

Abstract
Elucidation of molecular targets is very important for lead optimization during the drug development process. We describe a direct method to find targets of antitrypanosomal compounds against Trypanosoma brucei using a trypanosome overexpression library. As proof of concept, we treated the library with difluoromethylornithine and DDD85646 and identified their respective targets, ornithine decarboxylase and N-myristoyltransferase. The overexpression library could be a useful tool to study the modes of action of novel antitrypanosomal drug candidates.
TEXT
The development of drugs against tropical diseases has been greatly boosted by public-private partnerships such as the Medicines for Malaria Venture and the Drugs for Neglected Diseases Initiative. The majority of novel chemical lead compounds for malaria have been discovered through live-cell phenotypic screens (1, 2), and similar efforts are now under way for the kinetoplastid organisms, trypanosomes, and Leishmania spp. (3). Although such compounds can be taken into the clinic without any knowledge of their mechanisms of action, lead optimization and development are greatly facilitated by knowledge of the molecular target. Several strategies are available for this. Selection of drug-resistant mutants may yield organisms that have mutations in the target protein or alterations in drug activation or uptake (4, 5). Other possibilities include metabolomic studies of treated organisms (6) and affinity chromatography approaches (7). Alternatively, cultivation of Leishmania in the presence of increasing drug concentrations leads to episomal gene amplification. Thus, sequencing of the genes present on the episomes can reveal the direct drug target itself (8) or proteins that facilitate drug resistance by other means, such as increased drug extrusion (9). An alternative approach for Leishmania is transfection of cosmid libraries, amplification of which could cause overexpression in the same way (10).
African trypanosomes do not spontaneously form episomes in response to drug selection, and gene amplification is unusual. High-throughput screening of trypanosome RNA interference (RNAi) libraries has been extraordinarily useful in elucidating the mechanisms of drug uptake (11, 12). It would be difficult, however, to use this reduced-function approach to find the target of an antitrypanosomal drug. Drugs often act by inhibiting essential enzymes, and depletion of the target should be lethal even in the absence of drug. Although weak RNAi could in theory result in slowly growing cells with enhanced drug susceptibility, in practice the cells are likely to be dead even without added drug. Moreover, if a cell is already “unhealthy” due to impairment of one pathway, it may be generally more susceptible to drugs apart from the pathway targeted. Alternative methodologies, based on gain-of-function approaches, are therefore much more appropriate for direct determination of the targets of toxic compounds (13). If an antitrypanosomal drug inhibits the function of a single protein, then overexpression of that protein or a fragment containing the drug binding site could result in reduced drug sensitivity (14).
In the course of another project, we created a library of trypanosomes, each of which is capable of overexpressing a different protein fragment in a tetracycline-inducible fashion (15). The overexpression library was constructed using randomly sheared DNA (sizes of 0.7 to 3 kbp) ligated into a plasmid suitable for tetracycline-inducible expression (Fig. 1A). The plasmid was built such that each insert sequence should be expressed as a protein with a short foreign peptide sequence at the N terminus (lambda-N peptide). Authentic trypanosome proteins will be expressed as lambda-N fusion peptides if the insert sequence contains a trypanosome open reading frame (ORF) that is in frame and in the correct orientation. High-throughput sequencing results suggested that approximately one in 25 plasmids fulfilled this criterion, meaning that the library has at least 10-fold coverage of Trypanosoma brucei ORFs (15).
FIG 1.
