

Abstract
R4.0, a synthetic CCL5/RANTES-derived peptide, exerts potent anti-HIV-1 activity via its nonactivating interaction with CCR5, the major HIV-1 coreceptor. CCR5 chronic activation may promote undesirable inflammatory effects and enhance viral infection; thus, receptor antagonism is a necessary requisite. HIV-1 gp120, CCL5, and maraviroc dock on CCR5 by sharing two receptor sites: the N terminus and the second extracellular loop. In combination studies, R4.0, CCL5, and maraviroc exhibited concomitant interactions with CCR5 and promoted synergic inhibition of HIV-1 in acute-infection assays. Furthermore, various degrees of additive/synergic HIV-1 inhibition were observed when R4.0 was tested in combination with drugs and lead compounds directed toward different viral targets (gp120, gp41, reverse transcriptase, and protease). In combination with tenofovir, R4.0 provides cross-clade synergic inhibition of primary HIV-1 isolates. Remarkably, an in vitro-generated maraviroc-resistant R5 HIV-1 strain was inhibited by R4.0 comparably to the wild-type strain, suggesting the presence of viral resistance barriers similar to those reported for CCL5. Overall, R4.0 appears to be a promising lead peptide with potential for combination in anti-HIV-1 therapy and in microbicide development to prevent sexual HIV-1 transmission.
INTRODUCTION
More than 34 million people worldwide are presently living with human immunodeficiency virus type 1 (HIV-1) (1), the causative agent of the AIDS, a pandemic that has killed more than 25 million people in 3 decades. Despite the highly active antiretroviral therapy (HAART), three major burdens remain for HIV-1 infection. The HAART has prohibitive costs for the vast majority of HIV-1-infected people, there is no therapeutic route that allows the eradication of the virus after infection (2), and there are no preventive measures that may warrant safety to a significant percentage of the population at risk of infection. Efficacious preventive measures are of the utmost importance and, while a protective HIV-1 vaccine is still a remote perspective, although positive hints are emerging (3, 4), topical anti-HIV-1 microbicides represent a promising alternative, as well as an option complementary to a vaccine (5). Microbicides embrace several formulations and molecular targets, and the concept of combining two or more lead compounds to decrease the insurgence of drug resistance and to increase prevention efficacy is progressively becoming a common trend (6). The HIV-1 coreceptor CCR5, exclusively used in primary infection, represents an important microbicide target since it is present on the surfaces of CD4+ T lymphocytes and macrophages localized in the vaginal, rectal, and foreskin epithelia (7). Any HIV-1 inhibitor targeting CCR5, preferably an antagonist, may therefore be seen as a potential microbicide. Maraviroc (MVC) and certain full-length and short peptide derivatives of CCL5/RANTES may well suit the requirements for the development of an effective HIV-1 microbicide (8–10). CCR5 antagonism (receptor binding devoid of activating capacity) is a fundamental requisite to prevent mucosal inflammation, as inflammation may very likely result in the enhancement of HIV-1 transmission.
Extensive efforts are being pursued to engineer CCL5 derivatives with high anti-HIV-1 potency to act as entry inhibitors and CCR5 antagonists (11). In this context, a long-lasting project has been focused on the design of potent peptide-based HIV-1 entry inhibitors (12–14), with R4.0 as the most active representative (10). In view of its possible implementation in combination protocols, R4.0 was tested in various cellular assays for its compatibility with several HIV-1 inhibitors directed toward different viral/cellular targets. R4.0 was revealed to be fully suitable for combinations with all compounds tested, including CCR5-directed inhibitors, proving itself a valuable new compound in anti-HIV-1 therapy and microbicide development.
MATERIALS AND METHODS
Peptide synthesis.
Peptides R3.0 and R4.0 were synthesized by standard solid-phase protocols using Fmoc chemistry and purified by reverse-phase high-performance liquid chromatography (RP-HPLC) to >95% purity, as described previously (10). Peptides were dimerized by oxidation of the N-terminal cysteine residues and N-terminally acetylated. For stable dimerization, peptides were incubated overnight in 50% dimethyl sulfoxide (DMSO) (Sigma, St. Louis, MO) in water; DMSO was removed by freeze-drying, and the dimerized peptides were purified by RP-HPLC. The purity of the final stock was >97%; the Ellman test for free sulfhydryl groups was negative.
Drug supply and formulation.
MVC (Pfizer, New York, NY) and tenofovir disoproxil fumarate (TDF) (Gilead, Foster City, CA) tablets (150 mg and 300 mg, respectively) were used for in vitro assays. Both tablets were ground into a powder and dissolved in a known volume of sterile water. Then, the preparations were filtered to remove a small amount of insoluble material (tablet excipients) and stored in aliquots at −20°C. Two hundred milligrams of emtricitabine (FTC) powder contained in a capsule (Gilead) was dissolved in sterile water, filtered, and stored in aliquots at −20°C.
HIV-1 Env-mediated cell fusion assay.
Antiviral activity was evaluated using two cellular assays, an HIV-1 envelope-mediated cell fusion assay and an acute HIV-1 infection assay (10), both based on the prototype CCR5-using (R5) isolate HIV-1BaL. The cell fusion assay was performed using a modification of the test based on vaccinia virus technology, originally developed by Nussbaum and coworkers (15). In the modified assay, the effector cells were chronically infected PM1 cells, whereas the target cells were NIH 3T3 mouse fibroblasts or HeLa TZM-bl cells stably expressing human CCR5 and human CD4. Sixteen hours before the test, effector cells were infected with a vaccinia virus vector expressing bacteriophage T7 RNA polymerase, while target cells were infected with a vaccinia virus vector expressing the lacZ reporter gene under the control of the T7 promoter. All vaccinia virus infections were performed in Dulbecco's modified Eagle's medium (DMEM) (Lonza BioWhittaker, Valais, Switzerland) supplemented with 2.5% fetal bovine serum (FBS) (Lonza BioWhittaker). The cells were then washed with 2.5% DMEM, and the effector cells were mixed for 2 h with the target cells in the presence or absence of the inhibitors. Cell fusion was determined by measurement of β-galactosidase activity in nonionic detergent cell lysates as described previously (15).
