

Abstract
Novel synthetic endoperoxides are being evaluated as new components of artemisinin combination therapies (ACTs) to treat artemisinin-resistant Plasmodium falciparum malaria. We conducted blinded ex vivo activity testing of fully synthetic (OZ78 and OZ277) and semisynthetic (artemisone, artemiside, artesunate, and dihydroartemisinin) endoperoxides in the histidine-rich protein 2 enzyme-linked immunosorbent assay against 200 P. falciparum isolates from areas of artemisinin-resistant malaria in western and northern Cambodia in 2009 and 2010. The order of potency and geometric mean (GM) 50% inhibitory concentrations (IC50s) were as follows: artemisone (2.40 nM) > artesunate (8.49 nM) > dihydroartemisinin (11.26 nM) > artemiside (15.28 nM) > OZ277 (31.25 nM) > OZ78 (755.27 nM). Ex vivo activities of test endoperoxides positively correlated with dihydroartemisinin and artesunate. The isolates were over 2-fold less susceptible to dihydroartemisinin than the artemisinin-sensitive P. falciparum W2 clone and showed sensitivity comparable to those with test endoperoxides and artesunate, with isolate/W2 IC50 susceptibility ratios of <2.0. All isolates had P. falciparum chloroquine resistance transporter mutations, with negative correlations in sensitivity to endoperoxides and chloroquine. The activities of endoperoxides (artesunate, dihydroartemisinin, OZ277, and artemisone) significantly correlated with that of the ACT partner drug, mefloquine. Isolates had mutations associated with clinical resistance to mefloquine, with 35% prevalence of P. falciparum multidrug resistance gene 1 (pfmdr1) amplification and 84.5% occurrence of the pfmdr1 Y184F mutation. GM IC50s for mefloquine, lumefantrine, and endoperoxides (artesunate, dihydroartemisinin, OZ277, OZ78, and artemisone) correlated with pfmdr1 copy number. Given that current ACTs are failing potentially from reduced sensitivity to artemisinins and partner drugs, newly identified mutations associated with artemisinin resistance reported in the literature and pfmdr1 mutations should be examined for their combined contributions to emerging ACT resistance.
INTRODUCTION
New antimalarial drugs are urgently needed to address the major global public health problem of malaria. Despite containment and control measures, malaria caused by Plasmodium falciparum affects hundreds of millions of people and kills nearly 1 million each year (1). In the absence of a vaccine, drugs are a mainstay to treat and prevent malaria. Artemisinin and associated derivatives, isolated from the plant Artemisia annua and discovered through Chinese traditional medicine, are highly effective antimalarials that are distinguished as having the most rapid activity against P. falciparum parasites of all currently applied drugs. The unique endoperoxide bridge of the artemisinin chemical structure (Fig. 1) is believed to be responsible for the observed potent antimalarial activity (2, 3). Despite having a rapid mechanism of action, artemisinin resistance eventually emerged and was first detected after approximately 35 years of monotherapy use in patients from Thailand and Cambodia border regions (4). Through a clinical study of artesunate (AS) monotherapy efficacy in falciparum malaria patients from western Cambodia in 2006 and 2007, we were the first research group to identify clinical evidence of artemisinin resistance in treatment failure cases, manifested as significant delays in the time required to clear parasites from the blood relative to that for cured patients (5, 6). Subsequently, additional reports of prolonged parasite clearance times with artesunate monotherapy emerged at Mae Sot in western Thailand on the border with Myanmar (7, 8) and at Pursat in western Cambodia (9), suggesting that artemisinin resistance was spreading throughout the Greater Mekong region.
FIG 1.
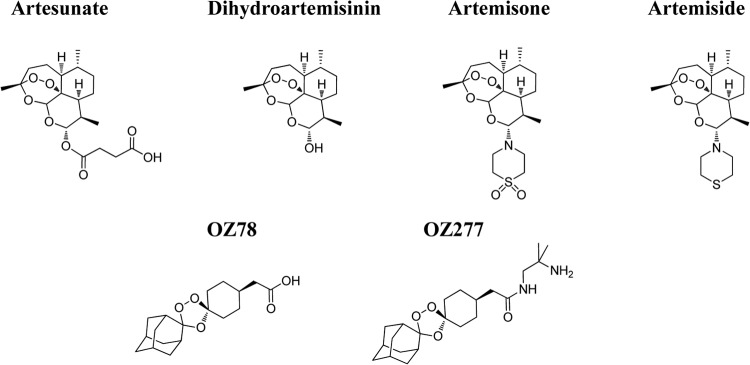
Chemical structures of antimalarial endoperoxides evaluated for activity against P. falciparum isolates in Cambodia.
In response to this emerging public health crisis, national malaria control programs implemented replacement of artemisinin monotherapy with artemisinin-based combination therapies (ACTs) comprising a fast-acting artemisinin-based drug paired with a slower-acting drug of another chemical class with a longer in vivo half-life. However, the current first-line ACTs used in Thailand and Cambodia are failing, as reported for artesunate-mefloquine (MQ) at the Thailand-Myanmar border (10) and dihydroartemisinin (DHA)-piperaquine in western Cambodia (11). Moreover, in a recent trial investigating the efficacy of a 3-day dosing regimen of the current first-line ACT in Cambodia (dihydroartemisinin-piperaquine) in falciparum malaria patients from northern provinces, we observed a dramatic rise in treatment failures, as indicated by the first 50 enrolled volunteers having a higher failure rate than in our study conducted at the same site only 3 years earlier (42-day-per-protocol efficacy of 90% in 2010 versus 64% in 2013) (12, 13). The alarming rise in ACT failures can be attributed to emerging parasite resistance to artemisinins, the partner drug, or both components. Thus, ex vivo drug susceptibility testing of isolates from regions of ACT resistance emergence, such as the Cambodia-Thailand border, can serve as an important drug development tool in evaluating the effectiveness of new candidates in replacing failing regimens.
In the present study, we evaluated a set of blinded test compounds from the Medicines for Malaria Venture (MMV) for ex vivo activity in the histidine-rich protein 2 enzyme-linked immunosorbent assay (HRP-2 ELISA) against 200 fresh P. falciparum clinical isolates from patients in western and northern Cambodia in 2009 and 2010. The blinded test compounds were evaluated for potential cross-susceptibility to 6 standard drugs (dihydroartemisinin [DHA], artesunate [AS], chloroquine [CQ], quinine [QN], mefloquine [MQ], and lumefantrine [LUM]) evaluated in parallel. After data analysis was complete, the test compounds were unblinded as endoperoxide derivatives of two types: (i) fully synthetic ozonide endoperoxides (OZ78 and OZ277) and (ii) semisynthetic endoperoxides (artemisone, artemiside, AS, and DHA). Because P. falciparum multidrug resistance gene 1 (pfmdr1) amplification is associated with cases of treatment failures in patients administered MQ or AS-MQ (14, 15) and there is in vitro evidence of pfmdr1 amplification resulting in artemisinin resistance (16, 17), we investigated correlations between pfmdr1 amplification and 50% inhibitory concentration (IC50) results to assess the possibility of the tested endoperoxides being susceptible to a pfmdr1-driven mechanism of resistance. This study represents the first-ever investigation into the ex vivo activity of synthetic endoperoxides undergoing clinical development as components of new combination therapies (18–20) against isolates from Southeast Asia in a region of known artemisinin resistance.
MATERIALS AND METHODS
Compounds.