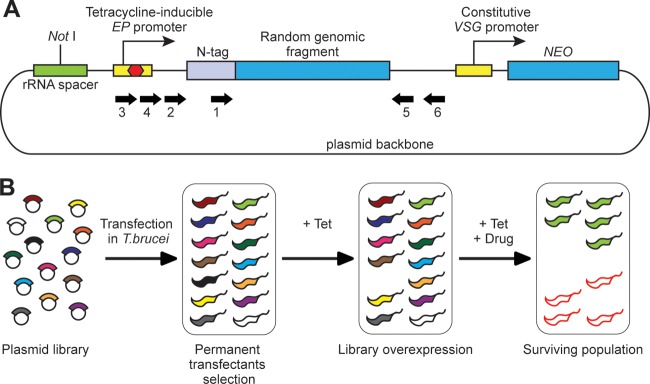
Overexpression approach for drug target discovery in T. brucei. (A) Schematic representation (not in scale) of the overexpression plasmid pHD2542 containing a random genomic fragment. The library insert is expressed with an N-terminal tag, the lambda peptide (N-tag). Expression is controlled by a tetracycline-inducible EP procyclin promoter, which is known to be able to drive overexpression in both bloodstream- and procyclic-form trypanosomes (26). The transfected cells are selected through resistance to neomycin (NEO), stably expressed from a constitutive VSG promoter. The linearized plasmid is integrated into a silent rRNA spacer. The inserts were identified using specific primers (thick arrows). Primer sequences (all from 5′ to 3′) were as follows: primer 1 (illuminaF), TAGCCACCGGGCCC; primer 2 (SeqLib2), CTTGAAGACTTCAATTACACC; primer 3 (SeqLib3), CTATCAGTGATAGAGATCCC; primer 4 (SeqLib4), TTAACATGTTCTCGTCCC; primer 5 (illuminaR), CACACAAATGGATCCTCAGC; primer 6 (LibR), CTGTACGTAAATGTGTTGC. (B) Schematic representation of the approach for drug target screening. The plasmid library is transfected into bloodstream-form T. brucei, and permanent transfectants are selected through resistance to neomycin. Library overexpression is then induced with the addition of tetracycline (+ Tet), and only cells expressing nontoxic polypeptides survive. Following 24-h tetracycline induction, the studied drug is added (+ Drug), and a surviving population is selected after some days. The population is composed of cells overexpressing the target (green with black outlines) and other cells (red outlines) that acquired a growth advantage under drug pressure, presumably through genomic mutations.
We transfected the plasmid library DNA into bloodstream-form T. brucei 2T1 cells, taking advantage of endonuclease-facilitated recombination (16), to obtain over 3 million clones (15). Since this is a random shotgun library, most plasmids do not encode full-length trypanosome proteins, and the presence of the N-terminal peptide precludes targeting of proteins to the secretory pathway or mitochondria. Correct targeting may not be necessary, however, in order for a protein active site to bind a drug and confer resistance. Since none of the inserts is more than 3 kbp, and most are substantially smaller, the library may not encode functional multidrug resistance carriers.
To determine whether our trypanosome overexpression library could be used to identify drug targets, we cultured it in the presence of well-characterized inhibitors of essential enzymes (Fig. 1B). As a first proof of concept, difluoromethylornithine (DFMO) (purchased from Sigma) was chosen. DFMO is clinically used for the treatment of human African trypanosomiasis (HAT). It targets ornithine decarboxylase (ODC) (17), an enzyme involved in the production of trypanothione, which is essential for redox homeostasis in trypanosomes (18). It is known that overexpression of ODC can yield resistance in Leishmania donovani (19), and we showed that, in trypanosomes, it clearly confers a selective advantage to cells growing under DFMO pressure (Fig. 2).
FIG 2.
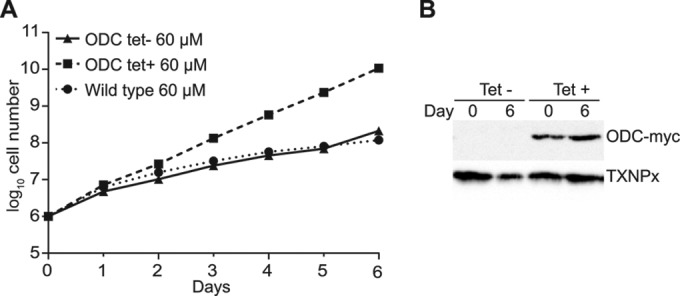
ODC overexpression supporting growth under DFMO pressure. (A) Cumulative growth curves for wild-type cells and induced (ODC tet+) and noninduced (ODC tet−) ODC-myc overexpression clones in the presence of 60 μM DFMO. (B) Protein samples collected at day 0 and day 6 and analyzed by Western blotting with anti-myc antibody. Tryparedoxin peroxidase (TXNPx) was used as a loading control.
