

Abstract
HIV-1 reverse transcriptase (RT)-associated RNase H activity is an essential function in viral genome retrotranscription. RNase H is a promising drug target for which no inhibitor is available for therapy. Diketo acid (DKA) derivatives are active site Mg2+-binding inhibitors of both HIV-1 RNase H and integrase (IN) activities. To investigate the DKA binding site of RNase H and the mechanism of action, six couples of ester and acid DKAs, derived from 6-[1-(4-fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester (RDS1643), were synthesized and tested on both RNase H and IN functions. Most of the ester derivatives showed selectivity for HIV-1 RNase H versus IN, while acids inhibited both functions. Molecular modeling and site-directed mutagenesis studies on the RNase H domain demonstrated different binding poses for ester and acid DKAs and proved that DKAs interact with residues (R448, N474, Q475, Y501, and R557) involved not in the catalytic motif but in highly conserved portions of the RNase H primer grip motif. The ester derivative RDS1759 selectively inhibited RNase H activity and viral replication in the low micromolar range, making contacts with residues Q475, N474, and Y501. Quantitative PCR studies and fluorescence-activated cell sorting (FACS) analyses showed that RDS1759 selectively inhibited reverse transcription in cell-based assays. Overall, we provide the first demonstration that RNase H inhibition by DKAs is due not only to their chelating properties but also to specific interactions with highly conserved amino acid residues in the RNase H domain, leading to effective targeting of HIV retrotranscription in cells and hence offering important insights for the rational design of RNase H inhibitors.
INTRODUCTION
Since the discovery of 3′-azidothymidine, the HIV-1-coded reverse transcriptase (RT) has been the main target for drug treatments that have successfully turned the lethal progression to AIDS into a chronic disease (1, 2). However, the emergence of side effects of long-term therapy and the selection of drug-resistant viral strains demand novel anti-HIV agents, possibly targeting viral functions not yet explored (3, 4). RT plays a crucial role in viral replication, carrying out the synthesis of integration-competent double-stranded DNA starting from the (+)-strand RNA viral genome. Retrotranscription proceeds through an RNA/DNA hybrid intermediate, whose RNA must be removed to allow (+)-strand DNA synthesis. This RNA removal is performed by the RT-associated RNase H function through a sequence of highly specific hydrolytic events. Since the RNase H function is essential for viral replication (5), it has been explored as a drug target, and a number of RNase H inhibitors (RHIs) have been reported (6–8). RHIs can be classified based on their binding sites, i.e., (i) RHIs that coordinate the two Mg2+ catalytic cofactors at the RNase H active site, such as N-hydroxyimides (9), hydroxytropolones (10), hydroxypyrimidines (11), naphthyridinones (12), nitrofuran-2-carboxylic acid carbamoylmethyl esters (13), hydroxyquinolinones (14), and thiocarbamates and triazoles (15), or (ii) allosteric RHIs, such as vinylogous ureas (16), thienopyrimidinones (17), hydrazones (18), anthraquinones (19), isatines (20, 21), and propenones (22).
Despite this large number of identified scaffolds, currently no RHI has successfully reached clinical approval. In fact, efforts aimed to develop Mg2+-binding RHIs have been hampered by the topology of the RNase H active site, which is relatively large and shallow in comparison with closely related virus-encoded polynucleotidyl transferases such as HIV-1 DNA polymerase and integrase (IN) (23, 24), hampering the identification of a suitable binding pocket near the catalytic region. In fact, in all cocrystallized structures of HIV-1 RT/RNase H and active site RHIs (12, 24, 25), the RHIs exhibited binding poses showing a large part of their structures extending out of the protein domain. Therefore, besides the coordination with the two catalytic Mg2+ ions, the inhibitors established very few contacts with RNase H binding site residues (26), providing poor information about the binding interactions. This lack of information hampered further optimization of lead RHIs to achieve greater potency and selectivity, which are required for detailed characterization of the mechanisms of action in cell-based assays.
Diketo acid (DKA) derivatives are among the first compounds reported to bind the Mg2+ cofactors in the active site of influenza virus endonuclease (27), HIV-1 IN (28), and HIV-1 RNase H (29, 30). Among them, 6-[1-(4-fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester (RDS1643) was the first ligand proven to be able to inhibit both HIV-1 RNase H in biochemical assays and viral replication in cell culture (30). Recently, a new series of pyrrolyl DKAs that are active against both HIV-1 IN and RNase H has been reported (31), with acidic compounds being more effective with IN and ester counterparts being active with both enzymes, with no particular difference. Moreover, a recent study on RHI effects on and binding to prototype foamy virus (PFV) RT showed a putative RDS1643 binding region in the PFV RNase H active site and suggested, given the high level of structural similarity between PFV and HIV-1 RNase H domains, the possibility that RDS1643 could show similar interactions with the HIV-1 RNase H domain (32).
Herein, we report the synthesis of six new ester/acid couples of pyrrolyl DKAs and assays aiming to identify the molecular determinants required for DKAs for selective interactions with the HIV-1 RT RNase H domain and to clarify whether the catalytic region of RNase H can offer additional anchor points that might be targeted by drugs. Subsequent molecular docking and site-directed mutagenesis studies allowed us to establish a well-defined interaction pattern for DKA derivatives with highly conserved amino acid residues in the RNase H active site, proving differences in binding orientations between ester and acid derivatives that allowed rationalization of their different inhibition profiles. Furthermore, we show that the derivative RDS1759 effectively and selectively inhibited HIV reverse transcription in cells. Overall, these findings provide relevant insights for rational drug design of RHIs.
MATERIALS AND METHODS
Chemistry.
Details concerning the synthesis and characterization procedures for all new compounds can be found in the supplemental material.
Site-directed mutagenesis.
Alanine substitutions were introduced into the p66 HIV-1 RT subunit, using the QuikChange protocol (Agilent Technologies, Santa Clara, CA), in a p(His)6-tagged p66/p51 HIV-1HXB2 RT-prot plasmid kindly provided by Stuart Le Grice (NCI Frederick).
Expression and purification of recombinant HIV-1 RTs.
(His)6-tagged p66/p51 HIV-1 RTs were expressed in Escherichia coli strain M15 containing the p6HRT-prot vector and grown to an optical density at 600 nm (OD600) of 0.7, and expression was induced for 4 h with isopropyl β-d-1-thiogalactopyranoside (IPTG) at 1.7 mM. Protein purification was carried out with a BioLogic LP system (Bio-Rad) with a combination of immobilized metal ion affinity chromatography and ion-exchange chromatography. Cell pellets were resuspended in lysis buffer (50 mM sodium phosphate [pH 7.8], 0.5 mg/ml lysozyme), the mixture was incubated on ice for 20 min, 0.3 M NaCl (final concentration) was added, and the mixture was sonicated and centrifuged for 1 h at 30,000 × g. The supernatant was loaded onto a Ni2+-Sepharose column that was preequilibrated with loading buffer (50 mM sodium phosphate [pH 7.8], 0.3 M NaCl, 10% glycerol, 10 mM imidazole) and was washed thoroughly with wash buffer (50 mM sodium phosphate [pH 6.0], 0.3 M NaCl, 10% glycerol, 80 mM imidazole). RT was subjected to gradient elution with elution buffer (wash buffer with 0.5 M imidazole). Fractions were collected, and protein purity was checked by SDS-PAGE and found to be greater than 90%. Enzyme-containing fractions were pooled, diluted 1:1 with dilution buffer (50 mM sodium phosphate [pH 7.0], 10% glycerol), and then loaded onto a HiTrap Heparin HP column (GE Healthcare Life Sciences) preequilibrated with 10 column volumes of loading buffer 2 (50 mM sodium phosphate [pH 7.0], 10% glycerol, 150 mM NaCl). The column was then washed with loading buffer 2, and RT was subjected to gradient elution with elution buffer 2 (50 mM sodium phosphate [pH 7.0], 10% glycerol, 150 mM NaCl). Purified protein was dialyzed and stored in buffer containing 50 mM Tris HCl (pH 7.0), 25 mM NaCl, 1 mM EDTA, and 50% glycerol. Catalytic activities and protein concentrations were determined. Enzyme-containing fractions were pooled, and aliquots were stored at −80°C.
