

Abstract
The intracellular protozoan parasite Trypanosoma cruzi is the etiologic agent of Chagas disease, a serious disorder that affects millions of people in Latin America. Cell invasion by T. cruzi and its intracellular replication are essential to the parasite's life cycle and for the development of Chagas disease. Here, we present evidence suggesting the involvement of the host's cyclooxygenase (COX) enzymes during T. cruzi invasion. Pharmacological antagonists for COX-1 (aspirin) and COX-2 (celecoxib) caused marked inhibition of T. cruzi infection when rat cardiac cells were pretreated with these nonsteroidal anti-inflammatory drugs (NSAIDs) for 60 min at 37°C before inoculation. This inhibition was associated with an increase in the production of NO and interleukin-1β and decreased production of transforming growth factor β (TGF-β) by cells. Taken together, these results indicate that COX-1 more than COX-2 is involved in the regulation of anti-T. cruzi activity in cardiac cells, and they provide a better understanding of the influence of TGF-β-interfering therapies on the innate inflammatory response to T. cruzi infection and may represent a very pertinent target for new therapeutic treatments of Chagas disease.
INTRODUCTION
Chagas disease, caused by Trypanosoma cruzi infection, remains an important neglected tropical disease and has emerged as an important global public health problem because many T. cruzi-infected people from Latin America immigrate to countries where the disease is not endemic (1). An estimated 14,000 people die annually from this disease worldwide (2). Clinically, T. cruzi infection causes acute myocarditis followed by chronic cardiomyopathy and vasculopathy in humans and in experimental models.
Studies from diverse laboratories using different host cell types and T. cruzi strains have demonstrated that this parasite can invade almost all nucleated cells, both phagocytic and nonphagocytic (3). Although T. cruzi trypomastigotes are broadly dispersed among many different organs in the mammalian host, cardiac tissue is an important target for this parasite, and the T. cruzi-cardiomyocyte interaction has been the subject of intense investigation (4–7).
During the T. cruzi-cardiomyocyte interaction, the parasite gains control of overall host cell gene expression, including expression of 353 genes related to the immune response, inflammation, cytoskeleton organization, cell-cell and cell-matrix interactions, apoptosis, the cell cycle, and response to oxidative stress. This information provides insights into how the parasite survives, replicates, and persists in the infected host and ultimately the clinical outcome of the infection (5).
T. cruzi induces upregulation of nitric oxide (NO) production in cardiomyocytes along with an upregulation in the levels of interleukin-6 (IL-6), IL-1β, tumor necrosis factor alpha (TNF-α), and transforming growth factor β (TGF-β) (8–13). The resulting acute myocarditis is characterized by an intense inflammatory response typified by upregulation of inflammatory mediators, such as cytokines, chemokines, inducible nitric oxide synthase (iNOS), and endothelin (7), and also eicosanoids (10), which are essential elements to the defensive reaction in cardiac tissue (4), and it can also result in cardiac hypertrophy (8, 9).
Many of the changes that occur during acute and chronic Chagas disease can be explained by the effects of arachidonic acid (AA)-derived lipids, such as leukotrienes, lipoxins, hydroxyeicosatetraenoic and hydroperoxyeicosatetraenoic acids, prostaglandins (PGs), and thromboxane (10). A recent study demonstrated that cardiac calcium-independent phospholipase A2γ (iPLA2γ) is responsible for AA and prostaglandin E2 (PGE2) release in T. cruzi infection (11). PGs are oxygenated lipid mediators formed from the ω6 essential fatty acid AA. The committed step in PG biosynthesis is the conversion of AA to PGH2, which is catalyzed by either PG endoperoxide H synthase-1 or -2, enzymes that are commonly known as cyclooxygenase-1 (COX-1) and -2 (COX-2), respectively (14, 15). Both COX-1 and COX-2 are nonselectively inhibited by nonsteroidal anti-inflammatory drugs (NSAIDs) such as aspirin (ASA) and ibuprofen, whereas COX-2 activity is selectively blocked by COX-2 inhibitors called coxibs (e.g., celecoxib) (16). The relevance of these enzymes and the bioactive lipids that they produce are not well understood in parasitic diseases, although the role of eicosanoids in the pathogenesis of Chagas disease is becoming better defined (10).
Given the increasing interest in the role of eicosanoids in T. cruzi infection, we investigated the effect of prostaglandin synthesis inhibition with ASA and celecoxib on the inflammatory response and cardiac myoblast invasion by T. cruzi. Our results showed that the internalization of the parasite was reduced when H9C2 cells were treated with ASA or celecoxib. This reduction was associated with an increase in the production of NO and IL-1β and reduction of TGF-β only by cells that were treated with ASA. Taken together, these results indicated that COX-1 is involved in the regulation of anti-T. cruzi activity by cardiac cells and that it participates in T. cruzi invasion of myoblasts. These results elucidate the influence of eicosanoids on the innate inflammatory response to T. cruzi infection as well as provide an alternate perspective of specific immune interventions.
MATERIALS AND METHODS
Chemicals, drugs, and reagents.
Dimethyl sulfoxide (DMSO), ASA, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenytetrazolium bromide (MTT) were purchased from Sigma-Aldrich, Brazil Ltda. Penicillin, streptomycin sulfate, gentamicin, Dulbecco's modified Eagle's medium (DMEM), and fetal bovine serum (FBS) were purchased from Gibco (Grand Island, NY). Celecoxib was purchased from Pfizer Pharmaceuticals. Forskolin and wortmannin were purchased from Santa Cruz Biotechnology, Inc.
Cardiac myoblast cultures.
The rat cardiac myoblast H9C2 cell line (ATCC CRL-1446) was maintained in DMEM supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate in a humidified incubator at 37°C in 5% CO2. The cells were plated onto 13-mm round glass coverslips and washed in warm phosphate-buffered saline (PBS) before the interaction assays. Additionally, 2 × 105 cells were plated onto 96-well dishes. One set of plates was used to quantify cytokines and the other set was for NO and iNOS detection.
Parasites.
Trypanosoma cruzi (Y strain) (17) was maintained by weekly intraperitoneal inoculation of Swiss mice with 2 × 105 trypomastigotes. To conduct our experiments, blood from previously infected mice was obtained by cardiac puncture with anticoagulant. The blood was centrifuged at 1,500 × g for 1 min and allowed to stand at 37°C for 60 min. The supernatant serum containing most of the T. cruzi trypomastigotes was centrifuged at 1,200 × g for 15 min. The sediment was resuspended in 1 ml of RPMI 1640 medium (Gibco, Grand Island, NY) containing 10% inactivated FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco, Grand Island, NY).
Blood trypomastigotes from 5 mice previously infected with strain Y were used to infect LLC-Mk2 cells (ATCC CCL-7; American Type Culture Collection, Rockville, MD), and trypomastigotes derived from the supernatants of T. cruzi-infected LLC-Mk2 cell cultures were used for subsequent experiments. T. cruzi-infected LLC-Mk2 cell cultures were grown in RPMI 1640 medium containing 10% inactivated FBS, 40 μg/ml gentamicin, 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco, Grand Island, NY). Subconfluent cultures of LLC-Mk2 cells were infected with 5 × 106 trypomastigotes. Free parasites were removed after 24 h, and cultures were maintained in 10% FBS–RPMI 1640. Five days postinfection, free trypomastigote forms could be found in the cell supernatants.
Treatment of myoblasts with drugs, including NSAIDs.
Before the experiments, previously washed H9C2 cells were incubated for 1 h at 37°C in a 5% CO2 atmosphere in the presence of different concentrations of ASA or celecoxib (2.5 mM, 1.25 mM, and 0.625 mM) to test the effects of the drugs on parasite internalization into the host cell. After incubation, the medium containing NSAIDS was removed and cells were allowed to interact with trypomastigote forms, added at a ratio of 5 parasites per cell. The interaction was allowed to proceed for 24 h at 37°C in a 5% CO2 atmosphere. The cells were then washed three times, fixed with Bouin's fixative, stained with Giemsa stain (Merck), and observed under a light microscope at 1,000× magnification. Other treatments included incubation with 10 μM forskolin for 20 min at 37°C and 200 nM wortmannin for 30 min at 37°C in the presence or absence of celecoxib. The internalization index was calculated by multiplying the percentage of infected cells by the mean number of parasites per infected cell (18). All the internalization indices were normalized.
