

Abstract
Diverse pathogens have evolved to survive and replicate in the endosomes or phagosomes of the host cells and establish persistent infection. Ehrlichiae are Gram-negative, intracellular bacteria that are transmitted by ticks. Ehrlichiae reside in the endosomes of the host phagocytic or endothelial cells and establish persistent infection in their vertebrate reservoir hosts. CD4+ T cells play a critical role in protection against phagosomal infections. In the present study, we investigated the expansion, maintenance, and functional status of antigen-specific CD4+ T cells during persistent Ehrlichia muris infection in wild-type and interleukin-10 (IL-10)-deficient mice. Our study indicated that early induction of IL-10 led to reduced inflammatory responses and impaired bacterial clearance during persistent Ehrlichia infection. Notably, we demonstrated that the functional production of gamma interferon (IFN-γ) by antigen-specific CD4+ T cells maintained during a persistent phagosomal infection progressively deteriorates. The functional loss of IFN-γ production by antigen-specific CD4+ T cells was reversed in the absence of IL-10. Furthermore, we demonstrated that transient blockade of IL-10 receptor during the T cell priming phase early in infection was sufficient to enhance the magnitude and the functional capacity of antigen-specific effector and memory CD4+ T cells, which translated into an enhanced recall response. Our findings provide new insights into the functional status of antigen-specific CD4+ T cells maintained during persistent phagosomal infection. The study supports the concept that a better understanding of the factors that influence the priming and differentiation of CD4+ T cells may provide a basis to induce a protective immune response against persistent infections.
INTRODUCTION
Several pathogens, including Ehrlichia, have evolved to survive in the endosomes or phagosomes of the host cells and establish persistent infection (1, 2). Ehrlichiae are pathogenic Gram-negative intracellular bacteria that are transmitted by ticks to humans and animals (3). Ehrlichiae reside in the endosomes of host phagocytic cells or endothelial cells and establish persistent infection in their reservoir vertebrate hosts despite the active host immune response (3). It is unclear whether Ehrlichia establishes persistent infection in humans, although occurrence of prolonged infection with Ehrlichia chaffeensis in human patients has been reported (4, 5). The reported high seroprevalence rates in humans from some regions in the absence of a high incidence of disease possibly suggest occurrence of subclinical and/or persistent infection (6–8). However, the mechanisms that contribute to persistence of ehrlichial infections are not well understood.
CD4+ T cells recognize peptide-major histocompatibility complex class II (peptide–MHC-II) complexes generated in the phagosomes and presented on the surface by antigen-presenting cells. CD4+ T cells play an important role in protection against intracellular pathogens, such as Ehrlichia, that replicate in the phagosomes of the host cells (1, 9, 10). CD4+ T cells proliferate and differentiate into Th1 effector cells following phagosomal infection and enhance the microbicidal activity of phagocytes (11). The cytokines gamma interferon (IFN-γ) and tumor necrosis factor alpha (TNF-α), secreted by polyfunctional Th1 cells, synergistically enhance the microbicidal activity of phagocytes through the induction of nitric oxide synthase (12). Although CD4+ T cells are effective in limiting phagosomal infections, these cells often fail to eliminate pathogens residing in the phagosomes, leading to establishment of persistent infection (13, 14). Formation of granulomas by phagosome-residing pathogens is thought to aid their persistence by limiting access of infected cells to activated T cells at the site of infection (15–17). Presentation of a small number of peptide–MHC-II complexes by the infected phagocytes within the granulomas limits production of IFN-γ and TNF-α by Th1 cells (18). In addition, Th1 cells at the site of persistent phagosomal infections acquire the capacity to produce the immunosuppressive cytokine interleukin-10 (IL-10), which dampens the microbicidal capacity of infected phagocytes at sites of persistent infections (19). Signaling through IL-10 receptor (IL-10R) suppresses production of IFN-γ and TNF-α and reduces expression of MHC-II and generation of nitric oxide by infected phagocytes, thereby limiting their microbicidal activity (20, 21). Phagocytes harboring pathogens in the phagosomes also produce IL-10, which suppresses T cells and inhibits phagosome-lysosome fusion (22, 23).
CD4+ T cells proliferate and expand in response to antigenic stimulation, resulting in generation of a peak of effector CD4+ T cells in about a week after acute infection, such as in the case of lymphocytic choriomeningitis virus (LCMV) or Listeria monocytogenes infection (24, 25). About 90% of the effector CD4+ T cells die by apoptosis, and the surviving population of memory phenotype cells consist of Th1 cells and follicular helper cell-like central memory cells (26). Unlike generation and maintenance of a stable population of memory CD8+ T cells following acute infection, the number of antigenic-specific CD4+ T cells gradually declines after the clearance of infection due to their lower rate of homeostatic proliferation driven by IL-15 than for memory CD8+ T cells (27). In contrast, in the case of pathogens that reside in the phagosomes and establish persistent infections, such as Mycobacterium, Leishmania, and Salmonella spp., a stable population of antigen-experienced Th1 cells is maintained due to antigen persistence (28–30). These antigen-experienced CD4+ T cells confer “concomitant immunity” against reinfection, but it requires antigen persistence (31).
Our previous study indicated that C57BL/6 mice infected with E. muris, which causes persistent infection, have increased expression of IL-10 in the spleen (32). In addition, our previous study indicated that elimination of persistent E. muris infection in mice leads to significant reduction in level of heterologous protection against Ixodes ovatus Ehrlichia, which was associated with decreased frequency of IFN-γ-producing antigen-specific CD4+ T cells (32). Mice infected with E. muris develop granulomas in the liver and lungs, and CD4+ T cells play an important role in protection against primary infection (10, 33). In the present study, we investigated the priming, expansion, and long-term maintenance of total polyclonal antigen-specific CD4+ T cells following E. muris infection in wild-type and IL-10-deficient (IL-10−/−) mice using the recently described surface markers CD49d and CD11a for the identification of antigen-experienced CD4+ T cells (34–36). Our studies indicated that the functional production of IFN-γ by antigen-specific CD4+ T cells maintained during a persistent phagosomal infection deteriorated progressively and that production of IL-10 early during the T cell priming phase negatively impacts antigen-specific CD4+ T cell expansion, maintenance, and functions during persistent phagosomal infection.
MATERIALS AND METHODS
Mice and bacteria.
Six- to 8-week-old female C57BL/6 mice and IL-10−/− mice (B6.129P2-Il10tm1Cgn/J) were obtained from the Jackson Laboratory (Bar Harbor, ME) and housed and cared for in the Animal Resource Center at the University of Texas Medical Branch (UTMB). All experiments were carried out in accordance with a protocol approved by the Institutional Animal Care and Use Committee. Ehrlichia muris strain AS145 was provided by Y. Rikihisa, Ohio State University, Columbus, OH. The bacterium was cultured in the canine macrophage-like cell line DH82. The bacterial inocula for infecting mice were prepared from the spleens of syngeneic mice infected with E. muris grown in DH82 cells by the intraperitoneal (i.p.) route as described previously (33). Briefly, spleens were collected aseptically from E. muris-infected mice on day 7 after inoculation. The spleens were homogenized, the resulting single-cell suspension was passed through 40-μm cell strainers, and the cells were suspended in freezing medium (10% dimethyl sulfoxide [DMSO] in fetal bovine serum [FBS]) for storage in liquid nitrogen at −196°C until further use.
Quantification of bacterial load.
Ehrlichial copy numbers in stocks and organs were determined by a quantitative real-time PCR method (37). Briefly, DNA was extracted from infected tissues, and a portion of the E. muris dsb gene (GenBank accession no. AY236484) and the host housekeeping gene, murine gapdh, were amplified and detected using the primers and the probes described previously (37). Plasmids carrying the dsb gene or gapdh gene were used to prepare standard curves. The number of copies of the dsb gene and gapdh gene and the number of host cells present in stocks were used to calculate the number of ehrlichiae present per milliliter of the stock. The bacterial loads in organs were expressed as the number of ehrlichiae (the number of copies of the E. muris dsb gene) present per 100 ng of total DNA. The limit of detection of the plasmid carrying the dsb gene by the PCR method used in the study was found to be 100 copies.
