

Abstract
It is now World Health Organization (WHO) policy that drug concentrations on day 7 be measured as part of routine assessment in antimalarial drug efficacy trials. The rationale is that this single pharmacological measure serves as a simple and practical predictor of treatment outcome for antimalarial drugs with long half-lives. Herein we review theoretical data and field studies and conclude that the day 7 drug concentration (d7c) actually appears to be a poor predictor of therapeutic outcome. This poor predictive capability combined with the fact that many routine antimalarial trials will have few or no failures means that there appears to be little justification for this WHO recommendation. Pharmacological studies have a huge potential to improve antimalarial dosing, and we propose study designs that use more-focused, sophisticated, and cost-effective ways of generating these data than the mass collection of single d7c concentrations.
INTRODUCTION
Providing effective antimalarial drugs is a cornerstone of public health policy in the majority of developing countries. Historically, the evolution of drug resistance undermined the effectiveness of first-line therapies (1–3), and failing drugs were retained for much too long (4). This led to a surveillance strategy of using regular monitoring to confirm the continued efficacy and effectiveness of local first-line therapies (5). Resistance is conventionally regarded as a binary trait where infections may be classified as “resistant” or “susceptible” depending on patient therapeutic outcome. However, it is becoming more widely recognized that drug resistance in malaria is not a strictly binary trait of resistance/susceptible but is a probabilistic trait with the therapeutic outcome depending on the interaction between three critical factors: (i) the level of parasite resistance (described by its pharmacodynamics [PD], such as half-maximal inhibitory concentration [IC50]), (ii) the amount of drug the patient takes and how she/he subsequently processes it (the pharmacokinetics [PK], such as drug distribution and elimination rate), and (iii) the levels of human immunity. The latter is usually ignored in antimalarial drug deliberations (but see references 6 and 7) on the basis that drugs should work even in nonimmune patients; this makes the prediction of therapeutic outcome a function solely of PK and PD. In principle, this approach should allow us to (crudely) distinguish drug “failure” from drug “resistance.” Treatment “failures” are the result of human factors such as low drug concentrations due to, for example, inadequate drug dosing, unnoticed vomiting, or natural human PK variation. Drug “resistance” occurs when infections survive treatment due to genetically encoded parasite factors such as reduced sensitivity. This realization led to suggestions (8, 9) that drug concentrations measured 7 days after treatment (day 7 drug concentration [d7c]) could be used to distinguish drug “failures” from drug “resistance.” Day 7 was justified for three main reasons (outlined in reference 10): (i) feasibility, as day 7 is one of the several days on which routine patient follow-up should be performed in antimalarial drug trials; (ii) pharmacodynamics, because, in theory, if d7c of slowly eliminated antimalarials are at least twice the minimum parasiticidal concentration, all the infecting parasites should be eliminated; (iii) pharmacokinetics, as in theory, day 7 drug exposure is determined only by variation in the elimination rate constant. Measurement of d7c in clinical trials of antimalarial drugs has been widely promoted and is now supported by well-resourced reference laboratories (11). The World Health Organization (WHO) (10) has also repeated these assertions stating that “Measurement of concentrations of longer-acting antimalarial drugs on day 7 following initiation of treatment should be considered a routine part of trials” (page 70) because “The drug concentration on day 7 is predictive of the outcome” (page 73). They further assert “Measurement of the blood, serum or plasma concentration of slowly eliminated antimalarial drugs (i.e., terminal elimination half-life >2 days) at a single time is simple and might be a better determinant of therapeutic response than the total AUC [area under the concentration-time curve]” (page 73).
We will argue that these assertions regarding the predictive ability of d7c are clearly contradicted by PK/PD simulations (see below) and by reanalysis of field data (see supplemental material) which suggest exactly the opposite, i.e., that d7c is actually a rather poor predictor of therapeutic outcome.
WHAT IS A ROC CURVE?
Receiver-operator characteristic (ROC) curves originated in radio technology but are now widely used in medical research to quantify the predictive value of a measurement, in our case, the ability of d7c to predict the therapeutic outcome (12). The x axis is 1 − specificity, and the y axis is sensitivity. It is clearer to relabel the x axis simply as “specificity” and reverse the axis, as in Fig. 1. Sensitivity is defined as the number of patients with “low” d7c who fail treatment divided by the total number of patients who fail treatment. Specificity is defined as the number of patients with “normal” d7c cured divided by the total number of patients cured.
FIG 1.

Graphical representation of a receiver-operator characteristic (ROC) curve. ROC curves are widely used in medical research to quantify the predictive value of a measurement as explained in the text. The black diagonal line is a ROC curve for a diagnostic with no predictive value, the blue line is an illustrative example of a good predictor, the red line is a ROC curve for day 7 drug concentrations obtained previously by simulation (18), and the black dashed line for point 4 illustrates how we estimate the area under the ROC curve from point estimates of sensitivity and sensitivity (see supplemental material). These point estimates correspond to the following sources: point 1 is from White et al. (45), point 2 is from Denis et al. (46), point 3 is from Price et al. (47), point 4 is from Ezzet et al. (30), point 5 is from Checchi et al. (48), and point 6 is from McGready et al. (49).