In addition, another modified fusion assay was used, as previously reported (10). In this assay, primary CD4+ T lymphocytes purified from human peripheral blood mononuclear cells (PBMC) were used as target cells instead of NIH 3T3 or TZM-bl cells. Briefly, PBMC were isolated by Lympholyte cell separation medium (Cedarlane Laboratories Limited, Burlington, Canada) gradient centrifugation of buffy coat preparations from healthy blood donors. Afterwards, PBMC were stimulated with 500 U/ml recombinant human interleukin 2 (IL-2) (Chiron, Emeryville, CA) in complete RPMI medium (Lonza BioWhittaker) for 7 to 21 days to induce surface expression of CCR5. The day before the fusion assay, CD4+ T cells were purified from PBMC by negative selection using Dynabeads goat anti-mouse IgG (Invitrogen, Carlsbad, CA) and a cocktail of purified monoclonal antibodies (MAbs) against human CD19, CD16, CD56, CD8 (Immunotools, Friesoythe, Germany), and CD14 (AbD Serotec, Raleigh, NC). CD4+ T cells were then infected with the vaccinia virus vector expressing the lacZ reporter gene under the control of the T7 promoter. Infection was carried out in DMEM in the absence of FBS during the first 2 h, and then the cells were diluted using DMEM supplemented with 2.5% FBS. The following day, CD4+ T cells (target) were incubated with effector cells (PM1 cells chronically infected with HIV-1BaL) for 4 h in the absence or presence of inhibitors. After incubation, cells were lysed and cell fusion was determined as described above.
HIV-1 infection assay.
Acute HIV-1 infection was obtained by adding HIV-1BaL and the primary isolates 5513 and 98IN007 (50 50% tissue culture infective doses [TCID50]/well) to PM1 cells (2 × 104/well) in complete RPMI medium. PM1 is a unique CD4+ CCR5+ T cell clone susceptible to a wide variety of primary HIV isolates, including those exclusively using CCR5 as the coreceptor (16). Instead, acute-infection HIV-1 RU570 (Russian G isolate) and RU570 MVC-resistant strains were obtained by adding the viral stocks (50 TCDI50/well) to 1 × 105 cells/well of PBMC isolated from buffy coat cells as described above and activated for 4 days with 5 μg/ml of phytohemagglutinin (PHA). Experiments were performed in triplicate using 96-well round-bottom microtiter plates in the presence or absence of inhibitors. After incubation at 37°C for 16 h, the wells were washed twice, and complete medium, with or without the inhibitors, was added. After 48 h, 75% of the supernatant was removed for HIV-1 p24 antigen measurement and replaced by an equal volume of medium containing the inhibitors. Virus replication was assayed at day 4 postinfection by the p24 antigen enzyme-linked immunosorbent assay (ELISA). Supernatants were diluted in 1% Empigen BB detergent (Calbiochem, Gibbstown, NJ) to disrupt virions, added to a 96-well ELISA plate coated with anti-HIV-1 p24 polyclonal antibodies (Aalto Bio Reagents Ltd., Dublin, Ireland), and incubated for 2 h at room temperature. The plate was then washed three times in TBS buffer (1.5 M NaCl, 250 mM Tris, pH 7.5), and an alkaline phosphatase-conjugated anti-HIV-1 p24 monoclonal antibody (Aalto Bio Reagents Ltd.) was added for 1 h at room temperature. After the plates were washed three times with Tropix buffer (10 mM MgCl2, 200 mM Tris, pH 9.8), p24 was detected by adding the luminescence substrate CSPD Tropix (Applied Biosystems, Foster City, CA) and the signal was analyzed using a Mithras LB 940 luminometer (Berthold Technologies, Bad Wildbad, Germany). Levels of p24 were calculated by generating a standard curve with HIV-1 p24 antigen standards.
Combinations and statistical analysis.
Experimental design and analysis of synergy, additivity, or antagonism between different compounds were based on the combination index (CI) method of Chou and Talalay (17, 18). In an HIV-1 cell fusion assay and an HIV-1 infection assay, each drug was tested individually and in a fixed molar ratio (50% infective concentration [IC50] to IC50) combination over a range of 2-fold serial dilutions. The 50%, 75%, and 90% combination indexes (CI50, CI75, and CI90) to determine the effect of the interactions between the drugs were calculated using CalcuSyn software 2.0 (Biosoft, Ferguson, MO). A CI of <0.9 indicates synergy, a CI from 0.9 to 1.1 indicates additivity, and a CI of >1.1 indicates antagonism. Dose-response curves were fitted using GraphPad Prism version 5.04 (GraphPad Software, San Diego, CA) in order to calculate IC50s through nonlinear-regression analysis. All data are expressed as the means ± standard deviations (SD) of results from two independent experiments performed in triplicate. All P values were computed according to the Fischer rule.
Immunofluorescence microscopy and cytofluorimetric analysis.
For immunofluorescence microscopy, 1 × 105 CHO CD4 CCR5 cells were grown in 12-well plates on 18-mm glass coverslips (Zeus super) in complete DMEM. One day later, cells were washed twice in phosphate-buffered saline (PBS) and incubated at 37°C for 4 h in DMEM without FBS in the presence of 100 nM CCL5, 100 nM R4.0, and 100 nM MVC. Cells were washed twice in cold PBS, fixed with 3% fresh paraformaldehyde (Sigma) for 20 min, washed in PBS, and incubated with 50 mM ammonium chloride (Sigma) for 30 min at room temperature to quench free aldehydes. Then, fixed cells were incubated for 15 min at room temperature in PBS containing 5% bovine serum albumin (BSA) (Sigma) to saturate nonspecific binding sites, incubated for 20 min at 37°C with 5 μg/ml 3A9 anti-CCR5 antibody (BD Bioscience, San Jose, CA), washed three times with PBS containing 5% BSA, incubated for 20 min at 37°C with 2 μg/ml donkey anti-mouse IgG (H+L) Alexa Fluor 488 (Invitrogen, Carlsbad, CA), and washed again. Finally, the coverslips were mounted over Mowiol 4-88 (Sigma) and examined using a Zeiss Axiophot epifluorescence microscope (Carl Zeiss Microscopy LLC, Thornwood, NY) under a 63×/1.40 oil immersion objective equipped with a Hamamatsu digital charge-coupled device (CCD) camera (model C4742-95; Hamamatsu Photonics, Shizuoka, Japan).
Cytofluorimetric analysis for surface CCR5 expression was conducted on 1 × 105 CHO CD4 CCR5 cells incubated for 4 h at 37°C in DMEM without FBS in the presence of 100 nM CCL5, 100 nM R4.0, and 100 nM MVC. After the treatment, cells were washed twice with cold PBS containing 2% FBS and fixed with 2% fresh formaldehyde (Sigma) for 15 min. Then, the cells were incubated with the same primary and secondary antibodies used in immunofluorescence for 15 min at 4°C. After being washed, cells were analyzed using the Gallios flow cytometer (Beckman Coulter Inc., Brea, CA) and data were analyzed using the FlowJo software (Tree Star Inc., Ashland, OR).
RESULTS
R4.0 presents wide anti-HIV-1 compatibility with different inhibitors.