Mouse infection and antibiotic and antibody treatments.
Mice were infected with E. muris (∼1 × 104 bacterial genomes) by the i.p. route. For secondary-recall experiments, mice were challenged with E. muris (∼1 × 104 bacteria) by the i.p. route on day 70 after the primary infection. In some experiments, doxycycline antibiotic treatment (10 mg/kg of body weight) was given daily to mice by the i.p. route starting on day 10 after infection and continuing for 7 days. In order to block IL-10R, we treated mice with 500 μg of IL-10R-blocking antibody (clone 1B1.3a; BioXcell, West Lebanon, NH) by the i.p. route on days 0 and 5 after E. muris infection (38). Control mice received 500 μg of rat IgG1 isotype antibody (clone R3-34).
Splenocyte culture and ex vivo assay of immune responses.
Single-cell suspensions of spleens were prepared, and red blood cells (RBC) were lysed using RBC lysis buffer. Splenocytes were cultured ex vivo in complete medium (RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum, 10 mM HEPES buffer, 50 μM 2-mercaptoethanol, and antibiotics [penicillin at 100 U/ml and streptomycin at 100 μg/ml]) in 96-well plates (Costar, Corning, NY). The cells were cultured at a concentration of 5 × 105 cells per well. The cells were stimulated ex vivo for 5 h with 10 μg/ml of E. muris lysate antigen in the presence of the protein transport inhibitor brefeldin A (eBioscience, San Diego, CA) before being harvested for intracellular cytokine staining and analysis by flow cytometry.
Flow cytometric analysis.
Briefly, cells were stained with a fluorescent vital dye (Live/Dead Fixable Near-IR Dead Cell kit; Invitrogen, Carlsbad, CA) for 30 min on ice. Cells were then incubated with anti-Fc II/III receptor monoclonal antibodies (MAbs; (eBioscience) in fluorescence-activated cell sorting (FACS) buffer (Dulbecco's phosphate-buffered saline [PBS] without Mg2+ or Ca2+ containing 1% fetal calf serum [FCS] and 0.09% sodium azide) at 4°C for 15 min in order to block Fc receptors. Subsequently, cells were labeled with fluorochrome-conjugated MAbs (eBioscience) specific for mouse CD3, CD4, CD49d, CD11a, and PD-1 (programmed death 1) molecules. Cells were fixed, permeabilized, and stained for the intracellular cytokine IFN-γ using the BD Cytofix/Cytoperm fixation/permeabilization kit according to the manufacturer's instructions. Flow cytometric data were collected using an LSR II flow cytometer (BD Immunocytometry Systems, San Jose, CA). Live cells were gated based on the vital staining, and at least 200,000 live cells were analyzed. The data were analyzed with FlowJo 8.6 software (Tree Star Inc., Ashland, OR).
Purification of CD49dhi CD11ahi and CD49dlo CD11alo subpopulations.
Single-cell suspensions of spleen cells were prepared and the cells were washed and magnetically labeled with anti-CD4 microbeads (Miltenyi Biotec, Auburn, CA). After the washing, CD4+ T cells were obtained by positive selection using an AutoMACS Pro (Miltenyi Biotec). The CD4+ T cell-enriched population was stained with fluorochrome-labeled MAbs to CD3, CD4, CD11a, and CD49d, and CD4+ T cells were sorted into CD49dhi CD11ahi and CD49dlo CD11alo subsets by FACS using a BD FACSAria III. The purity of the sorted cells was more than 95% as assessed by FACS.
Serum cytokines.
Serum IL-10 concentration was measured using the mouse IL-10 enzyme-linked immunosorbent assay (ELISA) Ready-SET-Go kit (eBioscience). Selected serum cytokines were measured using multiplex bead-based assays (Bio-Rad Laboratories, Hercules, CA) by following the manufacturer's instructions. Plates were read and analyzed using the Bio-Plex array reader and Bio-Plex manager software.
Statistical analysis.
Analysis of the experimental data was carried out using the GraphPad Prism software version 5.01 for Windows (GraphPad Software, San Diego, CA). Unless indicated otherwise, data were square-root transformed and analyzed by a two-tailed unpaired student t test for comparison of two groups. Statistical significance was determined at 95% (P < 0.05), and asterisks in figures indicate levels of statistical significance (*, P = 0.01 to 0.05; **, P = 0.001 to 0.01; and ***, P < 0.001). Data presented are expressed as means plus standard deviations and are representative of results from two or three independent experiments.
RESULTS
IL-10 limits initial inflammatory responses and bacterial clearance.
We first analyzed the influence of IL-10 in the pathogenesis of Ehrlichia muris infection in C57BL/6 mice. After infection of mice with E. muris, there was an increase in serum IL-10 concentration, which peaked around day 8 postinfection (p.i.) and subsequently declined (Fig. 1A). We then addressed the influence of IL-10 on the course of the infection by infecting wild-type (WT) C57BL/6 mice and IL-10-deficient (IL-10−/−) mice with E. muris by the intraperitoneal route (i.p.) and observing the mice daily for the development of signs of disease. After infection with E. muris, WT mice did not develop any overt illness. In contrast, IL-10-deficient mice infected with E. muris developed overt disease signs by day 8 postinfection, including hunched posture, ruffled fur, and reluctance to move. However, IL-10-deficient mice infected with E. muris recovered from the illness and appeared healthy by day 15. We evaluated the concentrations of serum cytokines and chemokines and changes in body weight as an index of disease severity. The two strains of mice had comparable body weights at the time of E. muris inoculation. IL-10−/− mice infected with E. muris began losing body weight around day 6 postinfection and had significantly greater body weight loss until around day 35 postinfection than did WT mice infected with E. muris (Fig. 1B). IL-10−/− mice infected with E. muris had higher concentrations of the proinflammatory cytokines IL-6, IFN-γ, TNF-α, and IL-17 and the chemokines granulocyte colony-stimulating factor (G-CSF) and RANTES on day 9 postinfection than did infected WT mice (Fig. 1C). In addition, we observed increased infiltration of inflammatory cells in the liver, spleen, and lungs of IL-10−/− mice infected with E. muris on days 9 and 35 postinfection compared to that in infected WT mice (data shown for liver in Fig. 2).
FIG 1.

IL-10 limits the severity of illness and inflammatory cytokine and chemokine responses during acute Ehrlichia muris infection in mice. (A) IL-10 protein concentration at indicated time points in the serum of wild-type mice infected intraperitoneally with E. muris. Statistical analysis was performed by one-way analysis of variance (ANOVA) with Dunnett's post hoc test. (B) Percent weight loss in E. muris-infected WT and IL-10−/− mice at indicated time points postinfection. Statistical analysis was performed by a two-tailed paired t test. (C) Serum cytokine and chemokine concentrations of E. muris-infected WT (solid bars) and IL-10−/− (open bars) mice at day 9 postinfection. Data are the means for four or five mice per group and are representative of two independent experiments. Error bars represent the standard deviations. *, P = 0.01 to 0.05; **, P = 0.001 to 0.01; ***, P < 0.001.
FIG 2.