Dried drug-coated plates were prepared at the Armed Forces Research Institute of Medical Sciences (AFRIMS) laboratory in Bangkok as previously described (21). Standard malaria drugs included in the routine test panel were provided by the Walter Reed Army Institute of Research (WRAIR) (Silver Spring, MD, USA). Stock solutions (1 mg/ml) were prepared in 70% ethanol (EtOH) for DHA, AS, CQ, MQ, and QN and in a 100% ethanol-linoleic acid-Tween 80 (1:1:1) solution for LUM and were subsequently serially diluted to appropriate working concentrations in sterile distilled water. A range of 3-fold serial drug dilutions was dispensed in duplicate on standard 96-well microculture plates. The top row of each plate was reserved as a drug-free control to monitor growth of parasites in the absence of drug. The plates were dried overnight in a running biosafety cabinet and stored at 4°C until ready for use. Blinded test compounds and instructions for preparing stock and working solutions were provided by the Medicines for Malaria Venture (MMV), Geneva, Switzerland. At the conclusion of the study and after data analysis was complete, MMV identified the blinded compounds as OZ78, OZ277, artemisone, artemiside, AS, and DHA. Stock solutions (10 mg/ml) of test compounds were prepared in dimethyl sulfoxide (DMSO), and working solutions were subsequently made using three stepwise dilutions, i.e., two dilutions in 70% EtOH followed by a dilution in sterile distilled water, prior to applying to plates. This final working solution was used to prepare 10-fold serial dilutions in distilled water on plates to reach final concentrations in the range of 0.02 to 20,000 ng/ml for OZ78 and OZ277, 0.0002 to 200 ng/ml for artemisone, and 0.002 to 2,000 ng/ml for artemiside, AS, and DHA. Plates were dried overnight and stored at 4°C until ready for use. Quality control for dried drug plate integrity was conducted by evaluating the activity of the P. falciparum reference clone W2, which is resistant to CQ but susceptible to MQ and the artemisinins, before and after study conclusion to determine if transport and storage conditions in the field affect assay results (22). DHA was plated in duplicate on each plate containing dried blinded test compounds as a further internal plate quality control.
Clinical study sites and volunteers.
The fresh P. falciparum clinical isolates used to evaluate the ex vivo activities of the blinded test compounds and standard malaria drug panel were obtained from two different human use protocols approved by the WRAIR Institutional Review Board and the Cambodian National Ethics Committee for Health Research (NECHR). Both protocols complied with International Conference on Harmonization Good Clinical Practice (ICH-GCP) guidelines. A portion of the isolates included in this investigation (n = 72) were collected in 2009 from a randomized, open-label comparison of 3 regimens of AS monotherapy administered as a single oral dose of 2, 4, or 6 mg/kg/day for 7 days in otherwise healthy adult patients with acute falciparum malaria under protocol number WRAIR 1396 (23). Isolates from this trial were collected at the Tasanh Health Center, located in Battambang Province in western Cambodia, near the Thailand border and directly south of Pailin. A total of 166 isolates were also collected in 2009 and 2010 through the AFRIMS malaria drug resistance surveillance protocol in Cambodia under protocol number WRAIR 1576 (21), with a study population of individuals ≥13 years old living in areas with relatively high incidence rates of malaria in northern and western Cambodia. Individuals who gave a history of antimalarial drug use within the previous 7 days or who showed symptoms of severe or complicated malaria were excluded from participation.
Ex vivo drug susceptibility assay.
After obtaining informed consent from clinical study volunteers, venous blood was collected in sodium heparin tubes from malaria patients who were confirmed by microscopy to have P. falciparum monoinfection for ex vivo drug sensitivity testing. Within 6 h after phlebotomy, fresh isolates (without culture adaptation) were added to 96-well dried drug-coated plates and evaluated using the HRP-2 ELISA, as previously described (21, 24, 25). Prior to transfer to dried drug-coated plates, samples with a parasitemia of ≤0.5% were adjusted to 1.5% hematocrit in 0.5% Albumax RPMI, whereas those with >0.5% parasitemia were diluted to the plate parasitemia range of 0.2 to 0.5% and adjusted to a 1.5% hematocrit by adding 50% hematocrit human O+ red blood cells in 10% serum–RPMI 1640. Parasites were then incubated for 72 h at 37°C in a candle jar, after which plates were frozen and later thawed for analysis of parasite growth inhibition using the HRP-2 ELISA. ELISA plate reader optical density (OD) values translate to the HRP-2 concentration of the samples and thus serve as indicators of parasite growth in the presence and absence of test compound. Samples were excluded from data analysis if an OD ratio of <1.7 was obtained when comparing the average OD for the no-drug test wells (basal growth control) to the OD for the well containing the maximum tested drug concentration.
Molecular markers of malaria drug resistance.
From each consented study volunteer, blood samples in EDTA were drawn to conduct molecular analyses of malaria drug resistance. P. falciparum DNA was extracted from each clinical sample using the QIAamp DNA blood minikit blood and body fluid spin protocol (Qiagen, Valencia, CA, USA). DNA samples were evaluated for P. falciparum multidrug resistance 1 (pfmdr1) gene copy number using TaqMan real-time PCR (26). Relative quantification was used to determine pfmdr1 copy number calculated according to the formula copy number = (Eβtubulin)CT(βtubulin)/(Epfmdr1)CT(pfmdr1), as previously described (27). Genomic DNA of the P. falciparum 3D7 reference clone, which has a single copy of the pfmdr1 gene, was used as an internal control to calibrate the results. Samples were considered to have an amplified pfmdr1 gene if the copy number was >1.5. DNA samples were also genotyped at pfmdr1 codons 86, 184, 1034, and 1042 for single nucleotide polymorphism (SNP) analysis by real-time PCR on an ABI sequence detector 7000 (Applied Biosystems, Warrington, United Kingdom), as previously described (28). Samples were also probed for an additional pfmdr1 SNP (Y1246D) using a PCR-restriction fragment length polymorphism method (29). The clinical isolates were also screened for the K76T point mutation in the P. falciparum chloroquine resistance transporter gene 1 (pfcrt1) using real-time PCR (30). In addition, haplotyping to detect variants of pfcrt1 alleles 72 to 76 (wild-type CVMNK haplotype and two chloroquine resistance-associated alleles of the pfcrt1 gene, CVIET and SVMNT) was conducted using a multiplex real-time PCR method (31).
Statistical analysis.
HRP-2 OD values were plotted against drug concentrations, and the inhibitory concentrations resulting in 50% growth inhibition (IC50) values were estimated by nonlinear regression analysis using the ICEstimator program version 1.2 (32). Results were considered valid if curve fitting produced a sigmoidal concentration-response curve, with the calculated IC50 having a ratio of the higher limit of the 95% confidential interval (CI) to the lower limit of the 95% CI of <5. Statistical analyses were conducted using GraphPad Prism version 4 (GraphPad Software, Inc., La Jolla, CA, USA). Statistical differences of IC50s between groups were determined by the nonparametric Mann-Whitney U test. Spearman's rank correlation analyses were conducted to examine test compound drug cross-susceptibility patterns in clinical isolates and the influence of pfmdr1 amplification and baseline parasitemia on IC50 results.
RESULTS
Ex vivo drug susceptibilities of P. falciparum isolates from Cambodia.
A set of blinded test compounds and a panel of standard antimalarials were evaluated for activity in the HRP-2 ELISA in a total of 238 patients with P. falciparum monoinfection. Of these cases, IC50s were attained from best-fit sigmoidal concentration-response curves for each test compound for 200 isolates (84% assay success) from provinces of northern (n = 154, Oddar Meanchey and Preah Vihear Provinces) and western (n = 46, Battambang Province) Cambodia along the border with Thailand. Upon study completion after IC50 results were obtained, the test compounds were unblinded as endoperoxide derivatives of two types: (i) the fully synthetic ozonide endoperoxides OZ78 and OZ277 and (ii) the semisynthetic endoperoxides artemisone, artemiside, artesunate, and dihydroartemisinin (Fig. 1). The results suggest that the semisynthetic artemisinin derivatives were more potent against isolates than the synthetic ozonide endoperoxides (Table 1 and Fig. 2). In order of most to least potent, the drugs and their geometric mean (GM) IC50s were as follows: artemisone (2.40 nM) > artesunate (8.49 nM) > dihydroartemisinin (11.26 nM) > artemiside (15.28 nM) > OZ277 (31.25 nM) > OZ78 (755.27 nM).
TABLE 1.
