

Abstract
The opportunistic pathogen Staphylococcus aureus is one of the major causes of health care-associated infections. S. aureus is primarily an extracellular pathogen, but it was recently reported to invade and replicate in several host cell types. The ability of S. aureus to persist within cells has been implicated in resistance to antimicrobials and recurrent infections. However, few staphylococcal proteins that mediate intracellular survival have been identified. Here we examine if EsxA and EsxB, substrates of the ESAT-6-like secretion system (Ess), are important during intracellular S. aureus infection. The Esx proteins are required for staphylococcal virulence, but their functions during infection are unclear. While isogenic S. aureus esxA and esxB mutants were not defective for epithelial cell invasion in vitro, a significant increase in early/late apoptosis was observed in esxA mutant-infected cells compared to wild-type-infected cells. Impeding secretion of EsxA by deleting C-terminal residues of the protein also resulted in a significant increase of epithelial cell apoptosis. Furthermore, cells transfected with esxA showed an increased protection from apoptotic cell death. A double mutant lacking both EsxA and EsxB also induced increased apoptosis but, remarkably, was unable to escape from cells as efficiently as the single mutants or the wild type. Thus, using in vitro models of intracellular staphylococcal infection, we demonstrate that EsxA interferes with host cell apoptotic pathways and, together with EsxB, mediates the release of S. aureus from the host cell.
INTRODUCTION
Staphylococcus aureus is a Gram-positive coccus that causes infections ranging from superficial skin lesions to serious conditions such as pneumonia and endocarditis. S. aureus is also a major cause of hospital-acquired infections of surgical wounds and of indwelling medical devices. Staphylococcal infections, in particular systemic and chronic infections, place a major burden on health care systems worldwide (1, 2). Antibiotic resistance still remains a challenge in the management of staphylococcal infections, as methicillin-resistant S. aureus strains and strains with reduced susceptibility to vancomycin have complicated disease treatment (3, 4). During infection, S. aureus expresses a wide array of secreted and cell surface-associated virulence factors to evade immune responses by a variety of mechanisms, such as promoting adhesion to host cells, binding proteins in blood, and resisting immune cell attack (5–7).
In addition to its armor of virulence factors, the capacity of S. aureus to successfully evade host defenses was recently attributed to its ability to invade immune and nonimmune cells. S. aureus is mainly an extracellular pathogen, but an accumulating number of studies have shown that it can invade and replicate in many types of nonphagocytic host cells in vitro (8). Clinical studies have reported the presence of intracellular staphylococci from nasal epithelial cells, indicating that these may serve as a reservoir for recurrent infections (9, 10). Although the intracellular presence of S. aureus during in vivo staphylococcal infection remains unclear, a transient, intracellular lifestyle potentially provides protection against exposure to antibiotics and host immune responses, as well as a favorable environment for the formation of resistant variants (11, 12).
S. aureus possesses the Sec and Tat secretion systems, which presumably transport the majority of the known virulence factors (13, 14). A specialized ESAT-6 secretion system (Ess), similar to the Esx-1 secretion system described for Mycobacterium tuberculosis, was also identified in S. aureus (15). ESAT-6 homologs are also encoded in the genomes of other Gram-positive bacteria, including Bacillus subtilis, Bacillus anthracis, Clostridium acetobutylicum, and Listeria monocytogenes (16). Ess consists of 12 proteins, including EsxA and EsxB, which are similar to ESAT-6 and CFP-10 of M. tuberculosis. This region is highly conserved (89% to 94% sequence identity by BLASTn analysis) in the genomes of both community- and hospital-associated S. aureus strains. ESAT-6 (EsxA) and CFP-10 (EsxB) are well-characterized virulence factors of M. tuberculosis that are implicated in survival in macrophages, host cell lysis, and dissemination (17–19). For the staphylococcal Esx proteins, mutants that failed to secrete EsxA and EsxB displayed defects in S. aureus abscess formation in mice, suggesting that these proteins are important during staphylococcal disease (15). Other Ess proteins, such as EsaD, were reported to be important for staphylococcal virulence, while EsaC was required for persistent staphylococcal infection in mice (20, 21). Importantly, to date, no clear biological function has been attributed to the staphylococcal Esx proteins.
The precise structure of the Ess secretion apparatus is currently not known. Structural analysis of EsxA suggests that this protein may act as a chaperone or an adaptor protein to facilitate interactions with host receptor proteins (22). Codependent secretion of Ess substrates has been reported, similar to that observed for mycobacterial substrates (15). C-terminal residues are important for interaction of mycobacterial EsxB with other proteins of the apparatus and for secretion (23, 24). Recently, a C-terminal motif (YxxxD/E) of the Ess substrate EsxD was shown to be required for secretion of EsxA and EsaC (25).
In this study, we examined a potential intracellular role for staphylococcal EsxA and EsxB by employing an in vitro cellular model of S. aureus infection. We demonstrate here that EsxA interferes with S. aureus-induced apoptosis in human epithelial cells in vitro. This inhibitory effect is associated with the secretion of EsxA, which is mediated by C-terminal residues of the protein. Our data also suggest that EsxA and EsxB together affect the release of intracellular S. aureus from host cells.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
S. aureus strain USA300 (lac) was used for all experiments. Complemented S. aureus strains were grown in the presence of 10 μg/ml chloramphenicol. For infection experiments, bacteria were grown in tryptic soy broth (TSB) overnight (O/N) at 37°C, diluted 1/100 in fresh TSB, cultured until the exponential phase of growth (A600 = 0.6 to 0.7), and then rediluted 1/100 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS).
Construction of bacterial mutants.
For deletion of esxA and esxB, 2-kb DNA fragments flanking the esx genes were amplified by PCR and cloned into the Escherichia coli-S. aureus shuttle/suicide vector pKOR1 by using previously described methods (26). The esxA and esxB double mutant was obtained by deleting esxA from the ΔesxB mutant. All mutants were confirmed by PCR, using external primers targeting flanking regions, and by sequencing. For complementation of mutant strains, the full-length esxA and esxB genes were amplified and cloned into plasmid pOS1CK, which was generated by cloning the P1 constitutive promoter of the sarA gene into the pOS1 plasmid. For complementation with both genes, esxB was cloned downstream of esxA into the pOS1CKesxA construct. esxA variants with site-directed mutations were cloned into the episomal plasmid pOS1CK as described in the supplemental material. For generation of fluorescent bacteria, pOS1CK-GFP was cloned into the wild-type (WT) and ΔesxAB strains. Please see the supplemental material for details on all primers used for cloning.
