

Abstract
It is important to understand the relationship between antibiotic exposure and the selection of drug resistance in the context of therapy exposure. We sought to identify the ceftolozane-tazobactam exposure necessary to prevent the amplification of drug-resistant bacterial subpopulations in a hollow-fiber infection model. Two Pseudomonas aeruginosa challenge isolates were selected for study, a wild-type ATCC strain (ceftolozane-tazobactam MIC, 0.5 mg/liter) and a clinical isolate (ceftolozane-tazobactam MIC, 4 mg/liter). The experiment duration was 10 days, and the ceftolozane-tazobactam dose ratio (2:1) and dosing interval (every 8 h) were selected to approximate those expected to be used clinically. The studied ceftolozane-tazobactam dosing regimens ranged from 62.5/31.25 to 2,000/1,000 mg per dose in step fold dilutions. Negative-control arms included no treatment and tazobactam at 500 mg every 8 h. Positive-control arms included ceftolozane at 1 g every 8 h and piperacillin-tazobactam dosed at 4.5 g every 6 h. For the wild-type ATCC strain, resistance was not selected by any ceftolozane-tazobactam regimen evaluated. For the clinical isolate, an inverted-U-shaped function best described the relationship between the amplification of a drug-resistant subpopulation and drug exposure. The least (62.5/31.25 mg) and most (2,000/1,000 mg) intensive ceftolozane-tazobactam dosing regimens did not select for drug resistance. Drug resistance selection was observed with intermediately intensive dosing regimens (125/62.5 through 1,000/500 mg). For the intermediately intensive ceftolozane-tazobactam dosing regimens, the duration until the selection for drug resistance increased with dose regimen intensity. These data support the selection of ceftolozane-tazobactam dosing regimens that minimize the potential for on-therapy drug resistance selection.
INTRODUCTION
Antibiotic-resistant Pseudomonas aeruginosa is a serious clinical problem. The risk factors for the recovery of antibiotic-resistant P. aeruginosa from clinical culture specimens include hospitalization stays of more than 10 days, immunocompromised status, cardiopulmonary disease (1), and prior therapy with an antipseudomonal antibiotic (2).
Suboptimal antibiotic exposure may select antibiotic resistance on therapy. Tam et al. identified the relationship between garenoxacin exposure and antibiotic resistance amplification of P. aeruginosa using a hollow-fiber infection model. The identified relationship took the functional form of an inverted U, where at low and high garenoxacin exposures, resistance selection was minimized, while at intermediate exposures, it was higher (3). Tam et al. also demonstrated that the garenoxacin exposure necessary to prevent resistance amplification increased with duration of therapy. Similarly, using a hollow-fiber infection model, it was previously demonstrated for ceftolozane-tazobactam that the relationship between drug exposure and resistance amplification of CTX-M-15-producing Escherichia coli also took on the form of an inverted U (15). In these studies, ceftolozane-tazobactam dosing regimens greater than 750/375 mg every 8 h suppressed amplification of antibiotic-resistant bacterial subpopulations.
Ceftolozane-tazobactam is a combination of a novel cephalosporin with a β-lactamase inhibitor. Ceftolozane has potent in vitro (5) and in vivo (6) activities against many Enterobacteriaceae and P. aeruginosa. Given the morbidity and mortality associated with infections associated with antibiotic-resistant P. aeruginosa (7) and the paucity of currently available antibiotics to treat such infections, it is critical to understand the conditions under which selection of ceftolozane-tazobactam resistance can be minimized using dose ratio, dosing interval, and experimental duration that approximate those expected to be used clinically. Thus, the overarching goal of these studies was to identify the ceftolozane-tazobactam exposure necessary to prevent the selection of drug-resistant P. aeruginosa using an in vitro hollow-fiber infection model.
MATERIALS AND METHODS
Bacteria, antimicrobials, and β-lactamase inhibitor.
Ceftolozane and tazobactam were provided by Cubist Pharmaceuticals (Lexington, MA), while piperacillin was obtained from Sigma-Aldrich (St. Louis, MO). Two isolates of the challenge organism, P. aeruginosa, were selected for study based upon phenotypic characteristics. One was a wild-type strain (PAO1 BAA-47; American Type Culture Collection, Manassas, VA) with a MIC value that approximated the MIC50 and was piperacillin-tazobactam susceptible. The other was a clinical isolate (PAE 2638) with a MIC value that approximated the MIC90 and was piperacillin-tazobactam resistant. Previous genotypic studies with this isolate indicated that it exhibited elevated transcription levels of AmpC and efflux pump-encoding genes (mexA and mexX). In addition, this strain produced a narrow-spectrum β-lactamase enzyme (PSE-10) (data on file, Cubist Pharmaceuticals, Lexington, MA).
Media and in vitro susceptibility studies.
