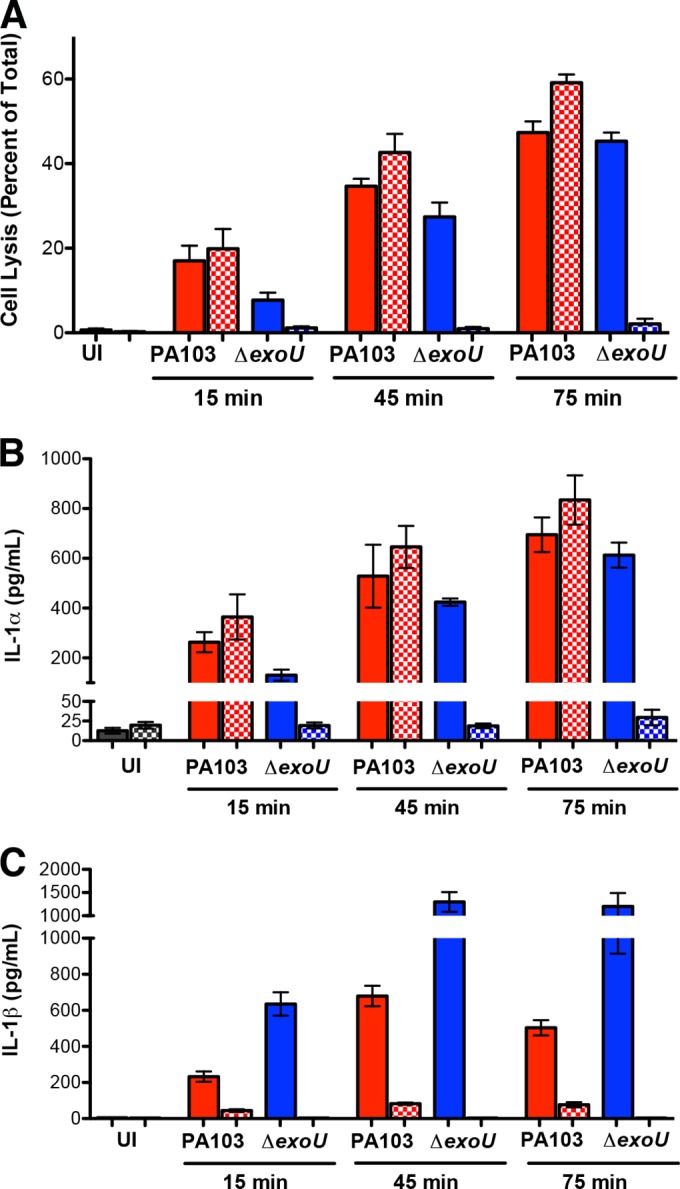
PA103-infected macrophages secrete IL-1α independently of caspase-1/11 activity. BMDMs harvested from C57BL/6 (solid bars) or casp1−/− casp11−/− (stippled bars) mice were primed with LPS (0.5 μg/ml) for 2.5 h and then infected with PA103 (red) or PA103 ΔexoU (blue) (MOI, 20) or inoculated with PBS (uninfected [UI]; gray), as indicated. Supernatants were assayed for LDH (as a measure of cell necrosis/pyroptosis), IL-1α, and IL-1β. Bars indicate the means ± SEMs for 4 to 6 samples assayed in two independent experiments.
TNF receptor signaling contributes to the host response to ExoU-positive P. aeruginosa bacteria.
IL-1R signaling contributes to the recognition of both ExoU-positive and ExoU-negative P. aeruginosa bacteria in the lung, but IL-1α and IL-1β play distinct and nonredundant roles during these infections, respectively. We also noted that Il1r−/− mice mount an attenuated response to PA103 infection that was absent in Myd88−/− mice (Fig. 1). We hypothesized that this other signal might involve tumor necrosis factor alpha (TNF-α), a cytokine that is produced following PA103 infection in a MyD88-dependent fashion (22). To test this hypothesis, we used etanercept, an agent that selectively binds and inhibits soluble TNF-α. We first confirmed that systemic administration of etanercept to mice could inhibit neutrophil recruitment following intranasal administration of TNF-α, provided that it was administered at doses comparable to those measured in BAL fluid after PA103 infection (see Fig. S3 in the supplemental material). We then treated Il1r−/− mice with etanercept or vehicle (PBS) prior to infection with PA103 bacteria. TNF-α blockade significantly attenuated neutrophil recruitment following infection, confirming a role for this cytokine in bacterial recognition (Fig. 6). A similar attenuation of the response was not observed in etanercept-treated Il1r−/− mice infected with PA103 ΔexoU (Fig. 6), suggesting that the relative importance of this cytokine is also dependent on the specific complement of T3SS effectors expressed by P. aeruginosa.