Eukaryotic cell culture.
The human lung epithelial cell line A549 (CCL-185) was obtained from the ATCC, cultured in DMEM supplemented with 10% heat-inactivated fetal calf serum (Biological Industries) (DMEM-10), and incubated at 37°C and 5% CO2. For cell passages, 100 μg/ml Primocin (Invivogen) was used to supplement the medium, whereas no antibiotics were used 24 h prior to and during infection assays.
S. aureus infection of eukaryotic cells.
Twenty-four hours before infection, 2 × 105 A549 cells/ml were seeded in a 24-well plate (Nunc, Wiesbaden, Germany) so as to have 80% cell confluence. S. aureus cultures grown as described above were diluted in DMEM-10 and added to A549 cells at a multiplicity of infection (MOI) of 7:1. After 2 h of incubation, extracellular bacteria were killed by adding 20 μg/ml lysostaphin (Sigma-Aldrich) for 30 min at 37°C. To remove lysostaphin and dead bacteria, cells were washed with growth medium once, and in the case of further incubation, fresh growth medium supplemented with 5 μg/ml lysostaphin was added to each well. Infected host cells were trypsinized and lysed with cold water in a final volume of 1 ml. Lysates were diluted with phosphate-buffered saline (PBS) and plated in serial dilutions on tryptic soy agar (TSA) plates to calculate the number of CFU/ml.
Confocal microscopy.
For fluorescence microscopy, cells were grown in chamber slides with a polylysine coating and infected with S. aureus as described above. After lysostaphin treatment, cells were rinsed once with warm culture medium and fixed for 15 min with 4% paraformaldehyde (PFA) at room temperature. For observation of intracellular infections by vancomycin-Bodipy FL staining (Molecular Probes), permeabilized cells were incubated with 2 μg/ml vancomycin–1% bovine serum albumin (BSA) in PBS at 4°C in the dark for 15 min and then washed once. Phalloidin (Molecular Probes) at a dilution of 1:40 was used for staining of F-actin. For analysis, 60 to 150 cells per field were counted, and at least 4 fields were counted for each experiment. For microcolony visualization, nonpermeabilized cells were incubated with rabbit anti-staphylococcus at a dilution of 1:1,000 (ProSci) and Alexa Fluor 488-labeled secondary antibodies at a dilution of 1:1,000, together with biotinylated wheat germ agglutinin (1:500) and streptavidin-Alexa Fluor 568 (1:1,000), for 15 min at room temperature. Images were acquired with Leica LSM700 and LSM710 confocal microscopes and were analyzed by use of Leica confocal software (Leica Microsystems, Heidelberg, Germany).
Preparation and analysis of bacterial fractions.
Bacterial lysates and supernatants were prepared as described previously (27), with modifications. Please refer to the supplemental material for details.
Flow cytometry analysis.
For analysis of apoptosis, cells were grown on 24-well plates and infected with S. aureus as described above. At various time points postinfection (p.i.), medium was aspirated and 1 × 107 cells were detached using cell dissociation buffer (Invitrogen). Cells were washed once at 3,000 × g, pelleted, resuspended in 50 μl Aqua Live-Dead (1:500; Invitrogen), and then incubated for 20 min in the dark at 4°C. Cells were pelleted at 3,000 × g, washed once with PBS, resuspended in 200 μl annexin V (eBioscience), and incubated for 20 min in the dark at 4°C. The cells were washed, fixed with 4% PFA, and resuspended in 150 μl PBS. Double staining was analyzed by use of a FACSCanto II flow cytometer using FlowJo software on a subpopulation of entire single cells selected from the initial population, based on morphology. Each experiment was repeated at least three times.
Expression of bacterial esxA in A549 cells.
We constructed pesxA-EYFP by cloning the full-length esxA gene upstream of and in frame with the enhanced yellow fluorescent protein (EYFP) gene of plasmid pEYFP-N1 (Clontech). For transfection of A549 cells, 8 × 105 cells/well were seeded in a 6-well plate O/N and transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocols. After 6 h, the medium was changed, and at 24 h posttransfection, cells were labeled with Aqua Live-Dead and annexin V. Cells were analyzed by flow cytometry, selecting for cells that were positive for EYFP.
Statistical analysis.
Nonparametric one-way analysis of variance (ANOVA) with Tukey's or Dunnett's multiple-comparison test at a 95% confidence interval was applied to data from multiple groups, and the two-tailed Mann-Whitney U test was used for data from two groups. P values of <0.05 were considered statistically significant.
RESULTS
EsxA modulates apoptosis in S. aureus-infected cells.
To study the potential roles of EsxA and EsxB, two proteins encoded by the Ess locus of S. aureus (15), in the invasion and survival of human epithelial cells, isogenic ΔesxA and ΔesxB single mutants were generated in S. aureus USA300. The absence of expression of esxA or esxB in the mutants was confirmed by quantitative reverse transcription-PCR (qRT-PCR) (see Fig. S1A in the supplemental material), and analysis of the growth phenotypes of the ΔesxA and ΔesxB mutants in TSB medium showed no growth defects (see Fig. S1B). A549 epithelial cells were used for infection assays because of their high infection efficiency with wild-type (WT) USA300 (50 to 60% of cells infected) at MOIs of 1:1 to 10:1. A549 cell invasion assays performed to compare the esx mutants to the WT did not reveal any major differences in internalization efficacy (Fig. 1). Intracellular trafficking studies of the esx mutants by confocal microscopy showed the presence of bacteria both in vacuoles and in the cytoplasm at 4 h p.i., and mostly in the cytoplasm at 6 h p.i. (data not shown). However, there were no significant differences in trafficking of the mutants compared to the WT. Interestingly, analysis of infected cells showed that, very often, the presence of intracellular bacteria in the cytoplasm was followed by actin reorganization, leading to rounding of infected cells. Compared with the WT, larger numbers of cells infected with the ΔesxA mutant showed increased accumulation of polymerized actin (see Fig. S2), a phenotype linked with the earlier steps of apoptosis (28). This observation suggested that EsxA may play a role in host cell apoptosis during staphylococcal infection.
