

Abstract
Staphylococcus aureus is a leading cause of hospital- and community-acquired infections, which exhibit broad resistance to various antibiotics. We recently disclosed the discovery of the oxadiazole class of antibiotics, which has in vitro and in vivo activities against methicillin-resistant S. aureus (MRSA). We report herein that MmpL, a putative member of the resistance, nodulation, and cell division (RND) family of proteins, contributes to oxadiazole resistance in the S. aureus strain COL. Through serial passages, we generated two S. aureus COL variants that showed diminished susceptibilities to an oxadiazole antibiotic. The MICs for the oxadiazole against one strain (designated S. aureus COLI) increased reproducibly 2-fold (to 4 μg/ml), while against the other strain (S. aureus COLR), they increased >4-fold (to >8 μg/ml, the limit of solubility). The COLR strain was derived from the COLI strain. Whole-genome sequencing revealed 31 mutations in S. aureus COLR, of which 29 were shared with COLI. Consistent with our previous finding that oxadiazole antibiotics inhibit cell wall biosynthesis, we found 13 mutations that occurred either in structural genes or in promoters of the genes of the cell wall stress stimulon. Two unique mutations in S. aureus COLR were substitutions in two genes that encode the putative thioredoxin (SACOL1794) and MmpL (SACOL2566). A role for mmpL in resistance to oxadiazoles was discerned from gene deletion and complementation experiments. To our knowledge, this is the first report that a cell wall-acting antibiotic selects for mutations in the cell wall stress stimulon and the first to implicate MmpL in resistance to antibiotics in S. aureus.
INTRODUCTION
Bacterial resistance to antibiotics poses a serious threat to human health and has emerged as a global health concern of the 21st century. At least 2 million individuals in the United States alone are infected annually by bacteria that are resistant to one or more antibiotics. A total of 23,000 deaths resulted directly from these infections (1). Staphylococcus aureus, a Gram-positive bacterium that colonizes the skin of approximately 30% of healthy individuals (2), is a major human pathogen and a leading cause of infections. These result in many illnesses ranging from minor skin infections to life-threatening diseases such as meningitis, pneumonia, and endocarditis (3). Staphylococcus aureus is extremely adaptive in response to antibiotic exposure (4). The emergence and spread of methicillin-resistant S. aureus (MRSA) (5), vancomycin-resistant S. aureus (VRSA) (6), and the recently reported linezolid resistance in S. aureus (7) presage a return to the preantibiotic era. The appearance of community-associated MRSA (CA-MRSA), an epidemic MRSA clone that often contains Panton-Valentine leukocidin (PVL) genes and is consequently more virulent than the traditional hospital-associated MRSA strains (2, 8, 9), exacerbates the concern and makes the discovery of new antibiotics urgent.
In this vein, we recently described a new class of non-β-lactam antibiotics, oxadiazoles, targeting penicillin-binding proteins (PBPs) of Gram-positive bacteria (Fig. 1) (10). The oxadiazoles act by inhibiting biosynthesis of the cell wall and demonstrate broad bactericidal activity against S. aureus, including vancomycin- and linezolid-resistant MRSA and other Gram-positive bacteria (10). Furthermore, the oxadiazoles show in vivo efficacy in a mouse model of infection and have 100% oral bioavailability (10). To explore the possibility of an emergence of resistance to this class of antibiotics, we serially passaged S. aureus COL (an MRSA strain) in tryptic soy broth (TSB) supplemented with increasing levels of the oxadiazole antibiotic (oxadiazole antibiotic 1; Fig. 1) and generated two strains with decreased susceptibility to it. Genomic sequencing identified a number of mutations in response to the antibiotic challenge. Further analyses revealed that MmpL, a putative member of the resistance, nodulation, and cell division (RND) family of proteins (11–13), is involved in the resistance to oxadiazole antibiotics.
FIG 1.
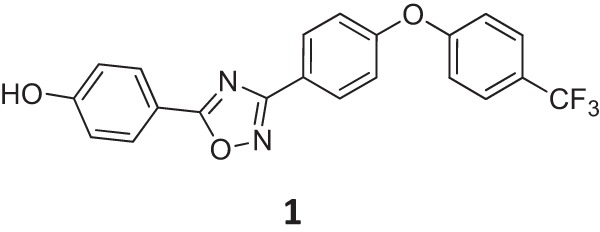
Chemical structure of oxadiazole antibiotic 1.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. The Staphylococcus aureus strains COL, RN4220, and RN9594 (RN4220 with the vector pCN40) were obtained from the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA). The plasmid pMAD was a kind gift from Vijay Pancholi. Deletion mutants and the complemented strains were constructed by the procedure described below. The S. aureus strains were grown on tryptic soy broth/agar (TSB/A) (BD Diagnostic Systems) or Mueller-Hinton broth (BD Diagnostic Systems). Escherichia coli strain DH5α was grown on Luria-Bertani medium (LB) (Fisher).
TABLE 1.