Experiments were performed in triplicate, and five independent experiments were completed. All the experiments included untreated, infected H9C2 cells as controls. Quantification was carried out via light microscopy, and a total of 500 cells were randomly counted.
Cell viability assay.
Viability of the cells obtained from the cultures before and after incubation experiments was determined in an MTT assay to show the mitochondrial activity of living cells. Briefly, the H9C2 cells were plated at 2 × 105 cells/well in a 96-well microplate for 24 h. After treatment, the cells were incubated with MTT (final concentration, 0.5 mg/ml) at 37°C for 4 h. The supernatant was aspirated, and DMSO was added to the wells. Insoluble crystals were dissolved by mixing, and the plates were read using a multiplate reader (Bio-Rad, Hercules, CA) at a test wavelength of 570 nm and a reference wavelength of 630 nm. The percentage of cell viability was calculated using the following formula, as previously described (19): percent cell viability = [(mean absorbance in test wells)/(mean absorbance in control wells)] × 100.
Detection of NO levels by high-sensitivity chemiluminescence.
NO levels were evaluated by employing a highly sensitive, previously described chemiluminescence system (20) with some modifications. In this method, NO reacts with hydrogen peroxide, resulting in peroxynitrite. In the presence of luminol, peroxynitrite produces triplet oxygen, which decays to singlet oxygen and emits photons; the photons are detected by using a luminometer system coupled to software.
To measure the NO/peroxynitrite level, supernatants of the H9C2 cell cultures were removed from incubation and immediately diluted in fresh sterile Na2CO3 buffer (2 mM; pH 8.5) that was previously degassed via N2 bubbling for 20 min, to eliminate the presence of molecular oxygen and oxidation of NO to nitrite/nitrate. The final reaction volume was 1 ml, with a cell concentration of 2 × 105 cells/ml.
The starting reagent was prepared by mixing equal volumes of luminol solution (4.39 μM dissolved in 1 M KOH) diluted 1:10 in desferrioxamine (36.58 μM), and H2O2 (2.44 μM) was added to 3 parts of degassed Na2CO3 buffer (2 mM; pH 8.5). This mixture was vortexed for 5 min before use. All the solutions were sterile, kept at 25°C in covered tubes, and protected from light. Finally, the samples were injected with 50 μl of starting reagent, and the reaction was performed in a Glomax luminometer (Promega) with an automatic reagent injector, employing a kinetic protocol that allowed 10 readings per second.
The total curve profile, integrated area, and curve were analyzed to determine the peroxynitrite levels. A standard curve was obtained for nitrite reduction at an acidic pH to determine the NO/peroxynitrite concentration. NO is generated by nitrite (NO2−) reduction at acidic pH, as previously described (19). In this reaction, 1.8 ml of 0.1 M H2SO4–0.1 M KI and 7.2 ml of 0.1 mM NaNO2 were used. The reaction immediately generates a solution of 400 pM NO at room temperature. This solution was diluted to final concentrations of 100 fM, 200 fM, 300 fM, and 400 fM NO in 2 mM Na2CO3 buffer, pH 8.5, that had been previously degassed using N2.
Immunocytochemistry labeling for iNOS.
Immunocytochemistry for iNOS was performed on coverslip-adherent cells by using the labeled streptavidin biotin method and a LSAB kit (Dako Japan, Kyoto, Japan) without microwave accentuation. The coverslips were incubated with 10% Triton X-100 solution for 1 h, washed 3 times in PBS, and treated for 40 min at room temperature with 10% bovine serum albumin. The coverslips were then incubated overnight at 4°C with primary antibody (anti-iNOS rabbit monoclonal antibody diluted 1:200; catalog number 610599; BD Biosciences), followed by secondary antibody treatment for 2 h at room temperature. Horseradish peroxidase activity was visualized by treatment with H2O2 and 3,3′-diaminobenzidine (DAB) for 5 min. At the last step, the sections were weakly counterstained with Harry's hematoxylin (Merck). Negative controls were prepared by omitting primary antibody. Intensity and localization of the immune reaction against primary antibody were examined on all coverslips with a photomicroscope (Olympus BX41; Olympus Optical Co., Ltd., Tokyo, Japan).
For image analysis, photomicroscopic color slides of representative areas (magnification, ×40) were digitally acquired. After conversion of the images into gray scale (Adobe Photoshop), iNOS-positive pixels and total pixels thresholds were determined and data were processed using the ImageJ software. Positive immunostained areas were calculated as the proportion (percentage) of positive pixels to total pixels.
ELISA for TNF-α, TGF-β, and IL-1β.
Culture supernatants from H9C2 cells in 96-well plates were untreated or treated with ASA or celecoxib, either infected or not infected with T. cruzi, and incubated for 24 h. Levels of TNF-α, TGF-β, and IL-1β in 100 μl medium were measured by using a commercial enzyme-linked immunosorbent assay (ELISA) kit (Ready-SET-Go!; eBioscience, San Diego, CA), according to the manufacturer's instructions.
Statistical analysis.
Statistical analysis was conducted via an analysis of variance with Bonferroni's multiple comparison test. Values are presented as means ± standard errors of the means. The results were considered significant when P was <0.05. Statistical analysis was performed with the GraphPad Prism 5.0 computer software (GraphPad Software, San Diego, CA).
RESULTS
ASA and celecoxib inhibit T. cruzi entry into H9C2 cells.
To determine whether COX-derived mediators are involved in T. cruzi entry into host cells, H9C2 cells were infected with trypomastigotes in the presence of ASA or celecoxib at various concentrations. The cells were treated with increasing amounts of NSAIDs for 1 h. After treatment, the medium containing the inhibitors was removed before exposure to the parasites in order to guarantee that the inhibitors only affected the host cell and not the parasites. After 24 h of incubation with parasites, which provided sufficient time for them to enter into cells, the free parasites were removed and the cells were stained with Giemsa stain.
Both inhibitors reduced the internalization of trypomastigotes into H9C2 cells at 2.5 mM, 1.25 mM, and 0.625 mM (ASA) as well as 1.25 mM, 0.625 mM, and 0.312 mM (celecoxib) (Fig. 1a and b). Thus, PGE2 synthesis inhibition by NSAIDs improves the myoblast response to T. cruzi infection. The cytotoxicity of inhibitors in the cells was evaluated in an MTT assay (Fig. 1c), and neither ASA nor celecoxib induced cell death. ASA irreversibly inhibited COX-1 by acetylation of a single serine residue on the enzyme, and this inactivation persisted for an extended period of time (≥24 h). Therefore, to guarantee a prolonged effect of ASA on H9C2 cells in the cultures, we used the highest concentration, since it was not cytotoxic.
FIG 1.