FIG 1.
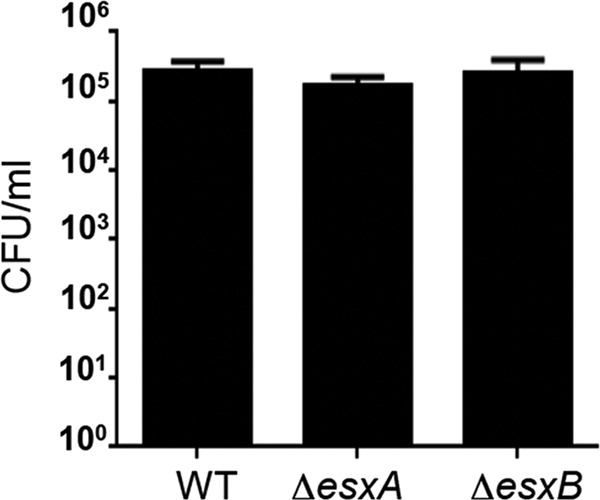
WT and mutant strains do not show any differences in cell invasion. A549 cells were infected with the WT and esx mutant strains as described in Materials and Methods. At 2 h p.i., extracellular bacteria were removed by lysostaphin treatment, cells were lysed, and intracellular bacteria were quantitated by colony counts.
To further investigate the potential role of EsxA in apoptosis, we performed flow cytometry at different time points after infection of cells with the WT and the esx mutants. Cells were stained with Aqua Live-Dead, which stains lysed cells (a marker of cell death), and annexin V, which specifically binds to the phosphatidylserine (PS) exposed on the plasma membranes of apoptotic cells. Staining with annexin V showed increased numbers of early and late apoptotic cells at 6 h p.i. for ΔesxA mutant-infected but not ΔesxB mutant-infected A549 cells (Fig. 2A and B). As shown in the scatterplots, the cells infected by the ΔesxA mutant consisted of increased subpopulations of early (bottom right quarter of the scatterplots) and/or late (top right quarter of the scatterplots) apoptotic cells compared to WT-infected cells. The increase in apoptosis observed for the mutant was reversed upon episomal expression of esxA in the ΔesxA mutant, i.e., with the ΔesxApOS1CKesxA strain (ΔesxA-esxA) (Fig. 2A and B). The differences in the numbers of apoptotic cells were found to decrease at later time points (8 h and 16 h), possibly because, during infection, cells containing intracellular bacteria die and detach from the monolayer (data not shown).
FIG 2.

EsxA modulates host cell apoptosis. (A) Flow cytometry analysis of S. aureus-infected A549 cells. Quantitation of apoptotic cells was performed by flow cytometry sorting of A549 cells infected with the WT, ΔesxA, ΔesxB, and ΔesxA-esxA strains at 6 h p.i. Cells were stained with Aqua Live-Dead (1:500) for the detection of lysed cells and with annexin V-fluorescein isothiocyanate (FITC) for the detection of apoptotic cells. Representative flow cytometry scatterplots depict the early apoptotic (bottom right quadrant), late apoptotic (top right quadrant), and dead (top left quadrant) cells at 6 h p.i. The numbers in the plots represent the percentage of gated A549 cell populations in each quadrant. (B) Percentages of early and late apoptotic cells in the parent population for mutant-infected cells in relation to that for WT-infected cells, which was set to 100%. *, significant difference by one-way ANOVA with Tukey's multiple-comparison test (P < 0.05). The data presented are the means and standard deviations (SD) of results from three independent experiments.
These data suggest that the EsxA protein may modulate S. aureus-induced apoptosis during epithelial cell infection in vitro.
C-terminal residues are important for secretion of EsxA: impeding EsxA secretion affects EsxA-mediated modulation of apoptosis.
Because EsxA is a secreted protein, we wanted to test if preventing secretion of EsxA could also result in increased apoptosis. As the signal sequences controlling EsxA secretion are not known, we first identified the residues that are required for secretion of the protein in vitro. Based on the homology between M. tuberculosis and S. aureus Esx proteins, we identified the amino acids that are conserved at the C termini of the staphylococcal proteins, a region thought to be important for secretion of Esx proteins in mycobacteria (23). Leucine90, serine91, and glycine95 were conserved in both the staphylococcal Esx proteins and mycobacterial EsxB (Fig. 3A).
FIG 3.

C-terminal residues are important for secretion of EsxA: impeding EsxA secretion affects EsxA-mediated modulation of apoptosis. (A) Alignment of aa sequences of M. tuberculosis and S. aureus EsxA and EsxB proteins. Residues shown in red are the highly conserved amino acid residues that were replaced with alanine, and the blue line indicates the 8 aa deleted in the truncated EsxA protein. (B) Immunoblot analysis of total extracts (TE) and supernatants (SN). Proteins in each fraction were precipitated with trichloroacetic acid (TCA), separated by SDS-PAGE, and detected by immunoblotting with anti-Esx, anti-RNA polymerase beta (loading and cell lysis control), and anti-hemolysin (loading control, supernatant). Loading was also normalized by determining the optical density at 600 nm (OD600) of the bacterial culture. The graph at the bottom shows the results of densitometry analysis performed using ImageJ software. (C) Flow cytometry analysis of apoptotic A549 cells infected with esxA mutants expressing native or mutant forms of EsxA. At 6 h p.i., cells were dissociated and stained with Aqua Live-Dead and annexin V-FITC. The graph presents the percentages of early and late apoptotic cells induced by the esxA mutant containing empty vector (ΔesxA-pOS1CK) or the different mutant forms (ΔesxA-esxAT and ΔesxA-esxALSG) in relation to the esxA mutant expressing native esxA (ΔesxA-esxA) (set at 100%). Data are the means and SD of results from three independent experiments. *, significant difference by one-way ANOVA with Tukey's multiple-comparison test (P < 0.05).