Susceptibility testing studies were performed using Clinical and Laboratory Standards Institute guidelines (8) for broth microdilution and agar dilution methods utilizing cation-adjusted Mueller-Hinton broth and Mueller-Hinton agar media (BD Laboratories, Franklin Lakes, NJ). Ceftolozane and piperacillin susceptibility tests were conducted alone and in combination with a fixed tazobactam concentration (4 mg/liter). All susceptibility studies were performed in triplicate over a 2-day period, and the MIC results presented as the modal values. Ceftolozane-tazobactam susceptibility tests were also conducted with and without the presence of 40 mg/liter of a broad-spectrum pump inhibitor (Phe-Arg-β-naphthylamide dihydrochloride) obtained from Sigma-Aldrich. P. aeruginosa ATCC 27853 was utilized as an internal control for all susceptibility testing.
Mutation frequency studies.
The frequency of mutation to drug resistance was determined for ceftolozane-tazobactam by plating 4 ml of log-phase growth suspension onto agar containing ceftolozane at concentrations of 1.5, 3, and 5 times the baseline MIC value and tazobactam at 4 mg/liter. Similarly, the frequency of mutation to drug resistance was estimated for piperacillin-tazobactam by plating 4 ml of log-phase growth suspension onto agar containing piperacillin at concentrations of 3 and 5 times the baseline MIC value and tazobactam at 4 mg/liter. The bacterial concentration within each suspension was determined by quantitative culture. The ratio of growth found on the drug-containing plates to that of the starting inoculum provided an estimate of the drug resistance frequency within a total population. This assay was performed in duplicate and from each hollow-fiber study. If growth was observed at 48 h, a subset of isolates were taken from each set of drug-containing plates and tested for a change in the MIC from the baseline to confirm decreased drug susceptibility.
Hollow-fiber infection model.
The hollow-fiber infection model has been used extensively for the examination of resistance prevention and was described previously (9, 10). In brief, this pharmacodynamic system allows pathogens to grow in a peripheral chamber of the hollow-fiber cartridge. The peripheral chamber is separated from the central compartment by semipermeable membranes. These membranes have pores with sizes that are large enough to allow nutrients, drugs, and bacterial metabolites to transverse freely into and out of the peripheral compartment but are too small for bacteria to leave the peripheral compartment. Fresh medium is pumped in and out of the central compartment using peristaltic pumps, while the challenge compounds (i.e., antibiotics) are pumped into the central compartment under computer control using multiple infusion pumps to simulate the different compound half-lives. Due to the high surface area-to-volume ratio, drug concentrations equilibrate rapidly in the peripheral compartments. Specimens for quantitative culture and drug concentration assay can be removed from the peripheral compartment through sampling ports. In the experiments described herein, we utilized cartridges from FiberCell systems Inc. (Frederick, MD) with a 12-ml volume.
Prevention of resistance amplification.
Each ceftolozane-tazobactam resistance amplification study included active and inactive control regimens. Ceftolozane-tazobactam regimens included a range of doses (125/62.5 to 2,000/1,000 mg) infused over 1 h every 8 h for 10 days, which varied by study isolate. Active control regimens included piperacillin-tazobactam at 4.5 g infused over 1 h every 6 h and ceftolozane at 1,000 mg infused over 1 h every 8 h, each for 10 days. Inactive control regimens included a no-treatment growth control and tazobactam at 500 mg infused over 1 h every 8 h for 10 days. Studies were conducted in duplicate for the clinical isolate and singularly for the wild-type strain.
For each study, the initial challenge isolate inoculum was prepared from an overnight culture in Trypticase soy agar with 5% sheep blood (BD Laboratories) at a temperature of 35°C. Colonies from each overnight culture were grown to mid-log phase and then suspended in an Erlenmeyer flask containing cation-adjusted Mueller-Hinton medium set in a water shaker bath (125 rotations per minute) at a temperature of 35°C. The bacterial concentration within the flask of Mueller-Hinton broth was determined by optical density using a previously confirmed growth curve for each challenge isolate. Subsequently, 12 ml of the bacterial suspension was inoculated into the extracapillary space of the hollow-fiber cartridges (FiberCell Systems) to achieve a final concentration of 1.0 × 108 CFU/ml.
Bacteria were exposed to fluctuating free-drug concentrations within the hollow-fiber cartridge that simulated the steady-state pharmacokinetics reported for healthy volunteers of 2.5 h for ceftolozane, 1 h for tazobactam, and 1 h for piperacillin (11, 12). Protein binding levels were assumed to be 20% for ceftolozane (Cubist, data on file) and 30% for tazobactam and piperacillin (11).
