

Abstract
Burkholderia pseudomallei, a facultative intracellular pathogen, causes severe infections and is inherently refractory to many antibiotics. Previous studies from our group have shown that interferon gamma (IFN-γ) interacts synergistically with the antibiotic ceftazidime to kill bacteria in infected macrophages. The present study aimed to identify the underlying mechanism of that interaction. We first showed that blocking reactive oxygen species (ROS) pathways reversed IFN-γ- and ceftazidime-mediated killing, which led to our hypothesis that IFN-γ-induced ROS interacted with ceftazidime to synergistically kill Burkholderia bacteria. Consistent with this hypothesis, we also observed that buthionine sulfoximine (BSO), another inducer of ROS, could substitute for IFN-γ to similarly potentiate the effect of ceftazidime on intracellular killing. Next, we observed that IFN-γ induced ROS-mediated killing of intracellular but not extracellular bacteria. On the other hand, ceftazidime effectively reduced extracellular bacteria but was not capable of intracellular killing when applied at 10 μg/ml. We investigated the exact role of IFN-γ-induced ROS responses on intracellular bacteria and notably observed a lack of actin polymerization associated with Burkholderia bacteria in IFN-γ-treated macrophages, which led to our finding that IFN-γ-induced ROS blocks vacuolar escape. Based on these results, we propose a model in which synergistically reduced bacterial burden is achieved primarily through separate and compartmentalized killing: intracellular killing by IFN-γ-induced ROS responses and extracellular killing by ceftazidime. Our findings suggest a means of enhancing antibiotic activity against Burkholderia bacteria through combination with drugs that induce ROS pathways or otherwise target intracellular spread and/or replication of bacteria.
INTRODUCTION
Burkholderia pseudomallei, the causative agent of melioidosis, is a Gram-negative, facultative intracellular pathogen that causes potentially lethal acute infections and occasionally chronic systemic infections in humans and animals (1–3). B. pseudomallei can invade and thrive inside professional phagocytes and nonphagocytic cells alike (4–6), and the stages of its intracellular pathogenesis are well defined (7–9). After uptake into a cell, Burkholderia bacteria escape from the phagosome into the cytoplasm, where they replicate, polymerize host cell actin, and spread to neighboring cells, usually causing cell fusion and multinucleated giant cell formation (5, 8–13).
Treatment of B. pseudomallei infection currently involves in-hospital administration of intravenous antibiotics followed by a lengthy eradication phase with oral antibiotics (2, 3, 14). With such an invasive and expensive treatment regimen, there is a great chance of noncompliance, which can lead to persistent infection. Therefore, there is a need to identify new treatments which may both accelerate recovery and make treatment administration more practical in areas where access to health care may be limited.
Recent melioidosis research has focused on understanding and characterizing the natural host immune responses to B. pseudomallei infection, specifically in regard to the effects of interferon gamma (IFN-γ). Studies show that IFN-γ is absolutely necessary for protection against acute melioidosis (15–17). There is also a known role for reactive oxygen species (ROS) responses due to IFN-γ stimulation in controlling early infection with B. pseudomallei (16, 18). While the effects of IFN-γ on infected macrophages have been studied in great detail (18–22), there are few studies that look at the combination of immune stimulation with antibiotic treatment, even though this scenario is realistically encountered in patients treated for melioidosis. Our lab has studied these immuno-antimicrobial interactions and has previously shown that treatment with IFN-γ can synergistically enhance the effect of ceftazidime treatment in controlling B. pseudomallei infection, both in vitro and in vivo, by significantly reducing intracellular bacterial burden (23). For this study, we used a combination of in vitro techniques to investigate the underlying mechanism of the interaction between IFN-γ and ceftazidime on killing of intracellular bacteria. From this, we developed a new model by which IFN-γ induces host antimicrobial pathways to control intracellular bacterial burden, and these pathways synergize with antibiotics to reduce overall bacterial burden of the macrophage environment.
MATERIALS AND METHODS
Biochemicals.