In an attempt to define targeting sequences responsible for mediating secretion by the Ess system, we studied effects of site-directed mutations and deletions at the C terminus of the EsxA protein. EsxA bearing mutations in 3 amino acids (aa) (L90A, S91A, and G95A) or with an 8-aa deletion was expressed episomally in the ΔesxA mutant to create the ΔesxApOS1CKesxA-LSG (ΔesxA-esxALSG) or ΔesxApOS1CKesxAtrunc (ΔesxA-esxAT) strain, respectively. Immunoblotting showed that the triple mutation in EsxA mildly reduced (by ∼25%) secretion of the protein into the culture supernatant (ΔesxA-esxALSG) compared with secretion of native EsxA (ΔesxA-esxA). A truncation of the C-terminal tail of EsxA comprising the last 8 aa severely impaired secretion of the protein (by ∼75%) (Fig. 3B). Similar amounts of the secreted protein hemolysin were detected in all samples. Probing for the abundant cytoplasmic protein RNA polymerase (RNAP) beta showed very minimal and similar levels of cell lysis in all supernatants (Fig. 3B).
To understand if hindering secretion of EsxA affects cellular apoptosis, we investigated whether the ΔesxA-esxALSG and ΔesxA-esxAT strains were able to reverse the increase in apoptosis induced by the ΔesxA mutant, as seen above with the ΔesxA mutant complemented with native EsxA (ΔesxA-esxA) (Fig. 2A and B). Flow cytometry analysis showed that the ΔesxA mutant expressing truncated EsxA (ΔesxA-esxAT) was able to induce more apoptotic cells than the mutant expressing the native EsxA protein (Fig. 3C) and had a profile similar to that of the control, i.e., the ΔesxA mutant transformed with empty plasmid (ΔesxA-pOS1CK). On the other hand, the ΔesxA-esxALSG strain did not show a similar increase in apoptosis, probably due to the minimal effects of the LSG mutations on EsxA secretion (Fig. 3B).
Thus, these data suggest that impeding the secretion of EsxA can affect the EsxA-mediated modulation of host cell apoptosis in epithelial cells in vitro.
EsxA delays apoptosis when expressed in the host cell.
In order to further confirm that EsxA interferes with host cell apoptotic pathways, A549 cells were transfected with a pEYFP plasmid expressing full-length EsxA or truncated EsxA (8 aa) as an N-terminal fusion with EYFP (pesxA-EYFP). Using flow cell cytometry, we measured the apoptotic subpopulations in the transfected cells (gated by expression of EYFP) by use of annexin V and Aqua Live-Dead staining. The expression of full-length esxA in the cytoplasm resulted in a 2.5-fold decrease of the number of apoptotic cells compared to that for control cells transfected with the empty vector pEYFP (Fig. 4A). These data are in agreement with the results described above for the esxA mutant. Expression of esxA lacking the C-terminal 8 aa, on the other hand, did not induce a significant decrease in apoptosis, suggesting that the C-terminal residues needed for secretion are not important for mediating apoptosis (see Fig. S3 in the supplemental material).
FIG 4.

EsxA interferes with apoptosis when expressed in the host cell. (A) Flow cytometry analysis of apoptotic A549 cells transfected with esxA. At 24 h posttransfection, cells were dissociated and then stained with Aqua Live-Dead and annexin V-allophycocyanin (APC). The percentage of apoptotic cells (early and late apoptotic cells) was measured in the subpopulation bearing EYFP, i.e., transfected cells. The value shown is a percentage of the level for cells transfected with the control vector. Scatterplots of the EYFP populations are shown in the bottom panels. Data are means and SD of results from three independent experiments. *, significant difference by two-tailed Mann-Whitney test. (B) Flow cytometry analysis of apoptosis after treatment with staurosporine. At 24 h posttransfection, A549 cells transfected with the plasmid pesxA-EYFP were treated with 2.5 mM staurosporine for 30 min, followed by Aqua Live-Dead and annexin V-APC staining. The percentage of apoptotic cells was measured in the subpopulation of transfected cells expressing EYFP. Data are the means and SD of results from three independent experiments. *, significant differences by one-way ANOVA with Dunnett's multiple-comparison test (P < 0.05).
Staurosporine, a potent inducer of apoptosis, is known to induce caspase-dependent and caspase-independent apoptotic pathways (29). We tested whether EsxA suppresses staurosporine-induced apoptosis. Twenty-four hours after transfection with the control plasmid pEYFP or pesxA-EYFP, cells were treated with 2.5 μM staurosporine for 30 min. Staurosporine-induced apoptosis, as measured by annexin V staining, was significantly reduced in cells transfected with pesxA-EYFP compared with the pEYFP-transfected cells (Fig. 4B; see Fig. S4 in the supplemental material).
In total, the data support a role for staphylococcal EsxA in interfering with cell apoptotic pathways.
Esx proteins affect escape of bacteria from host cells.
The mycobacterial counterparts of EsxA and EsxB are known to form a heterodimer and also function together to cause host cell lysis (30, 31). Recent data have shown that staphylococcal EsxA and EsxB can interact with different Ess substrates but do not interact directly with each other (22, 25). To understand the effects of EsxA and EsxB taken together, we constructed a double mutant of esxA and esxB. As observed for the single mutants, the ΔesxAB strain was similar to the WT for invasion of epithelial cells (data not shown). As observed for the ΔesxA strain (Fig. 2), the ΔesxAB strain induced more apoptosis in epithelial cells (see Fig. S5 in the supplemental material). Interestingly, colony counts from infected A549 cells showed a significant increase of the intracellular ΔesxAB strain at a later time point after cell invasion (16 h p.i.), while there were no significant differences for the ΔesxA and ΔesxB mutants compared to the WT (Fig. 5A). Examination of cells by confocal microscopy revealed the presence of more intracellular bacteria in epithelial cells infected with the ΔesxAB strain than in those infected with the WT (Fig. 5B). The differences between the WT and the double mutant were also confirmed using green fluorescent protein (GFP)-expressing S. aureus strains; cells containing more fluorescent bacteria were observed for the ΔesxAB mutant-infected than WT-infected cells (Fig. 5C).
FIG 5.