During the course of the 10-day experiment, 1-ml samples were taken from the extracapillary space at 0 and 5 h and days 1, 2, 3, 4, 6, 8, and 10. All samples were washed twice with sterile normal saline, serially diluted, and quantitatively cultured on drug-free Trypticase soy agar with 5% sheep blood agar plates to determine the effect of treatment on the total bacterial population. A portion of each sample was plated on Mueller-Hinton agar plates containing 1.5, 3, and/or 5 times the baseline MIC to ceftolozane alone or in combination with tazobactam (4 mg/liter) or piperacillin in combination with tazobactam (4 mg/liter) for the enumeration of the resistant subpopulation. Susceptibility studies were conducted for a subset of isolates found growing on the drug-containing plates on days 1, 3, 6, and 10 of each study. Susceptibility studies were conducted with and without the presence of 40 mg/liter of Phe-Arg-β-naphthylamide dihydrochloride.
Pharmacokinetic validation studies.
Over the first 48 h of each study, 1-ml specimens for drug assay were collected from the peripheral compartment at 1, 3, 5, 7, 9, 23, 25, 29, 31, 33, and 48 h and then immediately frozen at −80°C until assayed for drug concentration. Ceftolozane, piperacillin, and tazobactam concentrations were measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS) completed by Microconstants, San Diego, CA.
Analytical method.
All samples were assayed by LC-MS/MS (Waters, Milford, MA). The standard curve for ceftolozane was linear over a concentration range from 0.100 to 500 mg/liter. For tazobactam, the standard curve was linear over a concentration range from 0.100 to 100 mg/liter. The standard curve for piperacillin was linear over a concentration range from 0.500 to 500 mg/liter. The interday coefficient of variation (CV) for the ceftolozane, tazobactam, and piperacillin quality control samples ranged from 5.34 to 9.38, 1.93 to 7.42, and 1.15 to 5.55%, respectively. The lower limit of quantification was 0.1 mg/liter for all three compounds.
Pharmacokinetic-pharmacodynamic analysis.
Table 1 shows the predicted pharmacokinetic parameters and pharmacokinetic-pharmacodynamic measures for each study.
TABLE 1.
Ceftolozane-tazobactam free pharmacokinetic parameters and percent time above MICs for the wild-type and clinical isolatesa
| Parameter/measure | Value for single drug or combination |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ceftolozane at 1,000 mg | Ceftolozane/tazobactam at: |
Piperacillin-tazobactam at 4,000/500 mg | |||||||
| 62.5/31.25 mg | 125/62.5 mg | 250/125 mg | 500/250 mg | 750/375 mg | 1,000/500 mg | 2,000/1,000 mg | |||
| Cmaxb (mg/liter) | 49.2 | 2.16 | 5.6 | 13 | 27.2 | 39.7 | 49.2 | 111.4 | 209.3 |
| AUC0–24c (mg · h/liter) | 456.2 | 20 | 52.4 | 120.3 | 252.2 | 368.3 | 456.2 | 1,032.63 | 900.92 |
| % time above MIC for the WTd isolate | 100 | 80 | 100 | 100 | 100 | 100 | 100 | 100 | 92 |
| % time above MIC for the clinical isolate | 100 | 0 | 12.5 | 64 | 97.5 | 100 | 100 | 100 | Unknown |
The ceftolozane-tazobactam MICs for the wild-type isolate and the clinical isolate are 0.5 and 4 mg/liter, respectively. The piperacillin-tazobactam MIC for the wild-type isolate is 2 mg/liter.
Cmax, maximum concentration.
AUC0–24, area under the concentration-time curve from 0 to 24 h.
WT, wild type.
RESULTS
In vitro susceptibility studies.
Susceptibility test results are presented in Table 2. The MIC values for the quality control strain, P. aeruginosa ATCC 27853, were 2 to 4 mg/liter and 0.5 mg/liter for piperacillin-tazobactam and ceftolozane-tazobactam, respectively, both within the CLSI quality control range (13). The broth microdilution MIC values of ceftolozane were 0.5 and 4 mg/liter for PAO1 BAA-47 and PAE 2638, respectively, and were not potentiated by 4 mg/liter of tazobactam. The broth microdilution MIC values of piperacillin-tazobactam were 2 and >128 mg/liter for PAO1 BAA-47 and PAE 2638, respectively. Agar susceptibility test results were similar to those obtained using the broth microdilution method.
TABLE 2.
MICs for Pseudomonas aeruginosa isolates utilized in the hollow-fiber infection model studiesa
| Isolate | MIC (mg/liter) |
||||||
|---|---|---|---|---|---|---|---|
| Broth microtiter |
Agar dilution |
||||||
| TOL alone | TOL/TAZ (4 mg/liter) | PIP/TAZ (4 mg/liter) | TOL alone | TOL/TAZ (4 mg/liter) | TOL/TAZ (4 mg/liter) + pump inhibitorb | PIP/TAZ (4 mg/liter) | |
| PAO1 BAA-47 | 0.5 | 0.5 | 2 | 0.25 | 0.25 | 0.25 | 2 |
| PAE 2638 | 4 | 4 | >128 | 4 | 4 | 2 | >128 |
TOL, ceftolozane; TAZ, tazobactam; PIP, piperacillin.
Pump inhibitor was 40 mg/liter of Phe-Arg-β-naphthylamide dihydrochloride.