FIG 6.
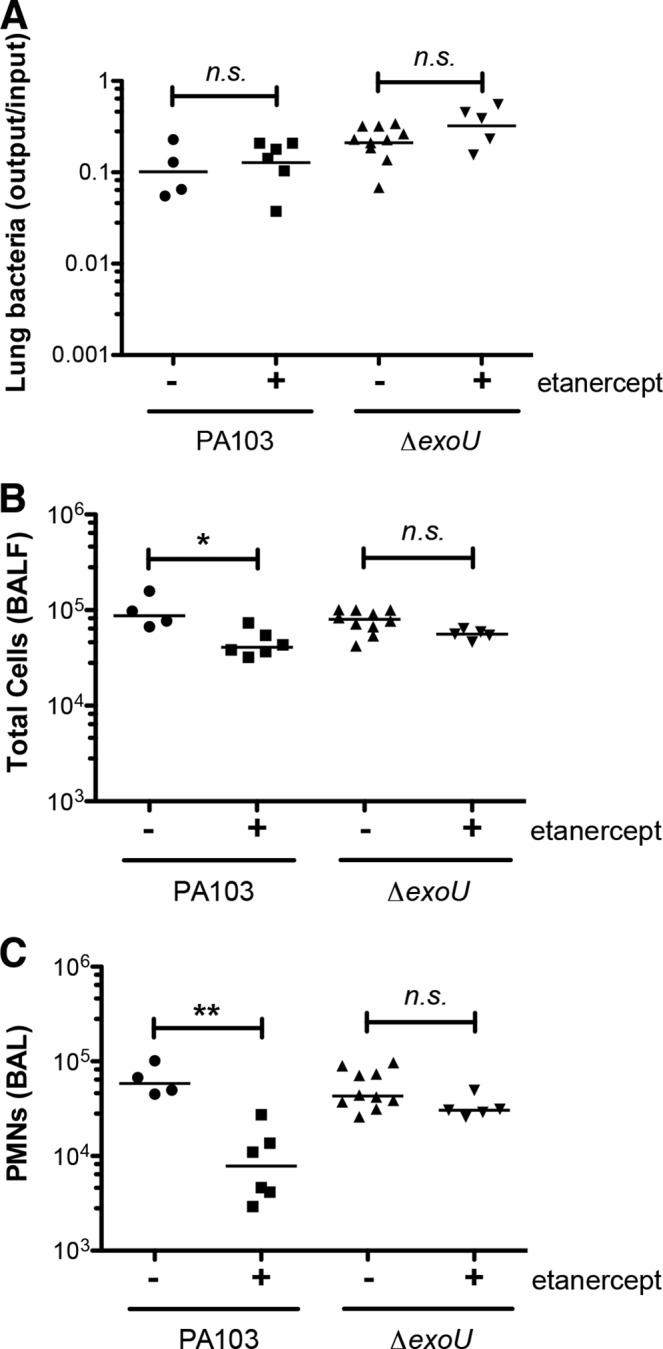
TNF-α signals specifically contribute to recognition of ExoU-producing P. aeruginosa bacteria. Il1r−/− mice were treated with etanercept or PBS vehicle (intraperitoneally) prior to intranasal infection with ca. 5 × 104 PA103 or isogenic ΔexoU bacteria and then sacrificed at 4 hpi. (A) Lung bacteria, expressed as the ratio of the number of CFU recovered/number of CFU in the inoculum; lines indicate the geometric mean for each group. (B) Total number of cells in BAL fluid; lines indicate the median. (C) Number of neutrophils in BAL fluid; lines indicate the median. Each symbol represents an individual animal. The statistical significance of pairwise comparisons was determined using analysis of variance (Kruskal-Wallis test), followed by Dunn's multiple-comparison test. n.s., not significant; *, P < 0.05; **, P < 0.01.