The ROC curve plotted as a continuous blue line in Fig. 1 is actually a dot plot rather than a true algebraic function. Each d7c cutoff value is assessed for sensitivity and specificity and plotted onto the coordinates; the software then links these points with a line. In this example, we assume that d7c lies between 0 and 100 ng/ml, and the assumption is that d7c is used to predict therapeutic failure. Each hypothetical d7c cutoff value is assessed and included on the plot (blue numbers): high cutoff values, such as 60 and 80, result in most patients being classified with “low” d7c, resulting in high sensitivity (the large group of “low” d7c patients includes most failures) but poor specificity (many cured patients will be in this class of “low” d7c), while low cutoff values, such as 10 and 30, have low sensitivity but higher specificity. In this example, the hypothetical cutoff value of 45 ng/ml seems the best compromise, but the choice of cutoff is an objective choice that depends on the weighting given to consequences of wrong classifications. Irrespective of the choice of cutoff value, the closer the ROC curve approaches the top left-hand corner of the graph (i.e., high sensitivity and high specificity), the better its diagnostic capability. For future reference, the enumerated black squares in Fig. 1 are the sensitivity and specificity of the cutoff values of d7c reported or extracted from the literature (see supplemental material); none of these points are remotely near the top left-hand corner of the plot, which would have indicated good predictive capability.
ROC curves allow an objective measure of the predictive capability of a diagnostic test (in this case, the ability of low d7c to predict treatment failure). In practice, d7c is often measured in large laboratory batches and only becomes available after the follow-up period. In this case, d7c serves as an explanation, rather than a predictor, of therapeutic outcome. As would be expected from a statistical analysis, ROC curve analysis is unaffected by these semantic differences and properly quantifies both d7c predictive and explanatory roles. The closer the area under the ROC curve (auROC) is to 1, the better the diagnostic test performs (an auROC of 1 implies the test is perfectly accurate). An area under the ROC curve of 0.5 indicates that the diagnostic test has no predictive value (i.e., the test is equivalent to relying on pure chance) and is represented by the solid black diagonal line in Fig. 1. The consensus for classifying the accuracy of a diagnostic test is the use of the “traditional” academic point system with 0.90 to 1 being excellent, 0.80 to 0.90 being good, 0.70 to 0.80 being fair, 0.60 to 0.70 being poor, and 0.50 to 0.60 being fail (we have been unable to find an academic citation for this “tradition,” but it can be found on websites [e.g., http://gim.unmc.edu/dxtests/roc3.htm]). The red line in Fig. 1 represents our simulated ROC analysis (6) for lumefantrine with an auROC of 0.615 (95% confidence interval [95% CI], 0.596 to 0.633). Using the classification above, this test “fails” as a diagnostic predictor but is consistent with field data (see Table S1 in the supplemental material).
HOW GOOD IS DAY 7 CONCENTRATION AS A PREDICTOR OF THERAPEUTIC OUTCOME?
Intuitively, d7c can act as a good predictor of therapeutic outcome only if it is the dominant parameter determining the outcome. There are a large number of interacting factors that ultimately determine the therapeutic outcome of treatment with a typical artemisinin combination therapy (ACT) (the currently recommended class of first-line antimalarials). ACTs typically contain two or three distinct drugs: the artemisinin parent drug (if given as artesunate or artemether), the artemisinin active metabolite dihydroartemisinin (DHA), and the partner drug (which may also have an active metabolite [e.g., amodiaquine]). The degree of parasite sensitivity to each drug (its PD profile) is typically described by Michaelis-Menten dynamics defined by three factors: IC50, maximal kill rate, and slope of the dose-response curve. The PK profile of each drug is described by three main factors: bioavailability, volume of distribution, and elimination rate (plus a series of absorption and conversion rates and distribution across separate physiological compartments [10] that we ignore in the interest of simplicity). This results in six main PK/PD parameters per drug, and hence 12 to 18 for a combination therapy, all of which contribute to the therapeutic outcome. Each of these parameters shows substantial variability (10): human PK typically varies over a 3-fold range (discussed further in reference 10), while parasite IC50 for a drug typically varies 50- to 1,000-fold (e.g., Fig. 3 of reference 13). Human immunity also plays a substantial role in the outcome (14, 15). Another determinant of therapeutic success that is often overlooked is multiclonal infections. Malaria infections consisting of several genetically distinct clones are commonly observed (up to around 8 clones per infection in higher transmission areas [16]). The clones are likely to vary in PD, and therapy must clear all the infections, including the most resistant. Hence, increasing the number of clones (quantified as a patient's multiplicity of infection [MOI]) will increase the failure rate (17). Consequently, higher MOI introduces another factor contributing to the therapeutic outcome (17). In summary, the substantial variation in PK/PD, human immunity levels, and MOI will obscure the relationship between d7c and therapeutic outcome. Therefore, we decided to review the evidence base for using d7c as a predictor of therapeutic outcome and used two strategies: PK/PD modeling, and critical appraisal of clinical data previously invoked as support for the predictive ability of d7c.
The consensus method of quantifying the predictive capability of a diagnostic measure is by receiver-operator characteristic (ROC) analysis (12) as described above. We investigated the predictive ability of d7c by analyzing simulated data of antimalarial drug treatment outcome generated with the PK/PD model described previously (6, 18). PK/PD modeling has the advantage that we know, and can alter, the factors underlying treatment outcome. This “mechanism-based PK/PD modeling” was recently reviewed in reference 19, and it is widely used in infection biology. These PK/PD simulations have been applied by other investigators to malaria (20–27) and recently extended by us to incorporate factors such as multiple doses and drug conversion (6, 10, 28).
Our simulations showed that d7c is, as expected, generally a good proxy for drug exposure as measured by the area under the drug concentration-time curve (AUC): the correlation coefficients of d7c with AUC measured up to 100 days posttreatment were 0.98 for lumefantrine (LF), 0.94 for chloroquine (CQ), 0.93 for piperaquine (PPQ), and 0.92 for mefloquine (MQ) (details in Table S3.1 of the supplemental material of reference 6). Population attributable risk percentage (PAR%) simulations showed that between 3% (artesunate [AS] plus MQ) and 17% (dihydroartemisinin [DHA] plus PPQ) of failures could be avoided if adequate drug levels were achieved throughout the patient population (details in Table 6 of reference 6). The simulations also showed that a “low” d7c was associated with a statistically significantly increased odds of failing treatment (details in Tables 5 and of reference 6). Furthermore, a simulated clinical trial of AS-MQ suggested that a low d7c was a more important risk factor in treatment outcome (measured using the Wald statistic) than the patient's initial parasitemia, high malaria transmission intensity, and patient age of <5 years (details in Table 5 of reference 6).