Increased numbers of intracellular bacteria in epithelial cells infected with the ΔesxAB mutant. (A) Colony counts of intracellular bacteria with WT and single and double mutants of the Esx proteins in A549 epithelial cells infected for 16 h. An ∼3-fold increase can be seen for the double mutant compared to the WT. **, significant differences by one-way ANOVA with Tukey's multiple-comparison test (P < 0.001). Data are representative of three independent experiments. (B) Confocal microscopy of A549 cells infected with the WT or ΔesxAB strain after 16 h of infection. Intracellular bacteria were stained with vancomycin-Bodipy (green), and cells were stained with phalloidin (red). (C) Representative images (magnification, ×10) of A549 cells with intracellular fluorescent (GFP-expressing) WT and ΔesxAB strains. The white arrows indicate the intracellular bacteria. As described in Materials and Methods, assays were performed in the presence of lysostaphin to ensure that bacteria were intracellular.
To understand if the ΔesxAB strain accumulated within cells as a result of its inability to exit cells, we used a continuous infection assay in which infected A549 cell monolayers were first treated with lysostaphin to remove extracellular bacteria but thereafter incubated in medium without lysostaphin for different times. Bacteria were seen to exit cells and progressively form extracellular bacterial aggregates on the cell layer. After 9 h p.i., we observed the formation of several such S. aureus “microcolonies” by immunofluorescence staining of cells infected with the WT, ΔesxA, and ΔesxB strains, but very few for cells infected with the ΔesxAB double mutant (Fig. 6). Equal infection efficiencies in this continuous infection assay were confirmed for all strains by quantitating intracellular counts after 2 h (data not shown). Bacterial counts performed with the cell culture medium of WT- and ΔesxAB mutant-infected cells at an earlier time after infection (to minimize the effect of reinvasion of the WT bacteria that had exited) confirmed the decreased numbers of the ΔesxAB mutant (see Fig. S6 in the supplemental material). Eventually, at later times, the esxAB mutant was able to exit cells as seen for the WT.
FIG 6.

The staphylococcal EsxA and EsxB proteins may mediate bacterial release from infected cells. Confocal microscopy analysis was performed at 9 h 30 min p.i. on A549 cells continuously infected by WT and mutant strains. Extracellular bacterial microcolonies and bacteria exiting infected dying cells can be seen on the cell layer (white arrows). Very few microcolonies were observed for the ΔesxAB mutant. Bacteria were stained with anti-S. aureus (green), and cells were stained with wheat germ agglutinin (WGA) (red). All the images are representative of at least 3 independent infection experiments. The white arrows indicate bacterial microcolonies outside epithelial cells.
In order to rule out any possible defects in the agr system, hemolysis on blood agar and production of alpha-hemolysin were examined in the single and double mutants, and no defects were observed (see Fig. S7 in the supplemental material). In addition, sequencing of RNA-III and saeS-saeR revealed no secondary mutations (data not shown).
These results suggest that in our in vitro infection assay, EsxA and EsxB together modulate cellular escape of S. aureus.
DISCUSSION
The human pathogen S. aureus has been shown to invade and survive in a range of host cells in vitro (8, 12, 32). Despite several studies describing intracellular staphylococcal infection, there is very little understanding of the bacterial factors or mechanisms involved in intracellular survival of S. aureus. Our data show that EsxA, a protein secreted by the specialized staphylococcal secretion system Ess, modulates host cell survival by interfering with apoptotic pathways. To our knowledge, this is the first description of a staphylococcal factor that has an antiapoptotic function in epithelial cells. Our data also support intracellular functions for the staphylococcal Esx proteins in cellular models of infection.
The interplay between bacterial virulence factors and host cell proteins is of utmost importance for a successful bacterial infection, and staphylococcal infection is no exception to the rule. In order to persist in the hostile environment of the host cell, invading pathogens have developed various mechanisms to survive intracellularly, e.g., by inhibiting lysosomal killing or by fine-tuning cell mechanisms, such as apoptosis or autophagy (33, 34). It is known that apoptosis, a well-known mechanism of cell death, is induced by intracellular S. aureus (35–37). The staphylococcal toxin, alpha-hemolysin (Hla), was reported to induce apoptosis upon intracellular staphylococcal infection (37). The apoptotic pathways affected by S. aureus appear to depend on the strain and host cell type used, and several studies argue for or against its employment of molecules such as caspases (38–41) or calcium (42) to induce apoptosis in epithelial cells. On the other hand, as seen for many other bacteria, S. aureus may also be able to block apoptosis. Although an antiapoptotic effect of S. aureus has not yet been demonstrated, S. aureus was recently reported to induce antiapoptotic factors in epithelial cells (43). During staphylococcal infections, EsxA alone and/or with other Ess effectors may transiently block cell apoptosis induced by extracellular staphylococci or by other staphylococcal proteins to allow for intracellular replication of bacteria. Indeed, it has been shown for other pathogens, such as Helicobacter pylori and enteropathogenic E. coli (44, 45), that bacterial proteins can have both pro- and antiapoptotic activities and that bacterial antiapoptotic factors are active in the host cell cytoplasm. S. aureus, like other bacterial intracellular pathogens, is capable of manipulating the host cell to its advantage.
EsxA and EsxB, which are substrates of a novel ESAT-6-like secretion system (Ess), are encoded in the same locus but are separated by 6 other genes (15). EsxA is expressed as a single transcript and regulated by sigma factor B and sigma factor B-controlled SpoVG, while we do not know how EsxB is regulated (46). Secretion of EsxA and EsxB into the culture medium during growth in vitro was demonstrated previously, with mutation of EsxB affecting secretion of EsxA, and vice versa (15). Very recent work further reports that deletions in either gene also affect secretion of additional Ess substrates, EsxC and EsxD (25), and EsxA and EsxB were reported to interact with different Ess substrates, such as EsxC and EsxD, respectively (25). Hence, it is not surprising that EsxA and EsxB mutants behave differently in terms of modulating apoptosis. EsxA and EsxB may affect stability and/or secretion of specific subsets of Ess substrates inside cells.
C-terminal residues were reported previously to mediate secretion of EsxB (CFP-10) and other Esx locus-encoded substrates secreted by mycobacterial Esx-1 and Esx-1 paralogs (23, 24). We showed that the C-terminal tail of EsxA, which contains residues conserved in mycobacterial and staphylococcal Ess substrates (EsxA and EsxB), is important for secretion of EsxA in vitro. The effect of these residues on the secretion of other Ess substrates was not examined in this study, but we believe that they may have an effect, as reported recently (25). Also, secretion of EsxA was not completely abolished in this case; hence, there are likely to be other residues or interactions governing secretion. Defective secretion of EsxA mimics the increase in apoptosis seen for the ΔesxA mutant. This suggests that secretion of EsxA is important for EsxA-mediated apoptosis. However, we have yet to demonstrate directly the intracellular secretion of this protein.