DISCUSSION
Host responses to bacterial infection are multitiered and complex. Distinct bacterial features trigger the activation of different pathogen recognition receptors in a context-dependent manner, leading to an immune response that is proportional to the threat posed by a pathogen. Pathways that respond to cytosolic bacterial determinants, like those leading to inflammasome activation, result in the processing and release of highly proinflammatory IL-1β, a rapid response appropriate to pathogens that can breach cellular membranes via virulence factors such as the T3SS or pore-forming toxins. In this study, we examined the basis for IL-1R-dependent signaling following murine infection with a P. aeruginosa strain that translocates a T3SS effector, ExoU, that inhibits inflammasome activation in BMDMs. We found that IL-1β signaling was largely dispensable for immune recognition of ExoU-producing PA103 bacteria, but the absence of IL-1α significantly attenuated the immune response following infection. Conversely, a role for IL-1α could not be demonstrated in infections with a ΔexoU mutant, and innate responses in this setting required IL-1β and caspase-1.
Though IL-1α and IL-1β both signal by binding the same IL-1R receptor, the production of these cytokines differs in important ways. Bioactive IL-1β is generated by proteolytic cleavage of an inactive precursor; both inflammasome-associated caspase-1 and serine proteases can generate mature IL-1β (30–32). Though tissue-resident macrophages are the cells most likely to encounter and initially respond to pathogens via inflammasome activation, with time recruited neutrophils make up the majority of cells present at sites of inflammation. Thus, they may make a substantial contribution to IL-1β production via either caspase-1-independent (33, 35) or caspase-1-dependent pathways (36, 37), even if they secrete relatively small amounts of cytokine per cell (37).
In contrast, IL-1α is biologically active in its pro-IL-1α form, and the release of pro-IL-1α by dying/necrotic cells can trigger a sterile inflammatory response (10). ExoU translocation causes rapid necrotic cell death, and it is likely that this drives IL-1α release from cells infected by ExoU-producing P. aeruginosa bacteria. In contrast, infection with ΔexoU bacteria leads to IL-1α release from BMDMs via a caspase-1/caspase-11-dependent pathway, suggesting that P. aeruginosa can engage the same pathway as flagellin-deficient, T3SS- or T4SS-expressing pathogens like Yersinia enterocolitica or L. pneumophila (15, 16). We note, however, that the amount of IL-1α produced by ΔexoU mutant-infected BMDMs is far less than the amount of IL-1β simultaneously secreted by these cells; this strong IL-1β response may account for our observation that Il1a−/− animals show little change from wild-type controls in their response to PA103 ΔexoU infection.
Many pathogens trigger IL-1R-mediated host responses during infection. These include extracellular pathogens, such as Streptococcus pneumoniae, Staphylococcus aureus, and Klebsiella pneumoniae (38–40), as well as intracellular bacteria, e.g., Listeria monocytogenes and Mycobacterium tuberculosis (41–43). Many papers describe the interaction of these pathogens with inflammasomes and the subsequent generation of IL-1β signals; these are summarized in recent reviews (44, 45). In some infection models, IL-1α has been shown to make a nonredundant contribution to host defense in vivo, as reported for the control of M. tuberculosis (46, 47) and L. pneumophila (15, 17) following murine pulmonary infection and as shown by us here for ExoU-producing P. aeruginosa bacteria. Although the complex biology surrounding IL-1α production, secretion, and signaling makes it unlikely that a single mechanism will account for its induction by different pathogens during infection, we predict that it will be implicated in IL-1R-mediated responses to many bacterial and fungal pathogens, given the importance of IL-1R in initiating neutrophil recruitment and other immune responses in the infected host.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant R01 AI081825 (to B.I.K.) from the National Institute of Allergy and Infectious Diseases and by a Burroughs-Wellcome Fund Investigator in Pathogenesis of Infectious Diseases Award (to B.I.K.).
We are grateful to Yoichiro Iwakura and Richard Flavell for generously providing knockout mice. We thank Elise Lavoie and Carla Weibel for their help in establishing the P. aeruginosa infection model in Il1a−/− and Il1b−/− mice.