Bacterial strains and plasmids used in this study
| Plasmid or strain | Description | Reference/source |
|---|---|---|
| Plasmids | ||
| pMAD | E. coli-S. aureus shuttle vector containing a thermosensitive origin of replication, bgaB | 17 |
| pMADΔtrx | pMAD containing up- and downstream regions of sacol1794 | This study |
| pMADΔmmpL | pMAD containing up- and downstream regions of sacol2566 | This study |
| pCN40 | E. coli-S. aureus shuttle vector | 33 |
| pCNtrx | pCN40 harboring sacol1794 gene and its promoter | This study |
| pCNtrxP70Q | pCNtrx harboring mutated sacol1794 gene that resulted in P70Q substitution in its product | This study |
| pCNmmpL | pCN40 harboring sacol2566 gene and its promoter | This study |
| pCNmmpLT172I | pCNmmpL harboring mutated sacol2566 gene resulted in T172I substitution in its product | This study |
| S. aureus strains | ||
| COL | Hospital-acquired methicillin-resistant S. aureus parent strain | NARSA |
| COLR | COL expressing resistance to oxadiazole antibiotics | This study |
| COLI | COL expressing intermediate resistance to oxadiazole antibiotics | This study |
| COLΔtrx | COL lacking sacol1794 | This study |
| COLΔmmpL | COL lacking sacol2556 | This study |
| RN4220 | Restriction-deficient derivative of NCTC 8325-4 | 34 |
| RN9594 | RN4220 containing pCN40 | 33 |
| RN4220Δtrx | RN4220 lacking sacol1794 | This study |
| RN4220ΔtrxΩ | RN4220Δtrx strain containing pCN40 (vector-only control) | This study |
| RN4220ΔtrxΩtrx | RN4220Δtrx strain complemented by pCNtrx | This study |
| RN4220ΔtrxΩtrxP70Q | RN4220Δtrx strain complemented by pCNtrxP70Q | This study |
| RN4220ΔmmpL | RN4220 lacking sacol2556 | This study |
| RN4220ΔmmpLΩ | RN4220ΔmmpL strain containing pCN40 (vector only control) | This study |
| RN4220ΔmmpLΩmmpL | RN4220ΔmmpL strain complemented by pCNmmpL | This study |
| RN4220ΔmmpLΩmmpLT172I | RN4220ΔmmpL strain complemented by pCNmmpLT172I | This study |
| E. coli strain | ||
| DH5α | supE44 ΔlacU169(Φ80lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | 35 |
Selection for resistance.
The Staphylococcus aureus strain COL was used for the isolation of resistant strains. Bacteria were grown in the presence of increasing concentrations of the oxadiazole antibiotic for seven passages. The starting culture was grown in freshly prepared TSB supplemented with 1 μg/ml (0.5 MIC) of the oxadiazole antibiotic. For each subsequent passage, the concentration of the oxadiazole antibiotic was increased 2-fold, and the inoculum ratio was 1:100. The highest concentration used for the passages was 8 μg/ml (the solubility limit of the oxadiazole antibiotic), and the bacteria were passaged four times under this condition. The determination of MIC was performed to monitor changes in susceptibility to the oxadiazole antibiotic. Two single colonies were isolated from two passage cultures that displayed MIC values of 4 μg/ml and >8 μg/ml. The homogeneity of resistance of the two passage cultures was verified by plating the diluted cultures on plates containing TSA plus various concentrations of the oxadiazole antibiotic and counting bacterial colonies after 24 h of incubation at 37°C. The two isolates were designated strain COLI (MIC, 4 μg/ml) and strain COLR (MIC, >8 μg/ml) and were subjected to whole-genome sequencing and/or PCR-based DNA sequencing.
Antibiotic-susceptibility testing.
The MIC values of the antibiotics were determined using the broth microdilution method of the Clinical and Laboratory Standards Institute (14). Five commercially available antibiotics were tested: ampicillin (Sigma-Aldrich), ceftazidime (Sigma-Aldrich), imipenem (Merck), linezolid (AmplaChem), and vancomycin (Acros Organics). BBL Mueller-Hinton II broth was used for the MIC determination with a bacterial inoculum of 5 × 105 CFU/ml. The experiment was performed in triplicate, and the plates were incubated at 37°C for 16 to 20 h before the results were recorded.
Whole-genome sequencing.
The genomic DNA from the S. aureus strain COLR was sequenced to 90-fold coverage and compared to the genomic sequence of strain COL. The genome DNA was prepared with the DNeasy blood and tissue kit (Qiagen) and subjected to agarose gel to check the sample degradation. One microgram of genome DNA was processed with the TruSeq DNA LT sample preparation kit (Illumina, Inc.) to generate the DNA fragment library. The DNA fragment library was then loaded onto an MiSeq set to acquire 159-bp paired-end reads. The paired-end reads were filtered with a Q score (sequencing quality score) cutoff of 30 and aligned onto the reference genome of S. aureus COL using the Burrows-Wheeler aligner (BWA) with default parameters (15) and then sorted and indexed with SAMtools (sequence alignment/map) (16). SAMtools were used to determine single-nucleotide polymorphisms (SNPs) between S. aureus COL and COLR.
Confirmation of genomic mutations.
PCR-based sequencing was used to confirm the mutations predicted by the whole-genome sequencing. The DNA fragments harboring these predicted mutations were amplified by PCR. For a list of the primers used for each PCR, see Table S1 in the supplemental material. The PCR products were purified with a QIAquick PCR purification kit (Qiagen) and sequenced. Sequencing was performed at MCLAB, Inc., CA.