As discussed above, disrupting secretion of EsxA may affect EsxA-dependent Ess substrates. However, our transfection studies indicate that EsxA may directly mediate an antiapoptotic effect in the host cell cytoplasm. EsxA lacking the C-terminal 8 aa was not important for cellular effects, suggesting that any interactions with host or bacterial factors involve alternate regions of the protein. EsxA may function by either blocking one of the caspase-mediated pathways or imitating or inducing host antiapoptotic factors, such as Bcl2. Our preliminary studies suggest that EsxA effects may be caspase-3 independent, but further studies are required to understand how EsxA intercepts cell death pathways.
In other pathogens, an important step in bacterial dissemination, after intracellular replication, is bacterial exit from host cells. Previous studies have demonstrated that staphylococcal factors, such as phenol-soluble modulins and leukocidins, may affect bacterial escape from neutrophils (47, 48). The roles of these factors in nonimmune cells are not clear at present. While both ΔesxA and ΔesxAB mutants showed increased apoptosis, the exit of bacteria from epithelial cells in continuous in vitro invasion assays was affected only when both proteins were absent. Mycobacterial EsxA and EsxB function as a heterodimer (30), but recently, mycobacterial EsxA alone was demonstrated to mediate lipid membrane lysis (49). Staphylococcal EsxA was crystallized as a homodimer, and there is no current evidence suggesting that EsxA and EsxB directly interact (22). We propose the following two possible explanations for our results with the double mutant: (i) EsxA and EsxB directly mediate cell lysis, in association with host proteins, in a multiprotein complex; or (ii) EsxA and EsxB modulate secretion of specific subsets of other Ess effectors which mediate cell lysis, and deletion of both proteins results in total loss of all required effectors.
The orthologous Esx proteins in mycobacteria have been implicated in several aspects of pathogenesis, including survival of mycobacteria in macrophages, granuloma formation, induction of apoptosis and autophagy, phagosomal rupture, and host cell lysis (17, 50). Although other Gram-positive bacteria, such as Listeria monocytogenes, contain orthologs, the functions of these orthologs are unclear (51, 52). The EsxA proteins from S. aureus and M. tuberculosis demonstrate only 20.8% identity (15), although they show structural similarities (22). It is very intriguing that the Esx proteins seem to have an intracellular role in M. tuberculosis and S. aureus, two very diverse pathogens. The mechanisms by which they affect intracellular survival, however, appear to be different. Our data suggest that staphylococcal Esx proteins are not involved in avoiding phagosomal lysis, as indicated by a lack of differences in cytoplasmic versus vacuolar bacteria in infected epithelial cells, but may be involved in lysing the host cell. The staphylococcal proteins do not appear to affect the survival of the bacteria within the cell per se, as bacterial numbers by colony counts (Fig. 1 and 5) and by microscopy (data not shown) appeared to be unaltered. Such differences could also be attributed to the cell type under study, i.e., macrophages versus epithelial cells. Both pathogens, however, may use the host cell as a niche to replicate and disseminate, with a key role for Esx proteins. Indeed, for S. aureus, the relevance of intracellular bacteria to infection and the role of the Esx proteins in modulating this remain to be demonstrated.
The facultative intracellular pathogen S. aureus may use the host cell to escape unfavorable extracellular environments. This intracellular phase may play a key role in determining persistence of the bacterium within an infected tissue. Esx proteins have been implicated in persistence within abscesses in infected mice (20). Based on our data from an in vitro model of infection, we propose that by delaying host cell death and controlling bacterial exit from host cells, the Esx proteins may facilitate persistence and spread of the pathogen in the infected host. Future studies on how the intracellular effects of these proteins contribute to virulence will clarify their role during infection.
Supplementary Material
ACKNOWLEDGMENTS
We thank Enrico Malito and Mathew Bottomley for their help with alignment studies of the Esx proteins. We also thank Roberto Petracca and Sara Iozzi for their help with cloning.
This study was partially funded by the EIMID-IAPP project (grant GA 217768) of the EU FP7 People Programme (Marie Curie Actions).
Footnotes
Published ahead of print 21 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01576-14.
REFERENCES
- 1. Allegranzi B, Bagheri Nejad S, Combescure C, Graafmans W, Attar H, Donaldson L, Pittet D. 2011. Burden of endemic health-care-associated infection in developing countries: systematic review and meta-analysis. Lancet 377:228–241. 10.1016/S0140-6736(10)61458-4 [DOI] [PubMed] [Google Scholar]
- 2. Dulon M, Haamann F, Peters C, Schablon A, Nienhaus A. 2011. MRSA prevalence in European healthcare settings: a review. BMC Infect. Dis. 11:138. 10.1186/1471-2334-11-138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gould IM, David MZ, Esposito S, Garau J, Lina G, Mazzei T, Peters G. 2012. New insights into meticillin-resistant Staphylococcus aureus (MRSA) pathogenesis, treatment and resistance. Int. J. Antimicrob. Agents 39:96–104. 10.1016/j.ijantimicag.2011.09.028 [DOI] [PubMed] [Google Scholar]
- 4. Howden BP, Davies JK, Johnson PD, Stinear TP, Grayson ML. 2010. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin. Microbiol. Rev. 23:99–139. 10.1128/CMR.00042-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Edwards AM, Massey RC. 2011. How does Staphylococcus aureus escape the bloodstream? Trends Microbiol. 19:184–190. 10.1016/j.tim.2010.12.005 [DOI] [PubMed] [Google Scholar]
