

Abstract
The Mur ligases play an essential role in the biosynthesis of bacterial peptidoglycan and hence are attractive antibacterial targets. A screen of the AstraZeneca compound library led to the identification of compound A, a pyrazolopyrimidine, as a potent inhibitor of Escherichia coli and Pseudomonas aeruginosa MurC. However, cellular activity against E. coli or P. aeruginosa was not observed. Compound A was active against efflux pump mutants of both strains. Experiments using an E. coli tolC mutant revealed accumulation of the MurC substrate and a decrease in the level of product upon treatment with compound A, indicating inhibition of MurC enzyme in these cells. Such a modulation was not observed in the E. coli wild-type cells. Further, overexpression of MurC in the E. coli tolC mutant led to an increase in the compound A MIC by ≥16-fold, establishing a correlation between MurC inhibition and cellular activity. In addition, estimation of the intracellular compound A level showed an accumulation of the compound over time in the tolC mutant strain. A significant compound A level was not detected in the wild-type E. coli strain even upon treatment with high concentrations of the compound. Therefore, the lack of MIC and absence of MurC inhibition in wild-type E. coli were possibly due to suboptimal compound concentration as a consequence of a high efflux level and/or poor permeativity of compound A.
INTRODUCTION
Peptidoglycan, made up of sugars and amino acids, is a mesh-like structure that lies outside the cytoplasmic membrane of the bacterial cell. Chemically, peptidoglycan consists of alternating residues of β-(1,4)-linked N-acetylglucosamine and N-acetylmuramic acid with a three- or five-amino-acid peptide attached to the N-acetylmuramic acid. The attached peptide chains are cross-linked to each other and form the mesh-like structure of peptidoglycan (1, 2, 3). Multiple inhibitors of peptidoglycan biosynthesis leading to cell death have been identified, providing substantial evidence for the significance of the macromolecule in maintaining normal cellular physiology (2).
Clinically successful antibiotics that target peptidoglycan biosynthesis, including penicillin and vancomycin, inhibit the late stages of the process (4). While the success of the β-lactam class of compounds has firmly established the validity of this pathway for antibiotic discovery, increases in drug resistance have created an urgent need for compounds with novel modes of action (5). Discovery of new targets belonging to this well-understood and clinically validated cellular pathway is likely to fast-track development of novel antibiotics. Many of the enzymes catalyzing the early phases of the peptidoglycan pathway, e.g., MurA to MurF (6), are essential for bacterial survival and hence offer an opportunity to find inhibitors of peptidoglycan synthesis with distinct mechanisms of action. As an example, fosfomycin, a natural product isolated in 1969 from Streptomyces (7), has been shown to target MurA.
MurC, the third enzyme in the pathway, catalyzes the conversion of UDP-N-acetyl-muramic acid (UNAM) to UDP-N-acetyl-muramic acid-l-alanine (UNAM-Ala) (Fig. 1) and has been shown to be essential in Escherichia coli (8). Both small molecules and peptide MurC inhibitors, i.e., phosphinate substrate analogs (9), benzofuran acyl-sulfonamides (10), aminoquinoxazoline, C-1 compound (11), peptide inhibitors (12), and the 13-mer natural product feglymycin that inhibits both MurA and MurC (13), have been discovered and reported. Unfortunately, none of the small-molecule MurC inhibitors displayed significant antibacterial activity against either the wild-type (WT) or efflux-deficient strains of Gram-negative bacteria, except compound 3-(phenylethoxy) quinoxalin-2-amine (11), which showed very poor activity (MIC = 64 μg/ml) against a tolC and imp mutant strain. Feglymycin and novel peptides inhibiting MurC identified from a phage display approach were weakly active on Staphylococcus aureus but were inactive on Gram-negative bacteria. Therefore, whether inhibition of MurC would lead to cell death was not well understood.
FIG 1.
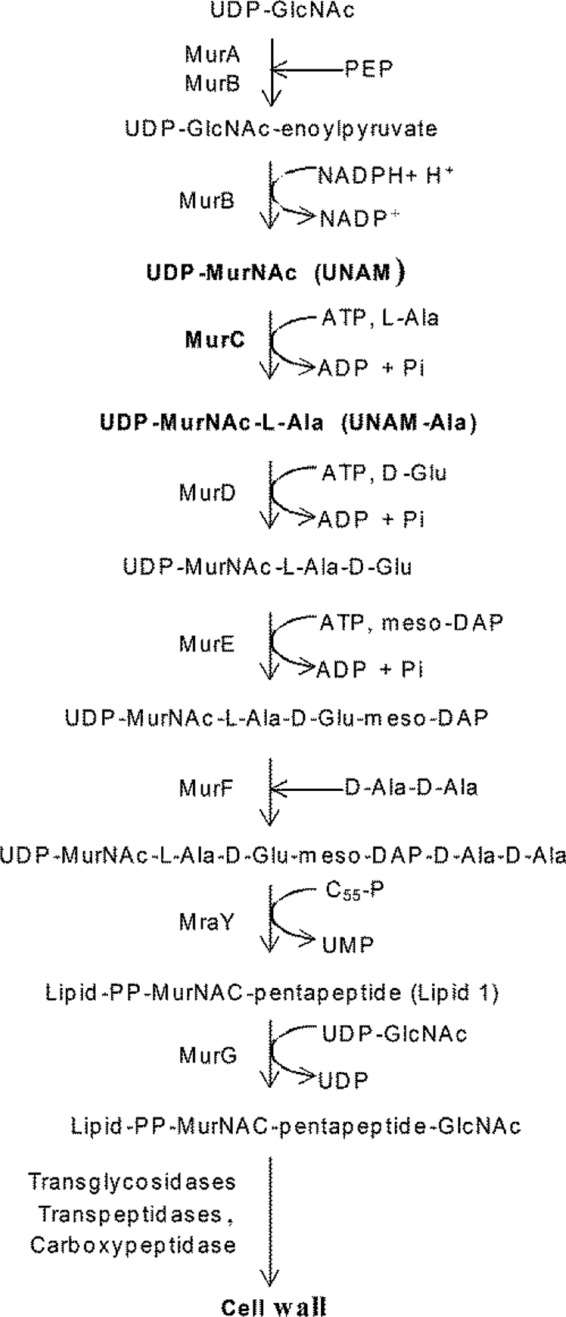
Steps of the peptidoglycan biosynthetic pathway. PEP, phosphoenolpyruvate; PP, phosphate-phosphate.