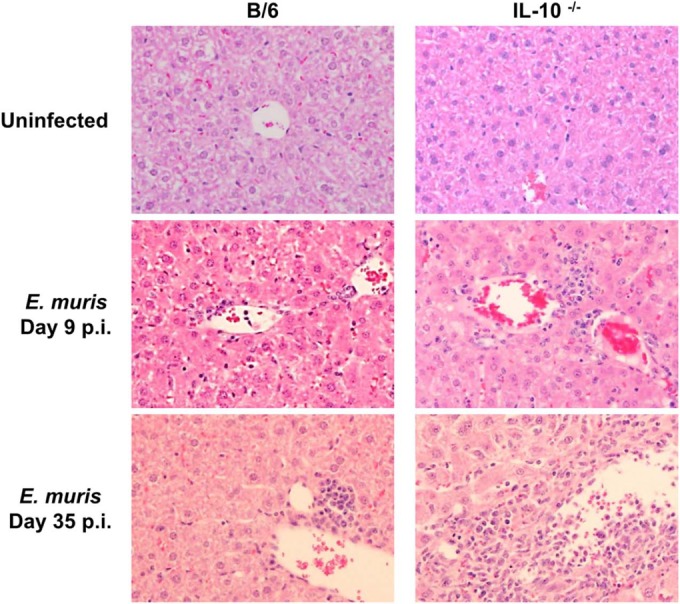
IL-10 restricts infiltration of inflammatory cells in the liver during Ehrlichia muris infection in mice. Representative liver sections stained with hematoxylin and eosin are shown for uninfected WT and IL-10−/− mice (top). The middle and bottom images show liver sections from E. muris-infected WT and IL-10−/− mice on days 9 and 35 postinfection, respectively. Data are representative of four mice per group and two independent experiments. Original magnification, ×200.
We then determined the bacterial burdens in different organs by quantitative real-time PCR. We found significantly reduced bacterial burdens in the blood, liver, and lungs of IL-10−/− mice infected with E. muris on day 9 postinfection compared to those in infected WT mice (Fig. 3A). The numbers of bacteria in the blood and lungs of infected WT mice were approximately 2.6 and 4.2 times higher, respectively, than in infected IL-10−/− mice on day 9 postinfection. We also found significantly higher numbers of bacteria in the liver and lungs of infected WT mice on day 35 postinfection than in infected IL-10−/− mice (Fig. 3B). The bacterial numbers in the lungs of WT mice infected with E. muris were significantly higher on day 90 postinfection than in infected IL-10−/− mice (Fig. 3C). The bacterial loads in each organ examined were below the limit of detection in infected IL-10−/− mice at day 90 postinfection. The data suggested that IL-10 controls initial inflammatory responses and impairs bacterial clearance during E. muris infection.
FIG 3.
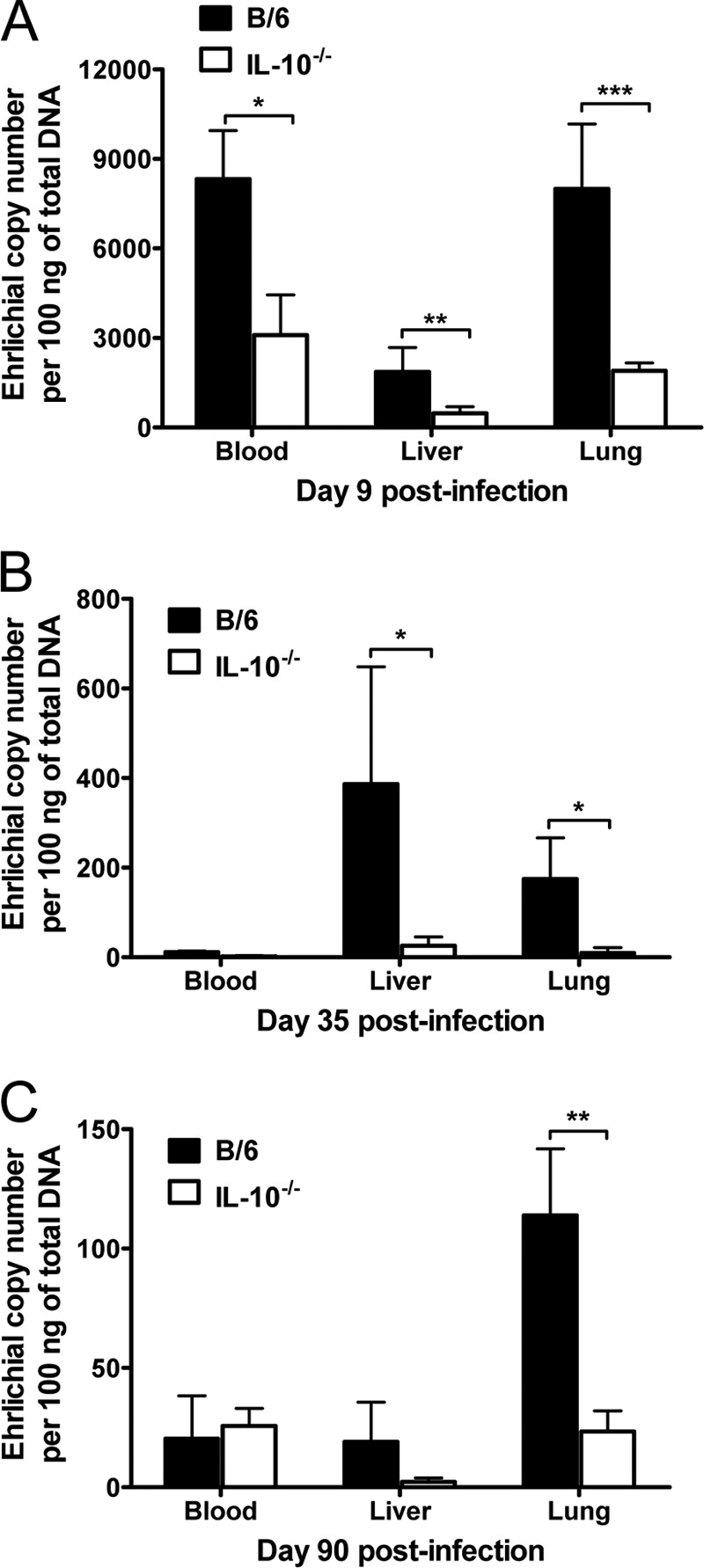
IL-10 impairs bacterial clearance during Ehrlichia muris infection in mice. Shown are representative bacterial loads in the blood, liver, and lungs of E. muris-infected WT and IL-10−/− mice on day 9 (A), day 35 (B), and day 90 (C) postinfection. The bacterial loads in organs and blood were expressed as the number of ehrlichiae (the number of copies of E. muris dsb gene) present per 100 ng of total DNA. Data are the means for five mice per group and are representative of three independent experiments. Error bars represent the standard deviations. *, P = 0.01 to 0.05; **, P = 0.001 to 0.01; ***, P < 0.001.
Coordinated upregulation of the surface markers CD49d and CD11a identifies Ehrlichia-specific CD4+ T cells.
To analyze the induction and maintenance of antigen-specific CD4+ T cells during persistent Ehrlichia infection, we first validated the use of the recently described surface markers CD49d and CD11a for identification of Ehrlichia-specific CD4+ T cell populations (34–36). Antigen-experienced CD4+ T cells upregulate surface expression of CD49d (alpha chain of the α4β1 integrin) and CD11a (alpha chain of the αLβ2 integrin) and retain higher expression of these markers into the memory phase. This approach has been used successfully for identification of antigen-specific CD4+ T cells during viral and Plasmodium infections in mice (34–36). A gating strategy for identification of antigen-experienced CD49dhi CD11ahi CD4+ T cells in the spleens of E. muris-infected mice is presented in Fig. 4A. We stimulated splenocytes ex vivo from uninfected naive or E. muris-infected mice (day 9) with E. muris lysate antigen and examined expression of the effector cytokine IFN-γ by the CD49dhi CD11ahi CD4+ T cell and CD49dlo CD11alo populations. A substantially greater percentage of CD4+ T cells exhibited the CD49dhi CD11ahi phenotype in E. muris-infected mice on day 9 postinfection than for uninfected naive controls (Fig. 4B and 5A). Furthermore, the CD49dhi CD11ahi population, but not the CD49dlo CD11alo population, of CD4+ T cells from E. muris-infected mice expressed the effector cytokine IFN-γ following ex vivo antigenic stimulation (Fig. 4B). Production of IFN-γ by these cells indicated that Ehrlichia-specific effector CD4+ T cells are CD49dhi CD11ahi. To evaluate the specificity of these markers during the memory phase, we purified CD49dhi CD11ahi CD4+ T cells and CD49dlo CD11alo CD4+ T cells from E. muris-infected mice, which had cleared persistent infection following a course of antibiotic treatment, on day 90 after infection. The purified CD4+ T cell subsets were labeled with carboxyfluorescein succinimidyl ester (CFSE) and stimulated in vitro with E. muris lysate antigen for 72 h in the presence of antigen-presenting cells (splenocytes from RAG−/− mice). Notably, CD49dhi CD11ahi cells, but not CD49dlo CD11alo cells, proliferated in response to antigenic stimulation (Fig. 4C). Thus, our data demonstrated that the CD49dhi CD11ahi phenotype reliably identified Ehrlichia-specific CD4+ T cells during the effector and memory phases.