Comparison of IC50 values of antimalarials against P. falciparum isolates and the W2 reference clone
| Test plate compounds | GM IC50, nM |
Isolate/W2 GM IC50 ratio | P valuec | |
|---|---|---|---|---|
| Isolates (range)a | W2 (95% CI)b | |||
| Blinded test compounds | ||||
| Artemisone | 2.4 (0.02–117.7) | 1.9 (1.5–2.4) | 1.3 | 0.004 |
| DHA internal control | 8.0 (1.5–38.1) | 5.0 (4.2–5.9) | 1.6 | <0.001 |
| AS | 8.5 (0.5–52.7) | 6.4 (5.0–8.1) | 1.3 | 0.019 |
| DHA | 11.2 (0.04–109.4) | 5.2 (3.8–7.1) | 2.2 | <0.001 |
| Artemiside | 15.3 (0.8–458.7) | 11.2 (8.7–14.3) | 1.4 | 0.022 |
| OZ277 | 31.3 (0.02–320.7) | 38.5 (30.2–49.1) | 0.8 | 0.726 |
| OZ78 | 755.7 (65.7–7,854) | 406.7 (319.8–517.2) | 1.9 | 0.003 |
| Standard drug panel | ||||
| AS | 5.8 (0.6–24.6) | 4.7 (4.4–5.1) | 1.2 | <0.001 |
| LUM | 7.4 (0.4–40.1) | 6.0 (4.9–7.3) | 1.2 | 0.002 |
| DHA | 9.0 (0.8–30.2) | 4.5 (4.1–4.9) | 2.0 | <0.001 |
| MQ | 58.3 (0.7–205.5) | 20.6 (18.2–23.2) | 2.8 | <0.001 |
| QN | 146.1 (2.2–439.2) | 149.0 (135.9–163.4) | 1.0 | 0.878 |
| CQ | 147.8 (4.7–786.0) | 258.2 (237.7–280.5) | 0.6 | <0.001 |
GM IC50s were determined for a total of 200 isolates.
GM IC50s against W2 in the blinded test compound panel were determined from n = 51, 52, 52, 48, 52, 51, and 48 independent experiments for artemisone, DHA internal control, AS, artemiside, OZ277, and OZ78, respectively. GM IC50s in the standard drug panel against W2 were determined from 123 independent experiments.
P values (Mann-Whitney U test) in bold indicate a significant difference between IC50s for each evaluated antimalarial against isolates and W2.
FIG 2.

Associations between IC50s and pfmdr1 amplification in P. falciparum clinical isolates. Ex vivo drug susceptibility (GM IC50s) for blinded endoperoxides and standard partner drugs against isolates (solid bars) with single (1 copy) and multiple (>1.5 copies) pfmdr1 copies were compared using the Mann-Whitney U test, with significant P values presented. Dashed lines indicate GM IC50s against the P. falciparum W2 clone. The solid line across the MQ, QN, and CQ results represents WHO drug resistance threshold values associated with clinical resistance to these drugs.
These fresh isolates were also evaluated for activity against a standard panel of six antimalarial drugs, i.e., dihydroartemisinin (DHA), artesunate (AS), chloroquine (CQ), mefloquine (MQ), quinine (QN), and lumefantrine (LUM), to characterize the drug susceptibility profile for this parasite population. The antimalarial susceptibilities of these isolates were compared to World Health Organization (WHO) threshold IC50s established for in vitro drug sensitivity assays using P. falciparum isolates that signify an association with clinical resistance: MQ (24 nM), CQ (85 nM), and QN (351 nM) (33, 34). The majority of Cambodian isolates evaluated appeared to be resistant to MQ and CQ, with GM IC50s of 58.27 nM and 147.8 nM, respectively, exceeding the WHO drug resistance thresholds. Given the lack of an established IC50 threshold for artemisinin or LUM resistance, the relative susceptibilities of the isolates against DHA, AS, and LUM were compared with IC50s against the P. falciparum W2 clone, which is of Southeast Asian origin and has been benchmarked thoroughly in our HRP-2 assay as a comparator reference line resistant to CQ but sensitive to MQ, LUM, and artemisinins (22). The isolates appeared to have reduced susceptibilities to DHA, AS, and LUM, as demonstrated by statistically significant higher GM IC50s versus the W2 clone results produced using the same dried drug plates (Table 1). Each plate with blinded test compounds included DHA as an internal control for dried drug plate integrity during the entire field study duration by evaluating activity against the P. falciparum W2 reference clone. This quality control check did not reveal any test plate integrity concerns, because internal control DHA yielded IC50 results within the acceptable standardized range for the W2 clone (0.8 to 7.7 nM) on all test plates (Table 1).
Comparative susceptibilities of clinical isolates and the P. falciparum W2 reference clone against endoperoxides and standard drugs.
The IC50s attained for test compounds and standard drugs against fresh P. falciparum isolates were compared to IC50 results against the P. falciparum W2 laboratory clone (Table 1). Nearly every test compound and drug evaluated, with the exception of OZ277, CQ, and QN, produced statistically significantly higher IC50s against isolates than W2. A susceptibility factor, calculated as the ratio of GM IC50s against isolates versus the W2 reference clone, was determined for each compound to evaluate the relative potential for reduced susceptibility against clinical isolates. Of all the compounds tested, MQ had the greatest IC50 ratio, 2.8, for activity against isolates versus W2, suggesting that the isolates were nearly 3 times less susceptible to MQ than the MQ-sensitive W2 clone. The isolates appeared to have similar CQ and QN GM IC50s, with slightly higher susceptibility to CQ but comparable sensitivity to QN compared to the CQ-resistant W2 clone. Susceptibility ratios were near 1.0 for artemisone, artemiside, AS, and OZ277, suggesting that the isolates and the artemisinin-sensitive W2 clone had comparable susceptibilities to each of these artemisinin derivatives. The isolates appeared to be approximately two times less susceptible to DHA and OZ78 than W2.
Cross-susceptibility profiling of endoperoxides against clinical isolates.
Spearman correlation analysis was used to evaluate drug cross-susceptibility patterns of the endoperoxide test compounds (Table 2). The activity of OZ78 was most strongly correlated with artemiside and AS, whereas a significant negative correlation in activity was noted for CQ. Relatively strong correlations in activity seemed to exist between OZ277 and artemisone, AS, and DHA. Artemisone and artemiside activities against isolates correlated significantly with each other and also with those of AS and DHA. The activities of MQ, QN, and LUM against isolates appeared to be weakly correlated with OZ277 and artemisone activities.
TABLE 2.
Correlation coefficients for antimalarial susceptibilities against P. falciparum clinical isolatesa
| Correlation | Coefficient | P value |
|---|---|---|
| OZ78 with: | ||
| Parasitemia | −0.065 | 0.365 |
| OZ277 | 0.077 | 0.276 |
| Artemisone | −0.010 | 0.891 |
| Artemiside | 0.257 | 0.0002 |
| Artesunate | 0.341 | <0.0001 |
| DHA | 0.191 | 0.007 |
| DHA internal control | 0.160 | 0.023 |
| Drug panel | ||
| DHA | 0.036 | 0.613 |
| Artesunate | 0.126 | 0.076 |
| Mefloquine | 0.114 | 0.109 |
| Quinine | −0.012 | 0.862 |
| Chloroquine | −0.270 | 0.0001 |
| Lumefantrine | 0.109 | 0.124 |
| OZ277 with: | ||
| Parasitemia | 0.217 | 0.002 |
| Artemisone | 0.566 | <0.0001 |
| Artemiside | 0.072 | 0.309 |
| Artesunate | 0.110 | 0.120 |
| DHA | 0.202 | 0.004 |
| DHA internal control | 0.253 | 0.0003 |
| Drug panel | ||
| DHA | 0.305 | <0.0001 |
| Artesunate | 0.320 | <0.0001 |
| Mefloquine | 0.172 | 0.015 |
| Quinine | 0.183 | 0.009 |
| Chloroquine | −0.038 | 0.592 |
| Lumefantrine | 0.215 | 0.002 |
| Artemisone with: | ||
| Parasitemia | 0.284 | <0.0001 |
| Artemiside | 0.180 | 0.011 |
| Artesunate | 0.324 | <0.0001 |
| DHA | 0.324 | <0.0001 |
| DHA internal control | 0.443 | <0.0001 |
| Drug panel | 0.475 | <0.0001 |
| DHA | ||
| Artesunate | 0.508 | <0.0001 |
| Mefloquine | 0.179 | 0.011 |
| Quinine | 0.157 | 0.027 |
| Chloroquine | −0.061 | 0.389 |
| Lumefantrine | 0.142 | 0.044 |
| Artemiside with: | ||
| Parasitemia | 0.2392 | 0.0007 |
| Artesunate | 0.4200 | <0.0001 |
| DHA | 0.3612 | <0.0001 |
| DHA internal control | 0.4685 | <0.0001 |
| Drug panel | ||
| DHA | 0.320 | <0.0001 |
| Artesunate | 0.425 | <0.0001 |
| Mefloquine | 0.086 | 0.226 |
| Quinine | 0.035 | 0.626 |
| Chloroquine | 0.042 | 0.553 |
| Lumefantrine | 0.069 | 0.335 |
| pfmdr1 copy no. with: | ||
| OZ78 | 0.238 | 0.001 |
| OZ277 | 0.265 | 0.0003 |
| Artemisone | 0.202 | 0.006 |
| Artemiside | −0.018 | 0.806 |
| Artesunate | 0.133 | 0.073 |
| DHA | 0.127 | 0.088 |
| DHA internal control | 0.136 | 0.067 |
| Drug panel | ||
| DHA | 0.186 | 0.012 |
| Artesunate | 0.241 | 0.001 |
| Mefloquine | 0.413 | <0.0001 |
| Quinine | 0.108 | 0.145 |
| Chloroquine | −0.083 | 0.263 |
| Lumefantrine | 0.373 | <0.0001 |
Spearman rank correlation was used to determine correlation coefficients and P values, with significant correlations indicated in bold.