We grew wild-type and ODC-overexpressing cells with a range of DFMO concentrations over 6 days, and we selected a concentration at which overexpression gave a growth advantage (Fig. 2). A culture of at least 4 × 107 cells (10-fold coverage) was then grown in HMI-11 medium (20) containing tetracycline (1 μg/ml) for 24 h before the initiation of drug selection; a separate similar culture was grown without tetracycline. DFMO at 60 μM was added on day 0 (Fig. 3A). The cells were grown to a maximum density of 2 × 106 cells/ml and were diluted to 2 × 105 cells/ml as necessary. The induced library initially grew slightly more slowly than noninduced cells (Fig. 3A), but the induced library grew faster from day 10, suggesting that some cells within the population had a growth advantage. Samples were collected at day 12 (Fig. 3A), and the library inserts present in the cells were amplified with primers 1 and 5 (Fig. 1A). The amplification pattern for the induced library (day 12, with tetracycline) was significantly less complex than that for the initial library (day 0) (Fig. 3B). The three major visible bands were sequenced using primer 4 (Fig. 1A). The upper band corresponded to the ODC ORF (Fig. 3B), lacking the codons for the first four amino acids, expressed in frame with the N-terminal tag. This confirmed that we could select the drug target using our library; the likelihood of randomly obtaining the expected gene, complete or in frame with the N-terminal tag, was less than one in 3.5 × 105. The two lower bands corresponded to three different sequences, none of which encoded bona fide trypanosome proteins; they contained intergenic regions or ORFs that were out of frame or in the antisense orientation. We cannot rule out the possibility that the short overexpressed peptides yielded drug resistance, but we think that it is more likely that the trypanosomes became resistant through more-conventional means, such as mutations in ODC or reduced drug uptake (11, 21).
FIG 3.
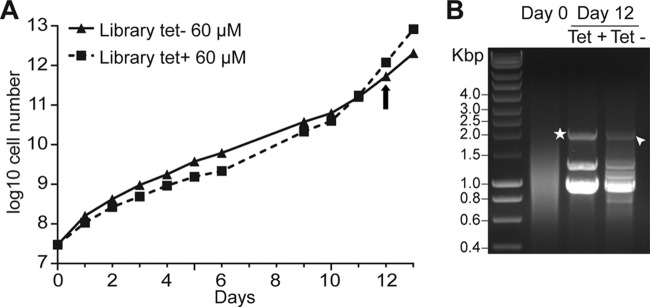
Selection of ODC-overexpressing cells from the library under DFMO pressure. (A) Cumulative growth curves for the trypanosome library in the presence of 60 μM DFMO. Samples of cells were collected at day 12 (arrow). (B) Library inserts amplified with primers 1 and 5 (Fig. 1), using genomic DNA as the template. Star, PCR product encoding ODC. The band indicated by the arrowhead did not contain the ODC gene.
The PCR pattern of the noninduced library (day 12, no tetracycline) was more complex than that of the induced library (day 12, tetracycline treated) but less complex than that of the initial library (day 0). The most enriched bands were sequenced, but the ODC gene was not contained in any of them. The loss of complexity in population without tetracycline was expected. If a small number of cells have a growth advantage due to genomic mutations, then only the plasmid inserts that are present in those cells will be detected after selection. This may explain why the background patterns in the lanes with and without tetracycline appear similar. However, the sizes of the bands are no indication of their identities, since the library contained 3 × 106 fragments within a limited size range; the band indicated by an arrowhead in Fig. 3B, lane Tet−, did not encode ODC.
Next, we decided to investigate whether this approach was suitable for cytocidal compounds, since these are preferable for treatment (22). For this, we used DDD85646 (a generous gift from Paul Wyatt), an N-myristoyltransferase (NMT) inhibitor (23, 24). NMT is an essential protein involved in protein myristoylation (25). The trypanosome library was grown as in the previous experiment, but in this experiment the cells were all induced with tetracycline. The cells were treated with DDD85646 concentrations ranging between 5 nM and 12.5 nM (the 3-day 50% effective concentration [EC50] is 2 nM [23]). Many cells were killed and in several cultures the numbers decreased below the detection limit, but in all cases the cultures recovered. At this point, samples were collected (Fig. 4A and B). As seen with DFMO, the patterns of amplified DNA (amplification with primers 6 and 3) (Fig. 1A) were considerably less complex than those for the initial library (Fig. 4C). The bands were reamplified with primers 4 and 5 (Fig. 1A) and directly sequenced with primer 2 (Fig. 1A) after gel purification. Only one of the bands, which was present under three conditions (5, 6, and 8.5 nM), encoded a bona fide trypanosome protein, and it contained the full-length NMT gene (Fig. 4C). This result showed that the library can be used to select for the targets of cytotoxic drugs.