Mutation frequency studies.
For PAO1 BAA-47, at 1.5, 3, and 5 times the baseline ceftolozane-tazobactam MIC value, the average density of the drug-resistant subpopulation was 1 CFU in 7.24 × 107, 1.15 × 108, and 2.69 × 108 CFU/ml of bacterial growth, respectively. For piperacillin against PAO1 BAA-47, at 3 and 5 times the baseline piperacillin-tazobactam MIC value, the average density of the drug-resistant subpopulation was 1 CFU in 1.62 × 107 and 4.26 × 107 CFU/ml of bacterial growth, respectively.
The ceftolozane-tazobactam MIC values of the PAO1 BAA-47 strain taken from the drug-containing plates ranged from 2 to 8 mg/liter, regardless of the concentration of drug found within the agar plate. The ceftolozane-tazobactam MIC values decreased in the presence of a broad-spectrum efflux pump inhibitor (Phe-Arg-β-naphthylamide dihydrochloride), dropping from 2 to 8 mg/liter to 1 to 2 mg/liter. Similarly, the piperacillin-tazobactam MIC value for the PAO1 BAA-47 strain taken from the drug-containing plates ranged from 32 to 64 mg/liter, regardless of the concentration of drug within the agar plate, and decreased to 8 to 16 mg/liter in the presence of the broad-spectrum efflux pump inhibitor.
For PAE 2638 at 1.5 times the baseline ceftolozane-tazobactam MIC value, the average density of the drug-resistant subpopulation was 1 CFU in 8.76 log10 of bacteria. At 3 and 5 times the baseline ceftolozane-tazobactam MIC value, mutation frequency could not be determined due to the lack of growth on the drug-containing plates. Thus, the mutation frequency for PAE 2638 at 3 and 5 times the baseline ceftolozane MIC value are less than the inoculum of 8.76 log10 or 5.75 × 108 CFU/ml. For piperacillin against PAE 2638, at 3 and 5 times the baseline piperacillin-tazobactam MIC value, mutation frequency studies were not carried out given that the baseline MIC value was >128 mg/liter.
The ceftolozane-tazobactam MIC value of PAE 2638 isolates taken from the drug-containing plates was 8 mg/liter, regardless of the drug concentration found within the agar plate. The ceftolozane-tazobactam MIC value decreased 1- to 2-fold in the presence of a broad-spectrum efflux pump inhibitor.
Pharmacokinetic validation studies.
Figure 1 shows the relationships between observed and targeted concentrations from all dosing regimens for ceftolozane, piperacillin, and tazobactam studied. The r2 values for these relationships were 0.972 for ceftolozane, 0.984 for tazobactam, and 0.978 for piperacillin. Thus, for all dosing regimens, the targeted ceftolozane, piperacillin, and tazobactam pharmacokinetic profiles were well simulated within the hollow-fiber infection model. For piperacillin and tazobactam, the line of best fit, through these data, did not differ from the line of identity (no regions of bias, with good precision). For ceftolozane, the precision was good and the intercept did not differ from zero, but the slope (0.89) differed from 1.0. This suggests that the actual concentrations achieved were approximately 11% below those expected but well within the variability observed in patient populations.
FIG 1.
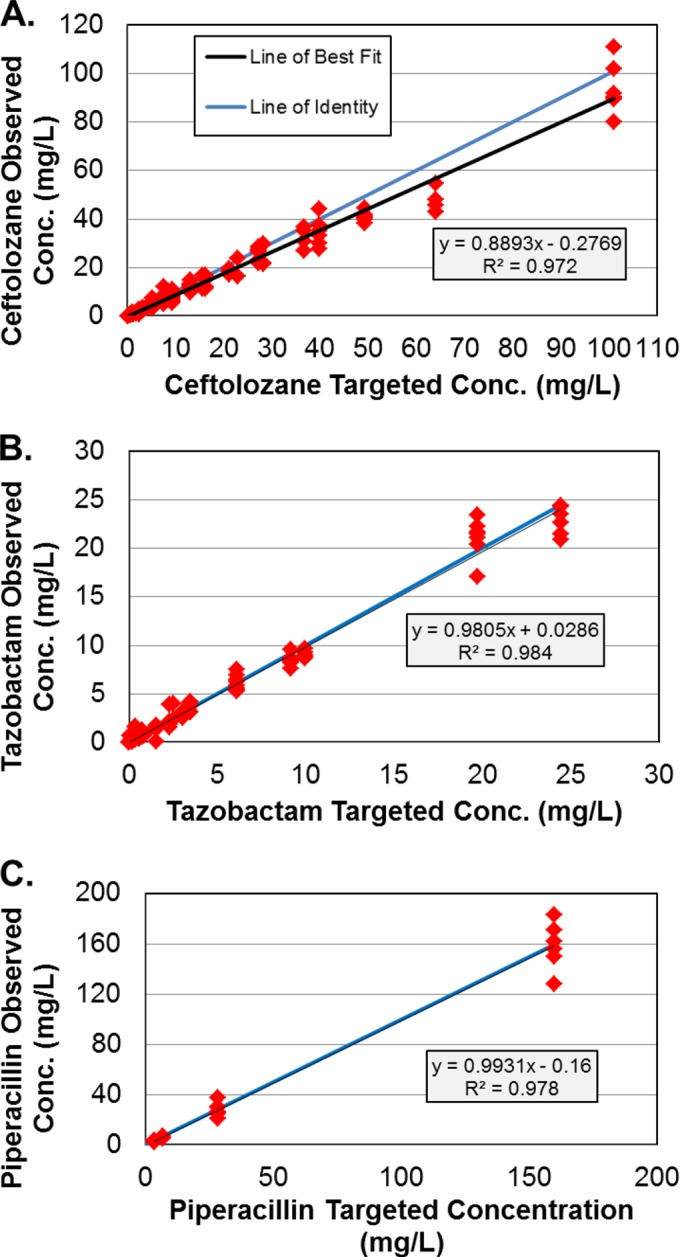
Relationships between the observed and targeted ceftolozane (A), tazobactam (B), and piperacillin (C) concentrations.