Expression and purification of recombinant HIV-1 IN.
HIV-1 IN was expressed essentially as reported previously (33). Briefly, His-tagged IN was produced in E. coli BL21-CodonPlus (DE3)-RIPL (Agilent, Santa Clara, CA) and purified under nondenaturing conditions. Protein production was induced to OD600 of 0.6 to 0.8 by adding IPTG to a concentration of 0.5 mM. Culture mixtures were incubated for 3 h at 37°C and then centrifuged at 1,100 × g for 20 min at 4°C. Cells were resuspended in buffer A (50 mM Tris-HCl [pH 8], 1 M NaCl, 4 mM β-mercaptoethanol) and lysed by passage through a French press. The lysate was centrifuged (30 min at 12,000 × g at 4°C), and the supernatant was filtered (pore size, 0.45 μm) and incubated with nickel-nitrilotriacetic acid-agarose beads (Qiagen, Venlo, The Netherlands) for at least 2 h at 4°C. The beads were washed with buffer A and then with buffer A supplemented with 80 mM imidazole. His-tagged proteins were then eluted from the beads with buffer A supplemented with 1 M imidazole and 50 μM zinc sulfate and were then dialyzed overnight against 20 mM Tris-HCl (pH 8), 1 M NaCl, 4 mM β-mercaptoethanol, 10% glycerol. Aliquots of the purification products were rapidly frozen and stored at −80°C.
HIV-1 DNA polymerase-independent RNase H activity determination.
Wild-type (wt) and mutant HIV RT-associated RNase H activity was measured as described previously (34). Briefly, RT-associated RNase H activity was measured in a 100-μl reaction volume containing 50 mM Tris-HCl (pH 7.8), 6 mM MgCl2, 1 mM dithiothreitol (DTT), 80 mM KCl, and 0.25 μM hybrid RNA/DNA (5′-GTTTTCTTTTCCCCCCTGAC-3′-fluorescein/5′-CAAAAGAAAAGGGGGGACUG-3′-dabcyl). The reaction mixture was incubated for 1 h at 37°C, the reaction was stopped with the addition of 50 μl of 0.5 M EDTA (pH 8.0), and products were quantified with a Victor 3 plate reader (Perkin) with excitation at 490 nm and emission at 528 nm. Different amounts of enzymes were used according to the linear ranges of the dose-response curves, i.e., 20 ng wt, 37.5 ng R448A, 62.5 ng N474A, 300 ng Q475A, 1 μg Y501A, and 75 ng R557A RTs. All experiments were performed at least three times.
HIV-1 RDDP activity determination.
The HIV-1 RT-associated RNA-dependent DNA polymerase (RDDP) activity was measured as described previously (34), in the presence of different amounts of enzymes according to the linear ranges of the dose-response curves, i.e., 20 ng wt, 30 ng R448A, 30 ng N474A, 100 ng Q475A, 30 ng Y501A, and 30 ng R557A RTs. All experiments were performed at least three times.
Evaluation of DNA polymerase-independent RNase H and RDDP kinetic efficiencies.
Kinetic analysis of the DNA polymerase-independent RNase H and RDDP activities was performed with Lineweaver-Burke plots, using SigmaPlot 10 software. Velocity (ν) was expressed as fmol/min.
HIV-1 IN activity determination.
The strand-transfer reaction was performed as described previously (35). Oligonucleotides HIV-1B (5′-TGTGGAAAATCTCTAGCA-3′) and HIV-1A (5′-ACTGCTAGAGATTTTCCACA-3′) (Eurogentec) were used for the strand-transfer reaction. HIV-1B was radiolabeled with T4 polynucleotide kinase (Biolabs) and [γ-32P]ATP (3,000 Ci/mmol; Amersham) and was purified on a Sephadex G-10 column (GE Healthcare). The strand-transfer reaction was carried out at 37°C in buffer containing 10 mM HEPES (pH 7.2), 1 mM DTT, and 7.5 mM MgCl2 or MnCl2, in the presence of 12.5 nM DNA substrate. Products were loaded on denaturing 18% acrylamide/urea gels. Gels were analyzed with a Molecular Dynamics Storm PhosphorImager, and results were quantified with ImageQuant 4.1 software.
Cells and viruses.
MT4 and 293T cells were cultured in RPMI 1640 medium and Dulbecco's modified Eagle's medium (DMEM), respectively. Both media were supplemented with 10% fetal calf serum (FCS). HIV-1 stocks were prepared by transfecting 293T cells with the HIV-1 molecular clone derived from pNL4-3 (Δenv viruses) (36); wt NLENG1-ES-IRES encodes the wt IN. Pseudotyping of Δenv viruses was performed by cotransfection of 293T cells with a vesicular stomatitis virus glycoprotein (VSV-G) plasmid using the calcium phosphate method. Viral supernatants were filtered (pore size, 0.45 μm) and frozen at −80°C. HIV-1 p24gag antigen contents in viral inocula were determined by enzyme-linked immunosorbent assay (Perkin-Elmer Life Sciences). A total of 120 ng of p24gag antigen per 106 cells, corresponding to a multiplicity of infection (MOI) of 0.3, was used for infection. When required, cells were treated with raltegravir (RAL) (100 nM) or efavirenz (EFV) (100 nM). Flow cytometric analysis was performed with a FACSCalibur flow cytometer (BD Bioscience), and results were analyzed using ImageQuant software.
Cytotoxicity assay.
The proliferation of MT4 cells was measured by seeding 1 × 104 cells/well on 96-well plates in 100 μl of RPMI 1640 medium with FCS (10%), penicillin (100 U/ml), and streptomycin (200 μg/ml), as described previously (34).
Molecular modeling.
The 3-dimensional structures of DKAs were generated with the Maestro Build Panel (Schrodinger, New York, NY), as described previously (37). The crystal structure of the full-length HIV-1 RT in complex with a naphthyridinone inhibitor bound to the RNase H active site (PDB accession no. 3LP1) was selected for docking studies. The missing residue R557, which is part of the RNase H active site, was modeled using the coordinates of the crystal structure with PDB accession no. 3K2P. The receptor was then prepared with the Protein Preparation Wizard of the Schrödinger 2012 molecular modeling package, as described previously (38). Docking studies were finally carried out with the grid-based program Glide 5.8 (Schrödinger) (38). For the grid generation, a box of 20 Å by 20 Å by 20 Å, centered on the active site Mg2+ ions, was created. The standard precision mode of the GlideScore function was used to score the binding poses obtained. The force field used for the docking was OPLS-2005 (39). All of the pictures were rendered with PyMOL (www.pymol.org) and Maestro (Schrödinger).
Quantification of viral DNA genomes.
Quantitative PCR (qPCR) was performed as described previously (40). Briefly, MT4 cells were infected and treated at time zero and 10 h postinfection (p.i.) in the presence of inhibitors RDS1759 and RDS1760 at concentrations of 10 μM, using as controls RAL and EFV at concentrations of 200 nM and 100 nM, respectively. Two million to five million cells were collected at each time point. Cells were washed in phosphate-buffered saline (PBS), and dry cell pellets were frozen at −80°C until use. DNA from infected cells was purified with a QIAamp DNA Blood minikit (Qiagen), according to the manufacturer's instructions. Quantification of total viral DNA, 2-long terminal repeat circle (2-LTRc), and integrated DNAs was performed with a LightCycler instrument (Roche Diagnostics) using the second-derivative-maximum method provided by the LightCycler quantification software (version 3.5; Roche Diagnostics). Amplification of the β-globin gene (two copies per diploid cell) was performed to normalize the results, using commercially available materials (control DNA kit; Roche Diagnostics). The quantification results for 2-LTRc, total HIV-1 DNA, and integrated HIV-1 DNA were expressed as copy numbers per μg DNA.
RESULTS
Inhibition of HIV-1 RT-associated DNA polymerase-independent RNase H activity.