The need for antiviral cocktails in HIV-1 infection therapy and prevention is dictated by the escape routes of the virus. Hence, it is mandatory to test new HIV-1 inhibitors for their additivity/synergy with existing drugs or other lead compounds. In this regard, the CCL5-derived peptide R4.0, the most active lead compound resulting from a long-lasting molecular evolution (10, 12–14), was subjected to an extensive combination survey. All compounds tested with their viral and cellular targets are illustrated in Fig. 1. An initial assessment of R4.0's additive, or even synergic, anti-HIV-1 features was carried out in cell fusion inhibition assays testing combinations with an array of different HIV-1 blockers. The anti-HIV-1 agents tested were cyanovirin-N (CV-N) and the MAb 2G12, recognizing the carbohydrate shield of gp120 (19, 20); T20 and the MAbs 2F5 and 4E10, recognizing gp41 (21–23); and MVC, a CCR5 antagonist (8) (Fig. 2). Combinations of R4.0 with T20 and, separately, MVC were also tested in cell fusion assays in which purified human CD4+ T cells were used as target cells, as previously reported (10). Although R4.0 blocked HIV-1 with low nanomolar potency, its IC50 improved when it was used in combination with all the different compounds, as illustrated with the dose-response curves of mixed inhibitors (Fig. 2). The calculated combination indexes (Table 1) indicated a synergic effect for the anti-HIV-1 MAbs and T20, with CI50s ranging from 0.43 to 0.81, and an additive effect for CV-N and MVC, with CI50s from 0.94 to 1.05. The strongest synergic interaction within this panel of inhibitors was observed between R4.0 and 4E10, with a CI50 value of 0.43, and the highest improvement in R4.0 antiviral potency was obtained using 2F5 and 4E10, with a 5-fold and a striking 18-fold IC50 increase, respectively.
FIG 1.

Schematic representation of HIV-1 infection of CD4+ T cells and the inhibitors used in this study. A survey of R4.0 combinations (double or triple) with different entry (CV-N, 2G12, MVC, and CCL5), fusion (T20, 2F5, and 4E10), and replication (FTC, TDF, and IDV) inhibitors yielded either full additivity or synergy. Arrows indicate viral and cellular inhibitor targets. R4.0 (not illustrated) targets CCR5.
FIG 2.

Anti-HIV-1BaL activity of the CCL5-derived peptide R4.0 in combination with different HIV-1 entry and fusion inhibitors. (A to F) R4.0-induced HIV-1 inhibition tested by Env-mediated cell fusion inhibition assays in combination with CV-N (A), 2G12 (B), T20 (C), 2F5 (D), 4E10 (E), and MVC (F). (G) HIV-1 inhibition induced by the CCL5-derived peptide R3.0 (10) tested in combination with MVC by cell fusion inhibition assays using purified human CD4+ T cells. (H to I) R4.0-induced HIV-1 inhibition tested by cell fusion inhibition assays using purified human CD4+ T cells in combination with T20 (H) or MVC (I). The resulting dose-response curves of mixed inhibitors are referred to as R4.0 or R3.0 and indicated as “mix” (P values < 0.0001). Values are the means ± SD of results from two independent experiments performed in triplicate.
TABLE 1.
CI values for R4.0 or CCL5 in combination with other inhibitors
| Drug(s) used in combination | Experimental condition | CI50 | CI75 | CI90 | CI50 effect |
|---|---|---|---|---|---|
| Drugs used with R4.0 | |||||
| CV-N | Fusion | 0.94 ± 0.12 | 0.90 ± 0.08 | 0.88 ± 0.27 | Additive |
| MVC | Fusion | 1.05 ± 0.03 | 1.00 ± 0.06 | 0.95 ± 0.08 | Additive |
| T20 | Fusion | 0.78 ± 0.08 | 0.79 ± 0.01 | 0.80 ± 0.06 | Synergy |
| 2G12 | Fusion | 0.78 ± 0.19 | 0.66 ± 0.01 | 0.61 ± 0.14 | Synergy |
| 4E10 | Fusion | 0.43 ± 0.09 | 0.51 ± 0.19 | 0.79 ± 0.04 | Synergy |
| 2F5 | Fusion | 0.81 ± 0.20 | 0.82 ± 0.10 | 1.01 ± 0.09 | Synergy |
| MVC | Fusion T cells | 1.05 ± 0.03 | 0.83 ± 0.06 | 0.66 ± 0.08 | Additive |
| T20 | Fusion T cells | 1.08 ± 0.10 | 0.99 ± 0.03 | 0.94 ± 0.06 | Additive |
| CCL5 | p24 | 0.90 ± 0.04 | 0.88 ± 0.13 | 0.86 ± 0.21 | Additive |
| CV-N | p24 | 0.92 ± 0.15 | 0.96 ± 0.11 | 1.01 ± 0.07 | Additive |
| MVC | p24 | 0.75 ± 0.19 | 0.81 ± 0.13 | 0.87 ± 0.05 | Synergy |
| FTC | p24 | 0.87 ± 0.03 | 0.78 ± 0.03 | 0.71 ± 0.08 | Synergy |
| TDF | p24 | 0.67 ± 0.01 | 0.73 ± 0.01 | 0.78 ± 0.01 | Synergy |
| IDV | p24 | 0.63 ± 0.08 | 0.67 ± 0.07 | 0.72 ± 0.06 | Synergy |
| TDF-IDV | p24 | 0.83 ± 0.23 | 0.82 ± 0.08 | 0.86 ± 0.09 | Synergy |
| TDF (5513) | p24 | 0.49 ± 0.03 | 0.54 ± 0.02 | 0.58 ± 0.01 | Synergy |
| TDF (98IN007) | p24 | 0.77 ± 0.04 | 0.80 ± 0.04 | 0.84 ± 0.03 | Synergy |
| MVC-CCL5 | p24 | 0.58 ± 0.16 | 0.65 ± 0.14 | 0.77 ± 0.18 | Synergy |
| Drugs used with CCL5 | |||||
| MVC | Fusion T cells | 0.66 ± 0.01 | 0.66 ± 0.04 | 0.69 ± 0.08 | Synergy |
| MVC | p24 | 0.91 ± 0.02 | 0.82 ± 0.01 | 0.78 ± 0.02 | Additive |
The possibility of combining R4.0 with CV-N and MVC was also examined in a more physiological assay in which PM1 cells were infected with HIV-1BaL (Fig. 3A and B). The results of these experiments are shown in Table 1 and revealed a reproducible additive effect for R4.0 and CV-N and a synergic effect for R4.0 and MCV, with CI50s of 0.92 and 0.75, respectively. In this assay, the R4.0–CV-N combination confirms the value obtained in the cell fusion assay, while the R4.0-MVC combination produces a stronger effect.
FIG 3.

Anti-HIV-1BaL activity of R4.0 in combination with HIV-1 inhibitors of different classes. (A to F) HIV-1 inhibition tested by acute-infection assays on the human CD4+ T cell clone PM1 and measured by a p24-based assay after 4 days of infection. R4.0 was tested in single combinations with CV-N (A), MVC (B), IDV (C), FTC (D), and TDF (E) and in a triple combination with TDF and IDV (F). The dose-response curves of mixed inhibitors (mix) are referred to as R4.0 (P values < 0.0001). Values are the means ± SD from two independent experiments performed in triplicate.