Resistance amplification prevention studies.
The change in total and drug-resistant population bacterial density over 10 days for the PAO1 BAA-47 strain is shown in Fig. 2. The bacteria grew well in the no-treatment growth control arms, reaching a bacterial density of 1.0 × 108 to >1.0 × 1010 CFU/ml by day 2 (Fig. 2A). The bacterial growth in the piperacillin-tazobactam control was similar to that of the no-treatment growth control arms and reached an average bacterial density of >1.0 × 1010 CFU/ml by day 2. The ceftolozane control arm produced a 4-log10 CFU/ml reduction in bacterial density over 2 days and prevented drug resistance amplification over the entirety of the 10-day study period (Fig. 2A). The range of ceftolozane-tazobactam dosing regimens evaluated proved effective at suppressing the growth of the total population and preventing the emergence of resistance for all dosing regimens studied (Fig. 2B). The ceftolozane-tazobactam 1,000/500-mg regimen was terminated on day 2 due to contamination of the hollow-fiber system.
FIG 2.

Emergence of resistance during drug administration versus PAO1 BAA-47. Panel A shows the negative-control (no treatment) and active-control (ceftolozane [TOL] at 1,000 mg every 8 h [Q8h], tazobactam [TAZ] at 500 mg Q8h, and piperacillin-tazobactam [PIP/TAZ] at 4.5 g every 6 h [Q6h]) study arms, while panel B shows ceftolozane-tazobactam regimens ranging from 500/250 to 2,000/1,000 mg Q8h. The TOL/TAZ treatment regimen of 1,000/500 mg Q8h was suspended after day 1 due to contamination of the system.
The change in total and drug-resistant population bacterial densities over 10 days for the PAE 2638 isolate are shown in Fig. 3. The bacteria grew well in the no-treatment growth control arms, reaching a bacterial density of 1.0 × 1010 CFU/ml by day 2 (Fig. 3A). Similarly, the bacterial growth in the piperacillin-tazobactam control reached an average bacterial density of 1.0 × 1010 CFU/ml by day 2.
FIG 3.

Emergence of resistance during drug administration versus clinical isolate PAE 2638. Panel A shows the negative-control (no treatment) and active-control (piperacillin-tazobactam at 4.5 g Q6h) study arms, while panel B shows ceftolozane-tazobactam (TOL/TAZ) regimens ranging from 62.5/31.25 to 2,000/1,000 mg delivered Q8h.
The ceftolozane-tazobactam dosing regimens investigated produced a full spectrum of drug effect (Fig. 3B). The least intensive ceftolozane-tazobactam regimen (62.5/31.25 mg) resulted in essentially no reduction in the total bacterial density over the first 2 days of therapy and emergence of a bacterial subpopulation displaying low-level resistance that remained present throughout the remaining 8 days of therapy. The most intensive ceftolozane-tazobactam regimen (2,000/1,000 mg) resulted in a complete reduction of the total bacterial population and prevention of resistance emergence over the entire 10-day study. The intermediately intensive ceftolozane-tazobactam regimens (125/62.5 to 1,000/500 mg) resulted in various reductions in total bacterial density and resistance emergence. The duration of time until the selection for drug resistance increased with dose regimen intensity. As shown in Fig. 4, the relationship between the change in bacterial density of the drug-resistant subpopulation and dose at day 10 took a hormetic or inverted-U form.
FIG 4.