Ceftazidime hydrate, N-acetyl l-cysteine (NAC), reduced l-glutathione (GSH), ammonium chloride (NH4Cl), aminoguanidine-hydrochloride, and l-buthionine sulfoximine (BSO) were purchased from Sigma-Aldrich (St. Louis, MO). Other reagents included recombinant murine IFN-γ (Peprotech, Rocky Hill, NJ), Griess reagents R1 and R2 (Cayman Chemical, Ann Arbor, MI), ketamine (Fort Dodge Animal Health, Overland Park, KS), and xylazine (Ben Venue Labs, Bedford, OH). For flow cytometry and fluorescence microscopy, we purchased the following biochemicals from Life Technologies (Carlsbad, CA): Alexa Fluor 488-conjugated phalloidin, ProLong Gold antifade mounting medium with 4′,6-diamidino-2-phenylindole (DAPI), 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA), monochlorobimane (mBCl), Alexa Fluor 488 streptavidin-conjugated antibody, and trypsin with EDTA. Other reagents included lysosome-associated membrane protein (LAMP) antibody (eBioscience, San Diego, CA), rabbit polyclonal anti-B. pseudomallei antibody (provided by D. Waag from USAMRIID), and goat anti-rabbit secondary IgG antibody conjugated to Cy3 (Jackson ImmunoResearch Labs, West Grove, PA).
Bacteria.
Burkholderia thailandensis E264 and B. pseudomallei 1026b strains were used for these studies (24, 25). Both strains were grown in Luria-Bertani broth at 37°C with rotary shaking for 16 h and then stored at −80°C with 15% glycerol until needed. Frozen vials of bacteria were thawed and diluted immediately prior to their use. Mid-logarithmic-phase bacteria were grown for approximately 3 h with rotary shaking from a 1:25 dilution of an overnight culture until an optical density at 600 nm of 0.5 to 0.8 was reached.
Cell lines.
RAW 264.7, J774A.1, and AMJ2 macrophage cell lines and murine fibroblast cell line L929 were purchased from American Type Tissue Collection (Manassas, VA). Cell lines were maintained in complete medium consisting of minimum essential medium (MEM; Life Technologies) supplemented with 10% fetal bovine serum (Atlas, Fort Collins, CO), 0.075% sodium bicarbonate (Acros Organics, New Jersey), 1× nonessential amino acids, 0.5× essential amino acids (Life Technologies), and 2 mM l-glutamine (Sigma-Aldrich). Antibiotic additions of 100 units/ml penicillin and 100 μg/ml streptomycin (Life Technologies) were added to media for maintenance of cell lines, while all experiments were conducted in antibiotic-free medium. All cells were maintained at 37°C with 5% CO2.
Mice.
Female BALB/c, C57BL/6, and ICR mice were used for these studies. The BALB/c and C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME). The ICR mice were purchased from Harlan Laboratories (Indianapolis, IN). All mice were between 6 to 12 weeks old at the time of their use and were housed under pathogen-free conditions. All animal studies were approved by the Institutional Animal Care and Use Committee at Colorado State University.
Primary bone marrow macrophage culture.
Bone marrow macrophages were generated as previously described (26). Briefly, femurs and tibias were aseptically removed from mice and flushed of their bone marrow using needles and syringes. Red blood cells were lysed, and the remaining white blood cells were plated in 24-well plates at a concentration of 2 × 106 cells/ml in complete medium (cMEM) as described above. The addition of 10% L929-conditioned medium provided necessary growth factors for differentiation of myeloid progenitor cells into the macrophage/monocyte lineage. Adherent cells were incubated at 37°C and 5% CO2 until macrophages reached moderate confluence in wells (approximately 8 to 12 days).
Resting macrophage infection assay.