Correlation analysis of baseline parasitemia as influencing IC50 results against isolates.
The potential effect of baseline percent parasitemia of isolates added to the dried drug plates on IC50s for all test compounds and standard drugs evaluated in the HRP-2 ELISA was examined using Spearman correlation analysis (Table 2). The range of baseline parasitemia added to the dried drug-coated plates was 0.001 to 2.2%, with a GM value of 0.19% parasitemia. The results of Spearman correlation analysis suggest that baseline parasitemia was weakly positively correlated with IC50 results for OZ277, artemisone, artemiside, artesunate, and DHA in the HRP-2 ELISA testing of fresh isolates (Table 2). No significant correlations were found between baseline parasitemia and IC50s for MQ, QN, or CQ (data not shown), whereas a significant negative correlation was determined for LUM testing of isolates (r = −0.2036; P = 0.0039). However, the relatively low Spearman coefficient values attained in this analysis suggest that baseline parasitemia of clinical isolates in the HRP-2 assay was a potential weak confounder of IC50 results for all drugs tested.
Molecular characterization of P. falciparum clinical isolates.
The isolates were investigated for pfmdr1 gene amplification and SNPs as general molecular markers of multidrug resistance. A GM of 1.26 pfmdr1 copies was attained from a total of 182 samples successfully evaluated for copy number. The majority of isolates had a single pfmdr1 copy (64.8%), whereas the remainder had 2 (25.8%), 3 (7.1%), and 4 (2.2%) copies. The results of pfmdr1 SNP analysis for 5 different mutations, N86Y, Y184F, S1034C, N1042D, and Y1246D, revealed that 84.5% of samples (n = 192) contained the Y184F mutation, whereas mutations were not detected at the other pfmdr1 positions evaluated.
Isolates were also analyzed for pfcrt1 mutations associated with clinical resistance to CQ. All samples (n = 197 isolates with successful results) had the K76T point mutation and were of the CVIET pfcrt1 mutant haplotype, strongly suggesting fixation of CQ resistance in this population.
Correlation analysis of endoperoxide activity against isolates and pfmdr1 amplification.
The P. falciparum isolates were also evaluated for associations between pfmdr1 copies (single versus multiple) and GM IC50s for each of the tested endoperoxides and standard antimalarials (Fig. 2). Isolates with multiple pfmdr1 copies had significantly higher IC50s against OZ78, OZ277, MQ, and LUM. In contrast, no significant differences in IC50s between isolates with single and multiple pfmdr1 copy numbers were observed for the other test compounds. Spearman rank correlation analysis suggests weak correlation between absolute pfmdr1 copy number and GM IC50s for OZ78, OZ277, and artemisone (Table 2) and slightly stronger associations for MQ and LUM (r = 0.413 [P < 0.0001] and r = 0.373 [P < 0.0001], respectively). The presence of the pfmdr1 184F mutation in isolates was not correlated with activities for any of the evaluated endoperoxides or standard drugs (results not shown).
Activity of compounds in a case of artesunate monotherapy treatment failure.
The isolate samples in this study included one case of artesunate monotherapy treatment failure. This patient recrudesced on study day 35 after oral administration of 2 mg/kg AS for 7 consecutive days, the lowest dose evaluated in our previous trial (23). In this case of artesunate treatment failure, DHA and AS IC50s on day zero (prior to AS administration) and the day of recrudescence, respectively, were as follows: blinded DHA, 109.4 and 36 nM; blinded AS, 28 and 6.35 nM; DHA internal control, 36.9 and 12.8 nM; DHA in standard drug panel, 27 and 27 nM; and AS in standard drug panel, 20.6 and 24.6 nM. These results suggest that this patient was infected with artemisinin-resistant parasites, since DHA and AS IC50s at day zero and on the day of treatment failure were elevated relative to GM IC50s determined for the isolates in total and for the P. falciparum W2 clone (Table 1). In addition, the IC50 results for DHA and AS in this artesunate treatment failure were among the highest values observed for these drugs for all 200 isolates included in this study. In particular, GM IC50s (interquartile ranges) for DHA and AS in the standard drug panel for all isolates were 9.0 (6.5 to 13.4) and 5.8 (4.0 to 8.4) nM, respectively, an order of magnitude lower than those observed in the AS failure case. When comparing results in this AS treatment failure case at time zero with the day of recrudescence, IC50s increased for OZ78 (137.7 to 4202 nM), artemisone (4.63 to 30.81 nM), artemiside (50.42 to 120.1 nM), MQ (56.61 to 138.82 nM), QN (354.8 to 388.1 nM), CQ (292.2 to 380.7 nM), and LUM (14.1 to 40 nM). In contrast, the IC50 of OZ277 on day 0 (175.3 nM) did not increase on the day of recrudescence (133.2 nM). The time zero sample had a single pfmdr1 copy number and was positive for the pfmdr1 184F mutation. The recurrent sample also contained the 184F mutation, but no comparative pfmdr1 copy number results are available.
DISCUSSION
This study is the first reported investigation into the ex vivo activity of novel synthetic endoperoxide antimalarials against clinical isolates at the Cambodia-Thailand border region, where artemisinin resistance first emerged and continues to pose a global public health crisis in treating and eliminating malaria. The isolates used in this study were collected within 3 years after initial discoveries of artemisinin resistance in clinical studies of artesunate monotherapy (5, 6, 35), with a portion of the isolates derived from an AS monotherapy trial conducted in 2009 at the Tasanh Health Center in Battambang Province (23) and the remaining isolates obtained in 2009 and 2010 during a malaria drug resistance surveillance study performed in northern (Oddar Meanchey and Preah Vihear Provinces) and western (Battambang Province) Cambodia (21). The ex vivo susceptibility results suggest that these isolates had reduced sensitivity to DHA and AS, as demonstrated by significantly higher GM IC50s against isolates relative to the artemisinin-sensitive P. falciparum W2 clone. Included in this investigation were two endoperoxide antimalarials, artemisone and OZ277, undergoing clinical development. Of all the evaluated endoperoxides, including AS and DHA, the most potent compound against isolates (GM IC50 of 2.4 nM) was artemisone, a semisynthetic endoperoxide with encouraging phase IIa clinical trial results indicative of safety and pharmacological profiles attractive as a component of a new ACT to treat P. falciparum malaria (18). Also included in the test panel was the fully synthetic 1,2,4-trioxolane OZ277, a drug known as arterolane maleate (RBx 11160) (19, 36–38), that is currently employed in India as an ACT in combination with piperaquine (39). A next-generation ozonide, OZ439, has a more desirable pharmacokinetic profile with slower elimination (20) and is undergoing clinical development as a candidate component of a new ACT for use in malarious areas, including the Greater Mekong subregion. None of the evaluated test endoperoxides (artemisone, artemiside, OZ277, and OZ78) or AS had GM IC50s against isolates that differed more than 2-fold from results attained against W2, while the comparator blinded DHA showed a 2.2-fold difference in activity, suggesting that the test endoperoxides performed at a level comparable to that of AS in a population of isolates with reduced artemisinin susceptibility.