The spectrum of different inhibitors tested in combination with R4.0 was further expanded in acute-infection assays testing the inhibition combination outside the entry/fusion landscape by investigating intracellular HIV-1 inhibition. Hence, indinavir (IDV), a protease inhibitor (24), and FTC and TDF, two reverse transcriptase inhibitors (25, 26), were tested (Fig. 3C, D, and E). IDV, FTC, and TDF inhibited HIV-1BaL replication with IC50s of 14.8 nM, 42 nM, and 10.2 nM, respectively. All the compounds synergized with R4.0, yielding CI50 values ranging from 0.63 to 0.87, as reported in Table 1.
In a further effort to make a prospective microbicide cocktail, a triple R4.0-IDV-TDF combination was used to inhibit acute HIV-1BaL infection/replication in PM1 cells (Fig. 3F). Encouragingly, the resulting inhibition corresponded to a synergic effect, with a CI50 value of 0.83, improving R4.0's antiviral potency about 3-fold.
Cobinding to CCR5 allows effective and expanded anti-HIV-1 combinations at the receptor sites.
The efficient simultaneous targeting of CCR5 using R4.0 and MVC (Fig. 2F and I and 3B) provides the basis to investigate this concept further and expands CCR5 targeting possibilities (Fig. 4). CCR5 binding and the proficient anti-HIV-1 combination of CCL5 and MVC have already been reported (27). The latter finding was confirmed in our experimental setup by testing the CCL5-MVC combination in acute-infection assays, in which we observed an additive effect, with a CI50 of 0.91, and in the human lymphocyte fusion assay, in which we observed a synergic effect, with a CI50 of 0.66 (Fig. 4A and B). Despite the likelihood for antagonism, a combination of R4.0 and CCL5 was attempted in acute-infection assays (Fig. 4C). Surprisingly, this combination yielded an additive effect, with a CI50 of 0.9. Even more surprising was the successful attempt to combine three CCR5 inhibitors, R4.0, MVC, and CCL5. The resulting synergy presents a CI50 value of 0.58, improving R4.0's antiviral potency 11-fold.
FIG 4.

Combination of CCR5 blockers to inhibit HIV-1BaL infection. (A) HIV-1 inhibition induced by the combination of CCL5 with MVC tested by an acute-infection assay as described in the legend of Fig. 2. (B) CCL5 combination with MVC tested by a cell fusion inhibition assay using purified human CD4+ T cells. (A and B) The dose-response curves of mixed inhibitors (mix) are referred to as CCL5 (P values < 0.0001). (C to D) R4.0 in combination with CCL5 (C) or in triple combination with CCL5 and MVC (D) was tested as described above for panel A. (C and D) The dose-response curves of mixed inhibitors (mix) are referred to as R4.0 (P values < 0.0001). Values are the means ± SD from two independent experiments performed in triplicate.
R4.0's antagonism toward CCR5 was previously assessed in a chemotaxis assay in which the peptide exerted an activity similar to that of MVC (10). This aspect was further verified here using CHO CD4 CCR5 cells. As expected, the surface expression level of CCR5 (stained using MAb 3A9) is reduced in CHO CD4 CCR5 cells treated with CCL5, while it is not affected when the cells are treated with MVC or R4.0 (Fig. 5A to D). The level of cell surface CCR5 was quantified also by flow cytometry, which showed a 40% CCR5 internalization with CCL5, as previously reported (28), and comparable antagonistic activity for MVC and R4.0 with no internalization of the receptor (Fig. 5E).
FIG 5.
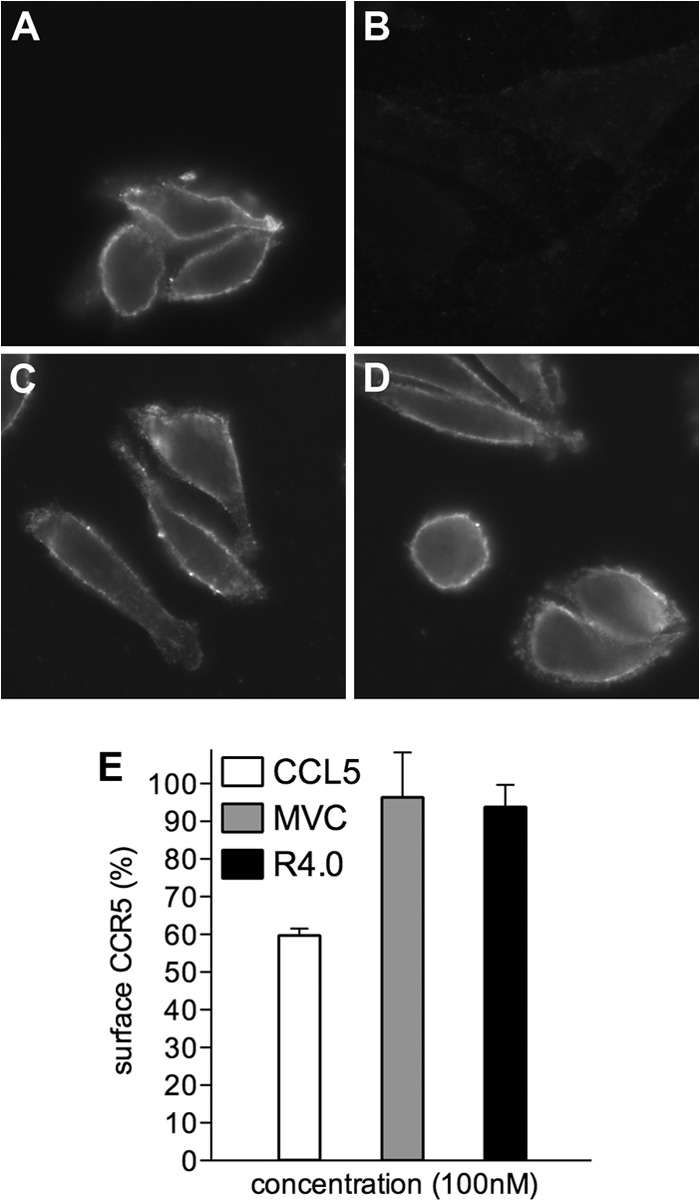
R4.0 acts as a CCR5 antagonist. (A to D) Immunofluorescence of CHO CD4 CCR5 cells labeled with the anti-CCR5 MAb 3A9 (A) or preincubated with 100 nM CCL5 (B), 100 nM MVC (C), or 100 nM R4.0 (D) and labeled with 3A9. (E) CHO CD4 CCR5 cell surface CCR5 measured by flow cytometry was set at 100% for cells treated with 3A9. The CCR5 cell surface expression of cells preincubated with CCL5, MVC, or R4.0 is shown as a mean percentage of the expression in control cells ± SD and is representative of two independent experiments.