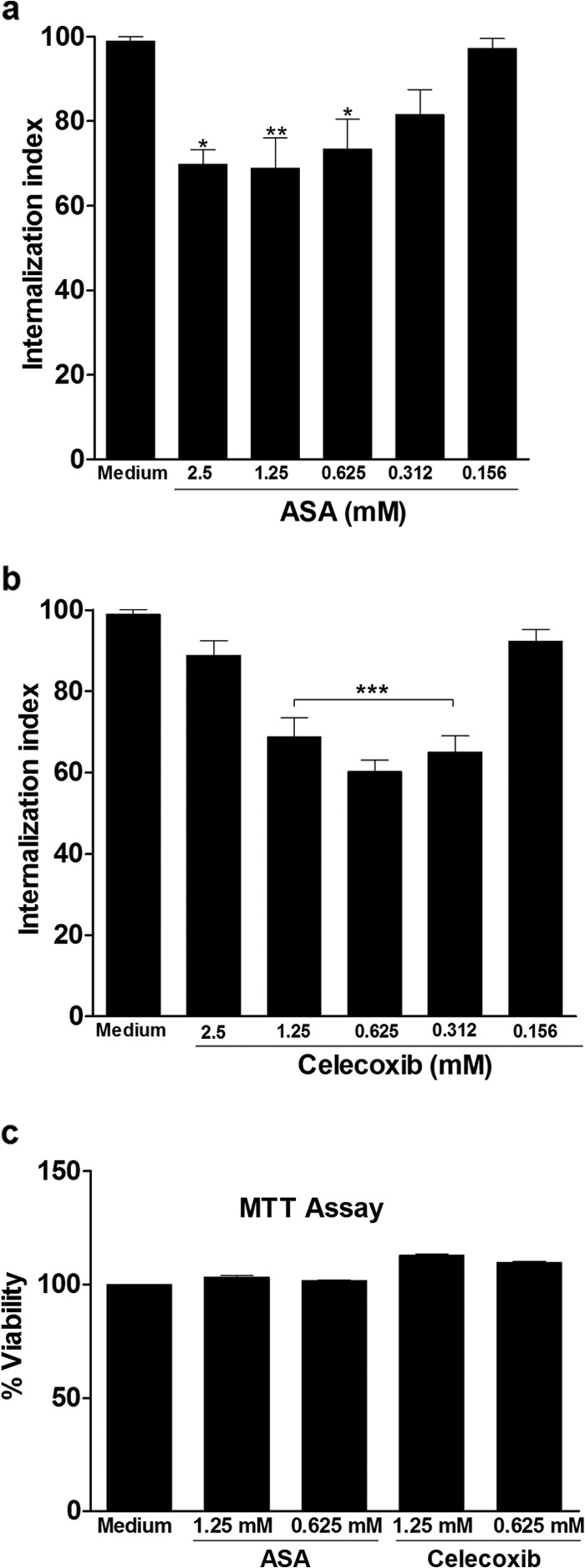
Aspirin and celecoxib inhibit T. cruzi entry into H9C2 cells. Internalization indices of the interaction process between macrophages treated for 1 h with increasing concentrations of ASA (a) or celecoxib (b) and exposed to T. cruzi (Y strain). After treatment with ASA or celecoxib, H9C2 cells interacted with a 5:1 parasite:cell ratio of trypomastigotes for 24 h, after which they were washed, fixed with Bouin's fixative, and stained with Giemsa stain. Quantification was carried out under a light microscope, where the number of intracellular parasites was counted in a total of least 500 cells. (c) The effects of ASA and celecoxib on cell viability. An MTT assay was conducted to measure cell viability in H9C2 cells after treatment with inhibitors at concentrations from 0.625 to 2.5 mM. Values are means ± standard errors of means of 10 experiments or two experiments (c). *, P < 0.05; **, P < 0.01; ***, P< 0.001 (compared to infected cells cultured in medium alone).
Effect of adenylyl cyclase activation on H9C2 cell invasion by T. cruzi.
Trypanosoma cruzi trypomastigotes trigger elevation in host cell cyclic AMP (cAMP) levels. Furthermore, parasite invasion is prevented by inhibition of host cell adenylyl cyclase and enhanced by stimulation of cAMP production (22). Here, we tested the effect of forskolin, an activator of adenylyl cyclase (23), on H9C2 cell invasion by T. cruzi after COX inhibition.
Cells were either untreated or treated with ASA (2.5 mM) or celecoxib (0.625 mM) for 1 h at 37°C in a 5% CO2 atmosphere. After washing in PBS, the cells were incubated with control (medium) or 10 μM forskolin for 30 min at 37°C. An equivalent amount of the carrier (DMSO) was added to untreated cells. After washing in PBS, control and forskolin-treated cells were incubated for 24 h with parasites at a cell ratio of 5:1.
The activation of adenylyl cyclase with forskolin did not affect H9C2 cell invasion by trypomastigote in the Y strain (P > 0.05) (Fig. 2) but reversed the effects of ASA and celecoxib (Fig. 2a and b).
FIG 2.
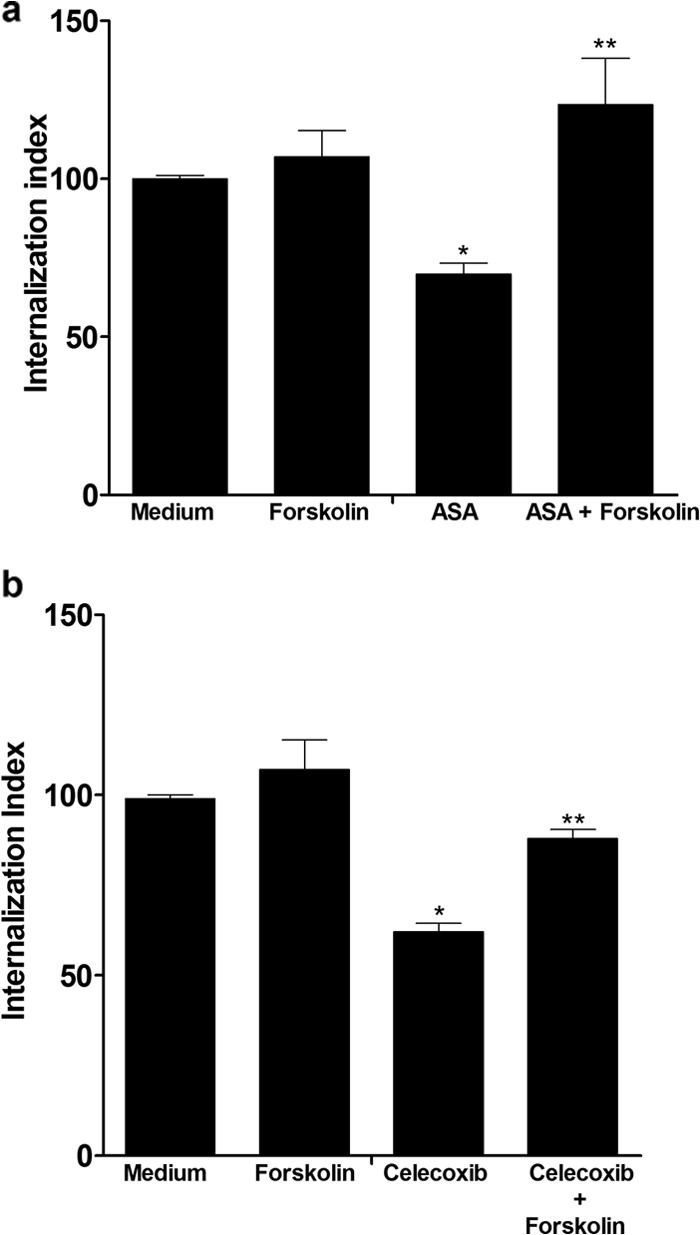
Activation of adenylyl cyclase with forskolin reverted the ASA and celecoxib effects on T. cruzi-infected H9C2 cells. Internalization indices were calculated for the interaction process between H9C2 cells treated for 1 h with ASA (2.5 mM) (a) or celecoxib (0.625 mM) (b) and exposed to T. cruzi (Y strain). Cells were pretreated or not with ASA (2.5 mM) or celecoxib (0.625 mM) for 1 h at 37°C in a 5% CO2 atmosphere. After washing in PBS, the cells were incubated with control (medium) or 10 μM forskolin for 30 min at 37°C. An equivalent amount of the carrier (DMSO) was added to untreated cells. After washing in PBS, control and forskolin-treated cells were incubated for 24 h with parasites at a parasite:cell ratio of 5:1. Values are the means ± standard errors of the means of three experiments. *, P < 0.001 (compared to infected cells cultured in medium alone); **, P < 0.05 (compared to cells cultured in ASA or celecoxib).
H9C2 cell PI3K activity is required for invasion by T. cruzi.
The involvement of phosphatidylinositol 3-kinase (PI3K) in T. cruzi host cell invasion has been examined using specific inhibitors, such as wortmannin (24). To investigate the role of PI3K in cells treated with NSAIDs, we administered wortmannin (200 nM) to cells for 30 min after NSAID treatment. An equivalent amount of the carrier (DMSO) was added to untreated cells.
The treatment of H9C2 cells with wortmannin significantly impaired T. cruzi invasion, independent of pretreatment with both NSAIDs (Fig. 3a and b). These data indicate that PI3 activity is involved in the entry into the cardiac cells used in this study, and this observation is consistent with previously published research demonstrating that both inhibitors cause downregulation in the PI3K/Akt pathway (25, 26).