FIG 4.
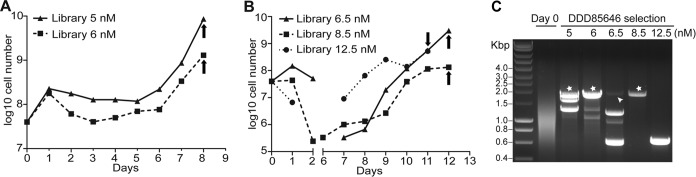
Selection of NMT-overexpressing cells under DDD85646 pressure. (A and B) Cumulative growth curves for the trypanosome library in the presence of a range of concentrations (5, 6, 6.5, 8.5, and 12.5 nM) of DDD85646. Arrows, times when samples of cells were collected. In panel B, the cells were below the detection limit from day 2 to day 6. To simplify the experiment, no culture without tetracycline was included. (C) Library inserts amplified with primers 1 and 5 (Fig. 1), using genomic DNA as the template. Stars, bands encoding N-myristoyltransferase. The band indicated by the arrowhead did not encode N-myristoyltransferase.
No other bands, including the band with similar length in the 6.5 nM sample (Fig. 4C, arrowhead), contained the NMT gene. Presumably, as for DFMO selection, the remaining trypanosomes had become resistant through genomic changes unrelated to the presence of the overexpressed fusion peptides. In some cases, the selected cells seemed to have attained a greater growth advantage than from NMT overexpression (e.g., 6.5 nM versus 8.5 nM) (Fig. 4B). If this approach is used to study drugs whose targets are not known, then it will be essential to test likely candidates individually through retransfection of expression plasmids into sensitive cells; this will show whether the encoded protein can confer resistance. If it cannot, then the parasite clone that harbored the sequence probably became resistant through other means, and the reason for this resistance can be analyzed.
In both studied cases, despite the calculated 10-fold coverage, only one of the bands contained the target gene. This is probably due to the fact that the library was prepared using random fragmentation. As a consequence, most clones expressed only part of the protein (e.g., C terminus). In the cases analyzed here, amino acids involved in folding and binding to the drug are spread along the entire sequence (17, 24). Therefore, we selected only expressed polypeptides that were long enough to ensure proper folding and drug binding and thus to confer a growth advantage during drug selection. This is a clear limitation of the random shotgun library. A further limitation is that, if the required open reading frame is longer than 3 kb, the library will not contain it. Our method will also probably not be useful for drugs that have a complex mechanism of action (for example, targeting multiple proteins or nonprotein targets).
In summary, selection of our trypanosome overexpression library successfully identified the targets of two drugs, one cytostatic and one cytocidal, that work by inhibiting a single enzyme. Therefore, this will be a very useful tool in target elucidation for the next generation of drugs against human and animal African trypanosomiasis. A similar approach could also be used for other kinetoplastid organisms, such as Trypanosoma cruzi and Leishmania.
ACKNOWLEDGMENTS
We thank Paul Wyatt (Drug Discovery Unit, University of Dundee) for the gift of DDD85646, the N-myristoyltransferase inhibitor. We thank Michael Barrett (University of Glasgow) and Alan Fairlamb (University of Dundee) for useful discussions.
Daniela Begolo and this project are funded by the European Commission FP7 Marie Curie Initial Training Network “ParaMet” (grant 290080).