Despite the clear association of d7c with overall drug exposure and treatment outcome, simulations show that d7c would have a very poor predictive capability when evaluated by their auROC curve (see above and Fig. 1), generally being in the range of 0.55 to 0.65 (Table 6 and Table S3.2 of reference 6). This was consistent with clinical data (see below). We also noted that even a d7c with a very poor predictive capability, quantified by its auROC, could still have a significant association with the outcome as quantified by its P value; for example, we predicted a P value of 0.001 associated with a d7c cutoff value (<15th percentile) for MQ (Table 5 of reference 6). This apparent discrepancy arises from differing roles of the P value and ROC curve. The ROC curve quantifies the extent to which d7c is a good (or bad) diagnostic predictor of the therapeutic outcome. In contrast, the P value simply tests a null hypothesis, i.e., that d7c has absolutely no association with the therapeutic outcome. The latter is, hopefully, unlikely, so it is entirely consistent that low P values can be associated with d7c whose ROC curves reveal a lack of any useful predictive value. The use of P values to identify “target” or “cutoff” values of d7c is therefore problematic and is discussed further in the supplemental material.
Clinical data were then collated and reviewed to ascertain whether they follow the patterns predicted by PK/PD modeling. We identified and read all the papers we could find that reported the use of d7c as a predictor of therapeutic success (see supplemental material). Disappointingly, no authors reported auROC, and all relied on the use of P values to justify a d7c cutoff which, as described above, tests the hypothesis of no association rather than quantifying predictive ability. Some papers reported success/failure rates associated with a d7c “threshold” which enabled us to make a crude reconstruction of an auROC curve (dashed black line in Fig. 1); the auROC values we extracted from these clinical reports lay in the range 0.6 to 0.7, which is disappointingly low but entirely consistent with the values obtained by PK/PD simulation described above and previously (6). In summary, the empirical evidence base for statements such as “drug concentration on day 7 is predictive of the outcome” (e.g., page 73 of reference 10) appears weak at best. In fact, our review of the literature reveals that most clinical data point toward d7c being an extremely poor predictor of therapeutic outcome.
POTENTIAL PITFALLS WHEN USING DAY 7 CONCENTRATION IN CLINICAL TRIALS
It is a current WHO recommendation that d7c be measured in antimalarial drug clinical trials to assess drug exposure. These data are being collected and reported so it is constructive to consider what role they can play in clinical trials and, equally important, to discuss the dangers that may arise from their uncritical use.
One potential use of d7c is to detect patients who poorly adhere to their drug regimens; once identified, such patients could be removed from the analysis, and drug cure rates calculated separately for completely and poorly adherent patients. Figure 2 shows our simulation of the effect of poor adherence on the d7c of PPQ, given as 3 daily doses with DHA, in 10,000 patients. As expected, the mean/median d7c declines as adherence decreases, but the large amount of natural variation in human PK means that d7c is unlikely to be diagnostic of poor adherence except in cases where patients take only the first of the three doses; obviously, the proportion of such patients should be very small to negligible in most clinical trials.
FIG 2.
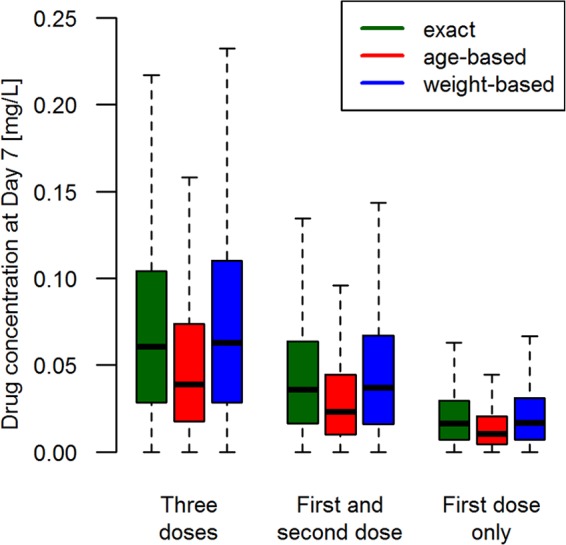
A simulation of how day 7 concentration (d7c) varies according to the level of patient adherence to the regimen. The figure shows the mean d7c of piperaquine (PPQ) in mg/liter simulated for 10,000 African patients given dihydroartemisinin (DHA)-PPQ once daily for 3 days using the PK/PD model described in references 18 and 28. The patients were either dosed with the exact dosage (mg/kg of body weight) (18 mg/kg per reference 50), dosed according to their age (per reference 51), or dosed according to their weight (per reference 52). To investigate patient adherence, it was assumed that patients took the full course, i.e., all three doses, only the first two doses, or the first dose only. Outliers are not displayed. The figure was produced using a published one-compartment model. A more-realistic two-compartment model parameterization based on the work of Staehli Hodel et al. (53) gave identical results except that the d7c in the central compartment is approximately 4-fold lower (see Fig. S1 in supplemental material). (The Fig. 2 parameter values were as follows. The elimination rate per day is 0.03, and the volume of distribution is 150 liters/kg.)