FIG 3.
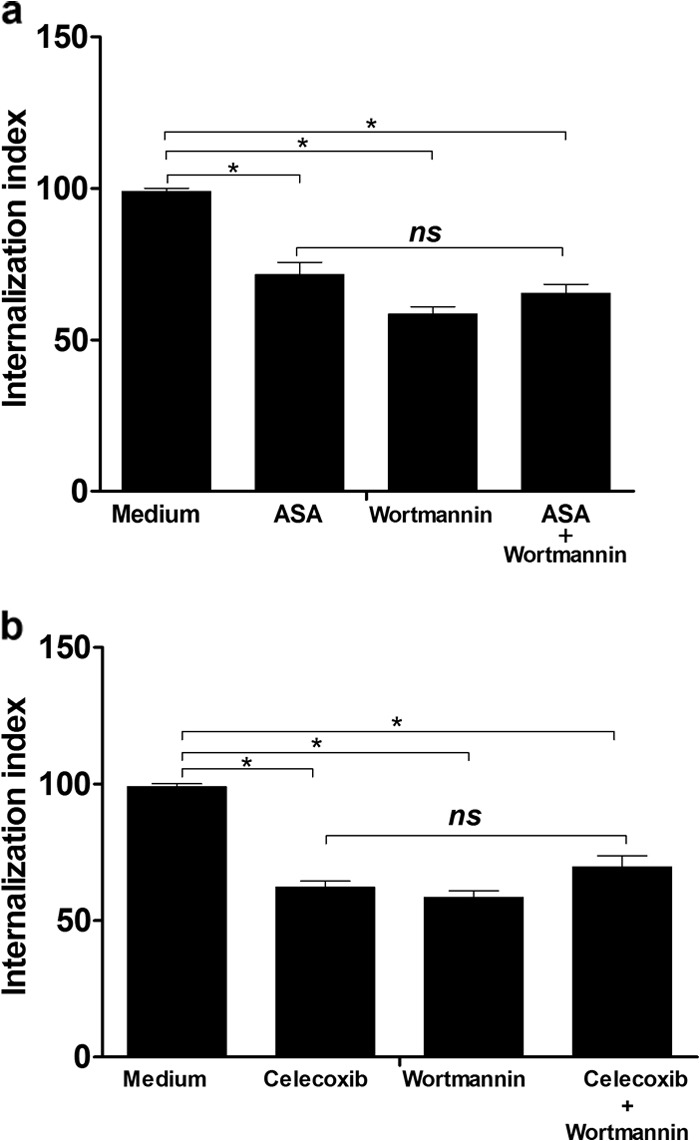
Treatment of H9C2 cells with wortmannin significantly diminished the infectivity of parasites independently of pretreatment with ASA or celecoxib. Internalization indices were calculated for the interaction process between H9C2 cells pretreated for 1 h with ASA (2.5 mM) (a) or celecoxib (0.625 mM) (b) and exposed to T. cruzi (Y strain). Cells were treated or not with ASA or celecoxib for 1 h at 37°C in a 5% CO2 atmosphere. After washing in PBS, the cells were incubated with medium (control) or wortmannin (200 nM) for 30 min at 37°C. An equivalent amount of the carrier (DMSO) was added to untreated cells. After washing in PBS, control and wortmannin-treated cells were incubated for 24 h with a parasite:cell ratio of 5:1. Values are the means ± standard errors of the means of three experiments. *, P < 0.001 (compared to infected cells cultured in medium alone); ns, not significant.
NSAIDs modulate the innate inflammatory response of H9C2 cells infected with T. cruzi.
We examined the effects on NO production and cytokines of various treatments that alter intracellular signal transducing pathways. Internalization of T. cruzi into H9C2 cells did not stimulate the release of TNF-α (Fig. 4a) or TGF-β (Fig. 4b), but it increased IL-1β production (Fig. 4c). Treatment with ASA (Fig. 4a) or celecoxib (Fig. 5a) did not affect TNF-α production by cardiac cells but increased IL-1β production (Fig. 4c and 5c), whereas ASA (Fig. 4b), but not celecoxib, inhibited TGF-β production by the cells (Fig. 5b).
FIG 4.
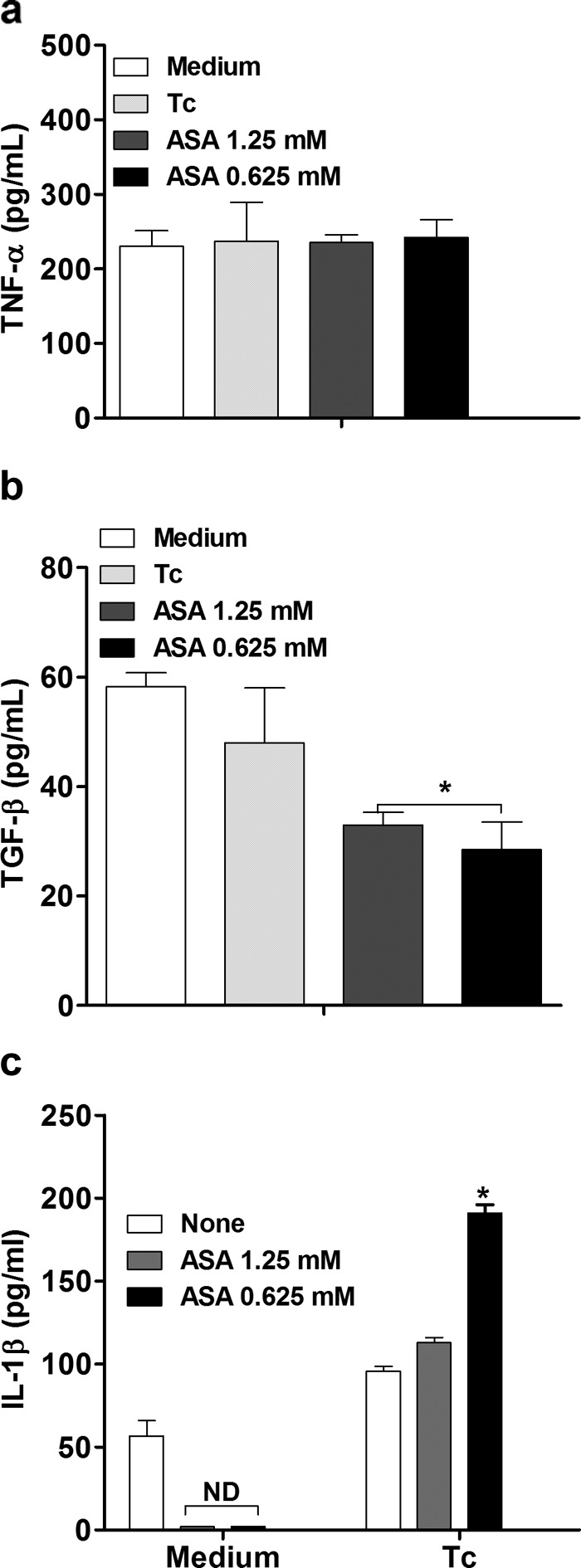
Effects of aspirin on TNF-α, TGF-β, and IL-1 β production in T. cruzi-infected H9C2 cells. Cells were treated for 1 h with ASA (0.625 and 1.25 mM) and exposed to T. cruzi (Y strain). After treatment, the cells interacted with a 5:1 trypomastigote:cell ratio for 24 h, after which they were cultured at 37°C in 5% CO2 during 24 h. TNF-α (a), TGF-β (b), and IL-1β (c) levels in supernatants were measured with a specific enzyme-linked immunosorbent assay. Results are the means ± standard errors of the means for duplicate determinations and are representative of two independent experiments. *, P < 0.05 (compared to cell culture in medium alone); ND, not detected.
FIG 5.