Macrophages were infected and treated as previously described (23). Briefly, macrophages were seeded into 24-well plates with cMEM (see above) and allowed to adhere overnight. B. thailandensis was added to macrophages at a multiplicity of infection of 5 and incubated for 1 h at 37°C in 5% CO2. After a 2-ml wash with phosphate-buffered saline (PBS), macrophages were exposed to 1 ml of high-dose kanamycin-sulfate (350 μg/ml) for 1 h to kill extracellular bacteria. After two 2-ml washes with PBS, macrophage treatments were diluted in MEM and applied to wells at a total volume of 0.5 ml/well and incubated for 18 h at 37°C in 5% CO2. After 18 h, treatment groups were at least 75% viable compared to the untreated control (K. Mosovsky and S. Dow, unpublished data). Extracellular bacteria were then washed off three times with PBS, and macrophages were lysed with 1 ml of sterile distilled water. Intracellular bacterial burden was then assessed by plating serial dilutions of the lysates on LB agar followed by colony counts 24 to 48 h after plating. For experiments that aimed to quantitate extracellular bacterial numbers after the 18-h treatment period, supernatants were first gently pipetted up and down to resuspend bacteria in wells without disturbing the adhered macrophages. Then, extracellular bacteria were quantitated by plating serial dilutions of the supernatants onto LB agar, followed by colony counts 24 to 48 h later.
Fluorescence microscopy.
At the indicated times, chamber slides containing B. thailandensis-infected macrophages were fixed with a solution of paraformaldehyde and permeabilized with 0.1% Triton X-100. Macrophages were incubated overnight at 4°C with rabbit anti-Burkholderia serum followed by a secondary goat anti-rabbit IgG antibody conjugated to Cy3. Phalloidin was added at 5 units/well for 30 min to stain actin filaments, and lysosome compartments were identified using a biotinylated anti-mouse LAMP antibody followed by an Alexa Fluor 488 streptavidin-conjugated antibody. Coverslips were applied with mounting medium containing DAPI to stain nuclei. Images were acquired with a Leica DM 4500B microscope (Leica Microsystems, Buffalo Grove, IL) fitted with a Retiga 2000R camera (QImaging, Surrey, BC, Canada) and by using QCapture Pro software (QImaging). Adobe Photoshop CS3 version 10.0.1 (Adobe, San Jose, CA) was used to create color overlay images as well as to make global manipulations to the linear parameters of black-point and individual color brightness for each experiment.
Preactivated macrophage infection assay.
In order to specifically study the intracellular antimicrobial effects of fully activated macrophages, we preactivated adhered macrophages with IFN-γ (10 ng/ml) or pretreated macrophages with ceftazidime (10 μg/ml) for 18 h prior to infection. Following the 18-h pretreatment, macrophages were infected with B. thailandensis E264 at a multiplicity of infection of 5. Since we expected rapid killing of bacteria in IFN-γ-preactivated macrophages, the infection time was only 30 min so that we could more quickly observe killing effects. Following the 30-min infection, macrophages were washed once with 2 ml of PBS, and high-dose kanamycin monosulfate (350 μg/ml) was applied to kill extracellular bacteria for 1 h. Macrophages were then washed twice with 2 ml of PBS, and cMEM was applied to the macrophages for up to 6 h. In order to ensure that only pretreatment effects were evaluated, no treatments were present during the infection or the 6-h assay. When NAC (50 mM) or GSH (50 mM) was used in the preactivated macrophage infection assay, these additional treatments were added for 3 h prior to infection and washed off with 1 ml PBS prior to the infection.
Extracellular bacterial killing assay.
In order to evaluate the effects of the treatments on bacterial killing alone, we used a macrophage-free bacterial killing assay consisting of bacteria and treatments only. Our goal was to simulate the exact extracellular environment of our resting macrophage infection model but in the absence of macrophages. Therefore, we used the same treatment concentrations, the same medium (cMEM) and volume of medium in wells (500 μl), and the same incubation conditions and times. Treatments were prepared ahead of time, diluted in cMEM, and added to 24-well plates at a total volume of 100 μl for treatments. Meanwhile, B. thailandensis was grown to mid-logarithmic phase from an overnight culture and diluted in cMEM, and 400 μl was added to the 24-well plate already containing the treatments. The initial density of bacteria was 1 × 107 CFU/well. Plates were incubated at 37°C with 5% CO2 for 18 h to simulate the same incubation conditions as our resting macrophage infection assay. After 18 h, well contents were resuspended by slowly pipetting up and down, and surviving bacteria were enumerated by plating serial dilutions of well contents on LB agar and counting colonies after 24 to 48 h.