One tempting way of using d7c data is to simply remove the patients failing treatment who have “low” d7c from a clinical trial; one obvious justification is that they may have been adhering poorly to the treatment regimen. However, removal of only these drug failures would overestimate the true drug efficacy, as it might for example exclude patients adhering fully to the treatment regimen with particularly extreme PK parameters. In some cases, this overestimation may keep the treatment efficacy above the 95% threshold of initiating policy change (29), thus removing the necessity to consider a replacement drug, an interpretation with potentially fatal consequences. This bias toward overestimating drug efficacy can be avoided by removing all patients with low d7c, but the ROC curve analysis and PK/PD simulation suggest that this will also remove a lot of drug successes: thus, the results would be unbiased, but sample size would fall and the confidence intervals around drug effectiveness would increase. This problem arises because d7c is such a poor predictor of outcome and is best illustrated using a trivial analogy. Suppose we thought, erroneously, that patients born on a Monday are more likely to fail treatment. Searching through records of patients failing treatment and removing all failures born on a Monday will reduce the number of failures and hence artificially increase the apparent drug efficacy; the correct strategy would be to remove all patients born on a Monday irrespective of their therapeutic outcome. This will eliminate the bias but reduce sample size analyzed in the clinical trial and hence increase the confidence interval(s) around the estimates of treatment efficacy.
There are considerable dangers in using d7c of a drug to identify a single d7c cutoff that can be used to distinguish between “adequate” and “inadequate” dosing and hence used as a “target” concentration. Informative threshold concentrations occur only with high values of auROC where the ROC curve is nonlinear and shows a clear cutoff point with high specificity and sensitivity (e.g., the hypothetical drug concentration cutoff of 45 ng/ml on the blue ROC in Fig. 1). Note that it would be inappropriate to use the concentration with the lowest P value as a cutoff, because as discussed above and in the supplemental material, the P value simply tests the assumption of no association and, critically, is affected by both the size of the effect and the sample size. Cutoffs between 175 and 600 ng/ml for LF have been suggested (for a review, see page 74 in reference 10) typically with P values of <0.001. The near-linear relationship between d7c and the chance of falling treatment means that virtually any “cutoff” value can be chosen and supported by statistics (P value, odds ratio, etc.) to support its selection. It is important to realize that unless the d7c is associated with a large auROC, the choice is largely arbitrary.
Drug dosage, regimen, and adherence (which jointly determine d7c) are the only factors we can actually control in malaria therapy; all the other PK and PD parameters are determined by human and malaria genetics, so it is important to assess the impact of changing the dosage. Analyses of clinical trials consistently show that the higher the dose given, and d7c attained, the better the chance of successful treatment; moreover, this relationship appears to be roughly linear (e.g., Fig. 5 of reference 30 and Fig. 4 of reference 31). On one level, this result is entirely expected and trivial (32): all antimalarials have had their dosages increased after their initial deployment, and Guinea-Bissau overcame its problem of CQ-resistant malaria by simply doubling the dosage of CQ given to patients (33, 34). The reason dosages are not routinely increased is because of concerns over toxicity (and no other country has followed Guinea-Bissau's policy of doubling CQ dosage); hence, the results emphasize the need for better toxicity data on antimalarial drugs which is, in our opinion, an underresearched area.
Finally, the WHO mandate that the drug efficacy must be >95% (35) so the expected failure rates will be low in the routine efficacy monitoring trials and any diagnostic ability of d7c will probably never be observed. It is pointless doing a ROC analysis when few or no treatment failures occur in a trial so it is not clear how these d7c can be properly incorporated except by large meta-analyses of numerous trials (e.g., pooled analysis of day 7 PPQ concentrations [31]). Several authors have already noted that d7c will be of little value when cure rates are very high. For example, White et al. (9) note that “Relationships between PK variables and cure rates are not evident when cure rates are very high. Such relationships are apparent only when resistance develops or doses are inadequate.” Most clinical trials of antimalarial drugs are now testing highly effective ACTs, often in noninferiority trials, to allow decisions to be made on factors such as cost, side effects, ease of regimens, length of posttreatment prophylaxis, and so on. It therefore follows that d7c collected in these trials will be largely superfluous until resistance starts to arise, in which case of course, the ACT would have to be replaced.
In summary, there are a number of tempting but ultimately incorrect ways of using d7c to interpret clinical trial data. It appears that d7c has no consistently clear predictive cutoff on a ROC curve so that subsequent analyses tend to draw the rather trivial conclusion that the more drug given to patients, the higher the subsequent d7c, and the more chance they have of being cured. The question therefore arises as to whether we can identify more informative and/or cost-effective ways of bringing PK/PD measures into current clinical trial methodology.
CAN WE GATHER MORE-INFORMATIVE PHARMACOLOGICAL DATA IN CLINICAL TRIALS?
Designing antimalarial therapies would be straightforward if all people and all parasites were identical. The enormous variation in parasite sensitivity to drugs, human variation in how a drug is absorbed, distributed, and metabolized, and how toxicity may occur makes the rational design of drug regimens enormously complex (36). It is this complexity that limits the use of d7c to a proxy for drug exposure. The subsequent realization that drug exposure is not the sole predictor of failure forces us to consider what other factors contribute to failure and how we can collect and integrate this information during routine clinical trials.