- 6. Foster TJ. 2009. Colonization and infection of the human host by staphylococci: adhesion, survival and immune evasion. Vet. Dermatol. 20:456–470. 10.1111/j.1365-3164.2009.00825.x [DOI] [PubMed] [Google Scholar]
- 7. Spaan AN, Surewaard BG, Nijland R, van Strijp JA. 2013. Neutrophils versus Staphylococcus aureus: a biological tug of war. Annu. Rev. Microbiol. 67:629–650. 10.1146/annurev-micro-092412-155746 [DOI] [PubMed] [Google Scholar]
- 8. Garzoni C, Kelley WL. 2009. Staphylococcus aureus: new evidence for intracellular persistence. Trends Microbiol. 17:59–65. 10.1016/j.tim.2008.11.005 [DOI] [PubMed] [Google Scholar]
- 9. Clement S, Vaudaux P, Francois P, Schrenzel J, Huggler E, Kampf S, Chaponnier C, Lew D, Lacroix JS. 2005. Evidence of an intracellular reservoir in the nasal mucosa of patients with recurrent Staphylococcus aureus rhinosinusitis. J. Infect. Dis. 192:1023–1028. 10.1086/432735 [DOI] [PubMed] [Google Scholar]
- 10. Sachse F, Becker K, von Eiff C, Metze D, Rudack C. 2010. Staphylococcus aureus invades the epithelium in nasal polyposis and induces IL-6 in nasal epithelial cells in vitro. Allergy 65:1430–1437. 10.1111/j.1398-9995.2010.02381.x [DOI] [PubMed] [Google Scholar]
- 11. Tuchscherr L, Medina E, Hussain M, Volker W, Heitmann V, Niemann S, Holzinger D, Roth J, Proctor RA, Becker K, Peters G, Loffler B. 2011. Staphylococcus aureus phenotype switching: an effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol. Med. 3:129–141. 10.1002/emmm.201000115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fraunholz M, Sinha B. 2012. Intracellular Staphylococcus aureus: live-in and let die. Front. Cell. Infect. Microbiol. 2:43. 10.3389/fcimb.2012.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Biswas L, Biswas R, Nerz C, Ohlsen K, Schlag M, Schafer T, Lamkemeyer T, Ziebandt AK, Hantke K, Rosenstein R, Gotz F. 2009. Role of the twin-arginine translocation pathway in Staphylococcus. J. Bacteriol. 191:5921–5929. 10.1128/JB.00642-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schneewind O, Missiakas DM. 2012. Protein secretion and surface display in Gram-positive bacteria. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367:1123–1139. 10.1098/rstb.2011.0210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burts ML, Williams WA, DeBord K, Missiakas DM. 2005. EsxA and EsxB are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. Proc. Natl. Acad. Sci. U. S. A. 102:1169–1174. 10.1073/pnas.0405620102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pallen MJ. 2002. The ESAT-6/WXG100 superfamily—and a new Gram-positive secretion system? Trends Microbiol. 10:209–212. 10.1016/S0966-842X(02)02345-4 [DOI] [PubMed] [Google Scholar]
- 17. Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, Eisenberg D, Russell RG, Derrick SC, Collins FM, Morris SL, King CH, Jacobs WR., Jr 2003. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc. Natl. Acad. Sci. U. S. A. 100:12420–12425. 10.1073/pnas.1635213100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Davis JM, Ramakrishnan L. 2009. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 136:37–49. 10.1016/j.cell.2008.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stanley SA, Raghavan S, Hwang WW, Cox JS. 2003. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc. Natl. Acad. Sci. U. S. A. 100:13001–13006. 10.1073/pnas.2235593100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burts ML, DeDent AC, Missiakas DM. 2008. EsaC substrate for the ESAT-6 secretion pathway and its role in persistent infections of Staphylococcus aureus. Mol. Microbiol. 69:736–746. 10.1111/j.1365-2958.2008.06324.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anderson M, Chen YH, Butler EK, Missiakas DM. 2011. EsaD, a secretion factor for the Ess pathway in Staphylococcus aureus. J. Bacteriol. 193:1583–1589. 10.1128/JB.01096-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sundaramoorthy R, Fyfe PK, Hunter WN. 2008. Structure of Staphylococcus aureus EsxA suggests a contribution to virulence by action as a transport chaperone and/or adaptor protein. J. Mol. Biol. 383:603–614. 10.1016/j.jmb.2008.08.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Champion PA, Stanley SA, Champion MM, Brown EJ, Cox JS. 2006. C-terminal signal sequence promotes virulence factor secretion in Mycobacterium tuberculosis. Science 313:1632–1636. 10.1126/science.1131167 [DOI] [PubMed] [Google Scholar]
- 24. Daleke MH, Ummels R, Bawono P, Heringa J, Vandenbroucke-Grauls CM, Luirink J, Bitter W. 2012. General secretion signal for the mycobacterial type VII secretion pathway. Proc. Natl. Acad. Sci. U. S. A. 109:11342–11347. 10.1073/pnas.1119453109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anderson M, Aly KA, Chen YH, Missiakas D. 2013. Secretion of atypical protein substrates by the ESAT-6 secretion system of Staphylococcus aureus. Mol. Microbiol. 90:734–743. 10.1111/mmi.12395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63. 10.1016/j.plasmid.2005.05.005 [DOI] [PubMed] [Google Scholar]
- 27. Frankel MB, Wojcik BM, DeDent AC, Missiakas DM, Schneewind O. 2010. ABI domain-containing proteins contribute to surface protein display and cell division in Staphylococcus aureus. Mol. Microbiol. 78:238–252. 10.1111/j.1365-2958.2010.07334.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Suarez-Huerta N, Mosselmans R, Dumont JE, Robaye B. 2000. Actin depolymerization and polymerization are required during apoptosis in endothelial cells. J. Cell Physiol. 184:239–245. [DOI] [PubMed] [Google Scholar]
- 29. Belmokhtar CA, Hillion J, Segal-Bendirdjian E. 2001. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 20:3354–3362. 10.1038/sj.onc.1204436 [DOI] [PubMed] [Google Scholar]