Flow cytometry.
Macrophages were treated in 24-well plates at a density of 4 × 105 cells/ml. After treatment, the cells were washed with PBS and detached with 0.25% trypsin with EDTA. Cells were incubated with either 5 μM carboxy-H2DCFDA for 30 min at 37°C or with 40 μM mBCl for 20 min at room temperature in the dark, as previously done (27). Data were collected on more than 50,000 cells per sample using a Gallios flow cytometer (Beckman Coulter, Brea, CA) and analyzed using FlowJo software version 7.6.5 (Tree Star, Ashland, OR).
Statistical analyses.
Means, standard errors of the means (SEM), and P values were determined and plotted using Prism software version 5.00 (GraphPad, La Jolla, CA). For comparisons of two groups, a two-tailed Student t test was used to determine statistically significant differences. For comparisons of three or more groups, the one-way analysis of variance (ANOVA) was used followed by Tukey's posttest for multiple comparisons. Grouped data were analyzed by two-way ANOVA, and statistical synergy was determined as before (28) from the interaction P value of a two-way ANOVA. All differences were considered statistically significant for P values of <0.05.
RESULTS
IFN-γ combination with ceftazidime significantly reduces intracellular bacterial burden in infected macrophages.
Previously, we found that the combination of IFN-γ with ceftazidime synergistically reduced the intracellular bacterial burden of B. pseudomallei-infected AMJ macrophages after 18 h of treatment (23). We confirmed the IFN-γ and ceftazidime synergistic interaction using a different macrophage cell line, RAW 264.7 (Fig. 1A), as well as primary bone marrow macrophages from C57BL/6, BALB/c, and ICR mice (Fig. 1B to D), all infected with B. thailandensis. For all macrophages tested, the combination of IFN-γ and ceftazidime significantly controlled the intracellular bacterial burden compared to ceftazidime or IFN-γ treatment alone and as much as 3 to 4 log10 units compared to that of untreated cells. Importantly, the IFN-γ and ceftazidime combination, compared to untreated controls, failed to reduce bacterial burden in the murine fibroblast cell line, L929 (see Fig. S1 in the supplemental material). Taken together, these results demonstrate that the synergistic interaction between IFN-γ and ceftazidime is exhibited by infected macrophages but not by cells incapable of phagocytosis.
FIG 1.

Ceftazidime and IFN-γ induce synergistic killing of intracellular bacteria in macrophages. Adherent RAW 264.7 cells (A) or primary bone marrow macrophages from C57BL/6 mice (B), BALB/c mice (C), or ICR mice (D) were infected with B. thailandensis and treated with ceftazidime (10 μg/ml for RAW 264.7 cells, 3 μg/ml for bone marrow macrophages), IFN-γ (10 ng/ml), or the combination of ceftazidime and IFN-γ for 18 h. Intracellular bacteria were then enumerated by plating macrophage lysates. Synergistic interactions between ceftazidime and IFN-γ were determined by two-way ANOVA (*, P < 0.01; **, P < 0.001; ***, P < 0.0001). Graphs are representative of data from two independent experiments with treatment groups run in triplicate.
Inhibitors of ROS pathway reverse synergistic interaction between IFN-γ and ceftazidime.