Site-directed deletions.
Primers for construction of deletion mutants are listed in Table 2. Markerless chromosomal in-frame trx or mmpL single-gene deletion mutants of S. aureus strains COL and RN4220 were constructed by homologous recombination (17). Two deletion vectors, pMADΔtrx and pMADΔmmpL, were constructed for allelic replacement. For the construction of pMADΔtrx, the primer pairs D1794UF/D1794UR and D1794DF/D1794DR were used to amplify chromosomal DNA approximately 0.6 kb upstream and 0.9 kb downstream of the gene trx. Similarly, for the construction of pMADΔmmpL, two DNA fragments, one 0.75 kb upstream and the other 0.95 kb downstream of the gene mmpL, were amplified using the primer pairs D2566UF/D2566UR and D2566DF/D2566DR, respectively. The upstream and downstream fragments were then digested by XhoI, mixed together, and ligated with T4 DNA ligase. The ligation product was inserted into the temperature-sensitive shuttle vector pMAD (17) and electroporated into E. coli strain DH5α. The resulting plasmid was then introduced into the S. aureus strain RN4220. For constructing deletion mutants in COL, pMADΔtrx and pMADΔmmpL extracted from RN4220 were separately transferred into COL. Allelic replacement was performed as described previously (17). The resulting mutants were identified by colony PCR and confirmed by DNA sequencing.
TABLE 2.
Primers used in this study for deletion of sacol1794 and sacol2566 and complementation
| Usage and name | Sequence (5′ to 3′)a | Restriction site |
|---|---|---|
| Deletion mutant | ||
| D1794UF | CCCGGATCCCACGCCATTCACACAGTTATCT | BamHI |
| D1794UR | CCCCTCGAGTTTCATTTTTATGTCTCCTTAATTAATGAT | XhoI |
| D1794DF | CCCCTCGAGGTGTAATTTAGACTAGAGAAAAACGGGGTA | XhoI |
| D1794DR | CCCCATGGCTAAACGCTGAATCACGTTTGGAT | NcoI |
| D2566UF | CCCGGATCCGGTAACGCTCATGAGTTTCTCATCTAC | BamHI |
| D2566UR | CCCCTCGAGTGCCAAAGTATATTGCCTCCTTTTAA | XhoI |
| D2566DF | CCCCTCGAGTAAAATAACATGTACATGCCTCCGC | XhoI |
| D2566DR | CCCCGGGGCGGTTGATACCCTTCCCCTA | SmaI |
| Complementation | ||
| Trx-F | CCCCCCGGATCCTCGGGTAATAACAACTATTATCTCTAAATAGTTATATATAATC | BamHI |
| Trx-R | CCGGCGCCTTACACGTATTGAGCTAAAAATGCATCTATC | NarI |
| MmpL-F | GGGGCATGCTTGTAGCAAGCTACTCGACAATGC | SphI |
| MmpL-R | GGGGGTACCTTATGATTTATTTCGTAGATTTTTTTCTAACAATTG | KpnI |
Restriction sites are underlined.
Complementation of deletions.
Primers for complementation are listed in Table 2. For complementation of trx deletion, the primer set Trx-F/Trx-R was used to amplify the DNA fragment containing the trx gene and its promoter by PCR using the genomic DNA of COL. Similarly, for complementation of mmpL deletion, the segment that includes the mmpL gene and its promoter was amplified by PCR using the primer set MmpL-F/MmpL-R. For complementation of trx or mmpL deletion with trxP70Q or mmpLT172I, the genomic DNA of COLR was used as the template. The PCR-amplified fragments were individually cloned into pCN40 and transferred into E. coli DH5α. The resulting constructs pCNtrx/pCN40trxP70Q and pCNmmpL/pCNmmpLT172I were confirmed by DNA sequencing and then separately transformed into RN4220Δtrx and RN4220ΔmmpL, respectively, for complementation. Plasmids were extracted from the complemented strain to verify their presence.
RESULTS AND DISCUSSION
Selection for resistance.
Through serial passage of the S. aureus COL in TSB supplemented with increasing concentrations of the oxadiazole antibiotic, we generated two variants that exhibited increased MIC values for the oxadiazole antibiotic. These strains were named COLI and COLR. The strain COLR showed a ≥4-fold-higher MIC for the oxadiazole antibiotic (MIC, >8 μg/ml, the limit of solubility of the antibiotic), whereas COLI showed a reproducible 2-fold increase in the MIC for the antibiotic (MIC, 4 μg/ml). We also evaluated if the two strains exhibited altered susceptibility to other antibiotics. We tested the β-lactam antibiotics ampicillin, ceftazidime, and imipenem, as well as the oxazolidinone linezolid and the glycopeptide antibiotic vancomycin. As shown in Table 3, neither COLR nor COLI displayed any changes in susceptibility to these antibiotics, compared to the wild-type COL. This indicates that the oxadiazole resistance mechanism might be distinct.
TABLE 3.