Lack of cellular activity is a significant challenge faced during early antibiotic discovery, especially with Gram-negative bacteria (14). One of the major reasons is the nature of the outer membrane of Gram-negative bacteria, which serves as an effective permeability barrier (15). In addition, susceptibility to antibiotics is evaded by efflux of compounds from the cells. Multidrug efflux pumps have emerged as important mechanisms of antimicrobial resistance in bacterial pathogens (16, 17). Thus, efflux pump mutants and inhibitors serve as valuable tools in drug discovery to understand the mechanism underlying the lack of cellular activity for novel scaffolds. This study investigated the differential activity displayed by compound A, a MurC inhibitor, against E. coli and an efflux pump mutant strain of E. coli (tolC mutant). The results demonstrate that MurC is an attractive target for discovery of novel antibacterials.
MATERIALS AND METHODS
MIC measurement.
MICs for E. coli W3110 (WT) and the E. coli tolC mutant were determined by the broth microdilution method (18). The experiment was performed at a cell density of ∼108 CFU/ml in Mueller-Hinton II cation-adjusted broth (MHB; BD-BBL). Cells were treated with the test compounds and incubated at 37°C for 18 to 24 h. The cell growth was monitored by measuring absorbance at 600 nm. Control cultures were treated with amounts of dimethyl sulfoxide (DMSO) equivalent to those present in treated cultures. The MIC was defined as the concentration of the compound that resulted in ≥80% inhibition of growth compared to that seen with untreated culture. Most studies determine MICs at a lower inoculum density (∼105 CFU/ml). However, the experiments related to metabolite and compound estimations described in this work were carried out at ∼108 CFU/ml (optical density at 600 nm [OD600] = 0.1). To understand and minimize the impact of the inoculum density, MICs were determined at ∼108 CFU/ml and compared with MICs estimated at ∼105 CFU/ml. MICs estimated at low and high inoculum densities were within a 2-fold range of differences for compound A. For comparison across various drug treatments, all treatments were carried out with respect to fold MIC values determined at ∼108 CFU/ml.
MurC biochemical assay.
The reaction was carried out in a 25-μl volume containing 25 mM Tris-HCl (pH 8.0), 10 mM ammonium sulfate, 1.25 mM dithiothreitol (DTT), 0.002% Brij 35, 10 mM MgCl2, 40 μM UNAM, 100 μM ATP, 40 μM l-alanine (l-alanine), and 15 nM enzyme in a 384-well microtiter plate (Corning catalog no. 3702) at 25°C for 50 min. Inorganic phosphate formed by the hydrolysis of ATP was detected by adding 25 μl of malachite green reagent adapted from a protocol published by Baykov et al. (19). The modified malachite green reagent comprised reagent A (0.12% [wt/vol] malachite green in 6 N H2SO4), reagent B (7.5% ammonium molybdate in water), reagent C (11% [wt/vol] Tween 20), and water mixed in a 10:2.5:1:40.5 proportion. A630 was measured after 30 min of incubation at 25°C using SpectraMax Plus (Molecular Devices). Percentages of inhibition were calculated and fitted to the four-parameter Hill's equation to determine the 50% inhibitory concentration (IC50) of the compounds.
Killing kinetics.
The killing kinetics was determined by enumerating survivors at various time points following compound exposure. E. coli tolC mutant cells (∼108 CFU/ml; A600 of 0.1) were treated with various concentrations (200 μM to 0.4 μM) of compound A in a 96-well microtiter plate (Nest 701001). At various time intervals (0 to 4 h), 20-μl volumes of treated cultures were serially diluted and plated on MH agar plates. The plates were incubated at 37°C for 24 h, and the CFU levels were determined.
Preparation of cell extracts for precursor and compound measurement. (i) For precursor measurement.