We considered several mechanisms by which IFN-γ, with no direct bactericidal effects itself, could trigger the synergistic interaction with ceftazidime. These included induction of nitric oxide production, increased uptake of ceftazidime, and induction of ROS responses in IFN-γ-stimulated macrophages. Increased nitric oxide production was eliminated as a potential mechanism of synergy after specific inhibitors of the pathway failed to reverse the combination therapy synergy (see Fig. S2 in the supplemental material). Furthermore, direct quantization of intracellular ceftazidime concentrations showed no increased uptake of ceftazidime due to IFN-γ stimulation (R. Troyer and S. Dow, unpublished data). We were then left to explore IFN-γ induction of ROS pathways as a mechanism of synergy.
We reasoned that if IFN-γ synergized with ceftazidime through a ROS-mediated pathway, blocking ROS pathways using specific inhibitors should abolish the synergy. We found that NAC, an antioxidant and cysteine precursor for GSH synthesis (29–31), reversed the synergistic killing of the combination therapy in a concentration-dependent manner (Fig. 2A). We also observed this effect using B. pseudomallei 1026b (E. Silva and S. Dow, unpublished data). This led us to hypothesize that NAC was functioning through increased production of GSH, a prominent cellular antioxidant and ROS scavenger in macrophages (29, 32–38). To test this hypothesis, we measured intracellular GSH concentrations using monochlorobimane (mBCl) and confirmed that treatment with NAC increased GSH content (Fig. 2B) and also decreased total ROS levels in IFN-γ and ceftazidime-treated macrophages (Fig. 2C). Finally, when GSH was substituted directly for NAC, we observed a similar titratable effect of reversing the IFN-γ and ceftazidime synergistic killing of intracellular B. thailandensis (Fig. 2D) and B. pseudomallei (Silva and Dow, unpublished). Based on these findings, we concluded that inhibitors of ROS pathways reversed the killing activity of IFN-γ and ceftazidime, consistent with a ROS-mediated mechanism of synergy.
FIG 2.

ROS pathway inhibitors reverse IFN-γ and ceftazidime synergy. NAC (A) and GSH (D) reverse the intracellular killing effect of IFN-γ (10 ng/ml) and ceftazidime (10 μg/ml) combination therapy on B. thailandensis-infected macrophages in a dose-dependent manner. Statistical differences compared to the group treated with IFN-γ and ceftazidime were assessed by one-way ANOVA with Tukey's posttest (*, P < 0.05). (B) Mean fluorescent intensity of mBCl after treatment of uninfected RAW cells with NAC (20 mM) for 2 h (***, P = 0.0004); (C) intracellular ROS levels (as detected by carboxy-H2DCFDA) in RAW 264.7 cells after 10 h of treatment with ceftazidime and IFN-γ, with or without NAC (20 mM); (E and F) histogram overlays of intracellular ROS responses as measured with carboxy-H2DCFDA by flow cytometry; (E) IFN-γ-elicited ROS responses from uninfected RAW 264.7 macrophages, various hours after stimulation; (F) intracellular ROS responses of B. thailandensis-infected RAW 264.7 macrophages 18 h after treatment with ceftazidime (10 μg/ml) or IFN-γ (10 ng/ml). Results are representative of data from at least two independent experiments.
IFN-γ induces ROS production in macrophages.
We confirmed increased levels of intracellular ROS due to IFN-γ stimulation in uninfected macrophages and found that IFN-γ increased ROS as early as 6 h poststimulation, with increasing levels through 24 h (Fig. 2E). Significantly, the time required for the start of ROS induction, 6 h, coincided with the time it took for the combination of IFN-γ and ceftazidime to begin control of intracellular bacterial burden, as seen in our previous study (23). We also observed increased ROS in bone marrow macrophages following IFN-γ stimulation (Silva and Dow, unpublished). In infected macrophages, IFN-γ but not ceftazidime increased intracellular ROS levels (Fig. 2F). Combined, these results indicated that uninfected and infected macrophages increased production of ROS due to stimulation with IFN-γ.
Inducers of ROS can interact with ceftazidime to increase killing of intracellular B. thailandensis.