Footnotes
Published ahead of print 28 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02218-14.
REFERENCES
- 1. Kazmierczak BI, Murray TS. 2013. Chronic versus acute Pseudomonas aeruginosa infection states, p 21–39 In Vasil ML, Darwin AJ. (ed), Regulation of bacterial virulence. ASM Press, Washington, DC [Google Scholar]
- 2. Lavoie EG, Wangdi T, Kazmierczak BI. 2011. Innate immune responses to Pseudomonas aeruginosa infection. Microb. Infect. 13:1133–1145. 10.1016/j.micinf.2011.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koh AY, Priebe GR, Ray C, van Rooijen N, Pier GB. 2009. Inescapable need for neutrophils as mediators of cellular innate immunity to acute Pseudomonas aeruginosa pneumonia. Infect. Immun. 77:5300–5310. 10.1128/IAI.00501-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wangdi T, Mijares LA, Kazmierczak BI. 2010. In vivo discrimination of T3SS-positive and -negative Pseudomonas aeruginosa via a caspase-1-dependent pathway. Infect. Immun. 78:4744–4753. 10.1128/IAI.00744-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dinarello CA. 2009. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27:519–550. 10.1146/annurev.immunol.021908.132612 [DOI] [PubMed] [Google Scholar]
- 6. Miao EA, Ernst RK, Dors M, Mao DP, Aderem A. 2008. Pseudomonas aeruginosa activates caspase 1 through IPAF. Proc. Natl. Acad. Sci. U. S. A. 105:2562–2567. 10.1073/pnas.0712183105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. 2007. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 204:2235–2245. 10.1084/jem.20071239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A. 2010. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. U. S. A. 107:3076–3080. 10.1073/pnas.0913087107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fettelschoss A, Kistowska M, LeibundGut-Landmann S, Beer H-D, Johansen P, Senti G, Contassot E, Bachmann MF, French LE, Oxenius A, Kundig TM. 2011. Inflammasome activation and IL-1β target IL-1α for secretion as opposed to surface expression. Proc. Natl. Acad. Sci. U. S. A. 108:18055–18060. 10.1073/pnas.1109176108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen C-J, Kono H, Golenbock D, Reed G, Akira S, Rock KL. 2007. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 13:851–856. 10.1038/nm1603 [DOI] [PubMed] [Google Scholar]
- 11. Carruth LM, Demczuk S, Mizel SB. 1991. Involvement of a calpain-like protease in the processing of the murine interleukin 1 alpha precursor. J. Biol. Chem. 266:12162–12167 [PubMed] [Google Scholar]
- 12. Kobayashi Y, Yamamoto K, Saido T, Kawasaki H, Oppenheim JJ, Matsushima K. 1990. Identification of calcium-activated neutral protease as a processing enzyme of human interleukin 1α. Proc. Natl. Acad. Sci. U. S. A. 87:5548–5552. 10.1073/pnas.87.14.5548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mosley B, Urdal DL, Prickett KS, Larsen A, Cosman D, Conlon PJ, Gillis S, Dower SK. 1987. The interleukin-1 receptor binds the human interleukin-1 alpha precursor but not the interleukin-1 beta precursor. J. Biol. Chem. 262:2941–2944 [PubMed] [Google Scholar]
- 14. Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, Quadroni M, Drexler SK, Tschopp J. 2012. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 36:388–400. 10.1016/j.immuni.2012.01.018 [DOI] [PubMed] [Google Scholar]
- 15. Casson CN, Copenhaver AM, Zwack EE, Nguyen HT, Strowig T, Javdan B, Bradley WP, Fung TC, Flavell RA, Brodsky IE, Shin S. 2013. Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog. 9:e1003400. 10.1371/journal.ppat.1003400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, Flavell RA, Zamboni DS, Roy CR. 2013. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc. Natl. Acad. Sci. U. S. A. 110:1851–1856. 10.1073/pnas.1211521110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barry KC, Fontana MF, Portman JL, Dugan AS, Vance RE. 2013. IL-1alpha signaling initiates the inflammatory response to virulent Legionella pneumophila in vivo. J. Immunol. 