MICs of various antibiotics against S. aureus COL, COLI, and COLR strains
| Strain | MIC (μg/ml) of: |
|||||
|---|---|---|---|---|---|---|
| Antibiotic 1 | Ampicillin | Ceftazidime | Imipenem | Linezolid | Vancomycin | |
| COL | 2 | 16 | 128 | 16 | 2 | 2 |
| COLI | 4 | 16 | 128 | 16 | 2 | 2 |
| COLR | >8 | 16 | 128 | 16 | 2 | 2 |
Mutations in COLR and COLI.
We prepared the genomic DNA from the strain COLR and performed whole-genome sequencing, which predicted 31 mutations in 30 loci, as listed in Table 4. All predicted mutations, except the putative mutation 30, were confirmed by PCR sequencing. We did not succeed in amplifying the DNA fragment containing mutation 30 by PCR. Among the 31 mutations, 1 mutation (mutation 1) caused a frameshift in the open reading frame that resulted in the addition of 54 amino acids to the unknown protein SACOL0081, 18 mutations (mutations 2 to 19) resulted in single amino acid substitutions, and 10 mutations (mutations 20 to 29) occurred in noncoding regions. Mutations 30 and 31 were synonymous. Note that 13 of the 31 mutations in coding regions (Table 5) involved structural genes or promoters of the genes of the cell wall stress stimulon, a core and accessory set of cell wall-associated genes that have been reported to be overexpressed after the exposure of S. aureus to cell wall-active agents (18–20). This observation is consistent with our earlier finding that oxadiazole antibiotics act by inhibiting cell wall biosynthesis in S. aureus (10). In addition, it suggests that these mutations were likely selected under the pressure of the oxadiazole antibiotic.
TABLE 4.
Mutations predicted by whole-genome sequencing analysis of COLR strain
| Mutation type and no. | Mutation | Effect of mutation | Mutated locus | Function |
|---|---|---|---|---|
| In coding regions | ||||
| 1 | Deletion A91211 | Frameshift | SACOL0081 | Unknown |
| 2 | G805360→A805360 | A188T | SACOL0781 | Osmoprotectant ABC transporter ATP-binding protein |
| 3 | T1045164→G1045164 | N57K | SACOL1036 | Protease |
| 4 | G1063047→T1063047 | T327N | SACOL1057 | V8 protease |
| 5 | G1148922→T1148922 | P54T | SACOL1140 | LPXTG cell wall surface anchor protein |
| 6 | T1440658→A1440658 | L20I | SACOL1428 | Aspartate kinase |
| 7 | A1691714→G1691714 | L37S | SACOL1662 | Acetyl-coenzyme A carboxylase, biotin carboxyl carrier protein |
| 8 | C1757265→T1757265 | D102N | SACOL1727 | Translation initiation factor IF-3 |
| 9 | G1842648→T1842648 | P70Q | SACOL1794 | Thioredoxin |
| 10 | T1918837→A1918837 | Q68H | SACOL1866 | Serine protease SplD |
| 11 | C2004708→A2004708 | D367E | SACOL1941 | RNase BN |
| 12 | A2241657→G2241657 | L8S | SACOL2162 | Hypothetical protein |
| 13 | A2286720→T2286720 | S4T | SACOL2204 | Hypothetical protein |
| 14 | G2470606→T2470606 | T5N | SACOL2410 | Amino acid ABC transporter ATP-binding protein |
| 15 | T2482181→G2482181 | Y109S | SACOL2423 | Hypothetical protein |
| 16 | A2504486→T2504486 | I16L | SACOL2445 | fmtA-like protein |
| 17 | G2628671→A2628671 | T172I | SACOL2566 | MmpL |
| 18 | G2629609→T2629609 | D73Y | SACOL2567 | Hypothetical protein |
| 19 | G2770884→T2770884 | N3K | SACOL2696 | Phosphoribosyl-ATP pyrophosphatase/phosphoribosyl-AMP cyclohydrolase |
| In noncoding regions | ||||
| 20 | Insertion C after T459969 | Intergenic region between divergently transcribed genes sacol0457 (hypothetical protein) and sacol0458 (xanthine phosphoribosyltransferase) | ||
| 21 | Insertion G after A1001654 | Intergenic region between sacol0994 (oligopeptide ABC transporter ATP-binding protein) and sacol0995 (oligopeptide ABC transporter oligopeptide-binding protein). | ||
| 22 | T1022860→G1022860 | sacol1014 (pseudogene) | ||
| 23 | A1227842→T1227842 | Intergenic region between 2 hypothetical proteins SACOL1218 and SACOL1219 | ||
| 24 | A1227909→G1227909 | Intergenic region between 2 hypothetical proteins SACOL1218 and SACOL1219 | ||
| 25 | Deletion A1468810 | sacol1454 (pseudogene) | ||
| 26 | Deletion A2068156 | sacol2007 (pseudogene) | ||
| 27 | Deletion A2241731 | Intergenic region between 2 hypothetical proteins SACOL2162 and SACOL2163 | ||
| 28 | A2286771→T2286771 | sacol2205 (pseudogene) | ||
| 29 | Deletion T2492157 | Intergenic region between 2 hypothetical proteins SACOL2433 and SACOL2434 | ||
| Synonymous | ||||
| 30 | T60946→C60946 | S1245S | SACOL0050 | Methicillin-resistant surface protein |
| 31 | G2561347→A2561347 | D535D | SACOL2505 | Cell wall surface anchor family protein |
TABLE 5.