Starting from the DKA prototype RDS1643 (30), we synthetized six ester/acid couples of pyrrolyl DKAs designed to define the structure-activity relationships within this series. In particular, we (i) put halogen, alkyl, or alkoxy substituents at different positions on the benzyl ring, (ii) shifted the diketo ester/acid chain from position 2 to position 3 of the pyrrole moiety, and (iii) added a further phenyl ring in position 4 of the pyrrole ring (Fig. 1). The newly synthesized derivatives were tested for their ability to inhibit the HIV-1 RT-associated DNA polymerase-independent RNase H and RNA-dependent DNA polymerase (RDDP) functions as well as IN strand-transfer activity in biochemical assays and viral replication in cell-based assays (Table 1). All of the DKAs inhibited RNase H activity, with the ester derivatives generally being more potent than their acid counterparts (Table 1). This effect was more evident for derivatives RDS1759, RDS2291, and RDS2400, which showed 50% inhibitory concentration (IC50) values for RNase H activity of 7.3, 8.2, and 11.2 μM, respectively, while being completely inactive as IN inhibitors at up to 100 μM. Conversely, all of the acid derivatives were more potent than their ester counterparts as IN inhibitors, as reported previously (31). Interestingly, some derivatives, independent of their acid/ester structure, also inhibited RDDP activity. In particular, RDS1822 was active with respect to both RT-associated activities and IN activity in the same range of concentrations.
FIG 1.
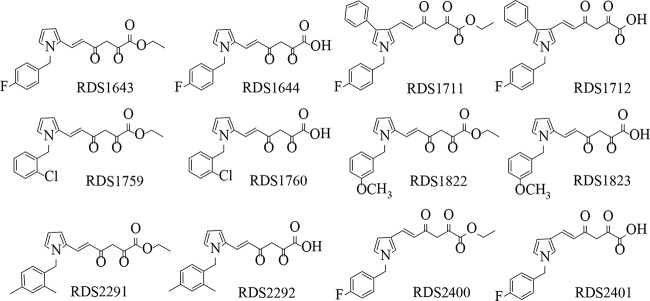
Chemical structures of newly synthesized DKA derivatives.
TABLE 1.
Biological effects of DKA derivatives on HIV-1 RT-associated RNase H and IN activities and HIV-1 replication
| Compound | RNase H IC50a (μM) | RDDP IC50b (μM) | IN IC50c (μM) | HIV-1 EC50d (μM) | MT4 CC50e (μM) | SIf |
|---|---|---|---|---|---|---|
| RDS1643 | 8.6 ± 1.3 | >50 | 21 ± 3 | 15.5 | >50 | >3.22 |
| RDS1644 | 16.1 ± 2.3 | >50 | 0.16 ± 0.03 | 0.29 | >50 | >172 |
| RDS1711 | 8.0 ± 0.2 | >50 | 21 ± 12 | 4.3 | 26.9 | 6.3 |
| RDS1712 | 7.7 ± 0.5 | >50 | 1.55 ± 0.15 | 17.2 | >50 | >2.9 |
| RDS1759 | 7.3 ± 0.1 | >50 | >100 | 2.10 | >50 | >23.8 |
| RDS1760 | 19.2 ± 0.8 | 40 ± 3 | 0.59 ± 0.77 | 15.4 | >50 | >3.2 |
| RDS1822 | 6.3 ± 1.8 | 31 ± 9 | 42 ± 28 | 3.4 | 14.1 | 4.14 |
| RDS1823 | 87 ± 5 | >50 | 0.59 ± 0.19 | 3.2 | >50 | >15.6 |
| RDS2291 | 8.2 ± 1.2 | 12.1 ± 2.5 | >100 | >15.4 | 15.4 | |
| RDS2292 | 19.3 ± 2.6 | 25 ± 2 | 26 ± 4 | >25.5 | 25.5 | |
| RDS2400 | 11.2 ± 1.1 | 26 ± 1 | >100 | >50 | >50 | |
| RDS2401 | 28 ± 5 | >50 | 28 ± 3 | >50 | >50 |
Compound concentration (mean ± standard deviation) required to inhibit HIV-1 RT-associated RNase H activity by 50%.
Compound concentration (mean ± standard deviation) required to inhibit HIV-1 RT RDDP activity by 50%.
Compound concentration (mean ± standard deviation) required to inhibit HIV-1 IN activity by 50%.
EC50, compound concentration required to decrease viral replication in MT-4 cells by 50%.
CC50, compound concentration required to reduce infected MT-4 cell viability by 50%.
SI, selectivity index (CC50/EC50 ratio).
To further investigate their modes of action, all of the derivatives were tested for their ability to bind Mg2+ ions, based on the DKA UV spectra recorded in the absence and in the presence of 6 mM MgCl2, and all were shown to be able to bind the ions (data not shown). Finally, cell-based assays revealed that four of the six new ester/acid couples were able to inhibit viral replication, with only the RDS2400/2401 and RDS2291/2292 derivatives being totally inactive (Table 1).
Molecular docking.
Molecular docking studies were performed to elucidate the binding modes of the newly synthesized DKA derivatives at the RNase H active site. The docking results suggested that the investigated ligands shared similar binding modes, although some differences in the interaction patterns established by ester and acid derivatives were observed. Among our set of RNase H inhibitors, we decided to consider primarily the binding modes of compounds RDS1643/RDS1644 and RDS1711/RDS1712, which, although not displaying the best RNase H/IN selectivity profiles, represented the most structurally different couples of ester and acid derivatives. In this view, the structural diversity of the selected compounds should result in different predicted ligand-enzyme interactions, so as to suggest the mutation of different residues and lead to experimental data with high information gain.
Docking of derivatives RDS1643/RDS1644 and RDS1711/RDS1712 suggested that both ester and acid DKAs coordinate the two catalytic Mg2+ ions; however, they adopt slightly different binding orientations due to the steric hindrance of the ethyl substituent of the ester derivatives (Fig. 2A and C). Besides the metal cofactor coordination, both RDS1643 and RDS1644 interact with a number of RNase H active site residues. In particular, the diketo ester branch of RDS1643 H-bonds with the N474 side chain and establishes lipophilic interactions with the A445 and I556 residues through its ethyl substituent, while the DKA moiety of RDS1644 forms a salt bridge with the R557 side chain. Additionally, the benzyl ring of both the ester and the acid forms parallel-displaced interactions with the Y501 side chain, while the pyrrole ring of RDS1643 can establish further lipophilic interactions with the C-α and C-β carbons of the Q475 residue. Finally, docking of the ester derivative RDS1711 (Fig. 2E) indicates that this compound is able to form an additional cation-π interaction with the R448 guanidinium group through its benzyl substituent, while the phenyl at position 4 on the pyrrole ring interacts with the Y501 side chain (Fig. 2E and F). Conversely, the corresponding acid RDS1712 is not predicted to interact with the R448 side chain, while its 4-phenyl substituent interacts with the Y501 side chain and its N-benzyl group contacts residue W535 (Fig. 2G and H). We also performed molecular docking of derivatives RDS1759, RDS2291, and RDS2400, obtaining the same docking patterns (see Fig. S2 in the supplemental material).
FIG 2.

Binding modes of DKA derivatives at the HIV-1 RNase H active site. (A, C, E, and G) Stick models of RDS1643 (A), RDS1644 (C), RDS1711 (E), and RDS1712 (G), represented as yellow, orange, light green, and dark green sticks, respectively. The receptor is shown as a gray cartoon. Amino acids involved in ligand binding are highlighted as sticks. The active site Mg2+ ions are represented as magenta spheres. (B, D, F, and H) Corresponding two-dimensional representations of DKA-RNase H interactions. Red and blue sticks represent oxygen and nitrogen atoms, respectively, and dashed lines represent hydrogen bonds.
Catalytic efficiency of mutant HIV-1 RTs.