FIG 4.

Upregulation of the surrogate surface markers CD49d and CD11a identifies Ehrlichia-specific CD4+ T cells. (A) Representative flow cytometric dot plots and gating strategy for identification of CD49dhi CD11ahi CD4+ T cells in the spleens of mice infected with E. muris. (B) Splenocytes from uninfected naive mice and E. muris-infected mice on day 9 postinfection were stimulated ex vivo with E. muris lysate antigen. The expression of CD49d and CD11a surface markers by CD4+ T cells and production of the effector cytokine IFN-γ by the CD49dhi CD11ahi CD4+ T cell and CD49dlo CD11alo CD4+ T cell populations were determined by flow cytometry. (C) Splenocytes were collected from E. muris-infected mice, which were treated with antibiotic to eliminate persistent infection, after day 90 postinfection. Then CD49dhi CD11ahi and CD49dlo CD11alo CD4+ T cell subsets were sorted, labeled with CFSE, and stimulated in vitro with E. muris lysate antigen for 72 h. Dilution of CFSE by CD4+ T cell subsets was analyzed by flow cytometry. Representative histograms indicate CFSE dilution profiles of CD49dhi CD11ahi and CD49dlo CD11alo CD4+ T cell subsets. Data in panel B are representative of five mice per group and two independent experiments. Data in panel C are representative of two independent experiments with splenocytes pooled from five or six E. muris-infected mice.
FIG 5.

Functional production of IFN-γ by antigen-specific CD4+ T cells maintained during persistent Ehrlichia infection progressively deteriorates. Frequencies of antigen-specific CD49dhiCD11ahi CD4+ T cells in the spleen and their capacity to secrete IFN-γ following ex vivo stimulation were analyzed in E. muris-infected WT and IL-10−/− mice on days 9, 35, and 90 postinfection. (A) Percentage of antigen-specific CD49dhi CD11ahi cells within the CD4+ T cell population in the spleens of E. muris-infected WT and IL-10−/− mice on the indicated days after infection. (B) Absolute numbers of antigen-specific CD49dhi CD11ahi CD4+ T cells in the spleens of E. muris-infected WT and IL-10−/− mice on the indicated days after infection. (C) Percentage of antigen-specific CD49dhi CD11ahi CD4+ T cells expressing IFN-γ after ex vivo antigenic stimulation in the spleens of E. muris-infected WT and IL-10−/− mice on the indicated days after infection. (D) Geometric mean fluorescence intensity of IFN-γ expressed by antigen-specific CD49dhi CD11ahi CD4+ T cells in the spleens of E. muris-infected WT and IL-10−/− mice. Numbers adjacent to outlined areas in flow cytometry plots (A and C) and bar graphs (B and D) represent the averages and standard deviations of four or five mice per group and are representative of two independent experiments. *, P = 0.01 to 0.05; **, P = 0.001 to 0.01; ***, P < 0.001.
Functional production of IFN-γ by antigen-specific CD4+ T cells maintained during persistent Ehrlichia infection progressively deteriorates.
To determine the expansion, function, and maintenance of antigen-specific T cells in the presence and absence of IL-10 during a persistent phagosomal infection, we analyzed the antigen-specific CD49dhi CD11ahi CD4+ T cells after E. muris infection in WT and IL-10−/− mice. In WT mice infected with E. muris, antigen-specific CD49dhi CD11ahi CD4+ T cells expanded by day 9 postinfection and were maintained through day 35 postinfection before contraction by day 90 postinfection (Fig. 5A). Similar kinetics, but of significantly greater magnitude, of antigen-specific CD49dhi CD11ahi CD4+ T cells was observed in IL-10−/− mice infected with E. muris (Fig. 5A). The total numbers of antigen-specific CD49dhi CD11ahi CD4+ T cells in the spleens of E. muris-infected IL-10−/− mice were approximately 4.3 and 1.6 times higher on days 9 and 35 postinfection, respectively, than in infected WT mice (Fig. 5B). We further evaluated the functional status of antigen-specific CD4+ T cells by determining their capacity to produce IFN-γ ex vivo in response to antigen stimulation. Significantly (P < 0.05) higher percentages of CD49dhi CD11ahi CD4+ T cells from infected IL-10−/− mice produced IFN-γ than from infected WT mice during the effector (day 9 p.i.) and memory (days 35 and 90 p.i.) phases (Fig. 5C). Notably, CD49dhi CD11ahi CD4+ T cells in infected WT mice progressively lost their capacity to produce the effector cytokine IFN-γ compared to their counterparts in IL-10 −/− mice (Fig. 5C). Furthermore, we assessed the quality of antigen-specific IFN-γ-producing CD49dhi CD11ahi CD4+ T cells by determining the geometric mean fluorescence intensity (GMFI) for IFN-γ signal, which indicates the amount of cytokine produced on a per-cell basis. The mean fluorescence intensity for IFN-γ signal was significantly lower in CD49dhi CD11ahi CD4+ T cells from infected WT mice than from IL-10−/− mice on days 9, 35, and 90 postinfection (Fig. 5D). Our data indicated that this specific functional attribute of antigen-specific CD4+ T cells (the capacity to express IFN-γ) progressively deteriorated in wild-type mice during persistent Ehrlichia infection. Similar to the case with CD4+ T cells in the spleen, we found significantly higher percentages of CD49dhi CD11ahi CD4+ T cells within the peripheral blood mononuclear cell (PBMC) population of E. muris-infected IL-10−/− mice than in that from infected WT mice on days 35 and 90 p.i. (Fig. 6).
FIG 6.

Increased percentage of antigen-specific CD4+ T cells within the peripheral blood mononuclear cell population in E. muris-infected IL-10-deficient mice. Shown are representative flow cytometry plots of antigen-specific CD49dhi CD11ahi CD4+ T cells within the peripheral blood mononuclear cell population in E. muris-infected WT and IL-10−/− mice on days 9, 35, and 90 postinfection. Data are representative of four or five mice per group and two independent experiments.
PD-1 expression is upregulated by Ehrlichia-specific effector CD4+ T cells in wild-type mice.
To understand the phenotypic heterogeneity of the antigen-specific CD4+ T cells, we assessed the expression of PD-1, an inhibitory molecule, on Ehrlichia-specific CD49dhi CD11ahi CD4+ T cells in both WT and IL-10−/− mice. We found upregulation of PD-1 expression on a substantial number of CD49dhi CD11ahi CD4+ T cells in both the spleen and within the PBMC population of E. muris-infected IL-10−/− and WT mice on day 9 postinfection compared to that in uninfected naive CD49dlo CD11alo CD4+ T cells, which indicates the effector nature of CD4+ T cells responding to antigen (Fig. 7A and B). However, the expression of PD-1 by the Ehrlichia-specific CD49dhi CD11ahi CD4+ T cells was higher in WT infected mice on day 9 postinfection than in infected IL-10−/− mice (Fig. 7C). Nonetheless, this difference was not observed on day 35 postinfection (Fig. 7C). Our data indicated that early upregulation of PD-1 by antigen-specific CD4+ T cells during E. muris infection in WT mice may contribute to their reduced expansion/activation and impaired effector function.