E. coli tolC mutant cells and E. coli WT cells at an A600 of 0.1 (∼108 CFU/ml) (100 ml) were incubated with compound A (test) or the equivalent concentration of DMSO (control) for 40 min or unless mentioned otherwise, with shaking. The cultures were subsequently chilled on ice, A600 was measured, and the cultures were plated for CFU estimation. Cultures were centrifuged, and cell pellets were treated with 2 ml of cold 5% trichloroacetic acid (TCA), mixed, and kept on ice for 30 min. The lysates were then centrifuged at 10,000 × g for 10 min, and the supernatants were extracted with an equal volume of diethylether thrice. The aqueous phases were lyophilized, dissolved in 1 ml of 5 mM ammonium acetate with 3% (vol/vol) acetic acid (reconstitution solution), and subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.
(ii) For compound measurement.
E. coli tolC mutant and E. coli WT cell cultures (A600 of 0.1) were divided into aliquots consisting of 1-ml fractions. Each intracellular-compound-estimation experiment involved three samples of 1 ml each, namely, “treated,” “untreated,” and “wash control” cultures. For treated cultures, cells were treated with the compound and allowed to grow for a defined time at 37°C with shaking at 180 rpm. For untreated control cultures, an equivalent amount of DMSO was added. For the wash control cultures, cells were chilled on ice for 15 min followed by addition of an equal amount of compound as described for the treated culture. Bovine serum albumin (BSA) was added to all the samples to reach a final concentration of 2% (wt/vol). Cells were harvested by centrifugation at 4°C and 13,000 rpm in a tabletop centrifuge, and pellets were washed with phosphate-buffered saline (PBS) containing 2% BSA followed by a second wash with PBS. Cells were resuspended in 200 μl of PBS and lysed by bead beating (using Sigma G-8772 425-to-600-μm-diameter glass beads) (420 rpm, 2 cycles). One ml of acetonitrile containing 10 ng/ml of carbamazepine (the process control) was added to the samples, and then the samples were centrifuged at 13,000 rpm for 5 min. The supernatant was collected and analyzed by LC-MS/MS.
Liquid chromatography and tandem mass spectrometry. (i) Measurement of precursors.
Measurement of UNAM and UNAM-Ala in E. coli tolC mutant and E. coli WT cell extracts was performed on an LC (Prominence UPLC XR; Shimadzu Corporation, Kyoto, Japan) coupled to a QQQ mass spectrometer (API3000; AB Sciex, Thornhill, Ontario, Canada). Details of the LC-MS method and mass spectrometer parameters have been provided as supplemental information (see Table S1A in the supplemental material). Azithromycin was used as a process control to compensate for any processing losses during sample preparation. The ratio of the precursor signal to the process control signal was used as the measure of the precursor level in the samples. The method was validated as described by Matuszewski et al. (20) with relevant modifications (see Table S2A). Basal concentrations of UNAM and UNAM-Ala in E. coli cells (untreated) were determined from the calibration curve generated using known precursor concentrations.
(ii) Measurement of compound A and rifampin concentrations.
Intracellular compound A and rifampin concentrations were measured using high-performance LC (HPLC) (Acquity UPLC; Waters, Milford, MA) coupled to a QQQ mass spectrometer (Xevo TQ-S; Waters, Milford, MA). Carbamazepine was used as a process control to compensate for any processing losses that occurred during the analysis. The concentration of the compound or of rifampin in the samples was determined from calibration curves generated from known concentrations of the compound or of rifampin, respectively. Method validation results for the measurement of compound A are presented in Table S2B in the supplemental material. The method was sufficiently sensitive to measure concentrations of 0.08 nM and above, and the results were linear from 0.08 to 50 nM.
RESULTS
High-throughput biochemical screen of AstraZeneca compounds for inhibitors of E. coli MurC enzyme identified pyrazolopyrimidines as representing a potent MurC enzyme inhibitor class. The understanding of the structure-activity relationship (SAR) for this series and its characterization and properties are described in a parallel manuscript (submitted for publication). Compound A, a representative pyrazolopyrimidine, inhibits MurC (IC50 of 12 nM) but did not inhibit E. coli MurD and MurE enzymes (IC50 of >100 μM; Fig. 2). Despite potent inhibition of MurC in vitro, compound A lacked activity (MIC, >200 μM) against Gram-negative bacteria (Fig. 2; the microbiological activity spectrum is shown in Table S3 in the supplemental material) such as E. coli and Pseudomonas aeruginosa but was found to have MICs against E. coli tolC mutant and P. aeruginosa ΔmexABCDXY efflux pump mutant strains of 12.5 and 6.25 μM, respectively (Fig. 2). The compound A MIC was also tested in the presence of efflux pump inhibitors. MICs of 25 and 50 μM against wild-type P. aeruginosa and E. coli, respectively, were observed in the presence of phenylalanine-arginine beta-naphthylamide (PAβN; see Table S4 in the supplemental material); no activity was observed with the other efflux inhibitors tested. Properties of compound A were shared by other active compounds in the series, which made it a realistic representative of this chemical class (see Table S5); hence, compound A was used to understand the differential activities of the compound (and series) against wild-type and efflux pump mutant E. coli strains. Differences in intracellular compound levels in the two cell types as a result of altered permeability and/or high efflux could be a possible explanation for the differential activities. Alternately, MurC might be inaccessible or might not be inhibited by small molecules in the cell; hence, the cellular activity observed for the E. coli tolC mutant would be unrelated to the inhibition of MurC in this scenario.