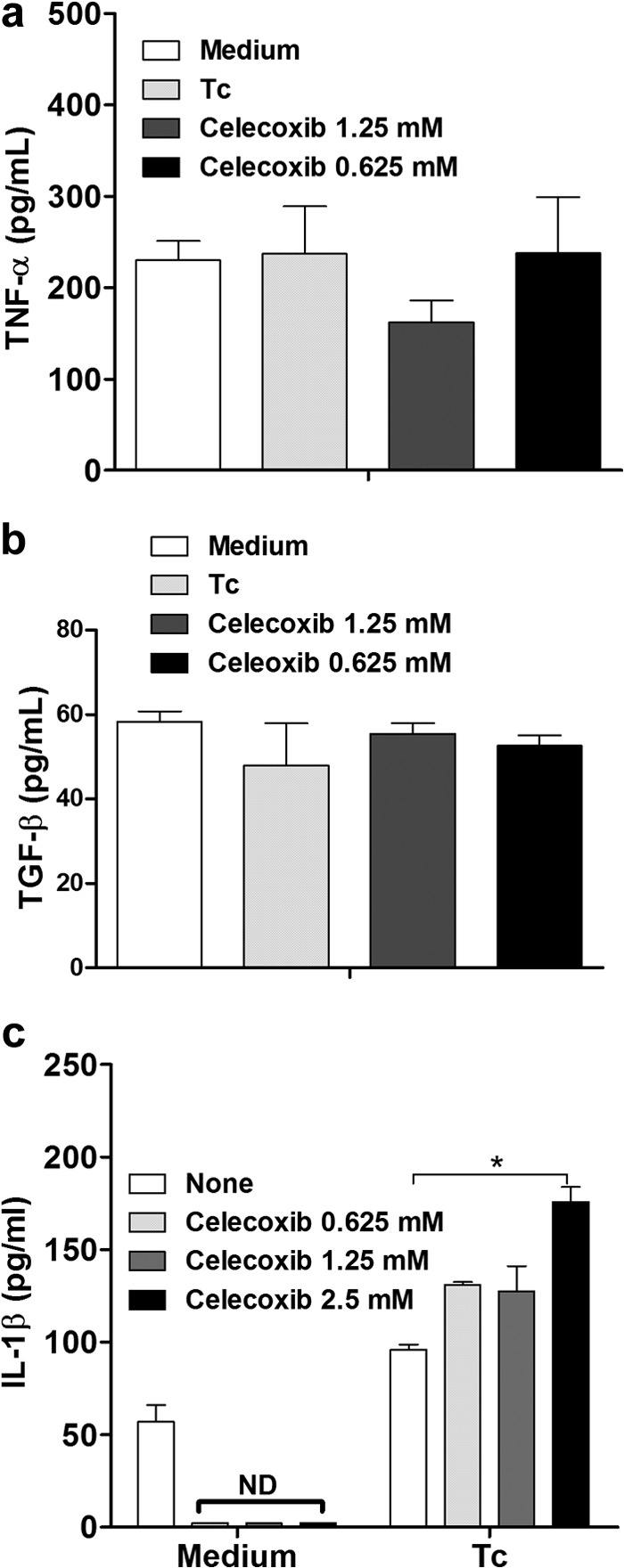
Effects of celecoxib on TNF-α, TGF-β, and IL-1β production in T. cruzi-infected H9C2 cells. Cells were treated for 1 h with celecoxib (0.625 and 1.25 mM) and exposed to T. cruzi (Y strain). After treatment, the cells interacted with a 5:1 trypomastigote:cell ratio for 24 h, after which they were cultured at 37°C in 5% CO2 during 24 h. TNF-α (a), TGF-β (b), and IL-1β (c) levels in supernatants were measured with a specific enzyme-linked immunosorbent assay. Results are the means ± standard errors of the means for duplicate determinations and are representative of two independent experiments. *, P < 0.05 (compared to cell culture in medium alone); ND, not detected.
The effect of both inhibitors on NO production was evaluated by detection of NO in T. cruzi-infected H9C2 supernatants using high-sensitivity chemiluminescence. Interesting, NO production in cardiac cells was diminished by T. cruzi and increased by prior treatment of cells with ASA (0.625 mM) and celecoxib (1.25 mM) (Fig. 6). The increase in NO production induced by ASA was concentration dependent. Additionally, we observed that ASA and celecoxib treatment stimulated iNOS expression in T. cruzi-infected H9C2 cells (Fig. 7). Interestingly, when cells were incubated with ASA in combination with forskolin, we observed a large number of internalized trypomastigotes (Fig. 2a), which was associated with increased TGF-β production by cells (Fig. 8).
FIG 6.
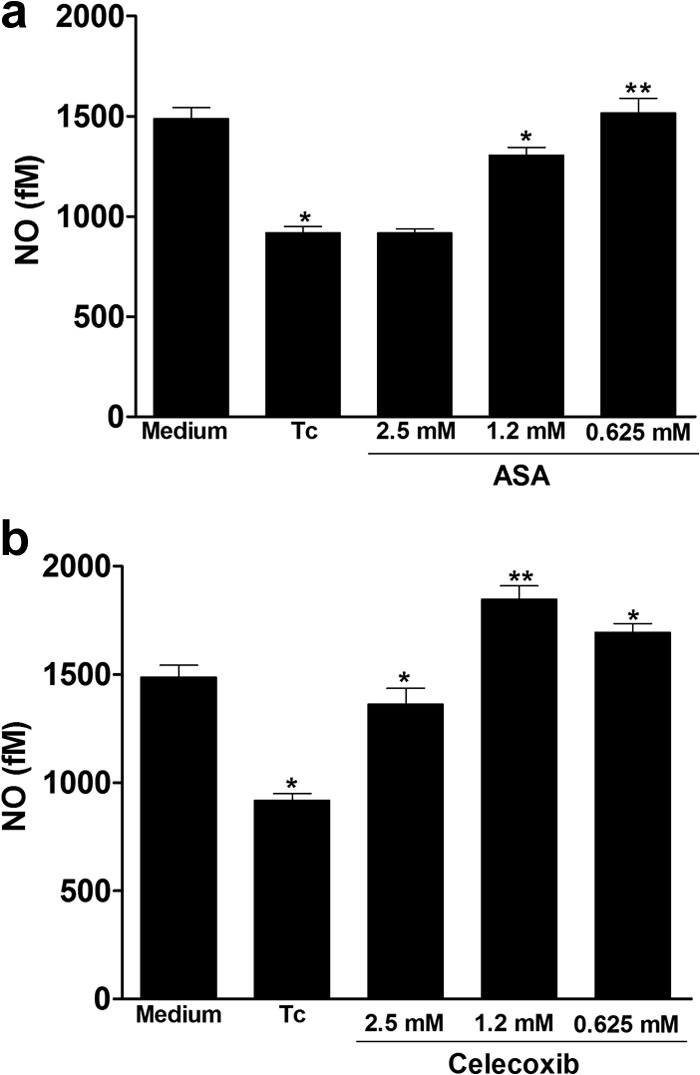
Effects of aspirin upon NO production in T. cruzi-infected H9C2 cells. Cells were treated for 1 h with ASA (0.625, 1.25, and 2.5 mM) (a) or celecoxib (0.625, 1.25 and 2.5 mM) (b) and exposed to T. cruzi (Y strain). After treatment with NSAIDs, cells interacted with a 5:1 trypomastigote:cell ratio for 24 h, after which they were washed and cultured at 37°C in 5% CO2 during 24 h. NO levels in supernatants were measured by high-sensitivity chemiluminescence. Results are the means ± standard errors of the means for duplicate determinations and are representative of two independent experiments. *, P < 0.05; **, P < 0.01 (compared to the untreated infected cell culture [Tc]).
FIG 7.