Based on the interaction patterns predicted by docking calculations, we selectively mutated into Ala residues R448, N474, Q475, Y501, and R557 within the sole p66 subunit of HIV-1 RT. It is important to note that the N474, Q475, and Y501 residues are highly conserved, playing crucial roles as part of the RNase H primer grip (31). In fact, amino acid substitutions of these residues have been reported to reduce the HIV-1 replication rate drastically (41) and to affect RNase H enzymatic activity strongly (42). To determine whether the selected mutated RTs were suitable for quantitative assays, we performed kinetic analyses of both their RNase H and RDDP catalytic efficiencies (Fig. 3). Consistent with previous observations (42), all of the RNase H primer grip-mutated RTs showed drastically reduced but still measurable catalytic efficiency for RNase H activity (Table 2). In contrast, the kcat/Km ratios of the R448A and R557A RTs showed no reductions. Finally, none of the amino acid-substituted RTs showed significant changes in the RT-associated RNA-dependent DNA polymerase (RDDP) function, compared with wt RT, with the sole exception of Q475A RT, which showed a kcat/Km ratio of 8.1 for RDDP function.
FIG 3.
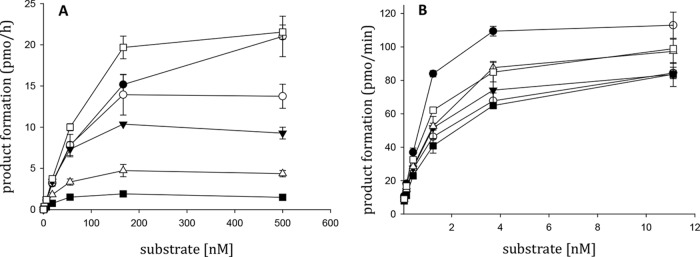
Comparison of the kinetics of polymerase-independent RNase H and RDDP activities of HIV-1 RT mutants. (A) The polymerase-independent RNase H cleavage activity for all HIV-1 RT mutants was measured in 100-μl reaction volumes containing increasing amounts of RNA/DNA hybrid substrate and fixed ratios of enzymes, according to the linear ranges of the dose-response curves. (B) The RDDP activity was measured in 25-μl volumes containing 100 μM poly(A)-oligo(dT), increasing concentrations of dTTP, and fixed ratios of enzymes, according to the linear ranges of the dose-response curves. The kinetic analyses were performed with Lineweaver-Burke plots of both RNase H and RDDP activities. □, wt RT; ○, R448A RT; △, N474A RT; ●, Q475A RT; ■, Y501A RT; ▼, R557A RT. Error bars represent standard deviations calculated from three independent determinations.
TABLE 2.
Comparison of kinetics of wt and mutant HIV-1 RT DNA polymerase-independent RNase H and RDDP activities
| RT | RNase H activity |
RDDP activity |
||||
|---|---|---|---|---|---|---|
| kcat (min−1) | Km (nM) | kcat/Km ratio | kcat (min−1) | Km (nM) | kcat/Km ratio | |
| wt | 112 ± 1 | 126 ± 5 | 0.89 | 23 ± 1 | 0.94 ± 0.03 | 25 |
| R557A | 25 ± 3 | 41 ± 1 | 0.60 | 19.2 ± 1.5 | 0.94 ± 0.10 | 20 |
| R448A | 44 ± 9 | 85 ± 22 | 0.52 | 14.6 ± 1.4 | 0.90 ± 0.01 | 16 |
| Q475A | 10.7 ± 1.9 | 100 ± 11 | 0.11 | 8.4 ± 0.5 | 1.04 ± 0.18 | 8 |
| N474A | 2.6 ± 0.5 | 30 ± 0.1 | 0.08 | 23 ± 1 | 1.19 ± 0.03 | 19 |
| Y501A | 0.25 ± 0.07 | 29 ± 9 | 0.008 | 20 ± 1 | 1.34 ± 0.03 | 15 |
Evaluation of effects of DKA derivatives on mutant RTs.
In order to experimentally verify the binding model suggested by computational studies, DKA derivatives were tested for their ability to inhibit the RNase H function of all RTs with amino acid substitutions, using the RHI β-thujaplicinol (BTP) (10) as a control (Table 3). Results showed drastic reductions of inhibitory potency (generally more than 1 order of magnitude) of all DKAs when tested on N474A RT RNase H, confirming the crucial role of this residue for DKA binding. The loss of the H-bond with the N474 side chain might explain the lower activities observed for the ester derivatives. However, the increases in the IC50 values observed also for the acid ligands suggest an additional functional/structural role for N474 at the RNase H active site. In the case of the R557A RT, only moderate increases in IC50 values were observed, which were greater for the acid derivatives than for their ester counterparts. Indeed, as predicted by docking calculations, acid derivatives make a salt bridge with the R557 side chain in the wt RT that cannot be established in the R557A mutant RT (Fig. 2D and H).
TABLE 3.
Inhibition of HIV-1 RT-associated RNase H activity of mutated HIV-1 RTs by DKAs
| Compound | IC50 (μM) (% activity)a |
||||
|---|---|---|---|---|---|
| R448A | R557A | N474A | Q475A | Y501A | |
| RDS1643 | 10.9 ± 2.1 | 17.0 ± 2.3 | 80 ± 6 | 92 ± 12 | >100 (86) |
| RDS1644 | 19.3 ± 1.6 | 45 ± 1 | >100 (55) | 5.5 ± 1.5 | 11.6 ± 5.1 |
| RDS1711 | 100 ± 2 | 50 ± 5 | >100 (100) | >100 (83) | >100 (100) |
| RDS1712 | 8.1 ± 0.3 | 19.6 ± 3.8 | >100 (62) | >100 (80) | >100 (80) |
| RDS1822 | 6.5 ± 3.5 | 17.4 ± 3.4 | 94 ± 10 | 71 ± 4 | >100 (76) |
| RDS1823 | 88 ± 6 | >100 (68) | 100 ± 2 | 7.4 ± 1.5 | 6.4 ± 1.6 |
| RDS2400 | 15.1 ± 2.9 | 25 ± 5 | 63 ± 12 | >100 (55) | >100 (70) |
| RDS2401 | 25 ± 2 | 61 ± 6 | >100 (65) | 28 ± 4 | 30 ± 7 |
| RDS1759 | 8.6 ± 0.1 | 16.2 ± 3.0 | 89 ± 6 | 96 ± 1 | >100 (100) |
| RDS1760 | 20 ± 2 | 63 ± 1 | >100 (79) | 6.3 ± 2.3 | >100 (80) |
| RDS2291 | 10.0 ± 2.0 | 20 ± 5 | >100 (56) | >100 (76) | >100 (85) |
| RDS2292 | 27 ± 3 | 58 ± 3 | >100 (84) | 3.4 ± 0.6 | >100 (100) |
| BTP | 0.15 ± 0.02 | 0.22 ± 0.05 | 0.09 ± 0.01 | 0.19 ± 0.03 | 0.07 ± 0.005 |
IC50, compound concentration (mean ± standard deviation) required to inhibit HIV-1 RT-associated RNase H activity by 50%. Numbers in parentheses indicate the percent enzyme activity measured in the presence of 100 μM compound.
When the DKAs were assayed with the Q475A RT, the decreases in potency shown by the ester derivatives were generally greater than those observed for the corresponding acids. In particular, the esters RDS1643, RDS1822, RDS2400, RDS1759, and RDS2291 exhibited IC50s at least 10 times higher than those for wt RT, while their acid counterparts exhibited 3- to 5-fold lower IC50s, with the sole exception of RDS1823, which showed an 11-fold IC50 decrease. According to docking results, the Q475A substitution significantly reduces the interaction surface accessible to ester derivatives within the RNase H active site. Conversely, the Q475A amino acid substitution can allow acid derivatives to interact more tightly with the Y501 side chain through the benzyl group (or through the 4-phenyl substituent for RDS1712).