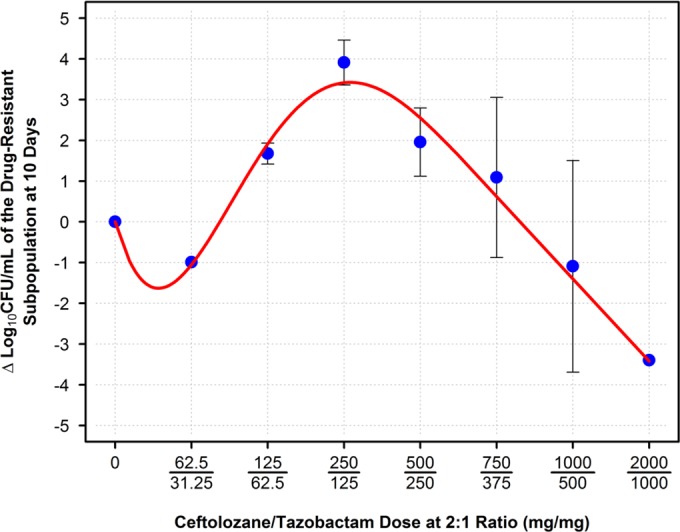
Relationship between ceftolozane-tazobactam (TOL/TAZ) dose and change in bacterial density of the average resistant subpopulation found on 3× MIC drug-containing plates.
MIC values were determined for a number of isolates grown on drug-containing plates for both isolates tested (data not shown). The susceptibilities of isolates taken from the no-treatment growth control ceftolozane-tazobactam drug-containing plates were found to be slightly lower than baseline MIC values, with inhibitory concentrations found to be 1 to 2 mg/liter for the PAO1 BAA-47 strain and 4 to 8 mg/liter for the PAE 2638 isolate. The PAO1 BAA-47 studies produced no resistant strains, as all treatment regimens were proven to be successful. For the PAE 2638 isolate, the intermediate regimens (125/62.5 to 1,000/500 mg) amplified an emergent resistant subpopulation whose MIC values started at 4 to 8 mg/liter on days 2 to 4 and increased to concentrations of 64 to >128 mg/liter by day 10. When tested in the presence of the pump inhibitor, the MIC value decreased 1- to 2-fold in dilution, regardless of the point during therapy at which they were collected. These small increases in MIC values are consistent with data generated by Hong et al., who determined that pseudomonal efflux pump overexpression does not affect susceptibility to ceftolozane (14).
DISCUSSION
The goal of these studies was to discriminate ceftolozane-tazobactam drug exposures that prevent the emergence of P. aeruginosa resistant subpopulations from those that do not in a hollow-fiber infection model. Dose ratio, dosing interval, and experimental duration were selected to approximate those expected to be used clinically. The ceftolozane-tazobactam dose range studies included the doses under investigation for complicated urinary tract infections and intra-abdominal infections (1,000/500 mg) as well as that for nosocomial pneumonia (2,000/1,000 mg).
For the wild-type ATCC strain, resistance was not selected by any ceftolozane-tazobactam dosing regimen evaluated. For this reason, future studies utilizing this strain were halted in favor of using resources to properly assess the resistance found in a clinically relevant isolate. For the clinical isolate, an inverted-U-shaped function best described the relationship between emergence of drug resistance and drug exposure. The least (62.5/31.25 mg) and most (2,000/1,000 mg) intensive ceftolozane-tazobactam dosing regimens did not select for drug resistance. Drug resistance selection was observed with intermediately intensive dosing regimens (125/62.5 through 1,000/500 mg). For the intermediately intensive ceftolozane-tazobactam dosing regimens, the duration of time until the selection for drug resistance increased with dose-regimen intensity.
Piperacillin-tazobactam was selected as a control arm because it has long been used successfully to treat infections associated with P. aeruginosa, such as complicated urinary tract infections and hospital-acquired bacterial pneumonia. As the piperacillin-tazobactam MIC of the clinical isolate was nonsusceptible (>128 mg/liter), piperacillin-tazobactam served as a control only for the studies involving the wild-type PAO1 strain (MIC, 2 mg/liter). In these studies, the piperacillin-tazobactam clinically used dose (4.5 g every 6 h) failed early in therapy, with the piperacillin-tazobactam-resistant subpopulation replacing the entire population by day 3 of therapy. These results are consistent with those of Felton et al. in a study which involved piperacillin-tazobactam, P. aeruginosa, and a high inoculum (1.0 × 108 CFU/ml) (4). The piperacillin-tazobactam MIC increased from a baseline value of 2 to 128 mg/liter on day 3 of therapy and decreased to 4 mg/liter in the presence of a broad-spectrum efflux pump inhibitor. The latter finding suggests that at least two resistance determinants (one being drug efflux) are responsible for the decrease in piperacillin-tazobactam susceptibility. The results of the current study are similar to those of a prior study involving ceftolozane-tazobactam and E. coli (15). In the prior study, the ceftolozane-tazobactam dose that suppressed the amplification of resistance was ≥750/375 mg, and the relationship between the amplification of a drug-resistant subpopulation and drug exposure took the form of an inverted U. There is one important difference between the results of the prior study and the current one that merit comment. While in both studies, the endpoint was amplification of a preexisting ceftolozane-tazobactam-resistant bacterial subpopulation, in the ceftolozane-tazobactam–E. coli studies, resistance amplification occurred early during therapy (day 1 to 2), while in the ceftolozane-tazobactam–P. aeruginosa study, amplification of the resistant subpopulation to concentrations greater than that found in the no-treatment control was held at bay until days 4 to 6. This time to resistance amplification correlates with the findings of Louie et al., who examined the time to initial resistance emergence of a resistant Pseudomonas subpopulation treated with the beta-lactam doripenem (16). In that study, the authors did not examine the cause of the delay in resistance amplification. In the studies described herein, the delay in amplification may have been due to the relatively low frequency of mutation to resistance to ceftolozane-tazobactam of the Pseudomonas isolates compared to that of the E. coli isolate we previously studied (15). Even though these two studies utilized the same initial concentration of bacteria, 1.0 × 108 CFU/ml, the relative mutation frequencies were 1 CFU in every 7 log10 of bacteria for the E. coli strain and 1 CFU in every 8 log10 of bacteria for the Pseudomonas strains. This difference in the initial concentrations of the drug-resistant subpopulations may be the key to delayed amplification of the drug-resistant subpopulation.