190:6329–6339. 10.4049/jimmunol.1300100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Galle M, Schotte P, Haegman M, Wullaert A, Yang HJ, Jin S, Beyaert R. 2008. The Pseudomonas aeruginosa type III secretion system plays a dual role in the regulation of caspase-1 mediated IL-1β maturation. J. Cell. Mol. Med. 12:1767–1776. 10.1111/j.1582-4934.2007.00190.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang J, Zhao Y, Shi J, Shao F. 2013. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc. Natl. Acad. Sci. U. S. A. 110:14408–14413. 10.1073/pnas.1306376110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. 2011. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477:596–600. 10.1038/nature10510 [DOI] [PubMed] [Google Scholar]
- 21. Rayamajhi M, Zak DE, Chavarria-Smith J, Vance RE, Miao EA. 2013. Cutting edge: mouse NAIP1 detects the type III secretion system needle protein. J. Immunol. 191:3986–3989. 10.4049/jimmunol.1301549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mijares LA, Wangdi T, Sokol C, Homer R, Medzhitov R, Kazmierczak BI. 2011. Airway epithelial MyD88 restores control of Pseudomonas aeruginosa murine infection via an IL-1-dependent pathway. J. Immunol. 186:7080–7088. 10.4049/jimmunol.1003687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Horai R, Asano M, Sudo K, Kanuka H, Suzuki M, Nishihara M, Takahashi M, Iwakura Y. 1998. Production of mice deficient in genes for interleukin (IL)-1α, IL-1β, IL-1α/β, and IL-1 receptor antagonist shows that IL-1β is crucial in turpentine-induced fever development and glucocorticoid secretion. J. Exp. Med. 187:1463–1475. 10.1084/jem.187.9.1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lara-Tejero M, Sutterwala FS, Ogura Y, Grant EP, Bertin J, Coyle AJ, Flavell RA, Galan JE. 2006. Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J. Exp. Med. 203:1407–1412. 10.1084/jem.20060206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu PV. 1966. The roles of various fractions of Pseudomonas aeruginosa in its pathogenesis: identity of the lethal toxins produced in vitro and in vivo. J. Infect. Dis. 116:481–489. 10.1093/infdis/116.4.481 [DOI] [PubMed] [Google Scholar]
- 26. Garrity-Ryan L, Kazmierczak B, Kowal R, Comolli J, Hauser A, Engel J. 2000. The arginine finger domain of ExoT is required for actin cytoskeleton disruption and inhibition of internalization of Pseudomonas aeruginosa by epithelial cells and macrophages. Infect. Immun. 68:7100–7113. 10.1128/IAI.68.12.7100-7113.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hauser AR, Kang PJ, Engel J. 1998. PepA, a novel secreted protein of Pseudomonas aeruginosa, is necessary for cytotoxicity and virulence. Mol. Microbiol. 27:807–818. 10.1046/j.1365-2958.1998.00727.x [DOI] [PubMed] [Google Scholar]
- 28. Finck-Barbancon V, Goranson J, Zhu L, Sawa T, Wiener-Kronish JP, Fleiszig SMJ, Wu C, Mende-Mueller L, Frank D. 1997. ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol. Microbiol. 25:547–557. 10.1046/j.1365-2958.1997.4891851.x [DOI] [PubMed] [Google Scholar]
- 29. Kenny EF, O'Neill LA. 2008. Signalling adaptors used by Toll-like receptors: an update. Cytokine 43:342–349. 10.1016/j.cyto.2008.07.010 [DOI] [PubMed] [Google Scholar]
- 30. Kono H, Orlowski GM, Patel Z, Rock KL. 2012. The IL-1-dependent sterile inflammatory response has a substantial caspase-1-independent component that requires cathepsin C. J. Immunol. 189:3734–3740. 10.4049/jimmunol.1200136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Netea MG, Simon A, van de Veerdonk F, Kullberg B-J, van der Meer JWM, Joosten LAB. 2010. IL-1β processing in host defense: beyond the inflammasomes. PLoS Pathog. 6:e10000661. 10.1371/journal.ppat.1000661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schonbeck U, Mach F, Libby P. 1998. Generation of biologically active IL-1 beta by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1 beta processing. J. Immunol. 161:3340–3346 [PubMed] [Google Scholar]