We have proven the utility of using the HRP-2 ex vivo drug susceptibility assay in conducting drug resistance surveillance and correlating elevated IC50s for artemisinins with artesunate monotherapy failures and prolonged parasite clearance time. We recently demonstrated that the HRP-2 ELISA is more sensitive than the malaria SYBR green I fluorescence assay in evaluating the activity of antimalarials against fresh P. falciparum clinical samples of relatively low parasitemia (<0.2%), as typically encountered in patients from Cambodia and Thailand, where malaria transmission rates are relatively low (24). Using the HRP-2 screen in our drug resistance surveillance efforts, we found that clinical isolates collected from western Cambodia during 2006 to 2010 showed a progressive decline in susceptibilities to all tested drugs, including the artemisinins (21). In the present investigation, DHA and AS IC50s in a case of AS monotherapy failure were among the highest observed values for these drugs against all isolates included in the study and were elevated relative to GM IC50s against the artemisinin-susceptible W2 clone. Moreover, we previously reported elevated DHA IC50s (14.2 nM) on day 0 (pretreatment) in two cases in western Cambodia of AS monotherapy failure in patients with prolonged parasite clearance times (PCTs) of 95 and 133 h, compared to cured cases with a lower GM DHA IC50 of 3.5 nM and a shorter median PCT of 52.2 h (6). These two failed patients had DHA IC50s that were nearly 10 times greater than those attained against W2. In another AS monotherapy trial in Cambodia, we found significantly higher median DHA IC50s for patients remaining parasite positive at 72 h (9.60 nM) than the median IC50 of 6.26 nM for patients clearing parasites within 72 h (23). We also reported previously that patients from Cambodia administered AS monotherapy with a 100% PCT of >72 h had a DHA median IC50 (9.6 nM) that was significantly greater than that observed in patients with a 100% PCT of ≤72 h (6.3 nM), while a median DHA IC50 of 3.9 nM was found for the W2 clone (22). Taken together, these findings suggest that elevated IC50s, relative to those for the W2 control clone, attained in the HRP-2 assay can serve as indicators of artemisinin-resistant isolates.
The results of our investigation also provide insight into the potential for endoperoxide antimalarials to show cross-resistance to MQ and other drugs susceptible to pfmdr1-mediated mechanisms of resistance. pfmdr1 amplification is a well-established molecular marker to detect and track MQ resistance and is associated with MQ treatment failure (14, 40), and in our study population, approximately 35% of the isolates had multiple pfmdr1 copies. The isolates had a relatively high prevalence (84.5%) of the pfmdr1 Y184F mutation, which appears to be selected for and spread by MQ drug pressure in western Cambodia (41) and has been reported to be associated with decreased susceptibility to AS, MQ, QN, and CQ in other recent drug resistance surveillance work conducted in Cambodia (42, 43). Our results suggest significant correlations in ex vivo activity against isolates between MQ and some evaluated endoperoxides (OZ277 and artemisone) as well as AS and DHA (data not shown). Moreover, isolates with multiple pfmdr1 copies had significantly reduced susceptibility to MQ, LUM, and the ozonide endoperoxides relative to single-copy isolates. Our findings are consistent with another recent ex vivo susceptibility study, which also documents an association between pfmdr1 amplification and elevated GM IC50s for MQ, AS, and DHA in isolates from western and northern Cambodia (44). Although our Spearman correlation analysis suggests a weaker correlation between pfmdr1 copy number and GM IC50s for the endoperoxides (AS, DHA, OZ277, OZ78, and artemisone) than for the partner drugs MQ and LUM, further investigation is warranted to investigate potential involvement of pfmdr1amplification in the development of resistance to endoperoxide antimalarials, especially given the extensive use of AS-MQ as first-line ACT in Thailand and Cambodia (4).
While pfmdr1 amplification is a hallmark of clinical resistance to MQ, the role of pfmdr1 mutations in selecting for artemisinin resistance is less understood. Extensive in vitro evidence suggests a role for pfmdr1 amplification in artemisinin resistance, such as indicated by a genetically modified P. falciparum clone expressing low levels of PFMDR1 being 2-fold more susceptible to artemisinins (45) and artemisinin pressure in vitro resulting in pfmdr1 amplification in laboratory strains (16, 17). However, consistent evidence suggesting a role for pfmdr1 amplification in conferring clinical resistance to the artemisinins is less clear than what is observed in vitro. We previously developed a linear regression model correlating the in vivo therapeutic response (fever and parasite clearance time) observed with artemisinin derivatives administered to Thai patients and in vitro sensitivity data, suggesting that in vitro cross-susceptibility of P. falciparum against MQ and artemisinins appears to have clinical relevance (46). We reasoned that this finding could be linked to a pfmdr1-driven mechanism of resistance, since the artemisinins did not show in vitro cross-sensitivity with CQ, a drug associated with a pfmdr1 deamplification resistance mechanism (47). This notion is further supported by our present study, in which the evaluated endoperoxides did not show cross-susceptibility with CQ but instead showed negative correlations between ex vivo activity for CQ and endoperoxides in isolates all shown to have molecular markers of CQ resistance. Moreover, our data suggest that isolates with multiple pfmdr1 copies are more susceptible to CQ, with a negative correlation found between CQ activity and pfmdr1 copy number, further suggesting a linkage between pfmdr1 amplification and enhanced CQ sensitivity. Although in vitro evidence is suggestive of pfmdr1 playing a role in conferring artemisinin resistance, studies aimed at correlating pfmdr1 molecular markers of resistance with artemisinin treatment outcomes in Cambodian and Thai patients administered AS monotherapy or AS-MQ did not reveal significant associations between delayed parasite clearance time and occurrence of pfmdr1 amplification or SNPs (35, 48). Recently, genome-wide association studies revealed PF3D7_1343700 kelch propeller domain (K13-propeller) mutant alleles that appear to be associated with prolonged in vivo parasite clearance half-lives in Cambodian patients treated with ACTs (49). Given that parasite populations are exposed extensively to artemisinins and partner drugs such as MQ in regions where malaria is endemic, further studies should examine the potential interplay between mutations in pfmdr1 and K13-propeller alleles to understand the complex mechanisms of resistance emergence and also to further define haplotypes associated with endoperoxide antimalarial resistance.
The results reported here for activities of the endoperoxide derivatives against Cambodian isolates can be compared with findings from previous studies in isolates from other countries where malaria is endemic. However, none of these other investigations evaluated the endoperoxides against artemisinin-resistant isolates, as we report, in Cambodia. For example, in an earlier study (50), OZ277 was reported to be active in the [3H]hypoxanthine incorporation assay against P. falciparum isolates from Tanzania and Colombia collected in 2004 and 2005 that were shown to be CQ resistant but susceptible to AS and MQ. In a more recent study, OZ277 and other endoperoxides were evaluated for ex vivo activity against isolates from Papua Indonesia (51). Although this investigation used the microscopy-based WHO schizont maturation assay, trends in IC50s (OZ277 > artemiside > artemisone) similar to those we found using the HRP-2 ELISA against isolates from Cambodia were observed. Comparable to our study, Marfurt et al. (51) used P. falciparum reference clones, including the Thai origin K1 clone, which has an artemisinin-sensitive and chloroquine-resistant profile like that of the W2 clone, as a quality control for dried drug plate integrity in field conditions and also for interpreting results from isolates. The IC50 results for OZ277, artemisone, and artemiside reported against K1 (20, 2.0, and 4.3 nM, respectively) are similar to our findings against W2 (38.5, 1.9, and 11.2 nM, respectively). However, the Papua Indonesia isolates had lower median IC50s against DHA and AS than the tested P. falciparum reference clones, suggesting that parasites from this region are not artemisinin resistant like the Cambodian isolates evaluated in our study.