The first requirement would be to move away from taking drug measurements at a single time point. Treatment is a dynamic process that requires repeat measurements either by intensive sampling or, more likely, through “sparse” sampling and appropriate population PK analysis (10). The use of sparse sampling and population PK modeling is highly informative, as it allows PK parameters such as absorption rates, volumes of distributions, elimination rates, and the number of physiological compartments to be determined in different human populations, alongside the intra- and interindividual variation of these parameters (37). PK/PD modeling has been successfully used in other organisms to optimize dosing strategies (38), and it seems reasonable to adopt the same approach for malaria. However, this requires the measured mean and associated variation in individual pharmacological parameters to determine treatment outcome. The d7c would be inadequate for such modeling, as it is a composite measure determined by several distinct PK parameters that need to be split into its constituent parts, i.e., dosage, bioavailability, volume of distribution, and elimination rate. Day 7 concentrations could still be measured in each patient (day 7 is a routine surveillance time point) but be augmented by additional sampling around this day to generate the sparse data sets required for PK analysis. PK parameters alone cannot address the issue of how to deal with natural variation in parasite drug sensitivity which typically ranges 10- to 100-fold even in the absence of “major” mutations controlling drug resistance (e.g., Fig. 3 of reference 13). This implies that PK data need to be accompanied by some indication of the drug sensitivity of local malaria population(s). This would be best achieved by in vitro measurement of fresh parasite isolates. These strategies have been worked out in some detail (reviewed in reference 39) and are now part of the World Wide Antimalarial Resistance Network (WWARN) depository system.
It would then be necessary to integrate these separate sources of data into a comprehensive framework linking the parameters to therapeutic outcome. We would suggest PK/PD modeling as a framework (6, 28) but are not dogmatic, provided that some sort of coherent framework is used to link parameters to the therapeutic outcome (19, 38). The application of such a framework has several major advantages.
Data can be combined from different trials, locations, and patient groups to calibrate these models. This greatly increases the scope and value of each data set including historical trials that may have collected PK or PD data for different reasons. In particular, it also allows current ACT trials, where few or no failures may occur, to contribute data useful for drug regimen optimization.
Clinical trials are run in highly controlled settings, making it problematic to extrapolate their efficacy estimated under near-ideal conditions into effectiveness under real-life conditions. PK/PD modeling can be used to make these extrapolations. Obvious applications are to investigate how robust the regimens are to poor adherence (40); it would be clearly unethical to give patients incomplete dosages in a clinical trial, but we know that nonadherence is routinely observed in the field (recently reviewed in reference 41). Other common exclusion criteria in clinical trials are patients who are severely ill or on comedication, both of which may substantially alter their PK. Comedication is important in many countries where malaria is endemic and where long-term medication for tuberculosis (TB) and HIV is relatively common; changes in PK caused by comedication can then be fed back into the PK/PD model to see whether and how the drug dosages given to these subgroups of patients should be altered. We do not argue that PK/PD modeling will necessarily give an exact dosing schedule for such people, but we do argue it can provide a dosing scheduling starting point for clinical trials in such patients.
Current trials cannot easily be used to predict future events, in particular the consequences of increasing drug resistance, which many commentators regard as inevitable (42). Once again, PK/PD modeling can investigate how robust regimens may be to small increases in the levels of parasite drug tolerance/resistance and hence their likely therapeutic life span (28). The PK/PD data can also be used to try and implement regimens that minimize the selection pressures for resistance.
CONCLUSIONS
The realization that d7c is apparently poorly predictive for the therapeutic outcome raises the obvious question of whether this PK component of clinical trials could be improved. Our first conclusion would be that the predictive capability of d7c should be evaluated and reported using ROC analysis. The collection of d7c data is not trivial. It requires correct blood sample collection, processing, storage, and transportation to a central reference laboratory, as well as collation of d7c with clinical outcome and possible stratification in the subsequent analysis. As argued above, the outcome of all this effort is likely to be the trivial conclusion that giving patients more drug improves their chance of treatment success. It is therefore arguable whether this approach represents the best use of resources and that better resource allocation might be achieved through a two-tier clinical trial framework using two distinct types of clinical trials to guide policy.
First, routine efficacy monitoring trials should be designed to check that local first-line drug(s) have remained effective. There seems little point in measuring d7c in such studies given that effectiveness is likely to be high so that few, if any, failures will occur. In light of the poor predictive ability of d7c and the difficulties inherent in pooling results from different studies (discussed in the supplemental material), we suggest that the WHO drops their recommendation that d7c should be collected in such studies and leave individual investigators to consider whether the costs of measuring d7c can be justified. One such justification would be to compare whether different populations vary in their drug absorption and/or subsequent metabolism. In this case, rather than relating the d7c to treatment outcome, investigators would examine whether d7c differs in “at risk” populations. Examples of “at risk” populations could be young or malnourished children or patients with comedication (e.g., antiretrovirals). However, these “at risk” populations are generally excluded from clinical trials so they may be better investigated as part of phase IV trials. One other justification would be as a crude measure of factors affecting PK: if the same population exhibits differing d7c over time, then it suggests, for example, that adherence to the drug regimen was altered as it is unlikely that human PK parameters will have changed.
Second, we recommend the periodic use of more-focused and specialized PK/PD trials, including a sophisticated PK component designed to generate the data required for long-term optimization of regimens and enrolling the full range of patient types. This PK component would necessitate an optimized, drug-specific, sampling schedule, which may or may not include measurement on day 7 (43). Furthermore, these trials should also be accompanied by culturing local parasite isolates to estimate PD parameters, thereby allowing investigation of changing circumstances, such as the possible evolution of drug resistance. However, conducting focused PK/PD trials will be challenging and likely to need external support. Analyzing one drug concentration sample point costs typically $20 or more. This does not include the cost of staff, consumables, sample storage, sample transport, acquisition or maintenance of analytical equipment, etc. The culturing component is also technically and logistically demanding, as the blood is sometimes collected in very remote areas and needs to reach the lab early enough that parasites are still viable (39).
We also require an ethical framework for performing the PK/PD trials. There are limits on how much blood can be drawn from any individual patient, particularly infants. Blood is routinely drawn at prescribed days of follow-up to check for parasitemia, but it is likely that at least some of the blood sample (for example for parasite culturing for IC50 analysis, repeat sampling for artemisinin PK) may be of no direct clinical benefit to the patient. This is not ethically impossible, but it does require some justification that individual patients will accrue future benefits from effective antimalarial drug provision that outweighs the inconveniences of providing additional blood sample not required for immediate clinical purposes. In summary, we need a consensus protocol for these strategic trials and a consensus that they can be deemed ethical.