The results with Y501A RT showed significant decreases in potency for all ester derivatives as well as for their acid counterparts, with the exception of compounds RDS1644 and RDS2401. This result could suggest that the Y501A substitution modifies the interactions with the adjacent Q475 by inducing a conformational rearrangement of the protein binding site that can be hardly noted in docking experiments with rigid protein models. Finally, the R448A substitution affected the potency of RNase H inhibition by RDS1711 (12-fold increase in its IC50) (Table 3), in agreement with the docking results for RDS1711 and RDS1712, showing that the former but not the latter can establish cation-π interactions with the R448 side chain of the wt RT through the benzyl group (Fig. 2C and D).
Characterization of mechanisms of DKA inhibition in cell-based assays.
Among the newly synthesized DKA derivatives, RDS1759 proved to be the sole compound able to selectively inhibit HIV-1 replication in cell-based assays and the RNase H function in biochemical assays without affecting either the RT-associated RDDP function (Table 1) or the IN strand-transfer function. Therefore, we chose the ester/acid DKA couple RDS1759/RDS1760 to investigate in more detail their mechanisms of action in cell-based assays, and fluorescence-activated cell sorting (FACS) and qPCR analyses were performed to detect which step of viral replication was targeted. MT4 cells were infected with the NLENG1-ES-IRES wt virus containing a green fluorescent protein (GFP) reporter system and were treated with 10 μM concentrations of RDS1759 and RDS1760 DKAs at the time of infection or 10 h postinfection (p.i.), since this time point is considered to occur at the end of the reverse transcription window (37). High mean fluorescence (HMF) and low mean fluorescence highlight expression from integrated viral DNA and unintegrated viral DNA, respectively, as described previously (36). Samples were collected and analyzed by FACS analysis at 48 and 72 h p.i., quantifying (i) the percentage of emitting GFP (eGFP) cells, indicating the amount of infected cells, which is reduced overall by reverse transcription inhibitors such as EFV; and (ii) the percentage of HMF, indicating the amount of integrated DNA, which is selectively affected by integration process inhibitors such as RAL (Fig. 4A to E). FACS analysis showed an RDS1759 inhibition pattern analogous to that observed for the nonnucleoside inhibitor EFV; in fact, RDS1759 reduced the percentage of eGFP cells by 68% and 33% at 48 and 72 h p.i., respectively (Fig. 4D). Surprisingly, inhibition of retrotranscription by RDS1759 was found to be time dependent, since it was significantly reduced at 72 h p.i. In order to confirm this mechanism of action, drugs were added at 10 h p.i., and results showed strong impairment of the effect of RDS1759 on the percentage of eGFP cells (only 30% inhibition). In contrast, RDS1760 showed an HIV-1 inhibition pattern similar to that of the IN inhibitor raltegravir (RAL), suggesting that RDS1760 mainly affects the integration step of the viral replication process.
FIG 4.

Inhibition of HIV-1 replication by DKAs. MT4 cells were infected with NLENG1-ES-IRES wt virus containing a GFP reporter system. (A to E) Samples were analyzed 48 h postinfection to quantify the amount of emitting GFP cells (eGFP) versus the GFP intensity of emission, categorized as low intensity (M1), indicating expression from unintegrated viral DNA, and high intensity (M2), indicating integrated viral DNA expressing GFP. (A) Infected MT4 cells. (B) MT4 cells treated with 100 nM EFV. (C) MT4 cells treated with 200 nM RAL. (D) MT4 cells treated with 10 μM RDS1759. (E) MT4 cells treated with 10 μM RDS1760. (F to I) Samples were analyzed 48 hours p.i. (gray) and 72 hours p.i. (black) to quantify the percentage of total (M1 and M2) emitting GFP cells (F and G) and the percentage of HMF (M2) (H and I) normalized to the percentage of the untreated control sample. Drugs (10 μM RDS1759, 10 μM RDS1760, 100 nM EFV, and 200 nM RAL) were added at the time of infection (F and H) or 10 h p.i. (G and I). The experiment was performed three times (error bars represent standard deviations).
Next, we used qPCR to determine the total viral DNA and 2-LTRc DNA amounts during the early phases of viral replication in the presence of the two DKAs. First, compounds were added at the time of infection and DNA samples were collected after 4, 6, 8, 10, 24, and 48 h (Fig. 5A and B). Second, DKAs were added at 10 h postinfection (p.i.) and DNA samples were collected after 24 and 48 h (Fig. 5C and D). 2-LTRc accumulation has been described when HIV-1 integration is impaired (43), and results showed that RDS1760 followed the RAL profile of 2-LTR accumulation after 24 and 48 h, when added either at the time of viral infection or at 10 h p.i. Moreover, RDS1760 caused a 30% decrease in the total amount of viral DNA, greater than the decrease observed with RAL treatment. Therefore, RDS1760 may partially inhibit reverse transcription also, although it appears to target mainly integration. Conversely, the derivative RDS1759 induced a strong reduction in the formation of total viral DNA and followed the EFV profile until the time point at 10 h. In fact, such reduction was partially reversed at 24 h. Consistent with the absence of IN inhibition, RDS1759 showed no accumulation of 2-LTRc DNA, compared to untreated control samples. These results clearly indicate that RDS1760 primarily acts on IN, while RDS1759 selectively inhibits reverse transcription in a time-dependent manner. To confirm this hypothesis, we determined the viral inhibition rate by RDS1759 at 24 and 48 h, observing an important shift in the 50% effective concentration (EC50) from 0.17 μM to 1.16 μM (see Fig. S2 in the supplemental material). This effect could be due either to cellular metabolism or to fast dissociation of the ligand from the target. To clearly distinguish between these two possibilities, we determined, at 24 and 48 h p.i., the amounts of integrated viral DNA with RDS1759 added 12 h before infection, at the time of infection, or 12 h p.i. and also added twice (at infection and 12 h p.i.), using RAL as a control (Fig. 6). RDS1759 addition 12 h before infection led to consistent decreases in viral inhibition, confirming the hypothesis of its cellular metabolism or degradation. In agreement with this hypothesis, the potency of RDS1759 was maximal when it was added at the time of infection or was added twice (at the time of infection and at 12 h p.i.); when it was added at 12 h p.i., inhibition also occurred but to a lesser extent. Taken together, these data demonstrate that RDS1759 efficiently targets HIV-1 reverse transcription.
FIG 5.

qPCR kinetics of total and 2-LTRc DNA forms during a single round of HIV replication in the presence of inhibitors. MT4 cells were infected with HIV-1 in the absence (●) or in the presence of 10 μM RDS1759 (○), 10 μM RDS1760 (▼), 500 nM RAL (△), or 100 nM EFV (■), added at infection (A and B) or 10 h p.i. (C and D). Samples were analyzed for total viral DNA and 2-LTRc at different p.i. time points. The experiment was performed three times (error bars represent standard deviations). All samples were standardized and quantified relative to known standards, as described in Materials and Methods.
FIG 6.
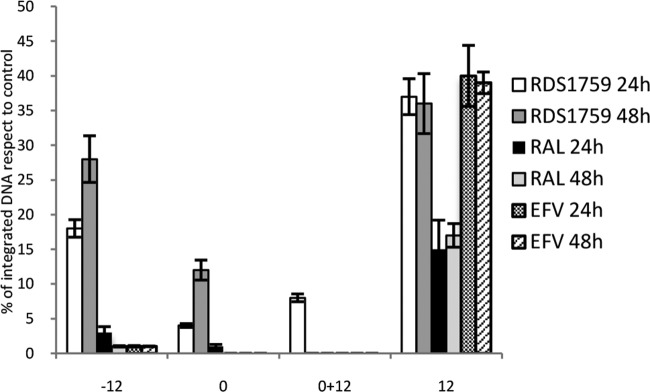
Effects of the time of RDS1759 addition on HIV-1 replication. HIV-1-infected MT4 cells were treated with 10 μM RDS1759, 200 nM RAL, or 100 nM EFV at different time points. The x axis shows the time (h) of compound addition. Samples were collected at 24 and 48 h, and integrated viral DNA was measured. All results were quantified by qPCR and normalized to values for infected untreated cells. The experiment was performed three times (error bars represent standard deviations).