Genes of cell wall stress stimulon mutated upon exposure to the oxadiazole antibiotic
| COL open reading frame | Gene function | Reference |
|---|---|---|
| SACOL0781 | Osmoprotectant ABC transporter ATP-binding protein | 18, 20 |
| SACOL1057 | Serine V8 protease | 18 |
| SACOL1140 | LPXTG cell wall surface anchor protein | 18, 19 |
| SACOL1662 | Acetyl-coenzyme A carboxylase, biotin carboxyl carrier protein | 18–20 |
| SACOL1727 | Translation initiation factor IF-3 | 19 |
| SACOL1794 | Thioredoxin | 19 |
| SACOL2162 | Predicted membrane-bound metal-dependent hydrolases | 19 |
| SACOL2410 | Amino acid ABC transporter ATP-binding protein | 18–20 |
| SACOL2445 | fmtA-like protein | 18–20 |
| SACOL0994a,c | Oligopeptide ABC transporter ATP-binding protein | 18–20 |
| SACOL0995a,c | Oligopeptide ABC transporter oligopeptide-binding protein | 18–20 |
| SACOL1218a | Hypothetical protein (predicted to be a lipoprotein) | 19 |
| SACOL0050b | Methicillin-resistant surface protein | 18–20 |
| SACOL2505b | Cell wall surface anchor family protein | 18–20 |
Mutation occurred in intergenic region.
Synonymous mutation.
Same mutations.
Of the 13 mutations in structural genes or promoters of the cell wall stress stimulon, 9 occurred in genes that belong to one of the two functional categories: the cell-envelope biogenesis (which includes sacol0050, sacol1140, sacol1662, sacol2445, and sacol2505) and transport and binding (which include sacol0781, sacol0094, sacol0095, and sacol2410). Other mutations occurred in genes that encoded the translation-initiation factor IF-3 (SACOL1727); the serine V8 protease (SACOL1057), which is a virulence factor of S. aureus and plays a role in S. aureus immune evasion (21); a putative thioredoxin (SACOL1794); and two hypothetical proteins (SACOL1218 and SACOL2162). SACOL2162 is a conserved hypothetical protein predicted to be a membrane-bound metal-dependent hydrolase, potentially linking it to the stress response induced by cell wall-acting antibiotics. The remaining 18 mutations occurred outside the cell wall stress stimulon. These affected genes encode two proteases (SACOL1036 and SAACOL1866), an aspartate kinase (SACOL1428), an RNase (SACOL1941), a bifunctional phosphoribosyl-ATP pyrophosphatase/phosphoribosyl-AMP cyclohydrolase that participates in histidine metabolism (SACOL2696,), a glycosyltransferase that participates in nucleotide metabolism (SACOL0458), a putative membrane protein MmpL (SACOL2566), nine hypothetical proteins (SACOL0081, SACOL2204, SACOL2423, SACOL2567, SACOL0457, SACOL1219, SACOL2163, SACOL2433, and SACOL24340), and four pseudogenes (sacol1014, sacol1454, sacol2007, and sacol2205). It is interesting to note that SACOL0081 is predicted to be a lipoprotein, which may potentially be implicated in the stress response to cell wall-acting antibiotics.
Since the strains COLR and COLI displayed distinct levels of resistance to the oxadiazole antibiotic, we sought to identify the mutations responsible for the higher level of resistance in COLR. We used the COLI genomic DNA as the template and amplified all 30 DNA fragments by PCR, as we did for COLR in PCR sequencing. These DNA fragments were then sequenced, and we found that COLR and COLI share 28 mutations. The two unique mutations in S. aureus COLR were the P70Q substitution in the putative thioredoxin Trx (SACOL1794) and the T172I substitution in MmpL (SACOL2566). This observation suggests a potential role for Trx and MmpL in the resistance to the oxadiazole antibiotic and a potential link between the two mutations and the higher level of resistance to oxadiazole antibiotics. Furthermore, the 28 shared mutations in COLR and COLI and the 2 unique mutations in COLR suggest a stepwise accumulation of the mutations, as the strain COLR was derived from the strain COLI. This argues that alteration in a single binding site, as in a target PBP, might not account for the emergence of resistance, and indeed, the collective effect of mutations at more than one site might be the outcome.
Role of MmpL and Trx in resistance to oxadiazole antibiotics.