Statisticians often recommend that researchers planning a study should first simulate, and then analyze, a realistic data set so that their eventual study design can avoid any likely pitfalls revealed in the simulations. For once we have heeded our own advice. The collection of PK data (d7c) was recently discussed in the WHO publication Global Report on Antimalarial Drug Efficacy and Drug Resistance: 2000–2010 (44), where it was noted that “The interpretation of the results of blood concentration studies for determining drug resistance is not, however, always straightforward.” We think that this is an understatement and that too little preparatory work has gone into identifying the best ways of analyzing the data. Part of the problem is that, of course, it is impossible to analyze the data until it has been generated, and methods of analysis typically have to be refined according to the data. In summary, we believe that simply collecting d7c is unlikely to be the best use of PK resources and that more-sophisticated PK elements of clinical trials be designed and rolled out. The resulting detailed PK data on mean values and distributions can then usefully contribute to the rationale design of robust and effective antimalarial drug regimens.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Bill and Melinda Gates Foundation (grant 37999.01) and the Medical Research Council (grant G1100522).
Footnotes
Published ahead of print 30 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02777-14.
REFERENCES
- 1.Olliaro P. 2005. Drug resistance hampers our capacity to roll back malaria. Clin. Infect. Dis. 41:S247–S257. 10.1086/430785 [DOI] [PubMed] [Google Scholar]
- 2.Peters W. 1985. The problem of drug resistance in malaria. Parasitology 90:705–715. 10.1017/S003118200005232X [DOI] [PubMed] [Google Scholar]
- 3.Wernsdorfer WH. 1994. Epidemiology of drug resistance in malaria. Acta Trop. 56:143–156. 10.1016/0001-706X(94)90060-4 [DOI] [PubMed] [Google Scholar]
- 4.Attaran A, Barnes KI, Curtis C, D'Alessandro U, Fanello CI, Galinski MR, Kokwaro G, Looareesuwan S, Makanga M, Mutabingwa TK, Talisuna A, Trape JF, Watkins WM. 2004. WHO, the Global Fund, and medical malpractice in malaria treatment. Lancet 363:237–240. 10.1016/S0140-6736(03)15330-5 [DOI] [PubMed] [Google Scholar]
- 5.World Health Organization. 2009. Methods for surveillance of antimalarial drug efficacy. World Health Organization, Geneva, Switzerland [Google Scholar]
- 6.Winter K, Hastings IM. 2011. Development, evaluation, and application of an in silico model for antimalarial drug treatment and failure. Antimicrob. Agents Chemother. 55:3380–3392. 10.1128/AAC.01712-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pongtavornpinyo W, Yeung S, Hastings I, Dondorp A, Day N, White N. 2008. Spread of anti-malarial drug resistance: mathematical model with implications for ACT drug policies. Malar. J. 7:229. 10.1186/1475-2875-7-229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barnes KI, Lindegardh N, Ogundahunsi O, Olliaro P, Plowe CV, Randrianarivelojosia M, Gbotosho GO, Watkins WM, Sibley CH, White NJ. 2007. World Antimalarial Resistance Network (WARN) IV: clinical pharmacology. Malar. J. 6:122. 10.1186/1475-2875-6-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White NJ, Stepniewska K, Barnes K, Price RN, Simpson J. 2008. Simplified antimalarial therapeutic monitoring: using the day-7 drug level? Trends Parasitol. 24:159–163. 10.1016/j.pt.2008.01.006 [DOI] [PubMed] [Google Scholar]
- 10.World Health Organization. 2011. Methods and techniques for assessing exposure to antimalarial drugs in clinical field studies. World Health Organization, Geneva, Switzerland [Google Scholar]
- 11.Lourens C, Watkins WM, Barnes KI, Sibley CH, Guerin PJ, White NJ, Lindegardh N. 2010. Implementation of a reference standard and proficiency testing programme by the World Wide Antimalarial Resistance Network (WWARN). Malar. J. 9:375. 10.1186/1475-2875-9-375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zweig MH, Campbell G. 1993. Receiver-operating characteristic (ROC) plots: a fundamental evaluation tool in clinical medicine. Clin. Chem. 39:561–577 [PubMed] [Google Scholar]
- 13.Mu J, Myers RA, Jiang H, Liu S, Ricklefs S, Waisberg M, Chotivanich K, Wilairatana P, Krudsood S, White NJ, Udomsangpetch R, Cui L, Ho M, Ou F, Li H, Song J, Li G, Wang X, Seila S, Sokunthea S, Socheat D, Sturdevant DE, Porcella SF, Fairhurst RM, Wellems TE, Awadalla P, Su XZ. 2010. Plasmodium falciparum genome-wide scans for positive selection, recombination hot spots and resistance to antimalarial drugs. Nat. Genet. 42:268–271. 10.1038/ng.528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langhorne J, Ndungu FM, Sponaas A-M, Marsh K. 2008. Immunity to malaria: more questions than answers. Nat. Immunol. 9:725–732. 10.1038/ni.f.205 [DOI] [PubMed] [Google Scholar]
- 15.Rogerson SJ, Wijesinghe RS, Meshnick SR. 2010. Host immunity as a determinant of treatment outcome in Plasmodium falciparum malaria. Lancet Infect. Dis. 10:51–59. 10.1016/S1473-3099(09)70322-6 [DOI] [PubMed] [Google Scholar]