DISCUSSION
DKAs have been reported to be the first active site agents that can effectively inhibit both HIV-1 RNase H and IN functions, or only one of them, in the low micromolar range and that can also inhibit HIV-1 replication in cell-based assays (30, 44). Despite these promising results, the binding modes of DKAs within the RNase H domain have not been further characterized, and no DKA derivative was further investigated to confirm whether it actually inhibited reverse transcription in cell-based assays. A recent study on DKA inhibition of both RNase H and IN functions showed that IN function was preferentially inhibited by acid analogs, while RNase H function was equally inhibited by both ester and acid derivatives (31). The different activity profiles of ester and acid DKAs for IN may arise from the different electrostatic properties of the HIV-1 RNase H and IN active sites. In particular, in the RNase H active site, four catalytic acid residues (D443, E478, D498, and D549) completely neutralize the four positive charges of the two active site Mg2+ ions. On the other hand, the IN active site is positively charged, since only three acidic residues (D64, D116, and D152) counterbalance the four positive charges of the two catalytic Mg2+ ions. With this perspective, negatively charged acidic compounds may bind more favorably to HIV-1 IN than to the RNase H active site (24). Starting from these results, in the present study we synthesized 6 new couples of diketo ester and DKA derivatives with various structural features, to be used as chemical tools in the characterization of RNase H binding modes. In agreement with previous studies (30, 31), among the designed molecules all of the acids were more potent against IN, while three esters (RDS1759, RDS2291, and RDS2400) proved to selectively inhibit RNase H activity, compared to IN. Interestingly, a few DKA derivatives were also able to inhibit RDDP activity. In particular, while most of these compounds (i.e., RDS2291, RDS2292, RDS2400, and RDS2401) did not inhibit viral replication, RDS1822 was active on both RT-associated activities and IN activity in the same range of concentrations and inhibited HIV replication. Further studies should be performed to characterize the RDDP inhibition by DKAs and to better define the RDS1822 mode of action on viral replication. Molecular modeling studies on the RT RNase H domain predicted that our DKAs would interact with a number of residues surrounding the RNase H active site, i.e., R448, N474, Q475, Y501, and R557, most of which are highly conserved (45) as part of the RNase H primer grip motif (41, 42, 46). This is a singular pattern that is interesting for the large number of residues involved and is in agreement with recent observations on the interaction of RDS1643 and the PFV domain of RNase H (32). In fact, all of the crystal structures of RT/RNase H prototypes solved in complexes with active site RHIs show the ligands coordinating the metal cofactors (25, 47) while exhibiting an orientation of binding that does not allow extended secondary interactions with amino acid side chains. The only exceptions are represented by hydroxytropolones and naphthyridinones (25, 48), with the latter being found to be sandwiched by a loop containing residues A538 and H539 on one side and N474 on the opposite side (12). Interestingly, our modeling studies propose different binding orientations for ester and acid DKAs in the RNase H domain, due to the steric hindrance of the alkyl substituent on the DKA branch of ester derivatives, which mainly influence the interactions with Q475 and R448. To confirm the calculation outcomes, all of the residues identified as significant for DKA binding were point mutated to alanine. It was reported previously that amino acid substitution of N474, Q475, and Y501 to Ala modifies RNase H cleavage specificity and can alter the Kd related to DNA polymerase substrates (42), reducing viral titration in cell-based assays up to 10-fold (41). In agreement with these studies, our evaluation of the catalytic efficiencies of both RNase H and RDDP enzymatic activities showed decreases in the kcat/Km ratios of RNase H activity for all RNase H primer grip mutants. However, these mutants fully conserved their efficiencies for RDDP function. Biochemical assays of our DKAs with each mutated RT confirm the interaction patterns indicated by docking studies, validating the PFV RNase H model (31) as a tool to characterize HIV-1 RHIs. Biochemical results also confirm the hypothesis of different binding orientations for esters and acids. In particular, in the presence of the Q475A substitution, the RNase H inhibitory potency is significantly decreased (with respect to wt RT) in the case of ester DKA derivatives, while it is increased in the case of their acid counterparts. Furthermore, in agreement with the docking-predicted interaction between R448 and ester RDS1711, this compound proved to be inactive with the R448A RT, while no change in IC50 was observed for the acid counterpart RDS1712.
The biochemical results for our new set of DKA derivatives revealed that the ester RDS1759 could selectively inhibit RNase H activity while being ineffective with both IN and RDDP functions, while its acid counterpart RDS1760 was shown to preferentially target HIV-1 IN. RDS1759 also inhibited HIV-1 replication in cell-based assays, with no evident toxic effects. Monitoring of early viral DNA product formation by FACS and qPCR showed that RDS1759 could selectively target the reverse transcription process in cell-based assays, although time-dependent decay of the inhibition of viral replication was observed. Preincubation in cell culture before viral infection compromised the efficacy of RDS1759 against HIV-1, while chronic exposure to the ligand completely restored the inhibition of viral replication. These results suggest that at least partial intracellular ligand inactivation might occur. It is noteworthy that the hypothesis that esterases may be involved, generating the acid counterpart, is not supported by the occurrence of late effects related to the inhibition of integration, such as 2-LTRc DNA accumulation, which still occur for the RDS1760 derivative at 48 h p.i. The more lipophilic nature of the ester DKA with respect to its acid counterpart might suggest instead better diffusion in other cellular compartments. Overall, while this time-dependent effect of RDS1759 prevented its use to select drug-resistant HIV-1 strains, the further development of RDS1759 analogs selective for RNase H inhibition and lacking this time-dependent effect will allow the selection of resistant virus to confirm the amino acid residues involved in the binding to RT.
In conclusion, RDS1759 is the first compound, to the best of our knowledge, that has been definitively proven to inhibit HIV-1 proliferation by inhibiting the genomic RNA hydrolysis by RT. Unlike previously reported RHIs, RNase H inhibition by this compound, as well as by the other DKA derivatives presented in this study, involves interactions not only with Mg2+ but also with highly conserved residues within the RNase H domain, thus offering important insights for rational optimization of RNase H active site inhibitors as new potential drugs for anti-HIV-1 treatment.
Supplementary Material
ACKNOWLEDGMENT
We thank the Italian Ministero dell'Istruzione, dell'Università e della Ricerca for financial support (grant PRIN 2010 no. 2010W2KM5L_003).
Footnotes
Published ahead of print 4 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03605-14.