One limitation of the studies described herein is the limited number of challenge isolates evaluated. While it is true that the MIC value of the two challenge isolates approximated the ceftolozane-tazobactam MIC50 and MIC90 for P. aeruginosa, studies with a larger number of isolates would be required to understand between-isolate pharmacodynamic variability; however, the observed efficacy and resistance prevention exhibited by ceftolozane-tazobactam relative to that observed for piperacillin-tazobactam is notable.
Finally, we successfully discriminated between the ceftolozane-tazobactam dosing regimens that prevented resistance emergence from those that did not. For the wild-type ATCC strain, resistance was not selected by any ceftolozane-tazobactam dosing regimen evaluated. For the clinical isolate, an inverted-U-shaped function best described the relationship between bacterial drug resistance emergence and drug exposure. Resistance greater than that found in the control did not develop for the lowest dose studied (62.5/31.25 mg) or for the two highest doses studied (1,000/500 mg and 2,000/1,000 mg), which are the doses currently being studied in clinical trials. For the 2,000/1,000-mg ceftolozane-tazobactam dosing regimen, resistance emergence did not occur and the total bacterial population was driven toward extinction by day 10. These results were dissimilar to that for piperacillin-tazobactam against the susceptible PAO1 strain, for which the drug-resistant subpopulation replaced the susceptible population by day 3. These data will be useful in the selection of ceftolozane-tazobactam dosing regimens to minimize the potential for on-therapy resistance emergence in seriously ill patients with infections associated with P. aeruginosa.
ACKNOWLEDGMENTS
We thank Kim A. Charpentier from ICPD (Latham, NY) and Lalitagauri M. Deshpande from JMI Laboratories (North Liberty, IA) for manuscript assistance and technical support.
This study was sponsored by Cubist Pharmaceuticals Inc., Lexington, MA.
The Institute for Clinical Pharmacodynamics (B.V., S.M.B., and P.G.A.) has received research support from Achaogen, Astellas, AstraZeneca, Basilea Pharmaceutica, Bayer HealthCare, Bristol-Meyers Squibb, Cempra Pharmaceuticals, Cerexa, Cubist Pharmaceuticals, Durata Pharmaceuticals, Fedora Pharmaceuticals, Forest Research Institute, Furiex Pharmaceuticals, GlaxoSmithKline, Meiji Seika Pharma, Nabriva Therapeutics, Nimbus, Pfizer, PolyMedix, Roche Bioscience, Rempex Pharmaceuticals, Rib-X Pharmaceuticals, Rock Therapeutics, Tetraphase Pharmaceuticals, and The Medicines Company.
JMI Laboratories, Inc. (R.E.M., M.C., and R.N.J.) received research and educational grants in 2009 to 2012 from American Proficiency Institute (API), Anacor, Astellas, AstraZeneca, Bayer, Basilea Pharmaceuticala, Cempra/Forest, Cerexa, Contrafect, Cubist, Daiichi, Dipexium, Durata, Enanta, Fedora, Furiex, GlaxoSmithKline, Johnson & Johnson (J&J; Ortho McNeil), LegoChem Biosciences Inc., Meiji Seika Pharma, Merck, Nabriva, Novartis, Pfizer (Wyeth), Rempex, Rib-X Pharmaceuticals, Seachaid, Shionogi, The Medicines Company, Theravance, and Thermo Fisher. JMI Laboratories serves as advisors/consultants for Astellas, Cubist, Pfizer, Cempra, Cerexa/Forest, J&J, and Theravance.
We have no speakers' bureaus or stock options to declare.
L.F. and J.S. are both employees and stockholders of Cubist Pharmaceuticals.