We next argued that if the IFN-γ effect was ROS mediated, then other prooxidant drugs that increased ROS should also interact with ceftazidime to enhance killing of intracellular Burkholderia bacteria. BSO specifically inhibits the rate-limiting step in GSH synthesis (29), depleting GSH content through prevention of biosynthesis and resulting in increased intracellular ROS as a result of decreased antioxidant activity (39–44). To address the role of ROS induction by non-IFN-γ pathways, BSO was combined with ceftazidime and/or IFN-γ in B. thailandensis-infected macrophages (Fig. 3A), and the effect on intracellular bacterial burden was assessed. The combination of BSO with ceftazidime produced the same degree of intracellular killing as the combination of IFN-γ with ceftazidime, an effect which was reversed by the addition of exogenous GSH (Fig. 3A). This effect of BSO was confirmed in B. pseudomallei (see Fig. S3 in the supplemental material). It should be noted that BSO treatment did not affect internalization of bacteria (Mosovsky and Dow, unpublished). We also confirmed that treatment with BSO increased intracellular concentrations of ROS in B. thailandensis-infected macrophages and further increased ROS levels above those triggered by IFN-γ alone (Fig. 3B). Analysis of these results corroborates the hypothesis that BSO was acting as a prooxidant in our macrophage infection model. Finally, we showed that treatment with BSO decreased intracellular GSH concentrations as predicted (Fig. 3C). These results demonstrated, therefore, that inducers of ROS could substitute for IFN-γ and interact with ceftazidime to synergistically reduce intracellular bacterial burden.
FIG 3.
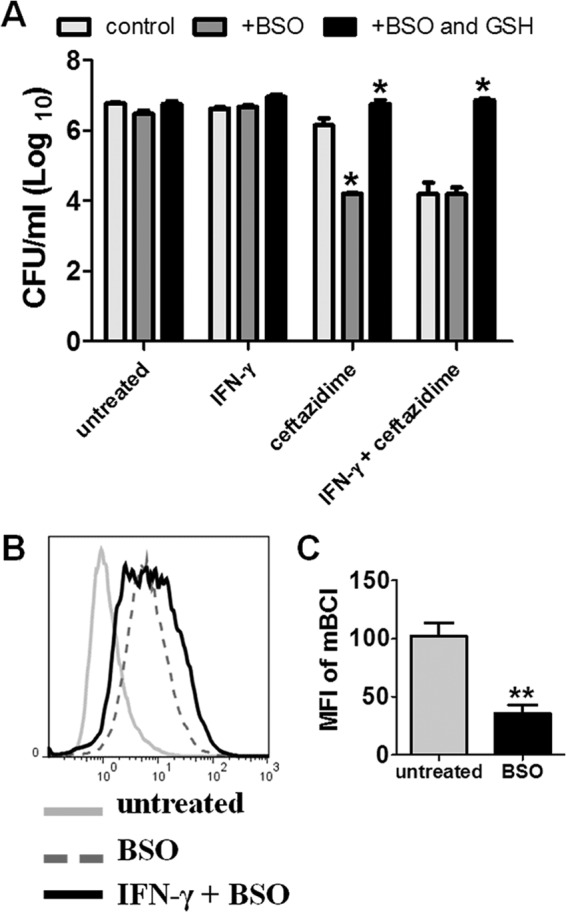
Glutathione depletion enhances antibiotic killing and induces intracellular ROS. (A) RAW 264.7 macrophages were infected with B. thailandensis and treated with ceftazidime (10 μg/ml), IFN-γ (10 ng/ml), or the combination of ceftazidime and IFN-γ for 18 h with or without BSO treatment (5 mM). In order to maximally deplete intracellular GSH, all BSO groups also received BSO treatment 18 to 24 h prior to infection. Intracellular bacteria were then enumerated by plating macrophage lysates. Significant differences between controls and BSO-treated groups were determined by two-way ANOVA (*, P < 0.05). (B) Histogram overlays of intracellular ROS after macrophage infection with B. thailandensis and treatment with BSO (5 mM) or the combination of IFN-γ and BSO for 12 h as measured by carboxy-H2DCFDA and flow cytometry; (C) mean fluorescent intensity of intracellular mBCl, indicating GSH content, after 18 h of treatment with 5 mM BSO (**, P = 0.0079). All data are representative of results from at least two independent experiments.