- 16.Arnot DE. 1998. Clone multiplicity of Plasmodium falciparum infections in individuals exposed to variable levels of disease transmission. Trans. R. Soc. Trop. Med. Hyg. 92:580–585 [DOI] [PubMed] [Google Scholar]
- 17.Jaki T, Parry A, Winter K, Hastings I. 2013. Analysing malaria drug trials on a per-individual or per-clone basis: a comparison of methods. Stat. Med. 32:3020–3038. 10.1002/sim.5706 [DOI] [PubMed] [Google Scholar]
- 18.Winter K, Hastings IM. 2011. Development, evaluation and application of an in silico model for antimalarial drug treatment failure. Antimicrob. Agents Chemother. 55:3380–3392. 10.1128/AAC.01712-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Czock D, Keller F. 2007. Mechanism-based pharmacokinetic-pharmacodynamic modeling of antimicrobial drug effects. J. Pharmacokinet. Pharmacodyn. 34:727–751. 10.1007/s10928-007-9069-x [DOI] [PubMed] [Google Scholar]
- 20.Austin D, White N, Anderson R. 1998. The dynamics of drug action on the within-host population growth of infectious agents: melding pharmacokinetics with pathogen population dynamics. J. Theor. Biol. 194:313–339. 10.1006/jtbi.1997.0438 [DOI] [PubMed] [Google Scholar]
- 21.Hoshen M, Na-Bangchang K, Stein W, Ginsburg H. 2000. Mathematical modelling of the chemotherapy of Plasmodium falciparum malaria with artesunate: postulation of ‘dormancy', a partial cytostatic effect of the drug, and its implication for treatment regimens. Parasitology 121:237–246. 10.1017/S0031182099006332 [DOI] [PubMed] [Google Scholar]
- 22.Hoshen M, Stein W, Ginsburg H. 1998. Modelling the chloroquine chemotherapy of falciparum malaria: the value of spacing a split dose. Parasitology 116:407–416. 10.1017/S0031182098002480 [DOI] [PubMed] [Google Scholar]
- 23.Hoshen MB, Stein WD, Ginsburg H. 2002. Mathematical modelling of malaria chemotherapy: combining artesunate and mefloquine. Parasitology 124:9–15. 10.1017/S0031182001008952 [DOI] [PubMed] [Google Scholar]
- 24.Hoshen MB, Stein WD, Ginsburg HD. 2001. Pharmacokinetic-pharmacodynamic modelling of the anti-malarial activity of mefloquine. Parasitology 123:337–346. 10.1017/S003118200100854X [DOI] [PubMed] [Google Scholar]
- 25.Saralamba S, Pan-Ngum W, Maude RJ, Lee SJ, Tarning J, Lindegardh N, Chotivanich K, Nosten F, Day NPJ, Socheat D, White NJ, Dondorp AM, White LJ. 2011. Intrahost modeling of artemisinin resistance in Plasmodium falciparum. Proc. Natl. Acad. Sci. U. S. A. 108:397–402. 10.1073/pnas.1006113108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simpson J, Watkins E, Price R, Aarons L, Kyle D, White N. 2000. Mefloquine pharmacokinetic-pharmacodynamic models: implications for dosing and resistance. Antimicrob. Agents Chemother. 44:3414–3424. 10.1128/AAC.44.12.3414-3424.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zaloumis S, Humberstone A, Charman SA, Price RN, Moehrle J, Gamo-Benito J, McCaw J, Jamsen KM, Smith K, Simpson JA. 2012. Assessing the utility of an anti-malarial pharmacokinetic-pharmacodynamic model for aiding drug clinical development. Malar. J. 11:303. 10.1186/1475-2875-11-303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kay K, Hastings IM. 2013. Improving pharmacokinetic-pharmacodynamic modeling to investigate anti-infective chemotherapy with application to the current generation of antimalarial drugs. PLoS Comput. Biol. 9:e1003151. 10.1371/journal.pcbi.1003151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kitua AY. 1999. Antimalarial drug policy: making systematic change. Lancet 354(Suppl):SIV32. 10.1016/S0140-6736(99)90375-6 [DOI] [PubMed] [Google Scholar]
- 30.Ezzet F, Mull R, Karbwang J. 1998. Population pharmacokinetics and therapeutic response of CGP 56697 (artemether + benflumetol) in malaria patients. Br. J. Clin. Pharmacol. 46:553–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.WorldWide Antimalarial Resistance Network (WWARN) DP Study Group. 2013. The effect of dosing regimens on the antimalarial efficacy of dihydroartemisinin-piperaquine: a pooled analysis of individual patient data. PLoS Med. 10:e1001564. 10.1371/journal.pmed.1001564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garner P. 2013. Artemisinin combination therapy: a good antimalarial, but is the dose right? PLoS Med. 10:1–3. 10.1371/journal.pmed.1001565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ursing J, Schmidt BA, Lebbad M, Kofoed P-E, Dias F, Gil JP, Rombo L. 2007. Chloroquine resistant P. falciparum prevalence is low and unchanged between 1990 and 2005 in Guinea-Bissau: an effect of high chloroquine dosage? Infect. Genet. Evol. 7:555–561. 10.1016/j.meegid.2007.03.006 [DOI] [PubMed] [Google Scholar]
- 34.Ursing J, Kofoed PE, Rodrigues A, Blessborn D, Thoft-Nielsen R, Björkman A, Rombo L. 2011. Similar efficacy and tolerability of double-dose chloroquine and artemether-lumefantrine for treatment of Plasmodium falciparum infection in Guinea-Bissau: a randomized trial. J. Infect. Dis. 203:109–116. 10.1093/infdis/jiq001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.World Health Organization. 2003. Assessment and monitoring of antimalarial drug efficacy for the treatment of uncomplicated falciparum malaria. WHO reference no. WHO/HTM/RBM/2003.50. World Health Organization, Geneva, Switzerland [Google Scholar]