In addition to traditional IC50 drug susceptibility assays, since the time of conducting our study, a new phenotypic microscopy-based assay referred to as the ring-stage survival assay (RSA) was developed to further identify infections with reduced susceptibility to the artemisinins (52, 53). Through the RSA, the survival rate of isolates is determined by comparing growth rates in the presence of a pharmacologically relevant DHA exposure (700 nM for 6 h) relative to those of the same isolates grown in parallel in the absence of drug. Ex vivo RSA survival rates were reported to be significantly correlated with in vivo parasite clearance half-lives from 30 falciparum malaria patients from Cambodia treated with DHA-piperaquine, suggesting that RSA results can be useful in detecting clinical cases of ACT failure associated with artemisinin resistance (53). Use of the RSA in conjunction with standard IC50 drug susceptibility assays, such as the HRP-2 ELISA, to investigate sensitivity against artemisinins and ACT partner drugs can prove useful in determining whether ACT clinical failure resulted from an infection with parasites resistant to artemisinins, the partner drug, or both components.
In summary, the results of this investigation, using the W2 clone as an artemisinin-sensitive comparator, provide evidence that next-generation synthetic endoperoxide antimalarials show activity comparable to that of artesunate against isolates with reduced artemisinin sensitivity from western and northern Cambodia. Ex vivo activity, however, is only one of many attributes important to selecting the most promising drug candidates to advance for further development. Clinical investigations evaluating the efficacy of new ACTs should apply a thorough approach of evaluating the interplay among ex vivo drug susceptibility, molecular markers of resistance, parasite clearance half-life, pharmacokinetics, and safety to help guide drug treatment policy and new ACT development decisions to combat multidrug-resistant malaria.
ACKNOWLEDGMENTS
This study was funded by the Medicines for Malaria Venture (MMV) and the Global Emerging Infections Surveillance (GEIS) Program, U.S. Department of Defense.
We are most grateful to Jonathan Vennerstrom, College of Pharmacy, University of Nebraska Medical Center, for providing OZ78 and OZ277 and to Richard Haynes, Department of Chemistry, Hong Kong University of Science and Technology, for providing artemisone and artemiside as blinded test compounds for use in this study and for reviewing the manuscript. We also thank members of Chris Plowe's laboratory at the University of Maryland for analyzing and providing results from pfmdr1 analysis for samples from the WRAIR 1396 clinical trial. We acknowledge the AFRIMS and Cambodian clinical and laboratory field teams for sample collection, microscopy, and other technical support.
The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the U.S. Department of the Army.
Footnotes
Published ahead of print 21 July 2014
REFERENCES
- 1.World Health Organization. 2012. World malaria report 2012. World Health Organization, Geneva, Switzerland [Google Scholar]
- 2.Dong Y, Chollet J, Matile H, Charman SA, Chiu FC, Charman WN, Scorneaux B, Urwyler H, Santo Tomas J, Scheurer C, Snyder C, Dorn A, Wang X, Karle JM, Tang Y, Wittlin S, Brun R, Vennerstrom JL. 2005. Spiro and dispiro-1,2,4-trioxolanes as antimalarial peroxides: charting a workable structure-activity relationship using simple prototypes. J. Med. Chem. 48:4953–4961. 10.1021/jm049040u [DOI] [PubMed] [Google Scholar]
- 3.Kaiser M, Wittlin S, Nehrbass-Stuedli A, Dong Y, Wang X, Hemphill A, Matile H, Brun R, Vennerstrom JL. 2007. Peroxide bond-dependent antiplasmodial specificity of artemisinin and OZ277 (RBx11160). Antimicrob. Agents Chemother. 51:2991–2993. 10.1128/AAC.00225-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wongsrichanalai C, Meshnick SR. 2008. Declining artesunate-mefloquine efficacy against falciparum malaria on the Cambodia-Thailand border. Emerg. Infect. Dis. 14:716–719. 10.3201/eid1405.071601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noedl H, Se Y, Schaecher K, Socheat D, Fukuda MM. 2008. Evidence of artemisinin-resistant malaria in western Cambodia. N. Engl. J. Med. 359:2619–2620. 10.1056/NEJMc0805011 [DOI] [PubMed] [Google Scholar]
- 6.Noedl H, Se Y, Sriwichai S, Schaecher K, Teja-Isavadharm P, Smith B, Rutvisuttinunt W, Bethell D, Surasri S, Fukuda MM, Socheat D, Lon C. 2010. Artemisinin resistance in Cambodia: a clinical trial designed to address an emerging problem in Southeast Asia. Clin. Infect. Dis. 51:e82–89. 10.1086/657120 [DOI] [PubMed] [Google Scholar]
- 7.Phyo AP, Nkhoma S, Stepniewska K, Ashley EA, Nair S, McGready R, ler Moo C, Al-Saai S, Dondorp AM, Lwin KM, Singhasivanon P, Day NP, White NJ, Anderson TJ, Nosten F. 2012. Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. Lancet 379:1960–1966. 10.1016/S0140-6736(12)60484-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrara VI, Lwin KM, Phyo AP, Ashley E, Wiladphaingern J, Sriprawat K, Rijken M, Boel M, McGready R, Proux S, Chu C, Singhasivanon P, White N, Nosten F. 2013. Malaria burden and artemisinin resistance in the mobile and migrant population on the Thai-Myanmar border, 1999-2011: an observational study. PLoS Med. 10:e1001398. 10.1371/journal.pmed.1001398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amaratunga C, Sreng S, Suon S, Phelps ES, Stepniewska K, Lim P, Zhou C, Mao S, Anderson JM, Lindegardh N, Jiang H, Song J, Su XZ, White NJ, Dondorp AM, Anderson TJ, Fay MP, Mu J, Duong S, Fairhurst RM. 2012. Artemisinin-resistant Plasmodium falciparum in Pursat Province, western Cambodia: a parasite clearance rate study. Lancet Infect. Dis. 12:851–858. 10.1016/S1473-3099(12)70181-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Na-Bangchang K, Muhamad P, Ruaengweerayut R, Chaijaroenkul W, Karbwang J. 2013. Identification of resistance of Plasmodium falciparum to artesunate-mefloquine combination in an area along the Thai-Myanmar border: integration of clinico-parasitological response, systemic drug exposure, and in vitro parasite sensitivity. Malar. J. 12:263. 10.1186/1475-2875-12-263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leang R, Barrette A, Bouth DM, Menard D, Abdur R, Duong S, Ringwald P. 2013. Efficacy of dihydroartemisinin-piperaquine for treatment of uncomplicated Plasmodium falciparum and Plasmodium vivax in Cambodia, 2008 to 2010. Antimicrob. Agents Chemother. 57:818–826. 10.1128/AAC.00686-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lon C, Manning JE, Vanachayangkul P, So M, Sea D, Se Y, Gosi P, Lanteri C, Chaorattanakawee S, Sriwichai S, Chann S, Kuntawunginn W, Buathong N, Nou S, Walsh DS, Tyner SD, Juliano JJ, Lin J, Spring M, Bethell D, Kaewkungwal J, Tang D, Chuor CM, Satharath P, Saunders D. 2014. Efficacy of two versus three-day regimens of dihydroartemisinin-piperaquine for uncomplicated malaria in military personnel in northern Cambodia: an open-label randomized trial. PLoS One 9:e93138. 10.1371/journal.pone.0093138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saunders DL, Vanachayangkul P, Lon C. 2014. Dihydroartemisin-piperaquine failure in Cambodia. N. Engl. J. Med. Lett. 371:484–485. 10.1056/NEJMc1403007 [DOI] [PubMed] [Google Scholar]