FIG 2.
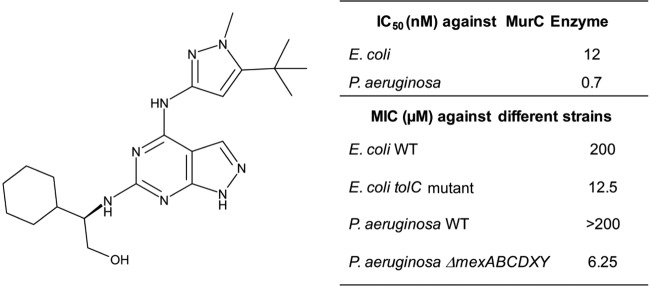
Structure and activity of compound A.
Time-dependent bactericidal activity for E. coli tolC mutant.
A kill kinetics experiment was carried out to identify treatment conditions that did not show significant cell killing in the assays to estimate intracellular precursor and compound A levels. Minimal cell death and lysis were pertinent with respect to preventing errors in the estimation of compound and precursor levels in the cell. To identify such a condition, an E. coli tolC mutant culture (∼108 CFU/ml) was treated with various concentrations (50, 100, and 200 μM) of compound A for 30, 60, 120, and 240 min. Increased cell killing was observed with increasing treatment time, and ∼1 and ∼2 log10 CFU/ml reductions were observed after compound exposures for 2 and 4 h, respectively (Fig. 3). We observed inhibition of cell growth but minimal cell death when cells were treated with 50 μM compound A for 30 min (Fig. 3). Hence, this treatment condition was chosen to assess modulation of cellular MurC substrate and product levels. Lack of kill curve information beyond 4 h was a limitation of the study; however, it was observed that ∼105 CFU/ml cells treated with compound A for 20 h showed an ∼3 log10 CFU/ml reduction at and above the MIC.
FIG 3.
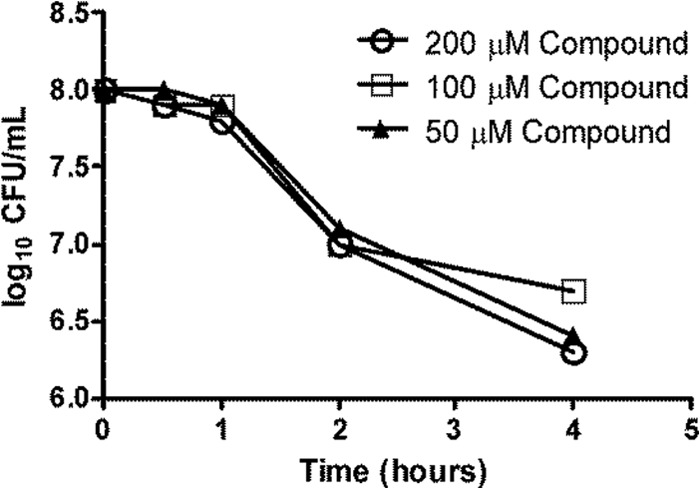
Killing kinetics of E. coli tolC mutant by compound A. An E. coli tolC mutant culture with a cell density of ∼108 CFU/ml was treated with different concentrations (50, 100, and 200 μM) of compound A for 30, 60, 120, and 240 min. After the exposure times, the cultures were appropriately diluted and plated for CFU estimation.
Accumulation of UNAM and decrease in UNAM-Ala levels in the E. coli tolC mutant upon compound treatment.
To understand whether compound A specifically inhibited MurC in the cells, we estimated changes in the levels of UNAM and UNAM-Ala in the E. coli tolC mutant upon treatment with various concentrations of the compound. A concentration-dependent increase in UNAM levels was observed, with a maximum of a ∼10-fold increase over the untreated culture levels, when the cells were treated with 12.5 μM compound A for 40 min (Fig. 4A). A decrease in UNAM-Ala levels was observed simultaneously, whereas CFU/ml levels remained unaltered under all treatment concentrations compared to the results seen under untreated conditions (Fig. 4A). Under these experimental conditions, the basal level of UNAM-Ala was ∼30-fold lower than that of UNAM. Due to low UNAM-Ala signals in untreated cells, treatment with even the lowest concentration of the compound reduced the UNAM-Ala signals to below the limit of detection of the assay.
FIG 4.