The S. aureus MmpL is a putative member of the resistance, nodulation, and cell division family of proteins (11, 12). This protein displays a high degree of sequence similarity to the MmpL proteins of Mycobacterium tuberculosis (25% to 30% identity, 43% to 45% similarity), which are proposed to function as scaffolds for the biosynthetic machinery of cell wall-associated lipids or to function in lipid secretion (13, 22, 23). The observation that MmpL was mutated in the strain COLR but not in strain COLI prompted us to examine the effect of mmpL deletion on the oxadiazole resistance of S. aureus COL. We constructed a markerless in-frame mmpL deletion mutant of strain COL and determined the MICs of a variety of antibiotics against it. We found that the deletion of mmpL in the strain COL resulted in a reproducible 2-fold decrease in susceptibility to the oxadiazole antibiotic but maintained the same level of susceptibility to the other antibiotics that we tested (Table 6). We next tried to complement the mmpL deletion in COL by pCN40mmpL, but this effort failed, which may be due to plasmid incompatibility, as S. aureus COL itself contains the plasmid pT181. As an alternative for complementation of mmpL deletion in S. aureus, we constructed a markerless in-frame mmpL deletion mutant in strain RN4220 and complemented the deletion mutant by pCNmmpL. As shown in Table 6, the deletion of mmpL in strain RN4220 also resulted in a reproducible 2-fold decrease in the MICs of the oxadiazole antibiotic. The role of MmpL in oxadiazole resistance was further confirmed by showing that the complemented strain, RN4220ΔmmpLΩmmpL, restored the susceptibility to the level of the wild-type strain (Table 6). To explore the effect of the T172I substitution in MmpL on oxadiazole resistance of S. aureus, we also complemented the deletion mutant RN4220ΔmmpL by pCNmmpLT172I. As shown in Table 6, the resulting complemented strain, RN4220ΔmmpLΩmmpLT172I, displayed an additional 2-fold increase in MICs of the oxadiazole antibiotic (overall, a 4-fold increase over the deletion mutant), compared to the wild-type S. aureus RN4220. On the other hand, the control strain, RN4220ΔmmpLΩ, which is the mmpL deletion mutant containing the empty vector pCN40, showed no changes in MIC, compared to the deletion mutant. Together these data implicate MmpL in resistance to oxadiazole antibiotics in S. aureus. Furthermore, this is the first report of a role for MmpL in antibiotic resistance to S. aureus. We have to add that certain MmpL proteins in M. tuberculosis have been implicated in resistance to cell wall-acting antibiotics such as cycloserine and vancomycin (24). To our knowledge, this is also the first report of a cell wall-acting antibiotic selecting for mutations in the cell wall stress stimulon.
TABLE 6.
MICs of different antibiotics against COL, RN4220, and their derivatives
| Strain | MIC (μg/ml) of: |
|||
|---|---|---|---|---|
| Antibiotic 1 | Ampicillin | Linezolid | Vancomycin | |
| COL | 2 | 16 | 2 | 2 |
| COLΔtrx | 2 | 16 | 2 | 2 |
| COLΔmmpL | 1 | 16 | 2 | 2 |
| RN4220 | 2 | 0.5 | 2 | 1 |
| RN4220Δtrx | 2 | 0.5 | 2 | 1 |
| RN4220ΔtrxΩ | 2 | 0.5 | 2 | 1 |
| RN4220ΔtrxΩtrx | 2 | 0.5 | 2 | 1 |
| RN4220ΔtrxΩtrxP70Q | 2 | 0.5 | 2 | 1 |
| RN4220ΔmmpL | 1 | 0.5 | 2 | 1 |
| RN4220ΔmmpLΩ | 1 | 0.5 | 2 | 1 |
| RN4220ΔmmpLΩmmpL | 2 | 0.5 | 2 | 1 |
| RN4220ΔmmpLΩmmpLT1721 | 4 | 0.5 | 2 | 1 |
We further explored a potential role for the thioredoxin gene trx (sacol1794) in oxadiazole resistance, which was also mutated in our sequencing analysis. Involvement of reactive-oxygen species in the bactericidal effect of antibiotics was proposed (25–27). On the other hand, it was reported that thioredoxin plays a protective role against oxidative stress in bacteria (28–32). In S. aureus COL, there are three putative thioredoxins, SACOL1794 (Trx), SACOL0875, and SACOL1155. Since trx was mutated in the strain COLR but not in strain COLI, we suspected that it might play a role in resistance to the oxadiazole antibiotics. Therefore, we constructed a markerless in-frame trx deletion mutant in strain COL and determined the MICs of a variety of antibiotics against the deletion mutant. However, we did not observe any changes in susceptibility to the antibiotics tested, including the oxadiazole antibiotic (Table 6). We also constructed a trx-deletion mutant of strain RN4220 and complemented this deletion mutant by pCNtrx and pCNtrxP70Q. As shown in Table 6, for all the antibiotics tested, there was no difference in antibiotic susceptibility among the wild-type RN4220, the deletion mutant RN4220Δtrx, and the two complemented strains RN4220ΔtrxΩtrx and RN4220ΔtrxΩtrxP70Q. We also examined if deletion of trx could affect S. aureus growth in TSB. Still, we did not observe any difference in growth rate between the deletion mutant and the wild-type strain (data not shown).
Thus, our experiments have demonstrated that the product of the mmpL gene contributes to the resistance to the oxadiazole antibiotic in S. aureus RN4220 and COL strains, while that of trx does not. However, the contributions of these genes to the resistance phenotype of the COLR strain are more difficult to interpret. Apart from mutations in both the mmpL and trx genes, the COLR strain has 28 additional mutations. The effect of one or a combination of these 28 additional mutations, shared between S. aureus strains COLI and COLR, is very likely responsible for the 2-fold increase in the MIC of the oxadiazole antibiotic against the strain COLI; hence, the simultaneous occurrences of mutations in both the mmpL and trx genes in the strain COLR likely further elevate the MIC by at least another 4-fold. We are aware that it is possible that COLI contains more mutations beyond the 28 additional mutations of COLR, as those mutations might have reverted during selection of COLR (COLR was derived from COLI), but this possibility is small, in our opinion. Given the presence of multiple mutations in the genome of COLR, it is plausible that the mmpL and trx genes work alone or in concert with other mutations to result in the higher level of resistance to the oxadiazole antibiotic observed in S. aureus COLR.