Validation of the R4.0-TDF combination on primary R5 HIV-1 strains.
In order to further test the possible implementation of R4.0 as a candidate for a microbicidal combination, two primary R5 HIV-1 isolates were tested, one belonging to clade B (5513) and one belonging to clade C (98IN007). In acute-infection assays, R4.0 inhibited HIV-15513 and HIV-198IN007, with IC50s of 56 nM and 196 nM, respectively. Both viruses are strongly inhibited by TDF (IC50s of 12.7 nM for HIV-15513 and 15.6 nM for HIV-198IN007). The combination with TDF increased the antiviral potency of R4.0 against the two primary viruses, showing a synergistic profile comparable to that of the R4.0-TDF combination observed for HIV-1BaL (Fig. 6A and B). The strongest synergy was obtained with HIV-15513 (CI50 of 0.49), while a CI50 of 0.77 was observed for HIV-198IN007. This result is particularly encouraging, since primary clade C and B strains represent a significant portion of HIV-1 R5 strains. In addition, TDF is widely used in microbicide development and clinical trials, it is a drug component of HAART, and, associated with FTC, it belongs to a recently approved oral preventive therapy (29).
FIG 6.

The R4.0 and TDF combination inhibits primary R5 HIV-1 strains in acute-infection assays. (A) Inhibition of clade C strain HIV-198IN007; (B) inhibition of clade B strain HIV-15513. The dose-response curves of mixed inhibitors (mix) are referred to as R4.0 (P values < 0.0001). Values are the means ± SD from two independent experiments performed in triplicate.
CCL5 and R4.0 entry inhibition of an MVC-resistant R5 HIV-1 strain strengthens the concepts of differential CCR5 binding site usage and of R4.0 being a barrier to virus drug resistance.
Indirect evidence of the additive/synergic effect between R4.0 or CCL5 and MVC was obtained using an MVC-resistant strain, RU570 (MVC-RU570). MVC-RU570 is an in vitro-generated variant of the Russian clade G primary isolate RU570, which became completely resistant to MVC after long-term cell culture of RU570 in the presence of MVC (30). The deletion of 3 amino acids within the V3 loop of RU570 appears to be responsible for the acquired resistance to MVC in that it provided the virus with the ability to utilize MVC-bound CCR5 for its entry process (30). PBMC from healthy donors were infected with the RU570 and MVC-RU570 strains to determine viral infections in the presence of MVC, CCL5, and R4.0. As illustrated in Table 2, MVC showed no antiviral activity against MVC-RU570, while CCL5 and R4.0 inhibited the virus, with IC50s of 3.1 and 495 nM, respectively. The superior anti-HIV-1 activity of CCL5 and R4.0 against MVC-RU570 compared to that against RU570 (3.6-fold increase for both CCL5 and R4.0) may be due to an altered gp120-CCR5 interaction with an increased reliance on the CCR5 N terminus in the resistant strain, as this was also previously described for other R5 viruses that became resistant to small chemical compounds (31–33). These data confirm that R4.0, like CCL5, may cooperate with MVC by binding to different CCR5 regions, strengthening the barrier against HIV-1 infection by lowering the emergence of virus resistance.
TABLE 2.
Antiviral activities of CCR5 inhibitors against the HIV-1 RU570 isolate and the MVC-resistant virusa
| HIV-1 inhibitor | IC50 for RU570 (nM) | IC50 for RU570-MVC (nM) |
|---|---|---|
| MVC | 6.8 ± 0.7 | >38,900 |
| CCL5 | 11.3 ± 2.1 | 3.1 ± 0.9 |
| R4.0 | 1,800 ± 97 | 495 ± 26 |
Values are the mean IC50s ± SD obtained with three different PBMC donors.
DISCUSSION
The development of new compounds and their combinations to enhance antiviral efficacy has a timely aspect in HIV-1 research. CCR5, the major HIV-1 coreceptor, is an important target for the development of anti-HIV-1 drugs and microbicides, a concept corroborated by the successful example of MVC, a CCR5-directed small chemical compound that was approved by the FDA in 2007 for its use in HIV-1-infected people. MVC, a CCR5 antagonist initially used as a systemic drug, is presently being investigated as a microbicide to prevent virus transmission during sexual intercourse. CCR5 is almost exclusively used by HIV-1 isolates involved in primary infection (34), and the presence of CD4+ T lymphocytes and macrophages within the human vaginal epithelium (35, 36) makes an HIV-1 blockade by CCR5 targeting a strategic option in microbicide development. Ideal CCR5-targeting anti-HIV-1 compounds should not activate the receptor but should instead act as CCR5 antagonists to prevent proinflammatory conditions and mucosal inflammation, which may enhance HIV-1 transmission. CCL5, a natural ligand of CCR5, is a potent HIV-1 inhibitor (37), and its engineering to enhance anti-HIV-1 activity has been a consistent source of efficient protein-based inhibitors (11, 38). An important proof of principle for the use of a CCL5 derivative as a microbicide came from PSC-RANTES. When applied in a monkey model, PSC-RANTES blocked HIV-1 vaginal transmission (39). However, despite its potent anti-HIV-1 activity, PSC-RANTES is a CCR5 agonist and may elicit inflammation and persistently eliminate CCR5 from the cell surface, its major mechanism of antiviral action (40). Conversely, a CCR5 antagonist such as R4.0, a CCL5/RANTES-derived peptide with potent anti-HIV-1 activity (10), should preserve CCR5 cell surface expression, possibly not altering its physiological function. Full-length CCL5 derivatives that block HIV-1 as CCR5 antagonists, such as C1C5-RANTES (41) and the potent compound 5p12-RANTES (9), are also available. Notably, C1C5-RANTES has been engineered in recombinant lactobacilli as a potential live microbicide (42), and several novel CCL5 variants have now been produced by using the same system (M. Secchi and L. Vangelista, unpublished data), taking advantage of the CCL5 expression proficiency of lactobacilli (43). Full-length CCL5 and peptide derivatives present similarities (lower HIV-1 resistance) but also mutually exclusive advantages and disadvantages, such as antiviral potency, a molecular size, resistance to proteolysis, and the risk of eliciting an antibody response.