- 33. Karmakar M, Sun Y, Hise AG, Rietsch A, Pearlman E. 2012. Cutting edge: IL-1beta processing during Pseudomonas aeruginosa infection is mediated by neutrophil serine proteases and is independent of NLRC4 and caspase-1. J. Immunol. 189:4231–4235. 10.4049/jimmunol.1201447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM. 2011. Non-canonical inflammasome activation targets caspase-11. Nature 479:117–121. 10.1038/nature10558 [DOI] [PubMed] [Google Scholar]
- 35. Schreiber A, Pham CT, Hu Y, Schneider W, Luft FC, Kettritz R. 2012. Neutrophil serine proteases promote IL-1β generation and injury in necrotizing crescentic glomerulonephritis. J. Am. Soc. Nephrol. 23:470–482. 10.1681/ASN.2010080892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mankan AK, Dau T, Jenne D, Hornung V. 2012. The NLRP3/ASC/caspase-1 axis regulates IL-1β processing in neutrophils. Eur. J. Immunol. 42:710–715. 10.1002/eji.201141921 [DOI] [PubMed] [Google Scholar]
- 37. Bakele M, Joos M, Burdi S, Allgaier N, Poschel S, Fehrenbacher B, Schaller M, Marcos V, Kummerle-Deschner J, Rieber N, Borregaard N, Yazdi A, Hector A, Hartl D. 2014. Localization and functionality of the inflammasome in neutrophils. J. Biol. Chem. 289:5320–5329. 10.1074/jbc.M113.505636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Marriott HM, Gascoyne KA, Gowda R, Geary I, Nicklin MJ, Iannelli F, Pozzi G, Mitchell TJ, Whyte MK, Sabroe I, Dockrell DH. 2012. Interleukin-1beta regulates CXCL8 release and influences disease outcome in response to Streptococcus pneumoniae, defining intercellular cooperation between pulmonary epithelial cells and macrophages. Infect. Immun. 80:1140–1149. 10.1128/IAI.05697-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cai S, Batra S, Wakamatsu N, Pacher P, Jeyaseelan S. 2012. NLRC4 inflammasome-mediated production of IL-1β modulates mucosal immunity in the lung against gram-negative bacterial infection. J. Immunol. 188:5623–5635. 10.4049/jimmunol.1200195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miller LS, Pietras EM, Uricchio LH, Hirano K, Rao S, Lin H, O'Connell RM, Iwakura Y, Cheung AL, Cheng G, Modlin RL. 2007. Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J. Immunol. 179:6933–6942. 10.4049/jimmunol.179.10.6933 [DOI] [PubMed] [Google Scholar]
- 41. Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, Kugler D, Hieny S, Caspar P, Nunez G, Schlueter D, Flavell RA, Sutterwala FS, Sher A. 2010. Caspase-1 independent IL-1β production is critical for host resistance to Mycobacterium tuberculosis and does not require TLR signaling in vivo. J. Immunol. 184:3326–3330. 10.4049/jimmunol.0904189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rogers HW, Sheehan KC, Brunt LM, Dower SK, Unanue ER, Schreiber RD. 1992. Interleukin 1 participates in the development of anti-Listeria responses in normal and SCID mice. Proc. Natl. Acad. Sci. U. S. A. 89:1011–1015. 10.1073/pnas.89.3.1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Havell EA, Moldawer LL, Helfgott D, Kilian PL, Sehgal PB. 1992. Type I IL-1 receptor blockade exacerbates murine listeriosis. J. Immunol. 148:1486–1492 [PubMed] [Google Scholar]
- 44. Broz P, Monack DM. 2013. Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 13:551–565. 10.1038/nri3479 [DOI] [PubMed] [Google Scholar]
- 45. Wen H, Miao EA, Ting JP. 2013. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity 39:432–441. 10.1016/j.immuni.2013.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guler R, Parihar SP, SPohn G, Johansen P, Brombacher F, Bachmann MF. 2011. Blocking IL-1a but not IL-1b increases susceptibility to chronic Mycobacterium tuberculosis infection in mice. Vaccine 29:1339–1346. 10.1016/j.vaccine.2010.10.045 [DOI] [PubMed] [Google Scholar]
- 47. Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, Oland S, Gordon S, Sher A. 2011. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity 35:1023–1034. 10.1016/j.immuni.2011.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.