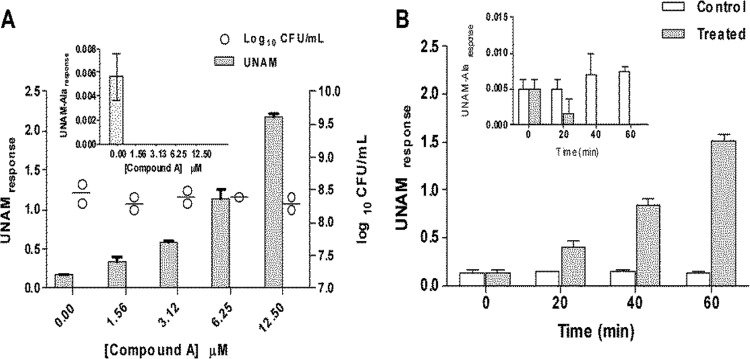
Modulation of UNAM and UNAM-Ala levels in the E. coli tolC mutant upon treatment with compound A. (A) E. coli tolC mutant cells treated with compound A at different concentrations (0, 1.56, 3.12, 6.25, and 12.5 μM) for 40 min showed an increase in UNAM levels with increasing compound concentrations. (Inset) UNAM-Ala levels decreased with increasing compound concentrations. The UNAM-Ala response was below the limit of detection at all the tested compound concentrations. (B) E. coli tolC mutant cells treated with 6.25 μM compound A for different time durations (0, 20, 40, and 60 min) showed an increase in UNAM levels with increasing time. (Inset) UNAM-Ala levels decreased with increasing treatment time. The UNAM-Ala response was below the limit of detection for cells treated for 40 and 60 min. The UNAM/UNAM-Ala response on the y axis is defined as the ratio of the area under the chromatogram of UNAM/UNAM-Ala (AUCUNAM/AUCUNAM-Ala) to the area under the chromatogram of the internal standard (AUCIS). Data represent the means of the results of triplicate measurements, with standard deviations shown as error bars.
To understand further the kinetics of the substrate and product modulation, cells were treated with 6.25 μM compound A for 20, 40, and 60 min. Increases in UNAM levels were observed in treated cells over time (Fig. 4B). A decrease in UNAM-Ala levels was also observed over time (Fig. 4B). The observed modulation of substrate and product levels upon compound treatment confirms that MurC activity was inhibited in E. coli tolC mutant cells. Moreover, overexpression of MurC in the E. coli tolC mutant leads to a ≥16-fold shift in the MIC, confirming that inhibition of MurC expression results in growth suppression (see Table S6 in the supplemental material). Finally, compound A specifically inhibited incorporation of radiolabeled DAP (diaminopimelic acid), a precursor in the peptidoglycan pathway, in E. coli tolC mutant cells (see Table S7 in the supplemental material). This reconfirms that the peptidoglycan synthesis pathway was inhibited in cells treated with compound A.
UNAM levels remained unchanged upon compound treatment in E. coli wild-type cells.
Results in E. coli tolC mutant cells (described above) demonstrated that in vivo inhibition of MurC results in the inhibition of cell growth. Since the biology characteristics of the targets of the wild-type and the mutant strains are likely to be similar, it was expected that compound A would inhibit MurC in wild-type E. coli as well. To investigate this idea, E. coli cells were treated with 6.25 μM compound A for 40 min and UNAM levels were measured. No significant change in the UNAM level was observed in treated cells with respect to untreated controls (Fig. 5). The levels remained unchanged even when the cells were treated with 30 μM compound for 40 min. In the same experiment, there was an increase in UNAM levels in E. coli tolC mutant cells treated with compound A compared to the untreated-control results (P < 0.05; analysis of variance [ANOVA]) (Fig. 5) as expected. Hence, MurC was not inhibited sufficiently in the wild-type cells, possibly due to suboptimal intracellular compound levels in these cells.
FIG 5.
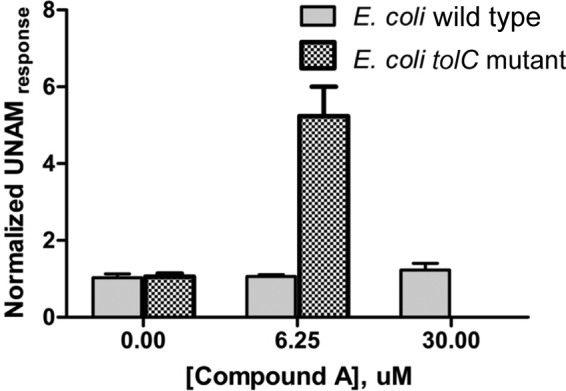
Effect of compound A on UNAM levels in E. coli tolC mutant and E. coli WT cells. E. coli tolC mutant cells treated with 6.25 μM compound A for 40 min showed a 5-fold increase in UNAM levels compared to untreated cells (P < 0.05). However, E. coli WT cells treated with 6.25 and 30 μM compound A for 40 min did not show any increase in UNAM levels compared to untreated cells. The normalized UNAM response was the UNAM response normalized to untreated culture.
Compound A was detected in the E. coli tolC mutant but not in the WT strain.
To assess compound A concentration in the E. coli WT strain versus that in the tolC mutant, cells were treated with 6.25 μM compound A for 5, 10, 20, and 40 min. Cell extracts were prepared as described in Materials and Methods. Wash control samples were used to measure nonspecific binding to the cell matrix. The difference between the concentrations of the wash control and treated samples was considered to be reflective of the intracellular concentration. Carbamazepine was used as a control across multiple independent experiments. The intracellular concentration of compound A in tolC mutant cells was significantly higher than in the E. coli WT cells (Fig. 6A) (P < 0.05 [ANOVA at the 20- and 40-min time points]). Moreover, an increase in compound A levels with time was observed in tolC mutant cells. This correlates well with the increased levels of UNAM observed in tolC mutant cells treated with a similar concentration of compound A (Fig. 4A). As a positive control, the rifampin level was estimated in WT and tolC mutant cells. Rifampin has similar MICs of 8 to 16 μg/ml for the two E. coli strains and in accordance showed similar intracellular compound levels in the two cell types (Fig. 6B). It is possible that the lipophilic nature of compound A contributes to the “higher” basal level observed compared to the levels determined with other drugs studied in our laboratory (21), leading to underestimation of the fold difference in compound levels between E. coli wild-type and E. coli tolC mutant cells.