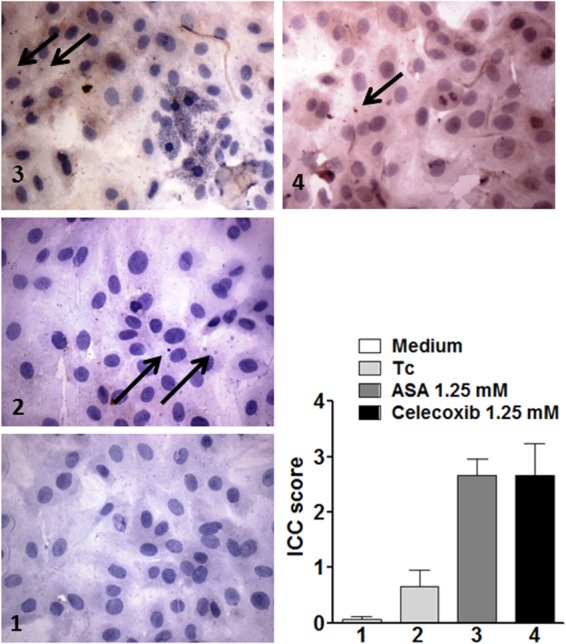
Effects of aspirin and celecoxib on iNOs expression in T. cruzi-infected H9C2 cells. Immunocytochemistry for iNOS was performed on coverslip-adherent cells by the labeled streptavidin biotin method with a LSAB kit (Dako Japan, Kyoto, Japan) without microwave accentuation. (1) Intracellular iNOS protein was detected by immunocytochemistry in uninfected (control) H9C2 cells. (2) T. cruzi provoked a discrete increase of iNOs expression. (3 and 4) ASA (3) and celecoxib (4) were effective inducers of iNOS expression in T. cruzi-infected H9C2 cells.
FIG 8.
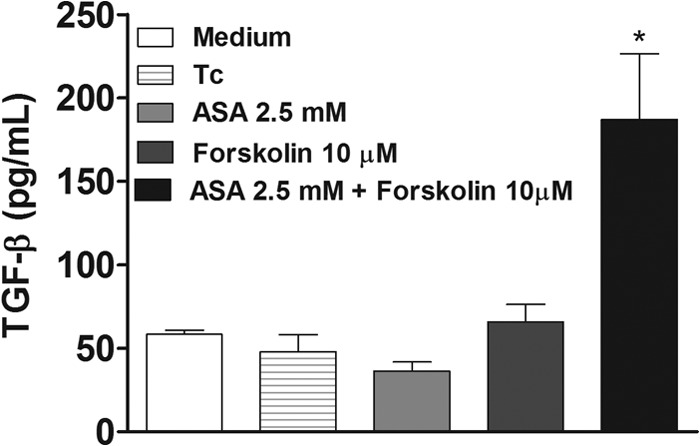
Aspirin in combination with 10 μM forskolin increased TGF-β production by T. cruzi-infected H9C2 cells. Cells were treated for 1 h with ASA (2.5 mM). After treatment, cells were washed and incubated with 10 μM forskolin for 20 min. After treatment, cells were washed again and allowed to interact with a 5:1 trypomastigote:cell ratio for 24 h at 37°C. TGF-β levels in supernatants were measured with a specific enzyme-linked immunosorbent assay. Results are means ± standard errors of the means for duplicate determinations and are representative of two independent experiments. *, P < 0.05 (compared to infected cells in cell culture medium, ASA, or forskolin alone).
DISCUSSION
Previous studies have shown that the release of eicosanoids during infection with T. cruzi regulates host responses and controls disease progression (10, 27–31). PGs, together with NO and TNF-α, participate in a complex circuit that controls lymphoproliferative and cytokine responses in T. cruzi infection (28). However, the involvement of COX-mediated PG production in the entry of T. cruzi into cardiac cells is largely unexplored.
The data shown herein demonstrate that treatment of rat cardiac cells with NSAIDs such as aspirin and celecoxib significantly inhibits internalization of T. cruzi trypomastigotes and strongly support the idea that the COX pathway plays a fundamental role in the process of parasite invasion. In fact, PGE2 production significantly increases in T. cruzi-infected macrophages compared with uninfected macrophages (35), and PGE2 synthesis inhibition by using aspirin synergistically enhances the activity of nifurtimox and benznidazole in infected RAW 264.7 cells (36).
The effects of aspirin on T. cruzi infection have been associated in part, with a switch to the AA pathway that is linked to acetylation of the COX-2 isoenzyme (37). This acetylation enables COX-2 to synthesize other lipid products derived from AA, some of them with anti-inflammatory properties, such as 15-epi-LXA4, known as an “aspirin-triggered lipoxin.” High levels of 15-epi-LXA4 were observed in T. cruzi-infected mice treated with the low doses of ASA, while high ASA doses decreased 15-epi-LXA4 levels (37). Importantly, 15-epi-LXA4 prevented parasitemia, mortality, and cardiac changes in vivo and restored the protective role in the treatment group that received a high dose of ASA (37). Additionally, polyamines seem to be crucial for the trypomastigote internalization process in at least some cellular types and in infection progression (38).
COX is related to an increase of ornithine decarboxylase (ODC) activity in T. cruzi-infected macrophages (33), which might increase the polyamine content in macrophages. Since T. cruzi uses these polyamines to synthesize trypanothione (an enzyme that participates in the hydroperoxide detoxification of T. cruzi), the inhibition of COX by ASA probably results in a reduction in polyamine levels caused by inhibition of ODC, indirectly contributing to decreased trypanothione synthesis in T. cruzi, as suggested by López-Muñoz and collaborators (36).
Trypomastigotes (the infective stages of T. cruzi) trigger elevations in host cell cAMP levels. This is a significant finding, because trypomastigotes are the T. cruzi life cycle stages that are capable of invading host cells through a Ca2+-dependent lysosome recruitment process, which involves parasite-mediated signaling (22). Elevation of intracellular Ca2+ levels has also been demonstrated in T. cruzi-infected cardiac cells (4).
Our results showed that treatment of H9C2 cells with the adenylyl cyclase activator forskolin (23) did not alter the infectivity of trypomastigotes (Y strain). The treatment of H9C2 cells with celecoxib (an inhibitor of COX-2) or ASA in combination with forskolin restored the infectivity of trypomastigotes in cardiac cells. This could have been due to the effects of NSAIDs through the inhibition of cAMP, as previously described (39).
The PI3K inhibitor wortmannin caused marked inhibition of T. cruzi infection when H9C2 cells were treated before inoculation. This inhibition was independent of pretreatment with aspirin or celecoxib. These findings suggest a role for host PI3K activities during the T. cruzi infection process into cardiac cells. Therefore, our findings are consistent with the hypothesis that PI3K inhibition results in an increase in COX-2 production (40). Further work will be required to test our hypothesis and to determine whether wortmannin promotes increases of PGs in T. cruzi-infected H9C2 cells.
Inhibition of COX activity may increase NO levels, thus restoring the antiparasitic activity of macrophages (38). Our results are consistent with this hypothesis. Additionally, we showed that iNOS expression in H9C2 cells increased with ASA or celecoxib treatment, which is also in agreement with our hypothesis.
Moreover, there is no evidence to support the hypothesis that low TGF-β production by H9C2 cells reduces T. cruzi infection. We attempted to determine whether TGF-β is involved in the effect of ASA or celecoxib on T. cruzi-infected cells. We did not find any effect of celecoxib on TGF-β production by H9C2 cells, but when we used ASA, we observed a decrease in TGF-β released by cells, indicating the role of TGF-β in ASA activity. In fact, TGF-β is required for the invasion of host cells by the parasite (41). T. cruzi infection induces the production of NO, which could contribute to parasite killing by host cells. TGF-β is a potent suppressor of NO production (42), and its inhibition caused decreased T. cruzi invasion in cardiomyocytes (43).
Finally, in T. cruzi-infected H9C2 cells, COX inhibition by ASA or celecoxib was related to the increase of IL-1β but not of TNF-α, which might explain in part the increase of antiparasitic activity of cardiac cells treated with NSAIDs. In fact, IL-1β is critical for the restriction of Leishmania amazonensis infection (44), and recently it was demonstrated that T. cruzi-infected macrophages treated with IL-1β released fewer trypomastigotes than untreated macrophages and that IL-1β triggered NO release by infected macrophages in a dose-dependent manner (45).
In conclusion, this is the first report, to our knowledge, showing the in vitro effect of NSAIDS (aspirin and celecoxib) on T. cruzi entry into rat cardiac cells, providing a better understanding of the influence of TGF-β-interfering therapies on the innate inflammatory response to T. cruzi infection and may represent a very pertinent target for new therapeutic treatments of Chagas disease.
ACKNOWLEDGMENTS
This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brasil (CNPq; 302097/2010-474792/2011-0), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and by the Fundação Araucaria (Convênio 419/2009).
We declare no conflict of interests.