Footnotes
Published ahead of print 21 July 2014
REFERENCES
- 1.Gamo FJ, Sanz LM, Vidal J, de Cozar C, Alvarez E, Lavandera JL, Vanderwall DE, Green DV, Kumar V, Hasan S, Brown JR, Peishoff CE, Cardon LR, Garcia-Bustos JF. 2010. Thousands of chemical starting points for antimalarial lead identification. Nature 465:305–310. 10.1038/nature09107 [DOI] [PubMed] [Google Scholar]
- 2.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, Smithson DC, Connelly M, Clark J, Zhu F, Jimenez-Diaz MB, Martinez MS, Wilson EB, Tripathi AK, Gut J, Sharlow ER, Bathurst I, El Mazouni F, Fowble JW, Forquer I, McGinley PL, Castro S, Angulo-Barturen I, Ferrer S, Rosenthal PJ, Derisi JL, Sullivan DJ, Lazo JS, Roos DS, Riscoe MK, Phillips MA, Rathod PK, Van Voorhis WC, Avery VM, Guy RK. 2010. Chemical genetics of Plasmodium falciparum. Nature 465:311–315. 10.1038/nature09099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Don R, Ioset JR. 2014. Screening strategies to identify new chemical diversity for drug development to treat kinetoplastid infections. Parasitology 141:140–146. 10.1017/S003118201300142X [DOI] [PubMed] [Google Scholar]
- 4.Spillman NJ, Allen RJ, McNamara CW, Yeung BK, Winzeler EA, Diagana TT, Kirk K. 2013. Na+ regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe 13:227–237. 10.1016/j.chom.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McNamara CW, Lee MC, Lim CS, Lim SH, Roland J, Nagle A, Simon O, Yeung BK, Chatterjee AK, McCormack SL, Manary MJ, Zeeman AM, Dechering KJ, Kumar TR, Henrich PP, Gagaring K, Ibanez M, Kato N, Kuhen KL, Fischli C, Rottmann M, Plouffe DM, Bursulaya B, Meister S, Rameh L, Trappe J, Haasen D, Timmerman M, Sauerwein RW, Suwanarusk R, Russell B, Renia L, Nosten F, Tully DC, Kocken CH, Glynne RJ, Bodenreider C, Fidock DA, Diagana TT, Winzeler EA. 2013. Targeting Plasmodium PI(4)K to eliminate malaria. Nature 504:248–253. 10.1038/nature12782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vincent IM, Creek DJ, Burgess K, Woods DJ, Burchmore RJ, Barrett MP. 2012. Untargeted metabolomics reveals a lack of synergy between nifurtimox and eflornithine against Trypanosoma brucei. PLoS Negl. Trop. Dis. 6:e1618. 10.1371/journal.pntd.0001618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mercer L, Bowling T, Perales J, Freeman J, Nguyen T, Bacchi C, Yarlett N, Don R, Jacobs R, Nare B. 2011. 2,4-Diaminopyrimidines as potent inhibitors of Trypanosoma brucei and identification of molecular targets by a chemical proteomics approach. PLoS Negl. Trop. Dis. 5:e956. 10.1371/journal.pntd.0000956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coderre JA, Beverley SM, Schimke RT, Santi DV. 1983. Overproduction of a bifunctional thymidylate synthetase-dihydrofolate reductase and DNA amplification in methotrexate-resistant Leishmania tropica. Proc. Natl. Acad. Sci. U. S. A. 80:2132–2136. 10.1073/pnas.80.8.2132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Legare D, Papadopoulou B, Roy G, Mukhopadhyay R, Haimeur A, Dey S, Grondin K, Brochu C, Rosen BP, Ouellette M. 1997. Efflux systems and increased trypanothione levels in arsenite-resistant Leishmania. Exp. Parasitol. 87:275–282. 10.1006/expr.1997.4222 [DOI] [PubMed] [Google Scholar]
- 10.Cotrim PC, Garrity LK, Beverley SM. 1999. Isolation of genes mediating resistance to inhibitors of nucleoside and ergosterol metabolism in Leishmania by overexpression/selection. J. Biol. Chem. 274:37723–37730. 10.1074/jbc.274.53.37723 [DOI] [PubMed] [Google Scholar]
- 11.Schumann Burkard G, Jutzi P, Roditi I. 2011. Genome-wide RNAi screens in bloodstream form trypanosomes identify drug transporters. Mol. Biochem. Parasitol. 175:91–94. 10.1016/j.molbiopara.2010.09.002 [DOI] [PubMed] [Google Scholar]