REFERENCES
- 1.Esposito F, Corona A, Tramontano E. 2012. HIV-1 reverse transcriptase still remains a new drug target: structure, function, classical inhibitors, and new inhibitors with innovative mechanisms of actions. Mol. Biol. Int. 2012:586401. 10.1155/2012/586401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsibris AMN, Hirsch MS. 2010. Antiretroviral therapy in the clinic. J. Virol. 84:5458–5464. 10.1128/JVI.02524-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asahchop EL, Wainberg MA, Sloan RD, Tremblay CL. 2012. Antiviral drug resistance and the need for development of new HIV-1 reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 56:5000–5008. 10.1128/AAC.00591-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang MW, Shafer RW. 2012. HIV-1 antiretroviral resistance: scientific principles and clinical applications. Drugs 72:e1–e25. 10.2165/11633630-000000000-00000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schatz O, Cromme FV, Naas T, Lindemann D, Mous J, Le Grice SFJ. 1990. Inactivation of the RNase H domain of HIV-1 reverse transcriptase blocks viral infectivity, p 293–404 In Papas T. (ed), Gene regulation and AIDS. Portfolio, Houston, TX [Google Scholar]
- 6.Tramontano E. 2006. HIV-1 RNase H: recent progress in an exciting, yet little explored, drug target. Mini Rev. Med. Chem. 6:727–737. 10.2174/138955706777435733 [DOI] [PubMed] [Google Scholar]
- 7.Tramontano E, Di Santo R. 2010. HIV-1 RT-associated RNase H function inhibitors: recent advances in drug development. Curr. Med. Chem. 17:2837–2853. 10.2174/092986710792065045 [DOI] [PubMed] [Google Scholar]
- 8.Corona A, Masaoka T, Tocco G, Tramontano E, Le Grice SF. 2013. Active site and allosteric inhibitors of the ribonuclease H activity of HIV reverse transcriptase. Future Med. Chem. 5:2127–2139. 10.4155/fmc.13.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klumpp K. 2003. Two-metal ion mechanism of RNA cleavage by HIV RNase H and mechanism-based design of selective HIV RNase H inhibitors. Nucleic Acids Res. 31:6852–6859. 10.1093/nar/gkg881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Budihas SR, Gorshkova I, Gaidamakov S, Wamiru A, Bona MK, Parniak MA, Crouch RJ, McMahon JB, Beutler JA, Le Grice SFJ. 2005. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones. Nucleic Acids Res. 33:1249–1256. 10.1093/nar/gki268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirschberg TA, Balakrishnan M, Squires NH, Barnes T, Brendza KM, Chen X, Eisenberg EJ, Jin W, Kutty N, Leavitt S, Liclican A, Liu Q, Liu X, Mak J, Perry JK, Wang M, Watkins WJ, Lansdon EB. 2009. RNase H active site inhibitors of human immunodeficiency virus type 1 reverse transcriptase: design, biochemical activity, and structural information. J. Med. Chem. 52:5781–5784. 10.1021/jm900597q [DOI] [PubMed] [Google Scholar]
- 12.Su H-P, Yan Y, Prasad GS, Smith RF, Daniels CL, Abeywickrema PD, Reid JC, Loughran HM, Kornienko M, Sharma S, Grobler JA, Xu B, Sardana V, Allison TJ, Williams PD, Darke PL, Hazuda DJ, Munshi S. 2010. Structural basis for the inhibition of RNase H activity of HIV-1 reverse transcriptase by RNase H active site-directed inhibitors. J. Virol. 84:7625–7633. 10.1128/JVI.00353-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuji H, Urano E, Futahashi Y, Hamatake M, Tatsumi J, Hoshino T, Morikawa Y, Yamamoto N, Komano J. 2009. Derivatives of 5-nitro-furan-2-carboxylic acid carbamoylmethyl ester inhibit RNase H activity associated with HIV-1 reverse transcriptase. J. Med. Chem. 52:1380–1387. 10.1021/jm801071m [DOI] [PubMed] [Google Scholar]
- 14.Suchaud V, Bailly F, Lion C, Tramontano E, Esposito F, Corona A, Christ F, Debyser Z, Cotelle P. 2012. Development of a series of 3-hydroxyquinolin-2(1H)-ones as selective inhibitors of HIV-1 reverse transcriptase associated RNase H activity. Bioorg. Med. Chem. Lett. 22:3988–3992. 10.1016/j.bmcl.2012.04.096 [DOI] [PubMed] [Google Scholar]
- 15.Di Grandi M, Olson M, Prashad AS, Bebernitz G, Luckay A, Mullen S, Hu Y, Krishnamurthy G, Pitts K, O'Connell J. 2010. Small molecule inhibitors of HIV RT ribonuclease H. Bioorg. Med. Chem. Lett. 20:398–402. 10.1016/j.bmcl.2009.10.043 [DOI] [PubMed] [Google Scholar]
- 16.Chung S, Wendeler M, Rausch JW, Beilhartz G, Gotte M, O'Keefe BR, Bermingham A, Beutler JA, Liu S, Zhuang X, Le Grice SFJ. 2010. Structure-activity analysis of vinylogous urea inhibitors of human immunodeficiency virus-encoded ribonuclease H. Antimicrob. Agents Chemother. 54:3913–3921. 10.1128/AAC.00434-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masaoka T, Chung S, Caboni P, Rausch JW, Wilson JA, Taskent-Sezgin H, Beutler JA, Tocco G, Le Grice SFJ. 2013. Exploiting drug-resistant enzymes as tools to identify thienopyrimidinone inhibitors of human immunodeficiency virus reverse transcriptase-associated ribonuclease H. J. Med. Chem. 56:5436–5445. 10.1021/jm400405z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Himmel DM, Sarafianos SG, Dharmasena S, Hossain MM, McCoy-Simandle K, Ilina T, Clark AD, Jr, Knight JL, Julias JG, Clark PK, Krogh-Jespersen K, Levy RM, Hughes SH, Parniak MA, Arnold E. 2006. HIV-1 reverse transcriptase structure with RNase H inhibitor dihydroxy benzoyl naphthyl hydrazone bound at a novel site. ACS Chem. Biol. 1:702–712. 10.1021/cb600303y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esposito F, Kharlamova T, Distinto S, Zinzula L, Cheng Y-C, Dutschman G, Floris G, Markt P, Corona A, Tramontano E. 2011. Alizarine derivatives as new dual inhibitors of the HIV-1 reverse transcriptase-associated DNA polymerase and RNase H activities effective also on the RNase H activity of non-nucleoside resistant reverse transcriptases. FEBS J. 278:1444–1457. 10.1111/j.1742-4658.2011.08057.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung S, Miller JT, Johnson BC, Hughes SH, Le Grice SFJ. 2012. Mutagenesis of human immunodeficiency virus reverse transcriptase p51 subunit defines residues contributing to vinylogous urea inhibition of ribonuclease H activity. J. Biol. Chem. 287:4066–4075. 10.1074/jbc.M111.314781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Distinto S, Maccioni E, Meleddu R, Corona A, Alcaro S, Tramontano E. 2013. Molecular aspects of the RT/drug interactions: perspective of dual inhibitors. Curr. Pharm. Des. 19:1850–1859. 10.2174/1381612811319100009 [DOI] [PubMed] [Google Scholar]
- 22.Meleddu R, Cannas V, Distinto S, Sarais G, Del Vecchio C, Esposito F, Bianco G, Corona A, Cottiglia F, Alcaro S, Parolin C, Artese A, Scalise D, Fresta M, Arridu A, Ortuso F, Maccioni E, Tramontano E. 2014. Design, synthesis, and biological evaluation of new 1,3-diarylpropenones as dual inhibitors of HIV-1 reverse transcriptase. ChemMedChem 9:1869–1879. 10.1002/cmdc.201402015 [DOI] [PubMed] [Google Scholar]
- 23.Rausch JW. 2013. Ribonuclease H inhibitors: structural and molecular biology, p 143–172 In Le Grice SFJ, Gotte M. (ed), Human immunodeficiency virus reverse transcriptase: a bench-to-bedside success. Springer, New York, NY [Google Scholar]
- 24.Esposito F, Tramontano E. 2014. Past and future: current drugs targeting HIV-1 integrase and reverse transcriptase-associated ribonuclease H activity: single and dual active site inhibitors. Antivir. Chem. Chemother. 23:129–144. 10.3851/IMP2690 [DOI] [PubMed] [Google Scholar]
- 25.Chung S, Himmel DM, Jiang J-K, Wojtak K, Bauman JD, Rausch JW, Wilson JA, Beutler JA, Thomas CJ, Arnold E, Le Grice SFJ. 2011. Synthesis, activity, and structural analysis of novel α-hydroxytropolone inhibitors of human immunodeficiency virus reverse transcriptase-associated ribonuclease H. J. Med. Chem. 54:4462–4473. 10.1021/jm2000757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klumpp K, Mirzadegan T. 2006. Recent progress in the design of small molecule inhibitors of HIV RNase H. Curr. Pharm. Des. 12:1909–1922. 10.2174/138161206776873653 [DOI] [PubMed] [Google Scholar]