P. Pinge-Filho, S. F. Yamada-Ogatta, M. C. Martins-Pinge, and A. D. Malvezi participated in research design. A. D. Malvezi, C. Panis, R. Valeriano da Silva, M. I. Lovo-Martins, N. G. Zanluqui, V. L. Tatakihara, and R. C. de Freitas conducted experiments. E. C. Neto, R. Cecchini, J. Bordignon, and S. Goldenberg contributed to the new reagents or analytical tools. A. D. Malvezi, C. Panis, R. Valeriano da Silva, M. I. Lovo-Martins, and P. Pinge-Filho performed the data analysis. A. D. Malvezi, J. Bordignon, M. C. Martins-Pinge, and P. Pinge-Filho contributed to the writing of the paper.
Footnotes
Published ahead of print 4 August 2014
REFERENCES
- 1.Pérez-Molina JA, Norman F, López-Vélez R. 2012. Chagas disease in non-endemic countries: epidemiology, clinical presentation and treatment. Curr. Infect. Dis. Rep. 14:263–274. 10.1007/s11908-012-0259-3 [DOI] [PubMed] [Google Scholar]
- 2.Schmunis GA, Yadon ZE. 2010. Chagas disease: a Latin American health problem becoming a world health problem. Acta Trop. 115:14–21. 10.1016/j.actatropica.2009.11.003 [DOI] [PubMed] [Google Scholar]
- 3.Romano PS, Cueto JA, Casassa AF, Vanrell MC, Gottlieb RA, Colombo MI. 2012. Molecular and cellular mechanisms involved in the Trypanosoma cruzi/host cell interplay. IUBMB Life 64:387–396. 10.1002/iub.1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calvet CM, Melo TG, Garzoni LR, Oliveira FOR, Jr, Silva Neto DT, Nazareth MNSL, Pereira MCS. 2012. Current understanding of the Trypanosoma cruzi cardiomyocyte interaction. Front. Immunol. 3:327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manque PA, Probst C, Pereira MCS, Rampazzo RCP, Ozaki LS, Pavoni DP, Silva Neto DT, Carvalho MR, Xu P, Serrano MG, Alves JMP, Nazareth MNSL, Meirelles L, Goldenberg S, Krieger MA, Buck GA. 2011. Trypanosoma cruzi infection induces a global host cell response in cardiomyocytes. Infect. Immun. 79:1855–1862. 10.1128/IAI.00643-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Machado FS, Dutra WO, Esper L, Gollob KJ, Teixeira MM, Factor SM, Weiss LM, Nagajyothi F, Tanowitz HB, Garg NJ. 2012. Current understanding of immunity to Trypanosoma cruzi infection and pathogenesis of Chagas disease. Semin. Immunopathol. 34:753–770. 10.1007/s00281-012-0351-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corral RS, Guerrero NA, Cuervo H, Gironès N, Fresno M. 2013. Trypanosoma cruzi infection and endothelin-1 cooperatively activate pathogenic inflammatory pathways in cardiomyocytes. PLoS Negl. Trop. Dis. 7:e2034. 10.1371/journal.pntd.0002034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petersen CA, Burleigh BA. 2003. Role for interleukin-1 beta in Trypanosoma cruzi-induced cardiomyocyte hypertrophy. Infect. Immun. 71:4441–4447. 10.1128/IAI.71.8.4441-4447.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petersen CA, Krumholz KA, Burleigh BA. 2005. Toll-like receptor 2 regulates interleukin-1β-dependent cardiomyocyte hypertrophy triggered by Trypanosoma cruzi. Infect. Immun. 73:6974–6980. 10.1128/IAI.73.10.6974-6980.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Machado FS, Mukherjee S, Weiss LM, Tanowitz HB, Ashton AW. 2011. Bioactive lipids in Trypanosoma cruzi infection. Adv. Parasitol. 76:1–31. 10.1016/B978-0-12-385895-5.00001-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma J, Eickhoff CS, Hoft DF, Ford DA, Gross RW, McHowat J. 2013. The absence of myocardial calcium-independent phospholipase A2γ results in impaired prostaglandin E2 production and decreased survival in mice with acute Trypanosoma cruzi Infection. Infect. Immun. 81:2278–2287. 10.1128/IAI.00497-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Machado FS, Martins GA, Aliberti JC, Mestriner FL, Cunha FQ, Silva JS. 2000. Trypanosoma cruzi-infected cardiomyocytes produce chemokines and cytokines that trigger potent nitric oxide-dependent trypanocidal activity. Circulation 102:3003–3008. 10.1161/01.CIR.102.24.3003 [DOI] [PubMed] [Google Scholar]
- 13.Machado FS, Souto JT, Rossi MA, Esper L, Tanowitz HB, Aliberti J, Silva J. 2008. Nitric oxide synthase-2 modulates chemokine production by Trypanosoma cruzi-infected cardiac myocytes. Microbes Infect. 10:1558–1566. 10.1016/j.micinf.2008.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rocca B, FitzGerald GA. 2002. Cyclooxygenases and prostaglandins: shaping up the immune response. Int. Immunopharmacol. 2:603–630. 10.1016/S1567-5769(01)00204-1 [DOI] [PubMed] [Google Scholar]
- 15.Park JY, Pillinger MH, Abramson SB. 2006. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin. Immunol. 119:229–240. 10.1016/j.clim.2006.01.016 [DOI] [PubMed] [Google Scholar]
- 16.Rimon G, Sidhu RS, Lauver DA, Lee JY, Sharma NP, Yuan C, Frieler RA, Trievel RC, Lucchesi BR, Smith WL. 2010. Coxibs interfere with the action of aspirin by binding tightly to one monomer of cyclooxygenase -1. Proc. Natl. Acad. Sci. U. S. A. 107:28–33. 10.1073/pnas.0909765106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silva LHP, Nussenzweig V. 1953. Sobre uma cepa de Trypanosoma cruzi altamente virulenta para o camundongo branco. Folia Clin. Biol. 20:191–203 [Google Scholar]
- 18.Barrias ES, Reignault LC, De Souza W, Carvalho TM. 2010. Dynasore, a dynamin inhibitor, inhibits Trypanosoma cruzi entry into peritoneal macrophages. PLoS One 5:e7764. 10.1371/journal.pone.0007764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ren G, Qiao HX, Yang J, Zhou CX. 2010. Protective effects of steroids from Allium chinense against H2O2-induced oxidative stress in rat cardiac H9C2 cells. Phytother. Res. 24:404–409. 10.1002/ptr.2964 [DOI] [PubMed] [Google Scholar]
- 20.Kikuchi K, Nagano T, Hayakawa H, Hirata Y, Hirobe M. 1993. Real time measurement of nitric oxide produced ex vivo by luminol-H2O2 chemiluminescence method. J. Biol. Chem. 268:23106–23110 [PubMed] [Google Scholar]
- 21.Pfeiffer S, Gorren ACF, Schmidt K, Werner ER, Hansert B, Bohle DS, Mayer B. 1997. Metabolic fate of peroxynitrite in aqueous solution: reaction with nitric oxide and pH-dependent decoposition to nitrite and oxygen in a 2:1 stoichiometry. J. Biol. Chem. 272:3465–3470. 10.1074/jbc.272.6.3465 [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez A, Martinez I, Chung A, Berlot CH, Andrews NW. 1999. cAMP regulates Ca2+-dependent exocytosis of lysosomes and lysosome-mediated cell invasion by trypanosomes. J. Biol. Chem. 274:16754–16759. 10.1074/jbc.274.24.16754 [DOI] [PubMed] [Google Scholar]
- 23.Seamon KB, Daly JW. 1981. Forskolin: a unique diterpene activator of cyclic AMP-generating systems. Cyclic Nucleotide Res. 7:201–224 [PubMed] [Google Scholar]