FIG 7.

Ehrlichia-specific effector CD4+ T cells express higher levels of PD-1 in wild-type mice than IL-10−/− mice. (A) PD-1 expression by the CD49dhi CD11ahi CD4+ T cell and CD49dlo CD11alo CD4+ T cell populations in the spleens of E. muris-infected WT and IL-10−/−mice on day 9 postinfection. (B) Percentage of CD49dhi CD11ahi CD4+ T cells expressing PD-1 within the peripheral blood mononuclear cell population in E. muris-infected WT and IL-10−/− mice on day 9 postinfection. (C) Expression of PD-1 by the antigen-specific CD49dhi CD11ahi CD4+ T cells in the spleens of E. muris-infected WT and IL-10−/− mice on days 9 and 35 postinfection. Data are representative of four mice per group and two independent experiments.
Blocking signaling through IL-10R during T cell priming enhances the magnitude and quality of memory CD4+ T cells and secondary recall response.
We reasoned that priming T cells in the absence of signaling through IL-10R early during infection would be enough to enhance the magnitude and quality of Ehrlichia-specific memory CD4+ T cells, as mice infected with E. muris had higher concentration of serum IL-10 at early time points during infection (Fig. 1A). Further, Ehrlichia-specific CD49dhi CD11ahi CD4+ T cells upregulated the inhibitory molecule PD-1 early during infection in WT mice compared to that in infected IL-10−/− mice (Fig. 7C). We infected mice with E. muris (∼1 × 104 bacterial genomes) and then treated them with an IL-10R blocking antibody on days 0 and 5 postinfection (Fig. 8A). Control mice were infected with E. muris and treated with an isotype control. In order to exclude the influence of persistent infection on the differentiation and function of memory CD4+ T cell responses, we treated both groups of mice with doxycycline for 7 days starting on day 10 postinfection. We determined bacterial burdens in the organs of antibiotic-treated mice on days 20 and 65 postinfection to assess the efficacy of the treatment. We did not detect bacterial genomes in the antibiotic-treated mice (data not shown), which was consistent with the previous reports of elimination of persistent E. muris infection in mice treated with doxycycline (32, 39). Unlike IL-10−/− mice infected with E. muris, infected WT mice treated with anti-IL-10 blocking antibody did not show overt signs of the disease before the initiation of antibiotic treatment on day 10 postinfection. We sacrificed mice on day 65 postinfection and analyzed CD4+ T cell responses by flow cytometry. Similar to the case with IL-10−/− mice, blocking signaling through IL-10R resulted in a significant (P < 0.05) increase in the percentage of CD49dhi CD11ahi CD4+ T cells compared to that in isotype-treated control mice (Fig. 8B). In addition, significantly higher percentages of CD49dhi CD11ahi CD4+ T cells produced greater quantities of IFN-γ in response to ex vivo antigenic stimulation in the spleens of IL-10R blocking-antibody-treated, E. muris-infected mice than for the isotype-treated, infected control mice (Fig. 8C and D).
FIG 8.

Transient IL-10R blockade early during E. muris infection enhances memory and secondary recall CD4+ T cell responses. (A) Schematic representation of anti-IL-10R antibody and antibiotic treatment and secondary challenge infection in E. muris-infected mice. E. muris-infected WT C57BL/6 mice were treated an IL-10R blocking MAb or an isotype control on days 0 and 5 postinfection. Both groups of mice were treated with a course of antibiotic for 7 days starting on day 10 after E. muris infection. Mice were challenged with a second dose of E. muris on day 70 after primary infection. CD4+ T cell responses were analyzed on day 65 after the primary infection and day 5 after the secondary infection. Tissue bacterial loads were determined after antibiotic treatment and day 5 after secondary challenge infection. (B to D) Percentage of antigen-specific CD49dhi CD11ahi cells within the CD4+ T cell population (B), production of IFN-γ after ex vivo antigenic stimulation (C), and mean fluorescence intensity (MFI) of IFN-γ (D) expressed by antigen-specific CD49dhi CD11ahi CD4+ T cells in the spleens of E. muris-infected WT mice treated with IL-10R blocking MAb or isotype control on day 65 postinfection. (E to G) Percentage of antigen-specific CD49dhi CD11ahi cells within the CD4+ T cell population (E), production of IFN-γ after ex vivo antigenic stimulation (F), and MFI of IFN-γ (G) expressed by antigen-specific CD49dhi CD11ahi CD4+ T cells in the spleens of E. muris-infected WT mice treated with IL-10R blocking MAb or isotype control on day 5 after secondary challenge infection. (H) Bacterial loads in the blood, liver, and lungs of E. muris-infected WT mice, treated with IL-10R blocking MAb or isotype control, on day 5 after secondary E. muris challenge. Bacterial loads were undetectable in the organs on days 20 and 65 postinfection after antibiotic treatment (data not shown). Data are representative of four or five mice per group and two independent experiments. *, P = 0.01 to 0.05; ***, P < 0.001.
We challenged the E. muris-infected mice with a second dose of E. muris (∼1 × 104 bacteria) on day 70 after the primary infection and assessed secondary recall responses on day 5 following rechallenge. We found a significant (P < 0.05) increase in the percentage of CD49dhi CD11ahi CD4+ T cells in the spleens of mice treated with IL-10R blocking antibody compared to that of isotype-treated control mice on day 5 after secondary challenge (Fig. 8E). Notably, significantly higher percentages of CD49dhi CD11ahi CD4+ T cells produced greater quantities of IFN-γ in response to ex vivo antigenic stimulation in the spleens of mice treated with IL-10R blocking antibody than for the isotype-treated control mice on day 5 after secondary challenge (Fig. 8F and G). Furthermore, the bacterial load in the lungs of mice treated with IL-10R blocking antibody was significantly lower than in the isotype-treated control mice on day 5 after secondary challenge (Fig. 8H). These findings suggest that transient blocking of signaling through IL-10R during T cell priming early in infection leads to generation of superior memory CD4+ T cells that mediate better recall responses against rechallenge.
DISCUSSION
CD4+ T cells play a critical role in protection against pathogens that reside in phagosomes (1). Herein, we demonstrate that the functional capacity of antigen-specific CD4+ T cells maintained during a persistent phagosomal bacterial infection progressively deteriorates and that transient blockade of signaling through IL-10R during T cell priming early in infection leads to generation and maintenance of superior-quality CD4+ T cells that mediate enhanced secondary recall responses. Using IL-10−/− mice, an IL-10R blocking antibody, and the recently identified surface markers CD49d and CD11a, which allow tracking of total polyclonal pathogen-specific CD4+ T cell responses, we demonstrated that IL-10 production reduced the initial inflammation after E. muris infection, impaired bacterial clearance, restricted Ehrlichia-specific CD4+ T cell expansion, and impaired effector cytokine IFN-γ production.
Various factors individually and in conjunction regulate immune responses against infections. IL-10 is one of the important factors in host defense that regulates host immune responses to pathogens (40). IL-10, a pleotropic cytokine, is produced by several cell populations and exerts diverse effects on numerous hematopoietic cell types (40). Induction of IL-10 during infections caused by phagosomal pathogens may impair pathogen clearance, thereby leading to pathogen persistence as reported for Leishmania major, Mycobacterium tuberculosis, and Salmonella (41–43). In the present study, we observed an increased production of IL-10 in mice infected with E. muris, as indicated by higher serum IL-10 concentration early during the infection. Furthermore, we also found that IL-10−/− mice infected with E. muris had increased inflammatory responses and reduced bacterial loads in different organs and blood compared to those in infected WT mice. These findings suggest that increased IL-10 production during the peak of E. muris infection leads to prevention of tissue injury caused by robust inflammatory responses but also interferes with bacterial clearance and development of adaptive immune responses.