FIG 6.
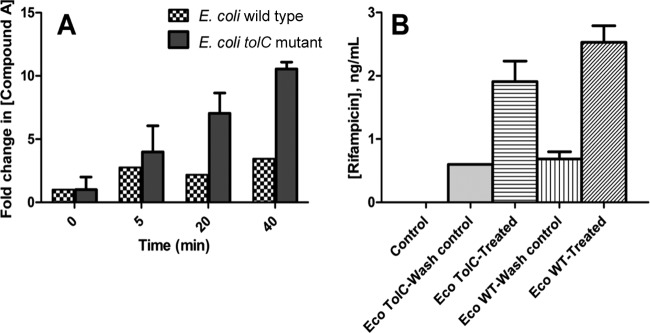
In summary, a compound concentration- and treatment time-dependent increase in MurC substrate levels and a reduction in MurC product levels were detected in the E. coli tolC mutant strain. Treatment with compound A led to the inhibition of peptidoglycan synthesis, and an upshift of MIC was observed upon target overexpression. Hence, inhibition of E. coli tolC mutant cells by compound A appears to be due to inhibition of MurC. Under similar experimental conditions, MurC substrate and product levels were unaltered in the E. coli WT. In parallel, a time-dependent increase in intracellular compound A levels in the E. coli tolC mutant was observed; however, such an increase in compound levels was not observed in the wild-type cells.
DISCUSSION
The MurA to MurF ATP-dependent ligases are enzymes involved in the early stages of peptidoglycan synthesis in bacteria. They are highly conserved, essential enzymes that are specific to bacteria. A significant amount of information regarding the structure and function of the enzymes is already available in the literature (22). In addition, the discovery of several compounds that inhibit one or more Mur enzymes in vitro demonstrates the suitability of these targets for use in the development of treatment drugs (23). The absence of a well-established link between inhibition of enzymes MurB to MurF in bacterial cells and cell death has caused a debate concerning whether the targets are vulnerable enough for the discovery of antibacterials (23). Indeed, Silver analyzed the antibacterial activity of the MurC to MurF enzyme inhibitors and concluded that there is no compelling evidence that the antibacterial activity of any of the MurC to MurF enzyme inhibitors can be ascribed to inhibition of these targets in the bacterial cell (24). It has also been hypothesized that the enzymes may be refractory to inhibition in vivo, since they are part of a multienzyme complex with possible channeling of reaction intermediates, likely making the individual active sites inaccessible to inhibitors (25). In this study, MurC inhibition leading to cell growth and peptidoglycan biosynthesis was demonstrated for pyrazolopyrimidine compound A in the efflux pump mutant strain of E. coli. Hence, MurC appears to be accessible to small-molecule inhibitors in bacterial cells and provides an opportunity to identify novel inhibitors with no preexisting resistance.
The MurC substrate and product were unaltered in wild-type E. coli cells, indicating a lack of target engagement. Measurement of the intracellular compound concentrations in both cell types revealed a minimal compound level in the wild type, whereas compound accumulation was observed in the E. coli tolC mutant. Weak E. coli and P. aeruginosa activity was observed in the presence of efflux pump inhibitor PAβN. PAβN, a well-studied peptidomimetic compound, is a broad-spectrum efflux pump inhibitor and significantly reduces the MIC of antibiotics such as fluoroquinolones and β-lactams for Gram-negative bacteria (26). It has also been implicated in permeabilizing the outer membrane of E. coli (27). The compound A MIC significantly improved for E. coli tolC mutant cells, and a compound concentration adequate to inhibit MurC enzyme was achieved. TolC is an outer membrane porin involved in the efflux of hydrophobic and amphipathic molecules (17, 28). It serves as a common outer membrane component of several multidrug efflux systems, including AcrAB, EmrAB, and MacAB. Inactivation of tolC in E. coli cells results in hypersensitivity to inhibitors, including bile salts, detergents, and hydrophobic antibiotics (29). This was initially attributed to the presence of defective lipopolysaccharide (LPS) in these cells (30); however, it was later demonstrated that TolC is required for the activity of E. coli efflux pumps (31). Compound A, being relatively hydrophobic, appears to be a good substrate for efflux pumps. Further optimization is required to address high efflux or low permeability for the chemical series. Combining a MurC inhibitor with a known efflux inhibitor or permeability enhancer is an interesting alternate path toward development of antibiotic therapy.
Estimation of cellular compound levels using LC-MS/MS is a relatively new approach in comparison to traditional methods using radioactive or fluorescently labeled compounds. The LC-MS/MS approach is sensitive and specific and does not require compound derivatization. Important conceptual work has been carried out in this area for mycobacteria (32–34). A previous study of Gram-negative bacteria by Cai et al. described the use of the LC-MS approach for estimation of ciprofloxacin accumulation in P. aeruginosa (35). That assay was performed with stationary-phase cultures, whereas logarithmically growing cultures were used in this study. In addition, wash and internal controls have been incorporated in each assay for assessment of nonspecific binding and process reproducibility, respectively. Finally, BSA was included in the assay to reduce nonspecific binding for compounds with high lipophilicity.