- 30. Renshaw PS, Lightbody KL, Veverka V, Muskett FW, Kelly G, Frenkiel TA, Gordon SV, Hewinson RG, Burke B, Norman J, Williamson RA, Carr MD. 2005. Structure and function of the complex formed by the tuberculosis virulence factors CFP-10 and ESAT-6. EMBO J. 24:2491–2498. 10.1038/sj.emboj.7600732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kinhikar AG, Verma I, Chandra D, Singh KK, Weldingh K, Andersen P, Hsu T, Jacobs WR, Jr, Laal S. 2010. Potential role for ESAT6 in dissemination of M. tuberculosis via human lung epithelial cells. Mol. Microbiol. 75:92–106. 10.1111/j.1365-2958.2009.06959.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kubica M, Guzik K, Koziel J, Zarebski M, Richter W, Gajkowska B, Golda A, Maciag-Gudowska A, Brix K, Shaw L, Foster T, Potempa J. 2008. A potential new pathway for Staphylococcus aureus dissemination: the silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS One 3:e1409. 10.1371/journal.pone.0001409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Galan JE, Cossart P. 2005. Host-pathogen interactions: a diversity of themes, a variety of molecular machines. Curr. Opin. Microbiol. 8:1–3. 10.1016/j.mib.2004.12.015 [DOI] [PubMed] [Google Scholar]
- 34. Mostowy S, Cossart P. 2012. Bacterial autophagy: restriction or promotion of bacterial replication? Trends Cell Biol. 22:283–291. 10.1016/j.tcb.2012.03.006 [DOI] [PubMed] [Google Scholar]
- 35. Wesson CA, Liou LE, Todd KM, Bohach GA, Trumble WR, Bayles KW. 1998. Staphylococcus aureus Agr and Sar global regulators influence internalization and induction of apoptosis. Infect. Immun. 66:5238–5243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kahl BC, Goulian M, van Wamel W, Herrmann M, Simon SM, Kaplan G, Peters G, Cheung AL. 2000. Staphylococcus aureus RN6390 replicates and induces apoptosis in a pulmonary epithelial cell line. Infect. Immun. 68:5385–5392. 10.1128/IAI.68.9.5385-5392.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Menzies BE, Kourteva I. 2000. Staphylococcus aureus alpha-toxin induces apoptosis in endothelial cells. FEMS Immunol. Med. Microbiol. 29:39–45. 10.1111/j.1574-695X.2000.tb01503.x [DOI] [PubMed] [Google Scholar]
- 38. Soong G, Chun J, Parker D, Prince A. 2012. Staphylococcus aureus activation of caspase 1/calpain signaling mediates invasion through human keratinocytes. J. Infect. Dis. 205:1571–1579. 10.1093/infdis/jis244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wesson CA, Deringer J, Liou LE, Bayles KW, Bohach GA, Trumble WR. 2000. Apoptosis induced by Staphylococcus aureus in epithelial cells utilizes a mechanism involving caspases 8 and 3. Infect. Immun. 68:2998–3001. 10.1128/IAI.68.5.2998-3001.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Imre G, Heering J, Takeda AN, Husmann M, Thiede B, zu Heringdorf DM, Green DR, van der Goot FG, Sinha B, Dotsch V, Rajalingam K. 2012. Caspase-2 is an initiator caspase responsible for pore-forming toxin-mediated apoptosis. EMBO J. 31:2615–2628. 10.1038/emboj.2012.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schnaith A, Kashkar H, Leggio SA, Addicks K, Kronke M, Krut O. 2007. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J. Biol. Chem. 282:2695–2706. 10.1074/jbc.M609784200 [DOI] [PubMed] [Google Scholar]
- 42. Eichstaedt S, Gabler K, Below S, Muller C, Kohler C, Engelmann S, Hildebrandt P, Volker U, Hecker M, Hildebrandt JP. 2009. Effects of Staphylococcus aureus-hemolysin A on calcium signalling in immortalized human airway epithelial cells. Cell Calcium 45:165–176. 10.1016/j.ceca.2008.09.001 [DOI] [PubMed] [Google Scholar]
- 43. Koziel J, Maciag-Gudowska A, Mikolajczyk T, Bzowska M, Sturdevant DE, Whitney AR, Shaw LN, DeLeo FR, Potempa J. 2009. Phagocytosis of Staphylococcus aureus by macrophages exerts cytoprotective effects manifested by the upregulation of antiapoptotic factors. PLoS One 4:e5210. 10.1371/journal.pone.0005210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oldani A, Cormont M, Hofman V, Chiozzi V, Oregioni O, Canonici A, Sciullo A, Sommi P, Fabbri A, Ricci V, Boquet P. 2009. Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog. 5:e1000603. 10.1371/journal.ppat.1000603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nougayrede JP, Foster GH, Donnenberg MS. 2007. Enteropathogenic Escherichia coli effector EspF interacts with host protein Abcf2. Cell. Microbiol. 9:680–693. 10.1111/j.1462-5822.2006.00820.x [DOI] [PubMed] [Google Scholar]
- 46. Schulthess B, Bloes DA, Berger-Bachi B. 2012. Opposing roles of sigmaB and sigmaB-controlled SpoVG in the global regulation of esxA in Staphylococcus aureus. BMC Microbiol. 12:17. 10.1186/1471-2180-12-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Geiger T, Francois P, Liebeke M, Fraunholz M, Goerke C, Krismer B, Schrenzel J, Lalk M, Wolz C. 2012. The stringent response of Staphylococcus aureus and its impact on survival after phagocytosis through the induction of intracellular PSMs expression. PLoS Pathog. 8:e1003016. 10.1371/journal.ppat.1003016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. DuMont AL, Yoong P, Surewaard BG, Benson MA, Nijland R, van Strijp JA, Torres VJ. 2013. Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infect. Immun. 81:1830–1841. 10.1128/IAI.00095-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. De Leon J, Jiang G, Ma Y, Rubin E, Fortune S, Sun J. 2012. Mycobacterium tuberculosis ESAT-6 exhibits a unique membrane-interacting activity that is not found in its ortholog from non-pathogenic Mycobacterium smegmatis. J. Biol. Chem. 287:44184–44191. 10.1074/jbc.M112.420869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Derrick SC, Morris SL. 2007. The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell. Microbiol. 9:1547–1555. 10.1111/j.1462-5822.2007.00892.x [DOI] [PubMed] [Google Scholar]
- 51. Way SS, Wilson CB. 2005. The Mycobacterium tuberculosis ESAT-6 homologue in Listeria monocytogenes is dispensable for growth in vitro and in vivo. Infect. Immun. 73:6151–6153. 10.1128/IAI.73.9.6151-6153.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gey Van Pittius NC, Gamieldien J, Hide W, Brown GD, Siezen RJ, Beyers AD. 2001. The ESAT-6 gene cluster of Mycobacterium tuberculosis and other high G+C Gram-positive bacteria. Genome Biol. 2:RESEARCH0044. 10.1186/gb-2001-2-10-research0044 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.