Generation and maintenance of CD4+ T cell responses and the factors that affect CD4+ T cell responses during infections remain incompletely understood (25, 28, 29). Identification and quantification of antigen-specific T cells induced after infection or vaccination requires prior knowledge of the antigen specificity of responding T cells or use of techniques such as enzyme-linked immunosorbent spot assay (ELISPOT), intracellular cytokine staining, or tetramers (29). Until recently, studies of CD4+ T cell responses to pathogens relied on responses to a few dominant antigens due to the lack of markers for identification and quantification of total polyclonal pathogen-specific T cell responses (34–36). In the present study, use of the surrogate markers CD49d and CD11a allowed us to track total Ehrlichia-specific CD4+ T cells during the effector and memory phases and to assess their functions in WT and IL-10−/− mice. We confirmed the specificity of these markers by purifying CD49dhi CD11ahi CD4+ T cells from E. muris-infected mice and measuring their proliferation in response to antigenic stimulation. Cytokines, costimulatory molecules, and coinhibitory molecules profoundly influence the development and functions of CD4+ T cells. Induction of CD4+ T cell responses during phagosomal infections is delayed compared to those induced following acute bacterial and viral infections. Activation of CD4+ T cells occurs in the draining lymph nodes in 1 to 2 weeks after phagosomal infection, which results in generation of a large number of effector cells that peak in about 2 to 3 weeks (28–30). In the present study, we observed expansion of Ehrlichia-specific CD4+ T cells in the spleen by day 9 postinfection, which was maintained through day 35 postinfection before contraction by day 90 postinfection. In addition, we observed that the functional production of IFN-γ by the antigen-specific CD4+ T cells progressively deteriorated in the spleen during persistent Ehrlichia infection. We found that an absence of IL-10 resulted in greater expansion of antigen-specific effector CD4+ cells in the spleen and blood during the initial phase of the infection as well as a higher magnitude and functional capacity of CD4+ T cells in the memory phase. Notably, antigen-specific CD4+ T cells produced significantly greater quantities of IFN-γ on a per-cell basis in IL-10−/− mice, as reflected by a higher MFI than for infected WT mice. It has been demonstrated that polyfunctional Th1 cells that produce multiple cytokines (IL-2, TNF-α, and IFN-γ) also produce greater quantities of IFN-γ than do cells that produce one or two cytokines (12). IL-10 inhibits IL-12 production, thereby reducing Th1 development and IFN-γ production (44). A previous study reported that mice deficient in IL-10 (IL-10−/−) and infected with Mycobacterium tuberculosis have increased clearance of bacteria from the lungs and spleen and have an increased percentage of IFN-γ+ CD4+ T cells in the lungs compared to those of infected wild-type mice (43). Antigen-specific CD4+ T cells may become more responsive to positive signals such as IFN-γ in the absence of the suppression mediated by IL-10, resulting in an increased number of effector cells (45). It was demonstrated that IFN-γ enhances the development of memory CD4+ T cells, and the absence of IFN-γ signaling leads to marked contraction of CD4+ T cells (46). IL-10 appears to exert its inhibitory effects on antigen-specific CD4+ T cells during priming, as antibody-mediated blockade of IL-10R early during bacterial infection was sufficient to enhance the effector and memory CD4+ T cell responses. This is consistent with the observation that E. muris-infected mice had increased concentrations of serum IL-10 early during the infection. A previous study indicated that inhibition of IL-10 signaling during Mycobacterium bovis bacillus Calmette-Guérin (BCG) vaccination resulted in enhanced Th1, Th17, innate lymphoid IFN-γ, and IL-17 responses and conferred significantly greater protection against aerosol M. tuberculosis challenge in mice (47). These findings suggest that induction of IL-10 during the initial phase of persistent phagosomal infection has a major role in determining the initial expansion of effector CD4+ T cells, their IFN-γ secreting capacity, and development of memory CD4+ T cell responses.
The cell source of IL-10 during E. muris infection in mice and the components of Ehrlichia that stimulate IL-10 production remain to be identified. Molecularly and metabolically distinct IL-10 producing immunoregulatory antigen-presenting cells that express increased levels of T cell-interacting molecules and negative regulatory factors develop during persistent viral infections and work to suppress T cell responses (48). A major future goal is to examine whether similar immunoregulatory antigen-presenting cells are also induced during persistent bacterial infections. Bacterial lipoproteins have been shown to inhibit pneumococcal protein antigen-induced proliferation of human memory CD4+ T cells in vitro through Toll-like receptor 2 (TLR-2), which was associated with increased IL-10 production (49). Anaplasma marginale, a related pathogen of ruminants, causes persistent high-load bacteremia and induces memory CD4+ T cell dysfunction (50).
The heterogeneity and effector function of T cells are also affected by T cell tolerance or exhaustion during infection. CD4+ T cells become dysfunctional or exhausted during certain chronic infections, which is associated with high surface expression of PD-1 (51, 52). Recently, Wei and coworkers demonstrated that the strength of PD-1 signaling differentially affects T-cell effector functions such as T cell expansion, cytotoxicity, and cytokine production (53). The study demonstrated that PD-1 ligation renders T cells less sensitive to T cell receptor (TCR)-generated signals and affects their functions (53). In present study, we observed that the expression of PD-1 by the antigen-specific effector CD49dhi CD11ahi CD4+ T cells in the spleen and blood of E. muris-infected WT mice was substantially higher early during the acute phase, but not at the persistent phase, of the infection than in the infected IL-10−/− mice. In contrast, a high level of PD-1 expression was observed on the virus-specific CD4+ T cells during chronic LCMV infection in both WT and IL-10−/− mice (38, 53). Increased expression of PD-1 by the antigen-specific effector CD4+ T cells in E. muris-infected WT mice was negatively correlated with their expansion, cytokine production, and subsequent differentiation into memory cells. Han et al. demonstrated that persistent antigen drives CD4+ T cell dysfunction independently of persistent inflammation, which was not associated with increased PD-1 expression, using an inducible model antigen with dendritic cells in a transgenic mouse system (54). The IL-10 and PD-1/PD-L1 pathways function independently to suppress T cell responses (55). The decreased expression of PD-1 by the antigen-specific effector CD4+ T cells in E. muris-infected IL-10−/− mice compared to that in infected WT mice could be due to lower bacterial burdens and/or other factors which need further investigation. Further characterization of Ehrlichia-specific CD4+ T cells, such as expression of inhibitory molecules (BTLA, CTLA-4, and CD200) and production of cytokines (IL-2 and TNF-α), is needed to establish their phenotypic and functional heterogeneity (56).
Induction of appropriate host responses early during infections is crucial for their effective resolution and subsequent development of memory immune responses that mediate protection against reinfection. Suboptimal host responses lead to either more severe disease or pathogen persistence, whereas excessive or inappropriate host responses lead to immune-mediated pathology or autoimmunity. In the present study, using the E. muris-C57BL/6 mouse model, we demonstrated that the functional capacity of antigen-specific CD4+ T cells maintained during a persistent phagosomal infection becomes progressively deteriorated, and transient blockade of signaling through IL-10R during T cell priming early in infection was sufficient to induce enhanced memory CD4+ T cell responses. A better understanding of the factors that influence the priming and differentiation of CD4+ T cells may provide a basis to induce long-term protective immune responses against persistent phagosomal infections.
ACKNOWLEDGMENTS
We thank David H. Walker and Linsey A. Yeager for critical reading of the manuscript and helpful suggestions. We thank Mark Griffin for assistance with FACS data collection.