In this report, we have focused on the investigation of the CCL5-derived peptide R4.0 through a combination survey with various classes of HIV-1 inhibitors. The possibility of implementing R4.0-based combinations, with the provision of virus- and cell-targeting compounds with full additivity or even synergy, has been proved. The antiviral activity of R4.0-TDF combinations on two primary R5 HIV-1 strains provided an important in vitro proof for cross-clade protection. All the HIV-1 inhibitors tested were suitable for R4.0 combination, and antagonism between the inhibitors has never been observed, including with a triple combination using IDV and TDF. The compounds tested in combination with R4.0 recognize HIV-1 gp120 (CV-N and 2G12), gp41 (T20, 2F5, and 4E10), reverse transcriptase (TDF and FTC), protease (IDV), and human CCR5 (MVC and CCL5). The most interesting information came from the evidence that simultaneous synergic CCR5 targeting by three different anti-HIV-1 ligands, as shown here with CCL5, MVC, and R4.0, is possible, a surprising and inspiring observation. The possibility of targeting CCR5 simultaneously with different compounds, such as MVC and CCL5, has already been reported (27); however, a triple CCR5-targeting combination further expands the mechanistic view and therapeutic perspective in this direction.
GPCRs may assume several conformations, providing the basis for allosteric regulation and differential receptor signaling (44). According to the observed synergic effect of a triple (R4.0-MVC-CCL5) anti-HIV-1 combination, the experimental evidence suggests that full CCR5 occupancy by these inhibitors occurs in such a manner that the net result does not comprise antagonism between the inhibitors. Considering the large surface of interaction between CCR5 and CCL5 (45), the conformational flexibility of the receptor, the relatively small targeting site by MVC, and the occupancy of only a portion of the CCL5 interaction area by R4.0, a scenario may be conceived in which most CCR5 molecules on the surface of the target cell are engaged by one or more of these ligands. A deeper explanation is inspired by a recent report investigating the CCR5 conformational preferences exerted by natural CCR5 ligands, 5p12-RANTES, and HIV-1 (46). While the virus does not discriminate between CCR5 conformations, CCR5-binding chemokines, including CCL5, have high affinity for nucleotide-free G protein-bound CCR5 and low affinity for CCR5 uncoupled from nucleotide-free G protein (46). Moreover, 5p12-RANTES seems to contribute its strong antiviral activity via an enhancement of affinity to nucleotide-free G protein-uncoupled CCR5, possibly the major CCR5 conformation portal for HIV-1 entry. It is therefore interesting to argue whether R4.0 reached a similar antiviral improvement through the modification of CCL5 regions other than the chemokine N terminus, i.e., the N-loop/β1-strand. This might be shown by the possibility of testing additive/synergic combinations such as R4.0-CCL5 and R4.0-MVC-CCL5 if we envisage CCL5 occupancy of nucleotide-free G protein-coupled CCR5 and R4.0 (or MVC-R4.0) occupancy of nucleotide-free G protein-uncoupled CCR5 totally shutting out HIV-1 entry. Interestingly, the recently reported three-dimensional structure of a complex between CCR5 and MVC (47) corroborates the hypothesis and experimental evidence discussed here. Hence, a triple R4.0-MVC-CCL5 combination would likely result in a complete shielding of the virus docking resources, regardless of a simultaneous triple occupancy per single molecule. The inhibition of an MVC-resistant virus by CCL5 and R4.0, together with the difficulty or even inability of R5 HIV-1 strains to evolve CCL5 resistance (48, 49), most likely mirrored by R4.0, further validates this type of approach.
In conclusion, our results provide a proof of principle for R4.0 usage in combination with several HIV-1 inhibitors, expand the view of CCR5 targeting, and corroborate the fight to limit virus resistance, important conceptual advancements toward the prevention of sexual transmission of HIV-1.
ACKNOWLEDGMENTS
This work was supported by Combined Highly Active Anti-Retroviral Microbicides (CHAARM) grant 242135 (FP7/2007-2013; EU, Brussels, Belgium). D.S. was supported by the Katholieke Universiteit Leuven (grants GOA 10/014 and PF 10/18).
We thank Renato Longhi (CNR-ICRM, Milan, Italy) for peptide synthesis; Francesca Sironi (San Raffaele Scientific Institute) for viral stock preparation; Davide De Battista for help with the cytofluorimetric analysis; Gabriella Scarlatti (San Raffaele Scientific Institute) for providing primary HIV-1 strains 98IN007 and 5513; Giuseppe Tambussi and Antonella Castagna (San Raffaele Scientific Institute) for providing MVC, FTC, and TDF; Alessandro Ambrosi (San Raffaele Scientific Institute) for help with statistical analysis; the Centre for AIDS Reagents, NIBSC, United Kingdom, for IDV (catalog no. ARP972) and TZM-bl cells (catalog no. ARP5011); the NIH AIDS Research and Reference Reagent Program for T20 (number 9845); Osel Inc., Mountain View, CA, USA, for CV-N; and Polymun Scientific, Austria, for MAbs 2G12, 4E10, and 2F5.
Footnotes
Published ahead of print 11 August 2014
REFERENCES
- 1.UNAIDS. 2012. Together we will end AIDS, 2012. UNAIDS, Geneva, Switzerland: http://www.unaids.org/en/resources/campaigns/togetherwewillendaids [Google Scholar]
- 2.Chun TW, Fauci AS. 2012. HIV reservoirs: pathogenesis and obstacles to viral eradication and cure. AIDS 26:1261–1268. 10.1097/QAD.0b013e328353f3f1 [DOI] [PubMed] [Google Scholar]
- 3.Picker LJ, Hansen SG, Lifson JD. 2012. New paradigms for HIV/AIDS vaccine development. Annu. Rev. Med. 63:95–111. 10.1146/annurev-med-042010-085643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anonymous. 2014. HIV immunity goes direct. Nat. Biotechnol. 32:397 (Editorial.) 10.1038/nbt.2907 [DOI] [PubMed] [Google Scholar]
- 5.Kelly CG, Shattock RJ. 2011. Specific microbicides in the prevention of HIV infection. J. Intern. Med. 270:509–519. 10.1111/j.1365-2796.2011.02454.x [DOI] [PubMed] [Google Scholar]