- 36.Duparc S, Lanza C, Ubben D, Borghini-Fuhrer I, Kellam L. 2012. Optimal dose finding for novel antimalarial combination therapy. Trop. Med. Int. Health 17:409–413. 10.1111/j.1365-3156.2012.02963.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simpson JA, Aarons L, White NJ. 2001. How can we do pharmacokinetic studies in the tropics? Trans. R. Soc. Trop. Med. Hyg. 95:347–351. 10.1016/S0035-9203(01)90178-6 [DOI] [PubMed] [Google Scholar]
- 38.Schuck EL, Derendorf H. 2005. Pharmacokinetic/pharmacodynamic evaluation of anti-infective agents. Expert Rev. Anti Infect Ther. 3:361–373. 10.1586/14787210.3.3.361 [DOI] [PubMed] [Google Scholar]
- 39.Noedl H, Wongsrichanalai C, Wernsdorfer WH. 2003. Malaria drug-sensitivity testing: new assays, new perspectives. Trends Parasitol. 19:175–181. 10.1016/S1471-4922(03)00028-X [DOI] [PubMed] [Google Scholar]
- 40.Hodel EM, Kay K, Hayes D, Terlouw D, Hastings I. 2014. Optimizing the programmatic deployment of the anti-malarials artemether-lumefantrine and dihydroartemisinin-piperaquine using pharmacological modelling. Malar. J. 13:138. 10.1186/1475-2875-13-138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruxvoort K, Goodman C, Kachur SP, Schellenberg D. 2014. How patients take malaria treatment: a systematic review of the literature on adherence to antimalarial drugs. PLoS One 9:e84555. 10.1371/journal.pone.0084555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sibley C, Barnes K, Watkins W, Plowe C. 2008. A network to monitor antimalarial drug resistance: a plan for moving forward. Trends Parasitol. 24:43–48. 10.1016/j.pt.2007.09.008 [DOI] [PubMed] [Google Scholar]
- 43.Jamsen KM, Duffull SB, Tarning J, Lindegardh N, White NJ, Simpson JA. 2012. Optimal designs for population pharmacokinetic studies of the partner drugs co-administered with artemisinin derivatives in patients with uncomplicated falciparum malaria. Malar. J. 11:143. 10.1186/1475-2875-11-143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.World Health Organization. 2010. Global report on antimalarial efficacy and drug resistance: 2000–2010.. World Health Organization, Geneva, Switzerland [Google Scholar]
- 45.White NJ, Van Vugt M, Ezzet F. 1999. Clinical pharmacokinetics and pharmacodynamics of artemether-lumefantrine. Clin. Pharmacokinet. 37:105–125. 10.2165/00003088-199937020-00002 [DOI] [PubMed] [Google Scholar]
- 46.Denis MB, Tsuyuoka R, Lim P, Lindegardh N, Yi P, Top SN, Socheat D, Fandeur T, Annerberg A, Christophel EM, Ringwald P. 2006. Efficacy of artemether-lumefantrine for the treatment of uncomplicated falciparum malaria in northwest Cambodia. Trop. Med. Int. Health 11:1800–1807. 10.1111/j.1365-3156.2006.01739.x [DOI] [PubMed] [Google Scholar]
- 47.Price RN, Hasugian AR, Ratcliff A, Siswantoro H, Purba HLE, Kenangalem E, Lindegardh N, Penttinen P, Laihad F, Ebsworth EP, Anstey NM, Tjitra E. 2007. Clinical and pharmacological determinants of the therapeutic response to dihydroartemisinin-piperaquine for drug-resistant malaria. Antimicrob. Agents Chemother. 51:4090–4097. 10.1128/AAC.00486-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Checchi F, Piola P, Fogg C, Bajunirwe F, Biraro S, Grandesso F, Ruzagira E, Babigumira J, Kigozi I, Kiguli J, Kyomuhendo J, Ferradini L, Taylor W, Guthmann J-P. 2006. Supervised versus unsupervised antimalarial treatment with six-dose artemether-lumefantrine: pharmacokinetic and dosage-related findings from a clinical trial in Uganda. Malar. J. 5:59. 10.1186/1475-2875-5-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McGready R, Tan SO, Ashley EA, Pimanpanarak M, Viladpai-nguen J, Phaiphun L, Wüstefeld K, Barends M, Laochan N, Keereecharoen L, Lindegardh N, Singhasivanon P, White NJ, Nosten F. 2008. A randomised controlled trial of artemether-lumefantrine versus artesunate for uncomplicated Plasmodium falciparum treatment in pregnancy. PLoS Med. 5:e253. 10.1371/journal.pmed.0050253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.World Health Organization. 2010. Guidelines for the treatment of malaria, 2nd ed. World Health Organization, Geneva, Switzerland [Google Scholar]
- 51.Hayes DJ. 2011. Developing age-based dosing regimens for antimalarials. Ph.D. dissertation. University of Liverpool, Liverpool, United Kingdom [Google Scholar]
- 52.Sigma-Tau Industrie Farmaceutiche Riunite S.p.A. 2011. Eurartesim product information. EMEA/H/C/001199 -II/0001/G. Sigma-Tau Industrie Farmaceutiche Riunite S.p.A., Rome, Italy [Google Scholar]
- 53.Staehli Hodel EM, Guidi M, Zanolari B, Mercier T, Duong S, Kabanywanyi A, Ariey F, Buclin T, Beck H-P, Decosterd L, Olliaro P, Genton B, Csajka C. 2013. Population pharmacokinetics of mefloquine, piperaquine and artemether-lumefantrine in Cambodian and Tanzanian malaria patients. Malar. J. 12:235. 10.1186/1475-2875-12-235 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.