- 14.Price RN, Uhlemann AC, Brockman A, McGready R, Ashley E, Phaipun L, Patel R, Laing K, Looareesuwan S, White NJ, Nosten F, Krishna S. 2004. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet 364:438–4347. 10.1016/S0140-6736(04)16767-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Brien C, Henrich PP, Passi N, Fidock DA. 2011. Recent clinical and molecular insights into emerging artemisinin resistance in Plasmodium falciparum. Curr. Opin. Infect. Dis. 24:570–577. 10.1097/QCO.0b013e32834cd3ed [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chavchich M, Gerena L, Peters J, Chen N, Cheng Q, Kyle DE. 2010. Role of pfmdr1 amplification and expression in induction of resistance to artemisinin derivatives in Plasmodium falciparum. Antimicrob. Agents Chemother. 54:2455–2464. 10.1128/AAC.00947-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen N, Chavchich M, Peters JM, Kyle DE, Gatton ML, Cheng Q. 2010. Deamplification of pfmdr1-containing amplicon on chromosome 5 in Plasmodium falciparum is associated with reduced resistance to artelinic acid in vitro. Antimicrob. Agents Chemother. 54:3395–3401. 10.1128/AAC.01421-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagelschmitz J, Barbara V, Wensing G, Roemer A, Fugmann B, Haynes RK, Kotecka BM, Rieckmann KH, Edstein MD. 2008. First assessment in humans of the safety, tolerability, pharmacokinetics, and ex vivo pharmacodynamic antimalarial activity of the new artemisinin derivative artemisone. Antimicrob. Agents Chemother. 52:3085–3091. 10.1128/AAC.01585-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olliaro P, Wells T. 2009. The global portfolio of new antimalarial medicines under development. Clin. Pharmacol. Ther. 85:584–595. 10.1038/clpt.2009.51 [DOI] [PubMed] [Google Scholar]
- 20.Charman SA, Arbe-Barnes S, Bathurst IC, Brun R, Campbell M, Charman WN, Chiu FCK, Chollet J, Craft JC, Creek DJ, Dong Y, Matile H, Maurer M, Morizzi J, Nguyen T, Papastogiannidis P, Scheurer C, Shackleford DM, Sriraghavan K, Stingelin L, Tang Y, Urwyler H, Wang X, White KL, Wittlin S, Zhou L, Vennerstrom JL. 2011. Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc. Natl. Acad. Sci. U. S. A. 108:4400–4405. 10.1073/pnas.1015762108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tyner SD, Lon C, Se Y, Bethell D, Socheat D, Noedl H, Sea D, Satimai W, Schaecher K, Rutvisuttinunt W, Fukuda MM, Chaorattanakawee S, Yingyuen K, Sundrakes S, Chaichana P, Saingam P, Buathong N, Sriwichai S, Chann S, Timmermans A, Saunders DL, Walsh DS. 2012. Ex vivo drug sensitivity profiles of Plasmodium falciparum field isolates from Cambodia and Thailand, 2005 to 2010, determined by a histidine-rich protein-2 assay. Malar. J. 11:198. 10.1186/1475-2875-11-198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rutvisuttinunt W, Chaorattanakawee S, Tyner SD, Teja-isavadharm P, Se Y, Yingyuen K, Chaichana P, Bethell D, Walsh DS, Lon C, Fukuda M, Socheat D, Noedl H, Schaeher K, Saunders DL. 2012. Optimizing the HRP-2 in vitro malaria drug susceptibility assay using a reference clone to improve comparisons of Plasmodium falciparum field isolates. Malar. J. 11:325. 10.1186/1475-2875-11-325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bethell D, Se Y, Lon C, Tyner S, Saunders D, Sriwichai S, Darapiseth S, Teja-Isavadharm P, Khemawoot P, Schaecher K, Ruttvisutinunt W, Lin J, Kuntawungin W, Gosi P, Timmermans A, Smith B, Socheat D, Fukuda MM. 2011. Artesunate dose escalation for the treatment of uncomplicated malaria in a region of reported artemisinin resistance: a randomized clinical trial. PLoS One 6:e19283. 10.1371/journal.pone.0019283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaorattanakawee S, Tyner SD, Lon C, Yingyuen K, Ruttvisutinunt W, Sundrakes S, Sai-gnam P, Johnson JD, Walsh DS, Saunders DL, Lanteri CA. 2013. Direct comparison of the histidine-rich protein-2 enzyme-linked immunosorbent assay (HRP-2 ELISA) and malaria SYBR green I fluorescence (MSF) drug sensitivity tests in Plasmodium falciparum reference clones and fresh ex vivo field isolates from Cambodia. Malar. J. 12:239. 10.1186/1475-2875-12-239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noedl H, Attlmayr B, Wernsdorfer WH, Kollaritsch H, Miller RS. 2004. A histidine-rich protein 2-based malaria drug sensitivity assay for field use. Am. J. Trop. Med. Hyg. 71:711–714 [PubMed] [Google Scholar]
- 26.Pickard AL, Wongsrichanalai C, Purfield A, Kamwendo D, Emery K, Zalewski C, Kawamoto F, Miller RS, Meshnick SR. 2003. Resistance to antimalarials in Southeast Asia and genetic polymorphisms in pfmdr1. Antimicrob. Agents Chemother. 47:2418–2423. 10.1128/AAC.47.8.2418-2423.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta Ct) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 28.Purfield A, Nelson A, Laoboonchai A, Congpuong K, McDaniel P, Miller RS, Welch K, Wongsrichanalai C, Meshnick SR. 2004. A new method for detection of pfmdr1 mutations in Plasmodium falciparum DNA using real-time PCR. Malar. J. 3:9. 10.1186/1475-2875-3-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lopes D, Rungsihirunrat K, Nogueira F, Seugorn A, Jose Pedro Gil Virgilio E, do Rosario Cravo P. 2002. Molecular characterization of drug-resistant Plasmodium falciparum from Thailand. Malar. J. 1:12. 10.1186/1475-2875-1-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson PE, Kazadi W, Kamwendo DD, Mwapasa V, Purfield A, Meshnick SR. 2005. Prevalence of pfcrt mutations in Congolese and Malawian Plasmodium falciparum isolates as determined by a new Taqman assay. Acta Trop. 93:97–106. 10.1016/j.actatropica.2004.09.010 [DOI] [PubMed] [Google Scholar]
- 31.Sutherland CJ, Haustein T, Gadalla N, Armstrong M, Doherty JF, Chiodini PL. 2007. Chloroquine-resistant Plasmodium falciparum infections among UK travellers returning with malaria after chloroquine prophylaxis. J. Antimicrob. Chemother. 59:1197–1199. 10.1093/jac/dkm104 [DOI] [PubMed] [Google Scholar]
- 32.Le Nagard H, Vincent C, Mentre F, Le Bras J. 2011. Online analysis of in vitro resistance to antimalarial drugs through nonlinear regression. Comput. Methods Programs Biomed. 104:10–18. 10.1016/j.cmpb.2010.08.003 [DOI] [PubMed] [Google Scholar]
- 33.Mbaisi A, Liyala P, Eyase F, Achilla R, Akala H, Wangui J, Mwangi J, Osuna F, Alam U, Smoak BL, Davis JM, Kyle DE, Coldren RL, Mason C, Waters NC. 2004. Drug susceptibility and genetic evaluation of Plasmodium falciparum isolates obtained in four distinct geographical regions of Kenya. Antimicrob. Agents Chemother. 48:3598–3601. 10.1128/AAC.48.9.3598-3601.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.World Health Organization. 2001. Mark III in vitro micro-test for the assessment of the response of Plasmodium falciparum to chloroquine, mefloquine, quinine, amodiaquine, sulfadoxine/pyrimethamine, and artemisinin. World Health Organization, Geneva, Switzerland [Google Scholar]