- 6.Goldberg DE, Siliciano RF, Jacobs WR., Jr 2012. Outwitting evolution: fighting drug-resistant TB, malaria, and HIV. Cell 148:1271–1283. 10.1016/j.cell.2012.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hladik F, McElrath MJ. 2008. Setting the stage: host invasion by HIV. Nat. Rev. Immunol. 8:447–457. 10.1038/nri2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilkin TJ, Gulick RM. 2012. CCR5 antagonism in HIV infection: current concepts and future opportunities. Annu. Rev. Med. 63:81–93. 10.1146/annurev-med-052010-145454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaertner H, Cerini F, Escola JM, Kuenzi G, Melotti A, Offord R, Rossitto-Borlat I, Nedellec R, Salkowitz J, Gorochov G, Mosier D, Hartley O. 2008. Highly potent, fully recombinant anti-HIV chemokines: reengineering a low-cost microbicide. Proc. Natl. Acad. Sci. U. S. A. 105:17706–17711. 10.1073/pnas.0805098105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Secchi M, Longhi R, Vassena L, Sironi F, Grzesiek S, Lusso P, Vangelista L. 2012. Enhancement of anti-HIV-1 activity by hot spot evolution of RANTES-derived peptides. Chem. Biol. 19:1579–1588. 10.1016/j.chembiol.2012.10.007 [DOI] [PubMed] [Google Scholar]
- 11.Vangelista L, Secchi M, Lusso P. 2008. Rational design of novel HIV-1 entry inhibitors by RANTES engineering. Vaccine 26:3008–3015. 10.1016/j.vaccine.2007.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nardese V, Longhi R, Polo S, Sironi F, Arcelloni C, Paroni R, DeSantis C, Sarmientos P, Rizzi M, Bolognesi M, Pavone V, Lusso P. 2001. Structural determinants of CCR5 recognition and HIV-1 blockade in RANTES. Nat. Struct. Biol. 8:611–615. 10.1038/89653 [DOI] [PubMed] [Google Scholar]
- 13.Vangelista L, Longhi R, Sironi F, Pavone V, Lusso P. 2006. Critical role of the N-loop and beta1-strand hydrophobic clusters of RANTES-derived peptides in anti-HIV activity. Biochem. Biophys. Res. Commun. 351:664–668. 10.1016/j.bbrc.2006.10.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lusso P, Vangelista L, Cimbro R, Secchi M, Sironi F, Longhi R, Faiella M, Maglio O, Pavone V. 2011. Molecular engineering of RANTES peptide mimetics with potent anti-HIV-1 activity. FASEB J. 25:1230–1243. 10.1096/fj.10-167627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nussbaum O, Broder CC, Berger EA. 1994. Fusogenic mechanisms of enveloped-virus glycoproteins analyzed by a novel recombinant vaccinia virus-based assay quantitating cell fusion-dependent reporter gene activation. J. Virol. 68:5411–5422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lusso P, Cocchi F, Balotta C, Markham PD, Louie A, Farci P, Pal R, Gallo RC, Reitz MS., Jr 1995. Growth of macrophage-tropic and primary human immunodeficiency virus type 1 (HIV-1) isolates in a unique CD4+ T-cell clone (PM1): failure to downregulate CD4 and to interfere with cell-line-tropic HIV-1. J. Virol. 69:3712–3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chou TC, Talalay P. 1984. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 22:27–55. 10.1016/0065-2571(84)90007-4 [DOI] [PubMed] [Google Scholar]
- 18.Chou TC. 1991. The median-effect principle and the combination index for quantization of synergism and antagonism, p 61–102 In Chou TC, Rideout DC. (ed), Synergism and antagonism in chemotherapy. Academic Press, San Diego, CA [Google Scholar]
- 19.Boyd MR, Gustafson KR, McMahon JB, Shoemaker RH, O'Keefe BR, Mori T, Gulakowski RJ, Wu L, Rivera MI, Laurencot CM, Currens MJ, Cardellina JH, II, Buckheit RW, Jr, Nara PL, Pannell LK, Sowder RC, II, Henderson LE. 1997. Discovery of cyanovirin-N, a novel human immunodeficiency virus-inactivating protein that binds viral surface envelope glycoprotein gp120: potential applications to microbicide development. Antimicrob. Agents Chemother. 41:1521–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trkola A, Purtscher M, Muster T, Ballaun C, Buchacher A, Sullivan N, Srinivasan K, Sodroski J, Moore JP, Katinger H. 1996. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J. Virol. 70:1100–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, Alldredge L, Hunter E, Lambert D, Bolognesi D, Matthews T, Johnson MR, Nowak MA, Shaw GM, Saag MS. 1998. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 4:1302–1307. 10.1038/3293 [DOI] [PubMed] [Google Scholar]
- 22.Purtscher M, Trkola A, Gruber G, Buchacher A, Predl R, Steindl F, Tauer C, Berger R, Barrett N, Jungbauer A, Katinger H. 1994. A broadly neutralizing human monoclonal antibody against gp41 of human immunodeficiency virus type 1. AIDS Res. Hum. Retroviruses 10:1651–1658. 10.1089/aid.1994.10.1651 [DOI] [PubMed] [Google Scholar]
- 23.Stiegler G, Kunert R, Purtscher M, Wolbank S, Voglauer R, Steindl F, Katinger H. 2001. A potent cross-clade neutralizing human monoclonal antibody against a novel epitope on gp41 of human immunodeficiency virus type 1. AIDS Res. Hum. Retroviruses 17:1757–1765. 10.1089/08892220152741450 [DOI] [PubMed] [Google Scholar]
- 24.Vacca JP, Dorsey BD, Schleif WA, Levin RB, McDaniel SL, Darke PL, Zugay J, Quintero JC, Blahy OM, Roth E, Sardana VV, Schlabach AJ, Graham PI, Condra JH, Gotlib L, Holloway MK, Lini J, Chen IW, Vastag K, Ostovic D, Anderson PS, Emini EA, Huff JR. 1994. L-735,524: an orally bioavailable human immunodeficiency virus type 1 protease inhibitor. Proc. Natl. Acad. Sci. U. S. A. 91:4096–4100. 10.1073/pnas.91.9.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balzarini J, Holy A, Jindrich J, Naesens L, Snoeck R, Schols D, De Clercq E. 1993. Differential antiherpesvirus and antiretrovirus effects of the (S) and (R) enantiomers of acyclic nucleoside phosphonates: potent and selective in vitro and in vivo antiretrovirus activities of (R)-9-(2-phosphonomethoxypropyl)-2,6-diaminopurine. Antimicrob. Agents Chemother. 37:332–338. 10.1128/AAC.37.2.332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schinazi RF, McMillan A, Cannon D, Mathis R, Lloyd RM, Peck A, Sommadossi JP, St Clair M, Wilson J, Furman PA, Painter G, Choi WB, Liotta DC. 1992. Selective inhibition of human immunodeficiency viruses by racemates and enantiomers of cis-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine. Antimicrob. Agents Chemother. 36:2423–2431. 10.1128/AAC.36.11.2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murga JD, Franti M, Pevear DC, Maddon PJ, Olson WC. 2006. Potent antiviral synergy between monoclonal antibody and small-molecule CCR5 inhibitors of human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 50:3289–3296. 10.1128/AAC.00699-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Escola JM, Kuenzi G, Gaertner H, Foti M, Hartley O. 2010. CC chemokine receptor 5 (CCR5) desensitization: cycling receptors accumulate in the trans-Golgi network. J. Biol. Chem. 285:41772–41780. 10.1074/jbc.M110.153460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Clercq E. 2013. The acyclic nucleoside phosphonates (ANPs): Antonín Holý's legacy. Med. Res. Rev. 10.1002/med.21283 [DOI] [PubMed] [Google Scholar]