Footnotes
Published ahead of print 28 July 2014
REFERENCES
- 1.Bhavnani SM, Hammel JP, Forrest A, Jones RN, Ambrose PG. 2003. Relationships between patient- and institution-specific variables and decreased antimicrobial susceptibility of Gram-negative pathogens. Clin. Infect. Dis. 37:344–350. 10.1086/375817 [DOI] [PubMed] [Google Scholar]
- 2.Harris AD, Perencevich E, Roghmann MC, Morris CM, Kaye KS, Johnson JA. 2002. Risk factors for piperacillin-tazobactam-resistant Pseudomonas aeruginosa among hospitalized patients. Antimicrob. Agents Chemother. 46:854–858. 10.1128/AAC.46.3.854-858.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tam VH, Louie A, Deziel MR, Liu W, Leary R, Drusano GL. 2005. Bacterial-population responses to drug-selective pressure: examination of garenoxacin's effect on Pseudomonas aeruginosa. J. Infect. Dis. 192:420–428. 10.1086/430611 [DOI] [PubMed] [Google Scholar]
- 4.Felton TW, Goodwin J, O'Connor L, Sharp A, Gregson L, Livermore J, Howard SJ, Neely MN, Hope WW. 2013. Impact of bolus dosing versus continuous infusion of piperacillin and tazobactam on the development of antimicrobial resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 57:5811–5819. 10.1128/AAC.00867-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sader HS, Rhomberg PR, Farrell DJ, Jones RN. 2011. Antimicrobial activity of CXA-101, a novel cephalosporin tested in combination with tazobactam against Enterobacteriaceae, Pseudomonas aeruginosa, and Bacteroides fragilis strains having various resistance phenotypes. Antimicrob. Agents Chemother. 55:2390–2394. 10.1128/AAC.01737-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Craig WA, Andes DR. 2013. In vivo activities of ceftolozane, a new cephalosporin, with and without tazobactam against Pseudomonas aeruginosa and Enterobacteriaceae, including strains with extended-spectrum β-lactamases, in the thighs of neutropenic mice. Antimicrob. Agents Chemother. 57:1577–1582. 10.1128/AAC.01590-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carmeli Y, Troillet N, Karchmer AW, Samore MH. 1999. Health and economic outcomes of antibiotic resistance in Pseudomonas aeruginosa. Arch. Intern. Med. 159:1127–1132. 10.1001/archinte.159.10.1127 [DOI] [PubMed] [Google Scholar]
- 8.Clinical and Laboratory Standards Institute. 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Approved standard, ninth ed. CLSI document M07-A9 Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 9.Louie A, Deziel MR, Liu WL, Drusano GL. 2007. Impact of resistance selection and mutant growth fitness on the relative efficacies of streptomycin and levofloxacin for plague therapy. Antimicrob. Agents Chemother. 51:2661–2667. 10.1128/AAC.00073-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Louie A, Castanheira M, Liu W, Grasso C, Jones RN, Williams G, Critchley I, Thye D, Brown D, VanScoy B, Kulawy R, Drusano GL. 2012. Pharmacodynamics of β-lactamase inhibition by NXL104 in combination with ceftaroline: examining organisms with multiple types of β-lactamases. Antimicrob. Agents Chemother. 56:258–270. 10.1128/AAC.05005-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wyeth Pharmaceuticals Inc. 2012. Zosyn package insert. Wyeth Pharmaceuticals Inc, Philadelphia, PA [Google Scholar]
- 12.Ge Y, Whitehouse M, Friedland I, Talbot G. 2010. Pharmacokinetics and safety of CXA-101, a new antipseudomonal cephalosporin, in healthy adult male and female subjects receiving single and multiple intravenous injections. Antimicrob. Agents Chemother. 54:3427–3431. 10.1128/AAC.01753-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clinical and Laboratory Standards Institute. 2013. Performance standards for antimicrobial susceptibility testing; twenty-third informational supplement. Approved standard, ninth ed. CLSI document M100-S23. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 14.Hong MC, Hsu DI, Bounthavong M. 2013. Ceftolozane/tazobactam: a novel antipseudomonal cephalosporin and β-lactamase-inhibitor combination. Infect. Drug Resist. 6:215–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.VanScoy B, Mendes RE, Castanheira M, McCauley J, Bhavnani SM, Forrest A, Jones RN, Ambrose PG. 2013. Relationship between ceftolozane-tazobactam exposure and drug resistance amplification in a hollow-fiber infection model. Antimicrob. Agents Chemother. 57:4134–4138. 10.1128/AAC.00461-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Louie A, Bied A, Fregeau C, VanScoy B, Brown D, Liu W, Bush K, Queenan AM, Morrow B, Khashab M, Kahn J, Nicholson S, Kulawy R, Drusano GL. 2010. Impact of different carbapenems and regimens of administration on resistance emergence for three isogenic Pseudomonas aeruginosa strains with differing mechanisms of resistance. Antimicrob. Agents Chemother. 54:2638–2645. 10.1128/AAC.01721-09 [DOI] [PMC free article] [PubMed] [Google Scholar]