IFN-γ, but not ceftazidime, mediates intracellular killing of bacteria through ROS responses.
After showing a major role for IFN-γ-induced ROS responses in the mechanism of synergy with ceftazidime, we next hypothesized that IFN-γ-induced ROS was directly responsible for killing intracellular bacteria. To investigate this hypothesis, we turned to a preactivated macrophage infection model to determine the intracellular killing potential of macrophages that were already fully activated by IFN-γ. Shortening the assay to 6 h instead of 18 h enabled us to more accurately describe killing in the intracellular compartment without confounding by overgrowth of extracellular bacteria. Macrophages were preactivated with IFN-γ for 18 h and then infected with B. thailandensis (see Materials and Methods for further details). Bacteria that had been taken up into the intracellular compartment during the initial infection were steadily killed over the entire 6-h assay (Fig. 4A). Conversely, Burkholderia bacteria actually replicated inside untreated macrophages over the same amount of time.
FIG 4.
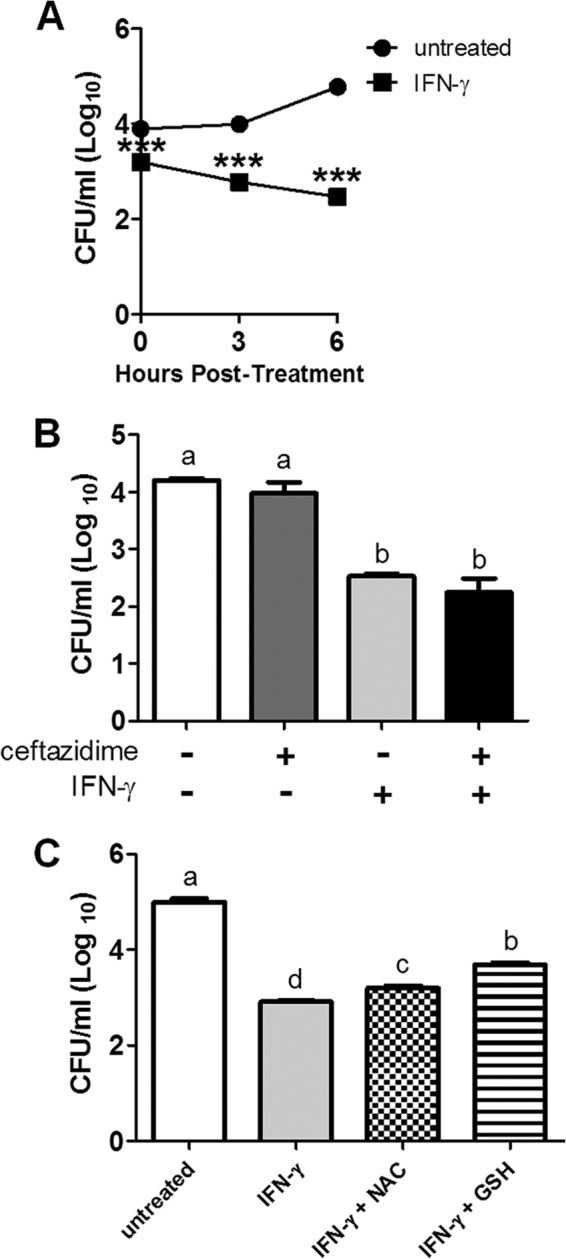
IFN-γ activation of macrophages kills intracellular bacteria. RAW 264.7 macrophages were preactivated with IFN-γ (10 ng/ml) for 18 h prior to infection with B. thailandensis. At the indicated times, lysates were plated to enumerate surviving intracellular bacteria. (A) Time course of intracellular killing due to preactivation with IFN-γ prior to infection. The difference in intracellular bacterial numbers at time t = 0 between untreated and preactivated macrophages was likely due to the time lapse between the initial start of infection and the end of the kanamycin treatment step. Significant differences compared to the untreated control were assessed at each time point by two-way ANOVA (***, P < 0.001). (B) Intracellular bacterial burden after 6 h of infection in macrophages pretreated with IFN-γ (as before) or with ceftazidime (10 μg/ml). Statistical differences were assessed by one-way ANOVA, a > b (P < 0.0001). (C) NAC (50 mM) or GSH (50 mM) were applied to macrophages for the last 3 h of the pretreatment regimen. Then all treatments were washed off and macrophages were infected with B. thailandensis. Intracellular bacterial burden was assessed 6 h after the end of the kanamycin step by plating lysates, a > b > c > d (P < 0.05). Data are representative of results from two independent experiments run in triplicate.