In summary, we generated two S. aureus COL derivatives (COLI and COLR) with decreased susceptibility to the oxadiazole antibiotic and found that exposure to oxadiazole antibiotics selected for mutations in the cell wall stress stimulon of S. aureus. Furthermore, we demonstrated that MmpL plays a role in the resistance to oxadiazole antibiotics, and the T172I substitution in MmpL contributes to a higher level of resistance. Given the predicted role of MmpL in cell wall assembly (as a member of the resistance, nodulation, and cell division family of proteins) and our previous finding that oxadiazole antibiotics act by inhibiting cell wall biosynthesis (10), the contribution of MmpL in oxadiazole resistance likely involves proteins that collectively influence cell wall assembly. The structural aspects of these events are not yet understood.
Supplementary Material
ACKNOWLEDGMENTS
We thank Vijay Pancholi, Department of Pathology, the Ohio State University, for supplying us with pMAD.
This work was supported by grant AI090818 from the National Institutes of Health (to M.C. and S.M.).
Footnotes
Published ahead of print 21 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03501-14.
REFERENCES
- 1.Centers for Disease Control and Prevention. 2013. Antibiotic resistance threats in the United States, 2013. Centers for Disease Control and Prevention, Atlanta, GA [Google Scholar]
- 2.DeLeo FR, Otto M, Kreiswirth BN, Chambers HF. 2010. Community-associated methicillin-resistant Staphylococcus aureus. Lancet 375:1557–1568. 10.1016/S0140-6736(09)61999-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diekema DJ, Pfaller MA, Schmitz FJ, Smayevsky J, Bell J, Jones RN, Beach M, SENTRY Participants Group 2001. Survey of infections due to Staphylococcus species: frequency of occurrence and antimicrobial susceptibility of isolates collected in the United States, Canada, Latin America, Europe, and the Western Pacific region for the SENTRY Antimicrobial Surveillance Program, 1997–1999. Clin. Infect. Dis. 32:S114–S132. 10.1086/320184 [DOI] [PubMed] [Google Scholar]
- 4.Fuda CC, Fisher JF, Mobashery S. 2005. Beta-lactam resistance in Staphylococcus aureus: the adaptive resistance of a plastic genome. Cell Mol. Life Sci. 62:2617–2633. 10.1007/s00018-005-5148-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The University of Chicago Medical Center. 2010. MRSA history timeline: the first half-century, 1959–2009. MRSA Research Center, The University of Chicago Medical Center, Chicago, IL [Google Scholar]
- 6.Bozdogan BÜ, Esel D, Whitener C, Browne FA, Appelbaum PC. 2003. Antibacterial susceptibility of a vancomycin-resistant Staphylococcus aureus strain isolated at the Hershey Medical Center. J. Antimicrob. Chemother. 52:864–868. 10.1093/jac/dkg457 [DOI] [PubMed] [Google Scholar]
- 7.Tsiodras S, Gold HS, Sakoulas G, Eliopoulos GM, Wennersten C, Venkataraman L, Moellering RC, Ferraro MJ. 2001. Linezolid resistance in a clinical isolate of Staphylococcus aureus. Lancet 358:207–208. 10.1016/S0140-6736(01)05410-1 [DOI] [PubMed] [Google Scholar]
- 8.Boyle-Vavra S, Daum RS. 2007. Community-acquired methicillin-resistant Staphylococcus aureus: the role of Panton-Valentine leukocidin. Lab. Invest. 87:3–9. 10.1038/labinvest.3700501 [DOI] [PubMed] [Google Scholar]
- 9.Maree CL, Daum RS, Boyle-Vavra S, Matayoshi K, Miller LG. 2007. Community-associated methicillin-resistant Staphylococcus aureus isolates and healthcare-associated infections. Emerg. Infect. Dis. 13:236–242. 10.3201/eid1302.060781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Daniel PI, Peng Z, Pi H, Testero SA, Ding D, Spink EE, Leemans E, Boudreau MA, Yamaguchi T, Schroeder VA, Wolter WR, Llarrull LI, Song W, Lastochkin E, Kumarasiri M, Antunes NT, Espahbodi M, Lichtenwalter K, Suckow MA, Vakulenko SB, Mobashery S, Chang M. 2014. Discovery of a new class of non-β-lactam inhibitors of penicillin-binding proteins with Gram-positive antibacterial activity. J. Am. Chem. Soc. 136:3664–3672. 10.1021/ja500053x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Domenech P, Reed MB, Barry CE., III 2005. Contribution of the Mycobacterium tuberculosis mmpL protein family to virulence and drug resistance. Infect. Immun. 73:3492–3501. 10.1128/IAI.73.6.3492-3501.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cuaron JA, Dulal S, Song Y, Singh AK, Montelongo CE, Yu W, Nagarajan V, Jayaswal RK, Wilkinson BJ, Gustafson JE. 2013. Tea tree oil-induced transcriptional alterations in Staphylococcus aureus. Phytother. Res. 27:390–396. 10.1002/ptr.4738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Truong-Bolduc QC, Dunman PM, Eidem T, Hooper DC. 2011. Transcriptional profiling analysis of the global regulator NorG, a GntR-like protein of Staphylococcus aureus. J. Bacteriol. 193:6207–6214. 