Conclusion.
In conclusion, a correlation between inhibition of enzyme and bacterial cell death has been demonstrated for MurC. Further, the relationship between compound levels and cellular activity against E. coli cells has been assessed and correlated. Since the target characteristics of wild-type and E. coli tolC pump mutant cells are expected to be similar, MurC is an attractive target for antibacterial discovery with no preexisting resistance.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Suresh Solapure, Vinayak Hosagrahara, Grant Walkup, and Boudewijn de Jonge for scientific discussions. We thank Shubha Sriram and Boudewijn de Jonge for carrying out the precursor incorporation experiments.
Support from the Biosciences and Chemistry Departments, AstraZeneca Bangalore, and AstraZeneca Boston is acknowledged.
V.H. performed IC50 measurements and designed and performed experiments to estimate levels of precursors and compounds in the cell, K.R.P. was responsible for LC-MS analysis, A.N. carried out extract preparations and CFU estimations, S.S. and S.G. performed MIC and kill kinetics experiments, V.R. was involved in the design and interpretation of microbiology experiments, P.M. and M. Chinnapattu carried out compound synthesis, P.S.H. was responsible for chemistry design and critical input, S.R. provided critical input related to the work and writing of the manuscript, V.H., K.R.P., and M. Chatterji wrote the manuscript with contributions from all coauthors, and M. Chatterji was responsible for the design and interpretation of all experiments.
Footnotes
Published ahead of print 11 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02890-14.
REFERENCES
- 1.Roger HJ, Perkins HR, Ward JB. 1980. Structure of peptidoglycan, p 190–214 In Microbial cell walls and membranes. Chapman and Hall, London, United Kingdom [Google Scholar]
- 2.Bugg TD, Walsh CT. 1992. Intracellular steps of bacterial cell wall peptidoglycan biosynthesis: enzymology, antibiotics, and antibiotic resistance. Nat. Prod. Rep. 9:199–215. 10.1039/np9920900199 [DOI] [PubMed] [Google Scholar]
- 3.Demchick P, Koch AL. 1996. The permeability of the wall fabric of Escherichia coli and Bacillus subtilis. J. Bacteriol. 178:768–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schneider T, Sahl HG. 2010. An oldie but a goodie – cell wall biosynthesis as antibiotic target pathway. Int. J. Med. Microbiol. 300:161–169. 10.1016/j.ijmm.2009.10.005 [DOI] [PubMed] [Google Scholar]
- 5.Fernandes P. 2006. Antibacterial discovery and development – the failure of success. Nat. Biotechnol. 24:1497–1503. 10.1038/nbt1206-1497 [DOI] [PubMed] [Google Scholar]
- 6.Kahne D, Leimkuhler C, Lu W, Walsh C. 2005. Glycopeptides and lipoglycopeptide antibiotics. Chem. Rev. 105:425–448. 10.1021/cr030103a [DOI] [PubMed] [Google Scholar]
- 7.Hendlin D, Stapley EO, Jackson M, Wallick H, Miller AK, Wolf FJ, Miller TW, Chaiet L, Kahan FM, Foltz EL, Woodruff HB, Mata JM, Hernandez S, Mochales S. 1969. Phosphonomycin, a new antibiotic produced by strains of streptomyces. Science 166:122–123. 10.1126/science.166.3901.122 [DOI] [PubMed] [Google Scholar]
- 8.Bocquel MT, Fairley M, Mengin-Lecreulx D, Parquet C, Realo E, Salah BK, Taburet Y, Harnois M, van Heijenoort J. 1997. Abstr. 97th Gen. Meet. Am. Soc. Microbiol., abstr H-2:285 [Google Scholar]
- 9.Reck F, Marmor S, Fisher S, Wuonola M. 2001. Inhibitors of the bacterial cell wall biosynthesis enzyme MurC. Bioorg. Med. Chem. Lett. 11:1451–1454. 10.1016/S0960-894X(01)00251-7 [DOI] [PubMed] [Google Scholar]
- 10.Ehmann DE, Demeritt JE, Hull KG, Fisher SL. 2004. Biochemical characterization of an Inhibitor of Escherichia coli UDP-N-acetylmuramyl-L-alanine ligase. Biochim. Biophys. Acta 1698:167–174. 10.1016/j.bbapap.2003.11.006 [DOI] [PubMed] [Google Scholar]
- 11.Zawadzke LE, Norcia M, Desbonnet CR, Wang H, Freeman-Cook K, Dougherty TJ. 2008. Identification of an inhibitor of MurC enzyme, which catalyzes an essential step in the peptidoglycan precursor synthesis pathway. Assay Drug Dev. Technol. 6:95–103. 10.1089/adt.2007.114 [DOI] [PubMed] [Google Scholar]
- 12.El Zoeiby A, Sanschagrin F, Darveau A, Brisson JR, Levesque RC. 2003. Identification of novel inhibitors of Pseudomonas aeruginosa MurC enzyme derived from phage-displayed peptide libraries. J. Antimicrob. Chemother. 51:531–543. 10.1093/jac/dkg010 [DOI] [PubMed] [Google Scholar]
- 13.Rausch S, Hänchen A, Denisiuk A, Löhken M, Schneider T, Süssmuth RD. 2011. Feglymycin is an inhibitor of the enzymes MurA and MurC of the peptidoglycan biosynthesis pathway. ChemBioChem 12:1171–1173. 10.1002/cbic.201100120 [DOI] [PubMed] [Google Scholar]