- 35.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NPJ, Lindegardh N, Socheat D, White NJ. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 361:455–467. 10.1056/NEJMoa0808859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vennerstrom JL, Arbe-Barnes S, Brun R, Charman SA, Chiu FCK, Chollet J, Dong Y, Dorn A, Hunziker D, Matile H, McIntosh K, Padmanilayam M, Santo Tomas J, Scheurer C, Scorneaux B, Tang Y, Urwyler U, Wittlin S, Charman WN. 2004. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature 430:900–904. 10.1038/nature02779 [DOI] [PubMed] [Google Scholar]
- 37.Valecha N, Looareesuwan S, Martensson A, Abdulla SM, Krudsood S, Tangpukdee N, Mohanty S, Mishra SK, Tyagi PK, Sharma SK, Moehrle J, Gautam A, Roy A, Paliwal JK, Kothari M, Saha N, Dash AP, Bjőrkman A. 2010. Arterolane, a new synthetic trioxolane for treatment of uncomplicated Plasmodium falciparum malaria: a phase II, multicenter, randomized, dose-finding clinical trial. Clin. Infect. Dis. 51:684–691. 10.1086/655831 [DOI] [PubMed] [Google Scholar]
- 38.Gautam A, Ahmed T, Sharma P, Varshney B, Kothari M, Saha N, Roy A, Moehrle JJ, Paliwal J. 2011. Pharmacokinetics and pharmacodynamics of arterolane maleate following multiple oral doses in adult patients with P. falciparum malaria. J. Clin. Pharmacol. 51:1519–1528. 10.1177/0091270010385578 [DOI] [PubMed] [Google Scholar]
- 39.Valecha N, Krudsood S, Tangpukdee N, Mohanty S, Sharma SK, Tyagi PK, Anvikar A, Mohanty R, Rao BS, Jha AC, Shahi B, Singh JPN, Roy A, Kaur P, Kothari M, Mehta S, Gautam A, Paliwal JK, Arora S, Saha N. 2012. Arterolane maleate plus piperaquine phosphate for treatment of uncomplicated Plasmodium falciparum malaria: a comparative, multicenter, randomized clinical trial. Clin. Infect. Dis. 55:663–671. 10.1093/cid/cis475 [DOI] [PubMed] [Google Scholar]
- 40.Lim P, Alker AP, Khim N, Shah NK, Incardona S, Doung S, Yi P, Bouth DM, Bouchier C, Puijalon OM, Meshnick SR, Wongsrichanalai C, Fandeur T, Le Bras J, Ringwald P, Ariey F. 2009. Pfmdr1 copy number and arteminisin derivatives combination therapy failure in falciparum malaria in Cambodia. Malar. J. 8:11. 10.1186/1475-2875-8-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vinayak S, Alam MT, Sem R, Shah NK, Susanti AI, Lim P, Muth S, Maguire JD, Rogers WO, Fandeur T, Barnwell JW, Escalante AA, Wongsrichanalai C, Ariey F, Meshnick SR, Udhayakumar V. 2010. Multiple genetic backgrounds of the amplified Plasmodium falciparum multidrug resistance (pfmdr1) gene and selective sweep of 184F mutation in Cambodia. J. Infect. Dis. 201:1551–1560. 10.1086/651949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khim N, Bouchier C, Ekala MT, Incardona S, Lim P, Legrand E, Jambou R, Doung S, Puijalon OM, Fandeur T. 2005. Countrywide survey shows very high prevalence of Plasmodium falciparum multilocus resistance genotypes in Cambodia. Antimicrob. Agents Chemother. 49:3147–3152. 10.1128/AAC.49.8.3147-3152.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lim P, Wongsrichanalai C, Chim P, Khim N, Kim S, Chy S, Sem R, Nhem S, Yi P, Duong S, Bouth DM, Genton B, Beck HP, Gobert JG, Rogers WO, Coppee JY, Fandeur T, Mercereau-Puijalon O, Ringwald P, Le Bras J, Ariey F. 2010. Decreased in vitro susceptibility of Plasmodium falciparum isolates to artesunate, mefloquine, chloroquine, and quinine in Cambodia from 2001 to 2007. Antimicrob. Agents Chemother. 54:2135–2142. 10.1128/AAC.01304-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lim P, Dek D, Try V, Eastman RT, Chy S, Sreng S, Suon S, Mao S, Sopha C, Sam B, Ashley EA, Miotto O, Dondorp AM, White NJ, Su X-Z, Char MC, Anderson JM, Amaratunga C, Menard D, Fairhurst RM. 2013. Ex vivo susceptibility of Plasmodium falciparum to antimalarial drugs in western, northern, and eastern Cambodia, 2011-2012: association with molecular markers. Antimicrob. Agents Chemother. 57:5277–5283. 10.1128/AAC.00687-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sidhu AB, Uhlemann AC, Valderramos SG, Valderramos JC, Krishna S, Fidock DA. 2006. Decreasing pfmdr1 copy number in Plasmodium falciparum malaria heightens susceptibility to mefloquine, lumefantrine, halofantrine, quinine, and artemisinin. J. Infect. Dis. 194:528–535. 10.1086/507115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noedl H, Wernsdorfer WH, Krudsood S, Wilairatana P, Viriyavejakul P, Kollaritsch H, Wiedermann G, Looareesuwan S. 2001. In vivo-in vitro model for the assessment of clinically relevant antimalarial cross-resistance. Am. J. Trop. Med. Hyg. 65:696–699 [DOI] [PubMed] [Google Scholar]
- 47.Barnes DA, Foote SJ, Galatis D, Kemp DJ, Cowman AF. 1992. Selection for high-level chloroquine resistance results in deamplification of the pfmdr1 gene and increased sensitivity to mefloquine in Plasmodium falciparum. EMBO J. 11:3067–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Imwong M, Dondorp AM, Nosten F, Yi P, Mungthin M, Hanchana S, Das D, Phyo AP, Lwin KM, Pukrittayakamee S, Lee SJ, Saisung S, Koecharoen K, Nguon C, Day NP, Socheat D, White NJ. 2010. Exploring the contribution of candidate genes to artemisinin resistance in Plasmodium falciparum. Antimicrob. Agents Chemother. 54:2886–2892. 10.1128/AAC.00032-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois Khim Kim A-CS, Duru V, Bouchier C, Ma L, Lim P, Leang R, Duong S, Sreng S, Suon S, Chuor CM, Bout DM, Menard S, Rogers WO, Genton B, Fandeur T, Miotto O, Ringwald P, Le Bras J, Berry A, Barale J-C, Fairhurst RM, Benoit-Vical F, Mercereau-Puijalon O, Menard D. 2014. A molecular marker of artemisinin resistant Plasmodium falciparum malaria. Nature 51:50–55. 10.1038/nature12876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Osorio L, Murillo C, Aponte S, Mayagaya V, Scheurer C, Brun R, Matile H, Wittlin S. 2007. In vitro susceptibility of P. falciparum populations from Colombia and Tanzania to a new synthetic peroxide (OZ277). Am. J. Trop. Med. Hyg. 76:1024–1026 [PubMed] [Google Scholar]
- 51.Marfurt J, Chalfein F, Prayoga P, Wabiser F, Wirjanata G, Sebayang B, Piera KA, Wittlin S, Haynes RK, Möhrle JJ, Anstey NM, Kenangalem E, Price RN. 2012. Comparative ex vivo activity of novel endoperoxides in multidrug-resistant Plasmodium falciparum and P. vivax. Antimicrob. Agents Chemother. 56:5258–5263. 10.1128/AAC.00283-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Witkowski B, Khim N, Chim P, Kim S, Ke S, Kloeung N, Chy S, Duong S, Leang R, Ringwald P, Dondorp AM, Tripura R, Benoit-Vical F, Berry A, Gorgette O, Ariey F, Barale J-C, Mercereau-Puijalon O, Menard D. 2013. Reduced artemisinin susceptibility of Plasmodium falciparum ring stages in western Cambodia. Antimicrob. Agents Chemother. 57:914–923. 10.1128/AAC.01868-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Witkowski B, Amaratunga C, Khim N, Sreng S, Chim P, Kim S, Lim P, Mao S, Sopha C, Sam B, Anderson JM, Duong S, Chuor CM, Taylor WRJ, Suon S, Mercereau-Puijalon O, Fairhurst RM, Menard D. 2013. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: in-vitro and ex-vivo drug-response studies. Lancet Infect. Dis. 13:1043–1049. 10.1016/S1473-3099(13)70252-4 [DOI] [PMC free article] [PubMed] [Google Scholar]