- 30.Westby M, Smith-Burchnell C, Mori J, Lewis M, Mosley M, Stockdale M, Dorr P, Ciaramella G, Perros M. 2007. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J. Virol. 81:2359–2371. 10.1128/JVI.02006-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogert RA, Ba L, Hou Y, Buontempo C, Qiu P, Duca J, Murgolo N, Buontempo P, Ralston R, Howe JA. 2009. Structure-function analysis of human immunodeficiency virus type 1 gp120 amino acid mutations associated with resistance to the CCR5 coreceptor antagonist vicriviroc. J. Virol. 83:12151–12163. 10.1128/JVI.01351-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roche M, Jakobsen MR, Sterjovski J, Ellett A, Posta F, Lee B, Jubb B, Westby M, Lewin SR, Ramsland PA, Churchill MJ, Gorry PR. 2011. HIV-1 escape from the CCR5 antagonist maraviroc associated with an altered and less-efficient mechanism of gp120-CCR5 engagement that attenuates macrophage tropism. J. Virol. 85:4330–4342. 10.1128/JVI.00106-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roche M, Salimi H, Duncan R, Wilkinson BL, Chikere K, Moore MS, Webb NE, Zappi H, Sterjovski J, Flynn JK, Ellett A, Gray LR, Lee B, Jubb B, Westby M, Ramsland PA, Lewin SR, Payne RJ, Churchill MJ, Gorry PR. 2013. A common mechanism of clinical HIV-1 resistance to the CCR5 antagonist maraviroc despite divergent resistance levels and lack of common gp120 resistance mutations. Retrovirology 10:43. 10.1186/1742-4690-10-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Margolis L, Shattock R. 2006. Selective transmission of CCR5-utilizing HIV-1: the ‘gatekeeper' problem resolved? Nat. Rev. Microbiol. 4:312–317. 10.1038/nrmicro1387 [DOI] [PubMed] [Google Scholar]
- 35.Hladik F, Sakchalathorn P, Ballweber L, Lentz G, Fialkow M, Eschenbach D, McElrath MJ. 2007. Initial events in establishing vaginal entry and infection by human immunodeficiency virus type-1. Immunity 26:257–270. 10.1016/j.immuni.2007.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen R, Richter HE, Clements RH, Novak L, Huff K, Bimczok D, Sankaran-Walters S, Dandekar S, Clapham PR, Smythies LE, Smith PD. 2009. Macrophages in vaginal but not intestinal mucosa are monocyte-like and permissive to human immunodeficiency virus type-1 infection. J. Virol. 83:3258–3267. 10.1128/JVI.01796-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. 1995. Identification of RANTES, MIP-1α, and MIP-1β as the major HIV-suppressive factors produced by CD8+ T cells. Science 270:1811–1815. 10.1126/science.270.5243.1811 [DOI] [PubMed] [Google Scholar]
- 38.Hartley O, Offord RE. 2005. Engineering chemokines to develop optimized HIV inhibitors. Curr. Protein Pept. Sci. 6:207–219. 10.2174/1389203054065400 [DOI] [PubMed] [Google Scholar]
- 39.Lederman MM, Veazey RS, Offord R, Mosier DE, Dufour J, Mefford M, Piatak M, Jr, Lifson JD, Salkowitz JR, Rodriguez B, Blauvelt A, Hartley O. 2004. Prevention of vaginal SHIV transmission in rhesus macaques through inhibition of CCR5. Science 306:485–487. 10.1126/science.1099288 [DOI] [PubMed] [Google Scholar]
- 40.Pastore C, Picchio GR, Galimi F, Fish R, Hartley O, Offord RE, Mosier DE. 2003. Two mechanisms for human immunodeficiency virus type 1 inhibition by N-terminal modifications of RANTES. Antimicrob. Agents Chemother. 47:509–517. 10.1128/AAC.47.2.509-517.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Polo S, Nardese V, De Santis C, Arcelloni C, Paroni R, Sironi F, Verani A, Rizzi M, Bolognesi M, Lusso P. 2000. Enhancement of the HIV-1 inhibitory activity of RANTES by modification of the N-terminal region: dissociation from CCR5 activation. Eur. J. Immunol. 30:3190–3198. [DOI] [PubMed] [Google Scholar]
- 42.Vangelista L, Secchi M, Liu X, Bachi A, Jia L, Xu Q, Lusso P. 2010. Engineering of Lactobacillus jensenii to secrete RANTES and a CCR5 antagonist analogue as live HIV-1 blockers. Antimicrob. Agents Chemother. 54:2994–3001. 10.1128/AAC.01492-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Secchi M, Xu Q, Lusso P, Vangelista L. 2009. The superior folding of a RANTES analogue expressed in lactobacilli as compared to mammalian cells reveals a promising system to screen new RANTES mutants. Protein Expr. Purif. 68:34–41. 10.1016/j.pep.2009.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langmead CJ, Christopoulos A. 2014. Functional and structural perspectives on allosteric modulation of GPCRs. Curr. Opin. Cell Biol. 27:94–101. 10.1016/j.ceb.2013.11.007 [DOI] [PubMed] [Google Scholar]
- 45.Duma L, Häussinger D, Rogowski M, Lusso P, Grzesiek S. 2007. Recognition of RANTES by extracellular parts of the CCR5 receptor. J. Mol. Biol. 365:1063–1075. 10.1016/j.jmb.2006.10.040 [DOI] [PubMed] [Google Scholar]
- 46.Colin P, Bénureau Y, Staropoli I, Wang Y, Gonzalez N, Alcami J, Hartley O, Brelot A, Arenzana-Seisdedos F, Lagane B. 2013. HIV-1 exploits CCR5 conformational heterogeneity to escape inhibition by chemokines. Proc. Natl. Acad. Sci. U. S. A. 110:9475–9480. 10.1073/pnas.1222205110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I, Li T, Ma L, Fenalti G, Li J, Zhang W, Xie X, Yang H, Jiang H, Cherezov V, Liu H, Stevens RC, Zhao Q, Wu B. 2013. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 341:1387–1390. 10.1126/science.1241475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nedellec R, Coetzer M, Lederman MM, Offord RE, Hartley O, Mosier DE. 2010. “Resistance” to PSC-RANTES revisited: two mutations in human immunodeficiency virus type 1 (HIV-1) SF162 or simian-human immunodeficiency virus (SHIV) SF162-p3 do not confer resistance. J. Virol. 84:5842–5845. 10.1128/JVI.01907-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nedellec R, Coetzer M, Lederman MM, Offord RE, Hartley O, Mosier DE. 2011. Resistance to the CCR5 inhibitor 5P12-RANTES requires a difficult evolution from CCR5 to CXCR4 coreceptor use. PLoS One 6:e22020. 10.1371/journal.pone.0022020 [DOI] [PMC free article] [PubMed] [Google Scholar]