- 14.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. 2007. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6:29–40. 10.1038/nrd2201 [DOI] [PubMed] [Google Scholar]
- 15.Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67:593–656. 10.1128/MMBR.67.4.593-656.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen GN. 2011. The outer membrane of Gram-negative bacteria and the cytoplasmic membrane, p 11–16 In Microbial biochemistry. Springer Science, New York, NY. 10.1007/978-90-481-9437-7_2 [DOI] [Google Scholar]
- 17.Poole K. 2005. Efflux-mediated antimicrobial resistance. J. Antimicrob. Chemother. 56:20–51. 10.1093/jac/dki171 [DOI] [PubMed] [Google Scholar]
- 18.Andrews JM. 2001. Determination of minimum inhibitory concentration. J. Antimicrob. Chemother. 48(Suppl 1):5–16. 10.1093/jac/48.suppl_1.5 [DOI] [PubMed] [Google Scholar]
- 19.Baykov AA, Evtushenko OA, Avaeva SM. 1988. A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay. Anal. Biochem. 171:266–270. 10.1016/0003-2697(88)90484-8 [DOI] [PubMed] [Google Scholar]
- 20.Matuszewski BK, Constanzer ML, Chavez-Eng CM. 2003. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 75:3019–3030. 10.1021/ac020361s [DOI] [PubMed] [Google Scholar]
- 21.Bhat J, Narayan A, Venkatraman J, Chatterji M. 2013. LC-MS based assay to measure intracellular compound level in Mycobacterium smegmatis: linking compound levels to cellular potency. J. Microbiol. Methods 94:152–158. 10.1016/j.mimet.2013.05.010 [DOI] [PubMed] [Google Scholar]
- 22.El Zoeiby A, Sanschagrin F, Levesque RC. 2003. Structure and function of the Mur enzymes: development of novel inhibitors. Mol. Microbiol. 47:1–12. 10.1046/j.1365-2958.2003.03289.x [DOI] [PubMed] [Google Scholar]
- 23.Silver LL. 2003. Novel inhibitors of bacterial cell wall synthesis. Curr. Opin. Microbiol. 6:431–438. 10.1016/j.mib.2003.08.004 [DOI] [PubMed] [Google Scholar]
- 24.Silver LL. 2006. Does the cell wall of bacteria remain a viable source of targets for novel antibiotics? Biochem. Pharmacol. 71:996–1005. 10.1016/j.bcp.2005.10.029 [DOI] [PubMed] [Google Scholar]
- 25.Silver LL. 2011. Challenges in antibacterial discovery. Clin. Microbiol. Rev. 24:71–108. 10.1128/CMR.00030-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zechini B, Versace I. 2009. Inhibitors of multidrug resistant efflux systems in bacteria. Recent Pat. Antiinfect. Drug Discov. 4:37–50. 10.2174/157489109787236256 [DOI] [PubMed] [Google Scholar]
- 27.Lamers RP, Cavallari JF, Burrows LL. 2013. The efflux inhibitor phenylalanine-arginine beta-naphthylamide (PAβN) permeabilizes the outer membrane of gram-negative bacteria. PLoS One 8:e60666. 10.1371/journal.pone.0060666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zgurskaya HI, Krishnamoorthy G, Ntreh A, Lu S. 2011. Mechanism and function of the outer membrane channel TolC in multidrug resistance and physiology of Enterobacteria. Front. Microbiol. 2:189. 10.3389/fmicb.2011.00189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whitney EN. 1971. The tolC locus in Escherichia coli K12. Genetics 67:39–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schnaitman CA, Klena J. 1993. Genetics of lipopolysaccharide biosynthesis in enteric bacteria. Microbiol. Rev. 57:655–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fralick JA. 1996. Evidence that TolC is required for functioning of the Mar/AcrAB efflux pump of Escherichia coli. J. Bacteriol. 178:5803–5805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarathy JP, Lee E, Dartois V. 2013. Polyamines inhibit porin-mediated fluoroquinolone uptake in mycobacteria. PLoS One 8:e65806. 10.1371/journal.pone.0065806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarathy J, Dartois V, Dick T, Gengenbacher M. 2013. Reduced drug uptake in phenotypically resistant nutrient-starved nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 57:1648–1653. 10.1128/AAC.02202-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chakraborty S, Gruber T, Barry CE, III, Boshoff HI, Rhee KY. 2013. Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science 339:88–91. 10.1126/science.1228980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai H, Rose K, Liang LH, Dunham S, Stover C. 2009. Development of liquid chromatography/mass-spectrometry-based accumulation assay in Pseudomonas aeruginosa. Anal. Biochem. 385:321–325. 10.1016/j.ab.2008.10.041 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.