10.1128/JB.05847-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clinical and Laboratory Standards Institute. 2006. Methods for dilution antimicrobial susceptibility rests for bacteria that grow aerobically; approved standard, 7th ed. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 15.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arnaud M, Chastanet A, Débarbouillé M. 2004. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 70:6887–6891. 10.1128/AEM.70.11.6887-6891.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sobral RG, Jones AE, Des Etages SG, Dougherty TJ, Peitzsch RM, Gaasterland T, Ludovice AM, de Lencastre H, Tomasz A. 2007. Extensive and genome-wide changes in the transcription profile of Staphylococcus aureus induced by modulating the transcription of the cell wall synthesis gene murF. J. Bacteriol. 189:2376–2391. 10.1128/JB.01439-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Utaida S, Dunman PM, Macapagal D, Murphy E, Projan SJ, Singh VK, Jayaswal RK, Wilkinson BJ. 2003. Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-active antibiotics reveals a cell-wall-stress stimulon. Microbiology 49:2719–2732. 10.1099/mic.0.26426-0 [DOI] [PubMed] [Google Scholar]
- 20.Kuroda M, Kuroda H, Oshima T, Takeuchi F, Mori H, Hiramatsu K. 2003. Two-component system VraSR positively modulates the regulation of cell wall biosynthesis pathway in Staphylococcus aureus. Mol. Microbiol. 49:807–821. 10.1046/j.1365-2958.2003.03599.x [DOI] [PubMed] [Google Scholar]
- 21.Jusko M, Potempa J, Kantyka T, Bielecka E, Miller HK, Kalinska M, Dubin G, Garred P, Shaw LN, Blom AM. 2014. Staphylococcal proteases aid in evasion of the human complement system. J. Innate Immun. 6:31–46. 10.1159/000351458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Varela C, Rittmann D, Singh A, Krumbach K, Bhatt K, Eggeling L, Besra GS, Bhatt A. 2012. MmpL genes are associated with mycolic acid metabolism in mycobacteria and corynebacteria. Chem. Biol. 19:498–506. 10.1016/j.chembiol.2012.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sondén B, Kocíncová D, Deshayes C, Euphrasie D, Rhayat L, Laval F, Frehel C, Daffé M, Etienne G, Reyrat JM. 2005. Gap, a mycobacterial specific integral membrane protein, is required for glycolipid transport to the cell surface. Mol. Microbiol. 58:426–440. 10.1111/j.1365-2958.2005.04847.x [DOI] [PubMed] [Google Scholar]
- 24.Domenech P, Reed MB, Barry CE., III 2005. Contribution of the Mycobacterium tuberculosis MmpL protein family to virulence and drug resistance. Infect. Immun. 73:3492–501. 10.1128/IAI.73.6.3492-3501.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810. 10.1016/j.cell.2007.06.049 [DOI] [PubMed] [Google Scholar]
- 26.Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC. 2012. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 336:315–319. 10.1126/science.1219192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Zhao X, Malik M, Drlica K. 2010. Contribution of reactive oxygen species to pathways of quinolone-mediated bacterial cell death. J. Antimicrob. Chemother. 65:520–524. 10.1093/jac/dkp486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dhamdhere G, Krishnamoorthy G, Zgurskaya HI. 2010. Interplay between drug efflux and antioxidants in Escherichia coli resistance to antibiotics. Antimicrob. Agents Chemother. 54:5366–5368. 10.1128/AAC.00719-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu J, Vlamis-Gardikas A, Kandasamy K, Zhao R, Gustafsson TN, Engstrand L, Hoffner S, Engman L, Holmgren A. 2013. Inhibition of bacterial thioredoxin reductase: an antibiotic mechanism targeting bacteria lacking glutathione. FASEB J. 27:1394–1403. 10.1096/fj.12-223305 [DOI] [PubMed] [Google Scholar]
- 30.Arnér E, Holmgren A. 2000. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 267:6102–6109. 10.1046/j.1432-1327.2000.01701.x [DOI] [PubMed] [Google Scholar]
- 31.Holmgren A. 1989. Thioredoxin and glutaredoxin systems. J. Biol. Chem. 264:13963–13966 [PubMed] [Google Scholar]
- 32.Gon S, Faulkner MJ, Beckwith J. 2006. In vivo requirement for glutaredoxins and thioredoxins in the reduction of the ribonucleotide reductases of Escherichia coli. Antioxid. Redox Signal. 8:735–742. 10.1089/ars.2006.8.735 [DOI] [PubMed] [Google Scholar]
- 33.Charpentier E, Anton AI, Barry P, Alfonso B, Fang Y, Novick RP. 2004. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl. Environ. Microbiol. 70:6076–6085. 10.1128/AEM.70.10.6076-6085.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kreiswirth BN, Lofdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, Novick RP. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712. 10.1038/305709a0 [DOI] [PubMed] [Google Scholar]
- 35.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580. 10.1016/S0022-2836(83)80284-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.