This work was supported by an NIAID Research Scholar Development (K22) award (AI089973) to N.R.T.
Footnotes
Published ahead of print 14 July 2014
REFERENCES
- 1. Tubo NJ, Jenkins MK. 2014. CD4+ T cells: guardians of the phagosome. Clin. Microbiol. Rev. 27:200–213. 10.1128/CMR.00097-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. MacMicking JD. 2005. Immune control of phagosomal bacteria by p47 GTPases. Curr. Opin. Microbiol. 8:74–82. 10.1016/j.mib.2004.12.012 [DOI] [PubMed] [Google Scholar]
- 3. Walker DH, Ismail N, Olano JP, McBride JW, Yu XJ, Feng HM. 2004. Ehrlichia chaffeensis: a prevalent, life-threatening, emerging pathogen. Trans. Am. Clin. Climatol. Assoc. 115:375–382 [PMC free article] [PubMed] [Google Scholar]
- 4. Dumler JS, Sutker WL, Walker DH. 1993. Persistent infection with Ehrlichia chaffeensis. Clin. Infect. Dis. 17:903–905. 10.1093/clinids/17.5.903 [DOI] [PubMed] [Google Scholar]
- 5. Roland WE, McDonald G, Caldwell CW, Everett ED. 1995. Ehrlichiosis—a cause of prolonged fever. Clin. Infect. Dis. 20:821–825. 10.1093/clinids/20.4.821 [DOI] [PubMed] [Google Scholar]
- 6. Marshall GS, Jacobs RF, Schutze GE, Paxton H, Buckingham SC, DeVincenzo JP, Jackson MA, San Joaquin VH, Standaert SM, Woods CR, and Tick-Borne Infections in Children Study Group. 2002. Ehrlichia chaffeensis seroprevalence among children in the southeast and south-central regions of the United States. Arch. Pediatr. Adolesc. Med. 156:166–170. 10.1001/archpedi.156.2.166 [DOI] [PubMed] [Google Scholar]
- 7. Standaert SM, Dawson JE, Schaffner W, Childs JE, Biggie KL, Singleton J, Jr, Gerhardt RR, Knight ML, Hutcheson RH. 1995. Ehrlichiosis in a golf-oriented retirement community. N. Engl. J. Med. 333:420–425. 10.1056/NEJM199508173330704 [DOI] [PubMed] [Google Scholar]
- 8. Yevich SJ, Sanchez JL, DeFraites RF, Rives CC, Dawson JE, Uhaa IJ, Johnson BJ, Fishbein DB. 1995. Seroepidemiology of infections due to spotted fever group rickettsiae and Ehrlichia species in military personnel exposed in areas of the United States where such infections are endemic. J. Infect. Dis. 171:1266–1273. 10.1093/infdis/171.5.1266 [DOI] [PubMed] [Google Scholar]
- 9. Bitsaktsis C, Huntington J, Winslow G. 2004. Production of IFN-gamma by CD4 T cells is essential for resolving ehrlichia infection. J. Immunol. 172:6894–6901. 10.4049/jimmunol.172.11.6894 [DOI] [PubMed] [Google Scholar]
- 10. Feng HM, Walker DH. 2004. Mechanisms of immunity to Ehrlichia muris: a model of monocytotropic ehrlichiosis. Infect. Immun. 72:966–971. 10.1128/IAI.72.2.966-971.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mogues T, Goodrich ME, Ryan L, LaCourse R, North RJ. 2001. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J. Exp. Med. 193:271–280. 10.1084/jem.193.3.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Darrah PA, Patel DT, De Luca PM, Lindsay RW, Davey DF, Flynn BJ, Hoff ST, Andersen P, Reed SG, Morris SL, Roederer M, Seder RA. 2007. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat. Med. 13:843–850. 10.1038/nm1592 [DOI] [PubMed] [Google Scholar]
- 13. Monack DM, Mueller A, Falkow S. 2004. Persistent bacterial infections: the interface of the pathogen and the host immune system. Nat. Rev. Microbiol. 2:747–776. 10.1038/nrmicro955 [DOI] [PubMed] [Google Scholar]
- 14. Ruby T, McLaughlin L, Gopinath S, Monack D. 2012. Salmonella's long-term relationship with its host. FEMS Microbiol. Rev. 36:600–615. 10.1111/j.1574-6976.2012.00332.x [DOI] [PubMed] [Google Scholar]
- 15. Guilloteau L, Buzoni-Gatel D, Blaise F, Bernard F, Pepin M. 1991. Phenotypic analysis of splenic lymphocytes and immunohistochemical study of hepatic granulomas after a murine infection with Salmonella abortusovis. Immunology 74:630–637 [PMC free article] [PubMed] [Google Scholar]
- 16. McElrath MJ, Murray HW, Cohn ZA. 1988. The dynamics of granuloma formation in experimental visceral leishmaniasis. J. Exp. Med. 167:1927–1937. 10.1084/jem.167.6.1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramakrishnan L. 2012. Revisiting the role of the granuloma in tuberculosis. Nat. Rev. Immunol. 12:352–366. 10.1038/nri3211 [DOI] [PubMed] [Google Scholar]
- 18. Egen JG, Rothfuchs AG, Feng CG, Horwitz MA, Sher A, Germain RN. 2011. Intravital imaging reveals limited antigen presentation and T cell effector function in mycobacterial granulomas. Immunity 34:807–819. 10.1016/j.immuni.2011.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. O'Garra A, Vieira P. 2007. T(H)1 cells control themselves by producing interleukin-10. Nat. Rev. Immunol. 7:425–428. 10.1038/nri2097 [DOI] [PubMed] [Google Scholar]
- 20. Ding L, Linsley PS, Huang LY, Germain RN, Shevach EM. 1993. IL-10 inhibits macrophage costimulatory activity by selectively inhibiting the up-regulation of B7 expression. J. Immunol. 151:1224–1234 [PubMed] [Google Scholar]
- 21. Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kuhn R, Muller W, Trinchieri G, Sher A. 1996. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J. Immunol. 157:798–805 [PubMed] [Google Scholar]
- 22. O'Leary S, O'Sullivan MP, Keane J. 2011. IL-10 blocks phagosome maturation in Mycobacterium tuberculosis-infected human macrophages. Am. J. Respir. Cell Mol. Biol. 45:172–180. 10.1165/rcmb.2010-0319OC [DOI] [PubMed] [Google Scholar]
- 23. Uchiya K, Groisman EA, Nikai T. 2004. Involvement of Salmonella pathogenicity island 2 in the up-regulation of interleukin-10 expression in macrophages: role of protein kinase A signal pathway. Infect. Immun. 72:1964–1973. 10.1128/IAI.72.4.1964-1973.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Homann D, Teyton L, Oldstone MB. 2001. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat. Med. 7:913–919. 10.1038/90950 [DOI] [PubMed] [Google Scholar]
- 25. Pepper M, Linehan JL, Pagan AJ, Zell T, Dileepan T, Cleary PP, Jenkins MK. 2010. Different routes of bacterial infection induce long-lived TH1 memory cells and short-lived TH17 cells. Nat. Immunol. 11:83–89. 10.1038/ni.1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pepper M, Pagan AJ, Igyarto BZ, Taylor JJ, Jenkins MK. 2011. Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity 35:583–595. 10.1016/j.immuni.2011.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Surh CD, Sprent J. 2008. Homeostasis of naive and memory T cells. Immunity 29:848–862. 10.1016/j.immuni.2008.11.002 [DOI] [PubMed] [Google Scholar]
- 28. Nelson RW, McLachlan JB, Kurtz JR, Jenkins MK. 2013. CD4+ T cell persistence and function after infection are maintained by low-level peptide:MHC class II presentation. J. Immunol. 190:2828–2834. 10.4049/jimmunol.1202183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pagán AJ, Peters NC, Debrabant A, Ribeiro-Gomes F, Pepper M, Karp CL, Jenkins MK, Sacks DL. 2013. Tracking antigen-specific CD4+ T cells throughout the course of chronic Leishmania major infection in resistant mice. Eur. J. Immunol. 43:427–438. 10.1002/eji.201242715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reiley WW, Shafiani S, Wittmer ST, Tucker-Heard G, Moon JJ, Jenkins MK, Urdahl KB, Winslow GM, Woodland DL. 2010. Distinct functions of antigen-specific CD4 T cells during murine Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. U. S. A. 107:19408–19413. 10.1073/pnas.1006298107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Uzonna JE, Wei G, Yurkowski D, Bretscher P. 2001. Immune elimination of Leishmania major in mice: implications for immune memory, vaccination, and reactivation disease. J. Immunol. 167:6967–6974. 10.4049/jimmunol.167.12.6967 [DOI] [PubMed] [Google Scholar]