- 12.Alsford S, Turner DJ, Obado SO, Sanchez-Flores A, Glover L, Berriman M, Hertz-Fowler C, Horn D. 2011. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 21:915–924. 10.1101/gr.115089.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alsford S, Kelly JM, Baker N, Horn D. 2013. Genetic dissection of drug resistance in trypanosomes. Parasitology 140:1478–1491. 10.1017/S003118201300022X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ranade RM, Gillespie JR, Shibata S, Verlinde CL, Fan E, Hol WG, Buckner FS. 2013. Induced resistance to methionyl-tRNA synthetase inhibitors in Trypanosoma brucei is due to overexpression of the target. Antimicrob. Agents Chemother. 57:3021–3028. 10.1128/AAC.02578-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erben ED, Fadda A, Lueong S, Hoheisel JD, Clayton C. 2014. A genome-wide tethering screen reveals novel potential post-transcriptional regulators in Trypanosoma brucei. PLoS Pathog. 10:e1004178. 10.1371/journal.ppat.1004178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glover L, Horn D. 2009. Site-specific DNA double-strand breaks greatly increase stable transformation efficiency in Trypanosoma brucei. Mol. Biochem. Parasitol. 166:194–197. 10.1016/j.molbiopara.2009.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grishin NV, Osterman AL, Brooks HB, Phillips MA, Goldsmith EJ. 1999. X-ray structure of ornithine decarboxylase from Trypanosoma brucei: the native structure and the structure in complex with α-difluoromethylornithine. Biochemistry 38:15174–15184. 10.1021/bi9915115 [DOI] [PubMed] [Google Scholar]
- 18.Fairlamb AH, Blackburn P, Ulrich P, Chait BT, Cerami A. 1985. Trypanothione: a novel bis(glutathionyl)spermidine cofactor for glutathione reductase in trypanosomatids. Science 227:1485–1487. 10.1126/science.3883489 [DOI] [PubMed] [Google Scholar]
- 19.Hanson S, Beverley SM, Wagner W, Ullman B. 1992. Unstable amplification of two extrachromosomal elements in alpha-difluoromethylornithine-resistant Leishmania donovani. Mol. Cell. Biol. 12:5499–5507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirumi H, Hirumi K. 1989. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 75:985–989. 10.2307/3282883 [DOI] [PubMed] [Google Scholar]
- 21.Vincent IM, Creek D, Watson DG, Kamleh MA, Woods DJ, Wong PE, Burchmore RJ, Barrett MP. 2010. A molecular mechanism for eflornithine resistance in African trypanosomes. PLoS Pathog. 6:e1001204. 10.1371/journal.ppat.1001204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilbert IH. 2014. Target-based drug discovery for human African trypanosomiasis: selection of molecular target and chemical matter. Parasitology 141:28–36. 10.1017/S0031182013001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frearson JA, Brand S, McElroy SP, Cleghorn LA, Smid O, Stojanovski L, Price HP, Guther ML, Torrie LS, Robinson DA, Hallyburton I, Mpamhanga CP, Brannigan JA, Wilkinson AJ, Hodgkinson M, Hui R, Qiu W, Raimi OG, van Aalten DM, Brenk R, Gilbert IH, Read KD, Fairlamb AH, Ferguson MA, Smith DF, Wyatt PG. 2010. N-Myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature 464:728–732. 10.1038/nature08893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brand S, Cleghorn LA, McElroy SP, Robinson DA, Smith VC, Hallyburton I, Harrison JR, Norcross NR, Spinks D, Bayliss T, Norval S, Stojanovski L, Torrie LS, Frearson JA, Brenk R, Fairlamb AH, Ferguson MA, Read KD, Wyatt PG, Gilbert IH. 2012. Discovery of a novel class of orally active trypanocidal N-myristoyltransferase inhibitors. J. Med. Chem. 55:140–152. 10.1021/jm201091t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price HP, Guther ML, Ferguson MA, Smith DF. 2010. Myristoyl-CoA:protein N-myristoyltransferase depletion in trypanosomes causes avirulence and endocytic defects. Mol. Biochem. Parasitol. 169:55–58. 10.1016/j.molbiopara.2009.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biebinger S, Wirtz LE, Lorenz P, Clayton C. 1997. Vectors for inducible expression of toxic gene products in bloodstream and procyclic Trypanosoma brucei. Mol. Biochem. Parasitol. 85:99–112. 10.1016/S0166-6851(96)02815-0 [DOI] [PubMed] [Google Scholar]