- 24.Wilkowsky SE, Barbieri MA, Stahl P, Isola ELD. 2001. Trypanosoma cruzi: phosphatidylinositol 3-Kinase and protein kinase B activation is assocated with parasite invasion. Exp. Cell Res. 264:211–218. 10.1006/excr.2000.5123 [DOI] [PubMed] [Google Scholar]
- 25.Liu B, Shi ZL, Feng J, Tao HM. 2008. Celecoxib, a cyclooxygenase-2 inhibitor, induces apoptosis in human osteosarcoma cell line MG-63 via down-regulation of PI3K/Akt. Cell Biol. Int. 32:494–501. 10.1016/j.cellbi.2007.10.008 [DOI] [PubMed] [Google Scholar]
- 26.Uddin S, Ahmed M, Hussain A, Assad L, Dayel F, Bavi P, Al-Kuraya KS, Munkarah A. 2010. Cyclooxygenase-2 inhibition inhibits PI3K/AKT kinase activity in epithelial ovarian cancer. Int. J. Cancer 126:382–394. 10.1002/ijc.24757 [DOI] [PubMed] [Google Scholar]
- 27.Celentano AM, Gorelik G, Solana ME, Sterin-Borda L, Borda E, González Cappa SM. 1995. PGE2 involvement in experimental infection with Trypanosoma cruzi subpopulations. Prostaglandins 49:141–153 [DOI] [PubMed] [Google Scholar]
- 28.Pinge-Filho P, Tadokoro CE, Abrahamsohn IA. 1999. Prostaglandins mediate suppression of lymphocyte proliferation and cytokine synthesis in acute Trypanosoma cruzi infection. Cell. Immunol. 193:90–98. 10.1006/cimm.1999.1463 [DOI] [PubMed] [Google Scholar]
- 29.Hideko Tatakihara VL, Cecchini R, Borges CL, Malvezi AD, Graça-de-Souza VK, Yamada-Ogatta SF, Rizzo LV, Pinge-Filho P. 2008. Effects of cyclooxygenase inhibitors on parasite burden, anemia and oxidative stress in murine Trypanosoma cruzi infection. FEMS Immunol. Med. Microbiol. 52:47–58. 10.1111/j.1574-695X.2007.00340.x [DOI] [PubMed] [Google Scholar]
- 30.Sterin-Borda L, Gorelik G, Goren N, Cappa SG, Celentano AM, Borda E. 1996. Lymphocyte muscarinic cholinergic activity and PGE2 involvement in experimental Trypanosoma cruzi infection. Clin. Immunol. Immunopathol. 81:122–128. 10.1006/clin.1996.0167 [DOI] [PubMed] [Google Scholar]
- 31.Mukherjee S, Machado FS, Huang H, Oz HS, Jelicks LA, Prado CM, Koba W, Fine EJ, Zhao D, Factor SM, Collado E, Weiss LM, Tanowitz HB, Ashto AW. 2011. Aspirin treatment of mice infected with Trypanosoma cruzi and implications for the pathogenesis of Chagas disease. PLoS One 6:e16959. 10.1371/journal.pone.0016959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abdalla GK, Faria GE, Silva KT, Castro ECC, Reis MA, Michelin MA. 2008. Trypanosoma cruzi: the role of PGE2 in immune response during the acute phase of experimental infection. Exp. Parasitol. 118:514–521. 10.1016/j.exppara.2007.11.003 [DOI] [PubMed] [Google Scholar]
- 33.Freire-de-Lima CG, Nascimento DO, Soares MBP, Bozza PT, Castro-Faria-Neto HC, de Mello FG, Dos Reis GA, Lopes MF. 2000. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature 403:199–203. 10.1038/35003208 [DOI] [PubMed] [Google Scholar]
- 34.Michelin MA, Silva JS, Cunha FQ. 2005. Inducible cyclooxygenase released prostaglandin mediates immunosuppression in acute phase of experimental Trypanosoma cruzi infection. Exp. Parasitol. 111:71–79. 10.1016/j.exppara.2005.05.001 [DOI] [PubMed] [Google Scholar]
- 35.Borges MM, Kloetzel JK, Andrade HF, Jr, Tadokoro CE, Pinge-Filho P, Abrahamsohn IA. 1998. Prostaglandin and nitric oxide regulate TNF-α production during Trypanosoma cruzi infection. Immunol. Lett. 63:1–8. 10.1016/S0165-2478(98)00034-0 [DOI] [PubMed] [Google Scholar]
- 36.López-Muñoz R, Faúndez M, Klein S, Escanilla S, Torres G, Lee-Liu D, Ferreira J, Kemmerling U, Orellana M, Morello A, Ferreira A, Maya JD. 2010. Trypanosoma cruzi: in vitro effect of aspirin with nifurtimox and benznidazole. Exp. Parasitol. 124:167–171. 10.1016/j.exppara.2009.09.005 [DOI] [PubMed] [Google Scholar]
- 37.Molina-Berríos A, Campos-Estrada C, Henriquez N, Faúndez M, Torres G, Castillo C, Escanilla S, Kemmerling U, Morello A, López-Muñoz RA, Maya JD. 2013. Protective role of acetylsalicylic acid in experimental Trypanosoma cruzi infection: evidence of a 15-epi-lipoxin A-mediated effect. PLoS Negl. Trop. Dis. 7:e2173. 10.1371/journal.pntd.0002173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kierszenbaum F, Wirth JJ, McCann PP, Sjoerdsma A. 1987. Impairment of macrophage function by inhibitors of ornithine decarboxylase activity. Infect. Immun. 55:2461–2464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moon HG, Kim YS, Choi JP, Choi DS, Yoon CM, Jeon SG, Gho YS, Kim YK. 2010. Aspirin attenuates the anti-inflammatory effects of theophylline via inhibition of cAMP production in mice with non-eosinophilic asthma. Exp. Mol. Med. 42:47–60. 10.3858/emm.2010.42.1.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monick MM, Robeff PK, Butler NS, Flaherty DM, Carter AB, Peterson MW, Hunninghake GW. 2002. Phosphatidylinositol 3-kinase activity negatively regulates stability of cyclooxygenase 2 mRNA. J. Biol. Chem. 277:32992–3000. 10.1074/jbc.M203218200 [DOI] [PubMed] [Google Scholar]
- 41.Araújo-Jorge TC, Waghabi MC, Soeiro Mde N, Keramidas M, Bailly S, Feige JJ. 2008. Pivotal role for TGF-β in infections heart disease: the case of Trypanosoma cruzi infection and consequent Chagasic myocardiopathy. Cytokine Growth Factor Rev. 19:405–413. 10.1016/j.cytogfr.2008.08.002 [DOI] [PubMed] [Google Scholar]
- 42.Gazzinelli RT, Oswald IP, Hieny S, James SL, Sher A. 1992. The microbicidal activity of interferon-gamma-treated macrophages against Trypanosoma cruzi involves an l-arginine-dependent, nitrogen oxide-mediated mechanism inhibitable by interleukin-10 and transforming growth factor-beta. Eur. J. Immunol. 22:2501–2506. 10.1002/eji.1830221006 [DOI] [PubMed] [Google Scholar]
- 43.Waghabi MC, Keramidas M, Calvet CM, Meuser M, de Nazaré Soeiro CM, Mendonça-Lima L, Araújo-Jorge TC, Feige JJ, Bailly S. 2007. SB-431542, a transforming growth factor beta inhibitor, impairs Trypanosoma cruzi infection in cardiomyocytes and parasite cycle completion. Antimicrob. Agents Chemother. 51:2905–2910. 10.1128/AAC.00022-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lima-Junior DS, Costa DL, Carregaro V, Cunha LD, Silva ALN, Mineo TWP, Gutierrez FRS, Bellio M, Bortoluci K, Flavell RA, Bozza MT, Silva JS, Zamboni D. 2013. Inflammasome-derived IL-1β production induces nitric oxide-mediated resistance to Leishmania. Nat. Med. 19:909–915. 10.1038/nm.3221 [DOI] [PubMed] [Google Scholar]
- 45.Silva GK, Costa RS, Silveira TN, Caetano BC, Horta CV, Gutierrez FPS, PMda Matta Andrade WA, Niz M, Gazzinelli RT, Zamboni DS, Silva JS. 2013. Apoptosis-associated speck–like protein containing a caspase recruitment domain inflammasomes mediate IL1-β response and host resistance to Trypanosoma cruzi infection. J. Immunol. 191:3373–3383. 10.4049/jimmunol.1203293 [DOI] [PubMed] [Google Scholar]