- 32. Thirumalapura NR, Crossley EC, Walker DH, Ismail N. 2009. Persistent infection contributes to heterologous protective immunity against fatal ehrlichiosis. Infect. Immun. 77:5682–5689. 10.1128/IAI.00720-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Olano JP, Wen G, Feng HM, McBride JW, Walker DH. 2004. Histologic, serologic, and molecular analysis of persistent ehrlichiosis in a murine model. Am. J. Pathol. 165:997–1006. 10.1016/S0002-9440(10)63361-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fang M, Siciliano NA, Hersperger AR, Roscoe F, Hu A, Ma X, Shamsedeen AR, Eisenlohr LC, Sigal LJ. 2012. Perforin-dependent CD4+ T-cell cytotoxicity contributes to control a murine poxvirus infection. Proc. Natl. Acad. Sci. U. S. A. 109:9983–9988. 10.1073/pnas.1202143109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McDermott DS, Varga SM. 2011. Quantifying antigen-specific CD4 T cells during a viral infection: CD4 T cell responses are larger than we think. J. Immunol. 187:5568–5576. 10.4049/jimmunol.1102104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT, Waldschmidt TJ, Crompton PD, Harty JT. 2012. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat. Immunol. 13:188–195. 10.1038/ni.2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stevenson HL, Jordan JM, Peerwani Z, Wang HQ, Walker DH, Ismail N. 2006. An intradermal environment promotes a protective type-1 response against lethal systemic monocytotropic ehrlichial infection. Infect. Immun. 74:4856–4864. 10.1128/IAI.00246-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. 2006. Interleukin-10 determines viral clearance or persistence in vivo. Nat. Med. 12:1301–1309. 10.1038/nm1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Racine R, McLaughlin M, Jones DD, Wittmer ST, MacNamara KC, Woodland DL, Winslow GM. 2011. IgM production by bone marrow plasmablasts contributes to long-term protection against intracellular bacterial infection. J. Immunol. 186:1011–1021. 10.4049/jimmunol.1002836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Couper KN, Blount DG, Riley EM. 2008. IL-10: the master regulator of immunity to infection. J. Immunol. 180:5771–5777. 10.4049/jimmunol.180.9.5771 [DOI] [PubMed] [Google Scholar]
- 41. Arai T, Hiromatsu K, Nishimura H, Kimura Y, Kobayashi N, Ishida H, Nimura Y, Yoshikai Y. 1995. Effects of in vivo administration of anti-IL-10 monoclonal antibody on the host defence mechanism against murine Salmonella infection. Immunology 85:381–388 [PMC free article] [PubMed] [Google Scholar]
- 42. Belkaid Y, Hoffmann KF, Mendez S, Kamhawi S, Udey MC, Wynn TA, Sacks DL. 2001. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J. Exp. Med. 194:1497–1506. 10.1084/jem.194.10.1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Redford PS, Boonstra A, Read S, Pitt J, Graham C, Stavropoulos E, Bancroft GJ, O'Garra A. 2010. Enhanced protection to Mycobacterium tuberculosis infection in IL-10-deficient mice is accompanied by early and enhanced Th1 responses in the lung. Eur. J. Immunol. 40:2200–2210. 10.1002/eji.201040433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou L, Nazarian AA, Smale ST. 2004. Interleukin-10 inhibits interleukin-12 p40 gene transcription by targeting a late event in the activation pathway. Mol. Cell. Biol. 24:2385–2396. 10.1128/MCB.24.6.2385-2396.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brooks DG, Walsh KB, Elsaesser H, Oldstone MB. 2010. IL-10 directly suppresses CD4 but not CD8 T cell effector and memory responses following acute viral infection. Proc. Natl. Acad. Sci. U. S. A. 107:3018–3023. 10.1073/pnas.0914500107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Whitmire JK, Eam B, Benning N, Whitton JL. 2007. Direct interferon-gamma signaling dramatically enhances CD4+ and CD8+ T cell memory. J. Immunol. 179:1190–1197. 10.4049/jimmunol.179.2.1190 [DOI] [PubMed] [Google Scholar]
- 47. Pitt JM, Stavropoulos E, Redford PS, Beebe AM, Bancroft GJ, Young DB, O'Garra A. 2012. Blockade of IL-10 signaling during bacillus Calmette-Guerin vaccination enhances and sustains Th1, Th17, and innate lymphoid IFN-gamma and IL-17 responses and increases protection to Mycobacterium tuberculosis infection. J. Immunol. 189:4079–4087. 10.4049/jimmunol.1201061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wilson EB, Kidani Y, Elsaesser H, Barnard J, Raff L, Karp CL, Bensinger S, Brooks DG. 2012. Emergence of distinct multiarmed immunoregulatory antigen-presenting cells during persistent viral infection. Cell Host Microbe 11:481–491. 10.1016/j.chom.2012.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang Q, Bagrade L, Clarke E, Paton JC, Nunez DA, Finn A. 2010. Bacterial lipoproteins differentially regulate human primary and memory CD4+ T and B cell responses to pneumococcal protein antigens through Toll-like receptor 2. J. Infect. Dis. 201:1753–1763. 10.1086/652495 [DOI] [PubMed] [Google Scholar]
- 50. Han S, Norimine J, Brayton KA, Palmer GH, Scoles GA, Brown WC. 2010. Anaplasma marginale infection with persistent high-load bacteremia induces a dysfunctional memory CD4+ T lymphocyte response but sustained high IgG titers. Clin. Vaccine Immunol. 17:1881–1890. 10.1128/CVI.00257-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Antoine P, Olislagers V, Huygens A, Lecomte S, Liesnard C, Donner C, Marchant A. 2012. Functional exhaustion of CD4+ T lymphocytes during primary cytomegalovirus infection. J. Immunol. 189:2665–2672. 10.4049/jimmunol.1101165 [DOI] [PubMed] [Google Scholar]
- 52. Brooks DG, Teyton L, Oldstone MB, McGavern DB. 2005. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. J. Virol. 79:10514–10527. 10.1128/JVI.79.16.10514-10527.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wei F, Zhong S, Ma Z, Kong H, Medvec A, Ahmed R, Freeman GJ, Krogsgaard M, Riley JL. 2013. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc. Natl. Acad. Sci. U. S. A. 110:E2480–E2489. 10.1073/pnas.1305394110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Han S, Asoyan A, Rabenstein H, Nakano N, Obst R. 2010. Role of antigen persistence and dose for CD4+ T-cell exhaustion and recovery. Proc. Natl. Acad. Sci. U. S. A. 107:20453–20458. 10.1073/pnas.1008437107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brooks DG, Ha SJ, Elsaesser H, Sharpe AH, Freeman GJ, Oldstone MB. 2008. IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc. Natl. Acad. Sci. U. S. A. 105:20428–20433. 10.1073/pnas.0811139106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, Wherry EJ. 2014. Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity 40:289–302. 10.1016/j.immuni.2014.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]