- 27.Tomassini J, Selnick H, Davies ME, Armstrong ME, Baldwin J, Bourgeois M, Hastings J, Hazuda D, Lewis J, McClements W. 1994. Inhibition of cap (m7GpppXm)-dependent endonuclease of influenza virus by 4-substituted 2,4-dioxobutanoic acid compounds. Antimicrob. Agents Chemother. 38:2827–2837. 10.1128/AAC.38.12.2827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wai JS, Egbertson MS, Payne LS, Fisher TE, Embrey MW, Tran LO, Melamed JY, Langford HM, Guare JP, Zhuang L, Grey VE, Vacca JP, Holloway MK, Naylor-Olsen AM, Hazuda DJ, Felock PJ, Wolfe AL, Stillmock KA, Schleif WA, Gabryelski LJ, Young SD. 2000. 4-Aryl-2,4-dioxobutanoic acid inhibitors of HIV-1 integrase and viral replication in cells. J. Med. Chem. 43:4923–4926. 10.1021/jm000176b [DOI] [PubMed] [Google Scholar]
- 29.Sluis-Cremer N, Arion D, Abram ME, Parniak MA. 2004. Proteolytic processing of an HIV-1 pol polyprotein precursor: insights into the mechanism of reverse transcriptase p66/p51 heterodimer formation. Int. J. Biochem. Cell Biol. 36:1836–1847. 10.1016/j.biocel.2004.02.020 [DOI] [PubMed] [Google Scholar]
- 30.Tramontano E, Esposito F, Badas R, Di Santo R, Costi R, La Colla P. 2005. 6-[1-(4-Fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester a novel diketo acid derivative which selectively inhibits the HIV-1 viral replication in cell culture and the ribonuclease H activity in vitro. Antiviral Res. 65:117–124. 10.1016/j.antiviral.2004.11.002 [DOI] [PubMed] [Google Scholar]
- 31.Costi R, Metifiot M, Esposito F, Cuzzucoli Crucitti G, Pescatori L, Messore A, Scipione L, Tortorella S, Zinzula L, Novellino E, Pommier Y, Tramontano E, Marchand C, Di Santo R. 2013. 6-(1-Benzyl-1H-pyrrol-2-yl)-2,4-dioxo-5-hexenoic acids as dual inhibitors of recombinant HIV-1 integrase and ribonuclease H, synthesized by a parallel synthesis approach. J. Med. Chem. 56:8588–8598. 10.1021/jm401040b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corona A, Schneider A, Schweimer K, Rösch P, Wöhrl BM, Tramontano E. 2014. Inhibition of foamy virus reverse transcriptase by human immunodeficiency virus type 1 ribonuclease H inhibitors. Antimicrob. Agents Chemother. 58:4086–4093. 10.1128/AAC.00056-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zamborlini A, Coiffic A, Beauclair G, Delelis O, Paris J, Koh Y, Magne F, Giron M-L, Tobaly-Tapiero J, Deprez E, Emiliani S, Engelman A, de Thé H, Saïb A. 2011. Impairment of human immunodeficiency virus type-1 integrase SUMOylation correlates with an early replication defect. J. Biol. Chem. 286:21013–21022. 10.1074/jbc.M110.189274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Esposito F, Sanna C, Del Vecchio C, Cannas V, Venditti A, Corona A, Bianco A, Serrilli AM, Guarcini L, Parolin C, Ballero M, Tramontano E. 2013. Hypericum hircinum L. components as new single-molecule inhibitors of both HIV-1 reverse transcriptase-associated DNA polymerase and ribonuclease H activities. Pathog. Dis. 68:116–124. 10.1111/2049-632X.12051 [DOI] [PubMed] [Google Scholar]
- 35.Delelis O, Parissi V, Leh H, Mbemba G, Petit C, Sonigo P, Deprez E, Mouscadet J-F. 2007. Efficient and specific internal cleavage of a retroviral palindromic DNA sequence by tetrameric HIV-1 integrase. PLoS One 2:e608. 10.1371/journal.pone.0000608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gelderblom HC, Vatakis DN, Burke SA, Lawrie SD, Bristol GC, Levy DN. 2008. Viral complementation allows HIV-1 replication without integration. Retrovirology 5:60. 10.1186/1742-4690-5-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arts EJ, Hazuda DJ. 2012. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2:a007161. 10.1101/cshperspect.a007161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Costi R, Métifiot M, Chung S, Cuzzucoli Crucitti G, Maddali K, Pescatori L, Messore A, Madia VN, Pupo G, Scipione L, Tortorella S, Di Leva FS, Cosconati S, Marinelli L, Novellino E, Le Grice SFJ, Corona A, Pommier Y, Marchand C, Di Santo R. 2014. Basic quinolinonyl diketo acid derivatives as inhibitors of HIV integrase and their activity against RNase H function of reverse transcriptase. J. Med. Chem. 57:3223–3234. 10.1021/jm5001503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jorgensen WL, Maxwell DS, Tirado-Rives J. 1996. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118:11225–11236. 10.1021/ja9621760 [DOI] [Google Scholar]
- 40.Munir S, Thierry S, Subra F, Deprez E, Delelis O. 2013. Quantitative analysis of the time-course of viral DNA forms during the HIV-1 life cycle. Retrovirology 10:87. 10.1186/1742-4690-10-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Julias JG, McWilliams MJ, Sarafianos SG, Arnold E, Hughes SH. 2002. Mutations in the RNase H domain of HIV-1 reverse transcriptase affect the initiation of DNA synthesis and the specificity of RNase H cleavage in vivo. Proc. Natl. Acad. Sci. U. S. A. 99:9515–9520. 10.1073/pnas.142123199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rausch JW, Lener D, Miller JT, Julias JG, Hughes SH, Le Grice SFJ. 2002. Altering the RNase H primer grip of human immunodeficiency virus reverse transcriptase modifies cleavage specificity. Biochemistry 41:4856–4865. 10.1021/bi015970t [DOI] [PubMed] [Google Scholar]
- 43.Delelis O, Thierry S, Subra F, Simon F, Malet I, Alloui C, Sayon S, Calvez V, Deprez E, Marcelin A-G, Tchertanov L, Mouscadet J-F. 2010. Impact of Y143 HIV-1 integrase mutations on resistance to raltegravir in vitro and in vivo. Antimicrob. Agents Chemother. 54:491–501. 10.1128/AAC.01075-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaw-Reid CA, Munshi V, Graham P, Wolfe A, Witmer M, Danzeisen R, Olsen DB, Carroll SS, Embrey M, Wai JS, Miller MD, Cole JL, Hazuda DJ. 2003. Inhibition of HIV-1 ribonuclease H by a novel diketo acid, 4-[5-(benzoylamino)thien-2-yl]-2,4-dioxobutanoic acid. J. Biol. Chem. 278:2777–2780. 10.1074/jbc.C200621200 [DOI] [PubMed] [Google Scholar]
- 45.Alcaro S, Artese A, Ceccherini-Silberstein F, Chiarella V, Dimonte S, Ortuso F, Perno CF. 2010. Computational analysis of human immunodeficiency virus (HIV) type-1 reverse transcriptase crystallographic models based on significant conserved residues found in highly active antiretroviral therapy (HAART)-treated patients. Curr. Med. Chem. 17:290–308. 10.2174/092986710790192695 [DOI] [PubMed] [Google Scholar]
- 46.Nikolenko GN, Palmer S, Maldarelli F, Mellors JW, Coffin JM, Pathak VK. 2005. Mechanism for nucleoside analog-mediated abrogation of HIV-1 replication: balance between RNase H activity and nucleotide excision. Proc. Natl. Acad. Sci. U. S. A. 102:2093–2098. 10.1073/pnas.0409823102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lansdon EB, Liu Q, Leavitt SA, Balakrishnan M, Perry JK, Lancaster-Moyer C, Kutty N, Liu X, Squires NH, Watkins WJ, Kirschberg TA. 2011. Structural and binding analysis of pyrimidinol carboxylic acid and N-hydroxy quinazolinedione HIV-1 RNase H inhibitors. Antimicrob. Agents Chemother. 55:2905–2915. 10.1128/AAC.01594-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su H-P, Yan Y, Prasad GS, Smith RF, Daniels CL, Abeywickrema PD, Reid JC, Loughran HM, Kornienko M, Sharma S, Grobler JA, Xu B, Sardana V, Allison TJ, Williams PD, Darke PL, Hazuda DJ, Munshi S. 2010. Structural basis for the inhibition of RNase H activity of HIV-1 reverse transcriptase by RNase H active site-directed inhibitors. J. Virol. 84:7625–7633. 10.1128/JVI.00353-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.