We had previously found that whole-cell intracellular concentrations of ceftazidime (either alone or without IFN-γ stimulation) were more than 50-fold below the MIC of ceftazidime (Troyer and Dow, unpublished). However, we speculated that ceftazidime might be more concentrated in specific compartments within the macrophage, and if this was the case, ceftazidime might still have a role in killing intracellular bacteria. We therefore pretreated macrophages for 18 h with IFN-γ (as before), ceftazidime, or the combination of both drugs. We then removed all treatments and infected the macrophages with B. thailandensis. After 6 h, we lysed the macrophages and determined surviving bacteria. Only macrophages pretreated with IFN-γ showed intracellular killing of B. thailandensis during the 6-h assay (Fig. 4B). Ceftazidime-pretreated macrophages were both unable to control intracellular replication and unable to enhance the killing effect seen with IFN-γ alone. These results suggest that over 18 h, ceftazidime is unable to accumulate in macrophages to an extent that could reduce or control intracellular bacterial numbers.
Finally, we showed that antioxidants NAC and GSH could both partially reverse the intracellular killing of B. thailandensis in IFN-γ-activated macrophages, suggesting a role for IFN-γ-induced ROS responses in the direct killing of intracellular bacteria (Fig. 4C).
B. thailandensis fails to escape the phagosome and polymerize actin inside IFN-γ-treated macrophages.
In order to understand how IFN-γ-induced ROS could increase killing of intracellular bacteria, we turned to fluorescence microscopy (Fig. 5). Microscopy revealed that B. thailandensis polymerized actin inside untreated or ceftazidime-treated macrophages (Fig. 5A and B) but failed to polymerize host cell actin in macrophages treated with IFN-γ (Fig. 5C and D). The failure to polymerize actin was recapitulated with BSO treatment as well (see Fig. S4 in the supplemental material), suggesting ROS involvement. Since Burkholderia bacteria polymerize host cell actin only after vacuolar escape into the cytoplasm (9–11), an investigation was prompted to determine whether bacteria inside IFN-γ-treated macrophages failed to escape the phagosome. We used fluorescence microscopy to quantitate the ratio of bacteria inside IFN-γ-treated macrophages that colocalized with lysosome-associated membrane protein 1 (LAMP-1) compared to that of untreated controls. Indeed, we found that IFN-γ-treated macrophages had a higher proportion of bacteria that colocalized with LAMP-1+-containing vacuoles than untreated controls, suggesting that bacteria failed to escape the phagosome in the IFN-γ-treated groups (Fig. 6). This explanation is consistent with the lack of actin tails on Burkholderia bacteria inside macrophages treated with IFN-γ or BSO.
FIG 5.

B. thailandensis fails to form actin tails inside IFN-γ-treated macrophages. RAW cells were infected with B. thailandensis and not treated (A) or treated with ceftazidime (10 μg/ml) (B), IFN-γ (10 ng/ml) (C), or ceftazidime and IFN-γ (D) for 12 h. Macrophages were then fixed, permeabilized, and stained with phalloidin to identify host cell actin (green), DAPI to stain nuclei (blue), and anti-Burkholderia serum followed by secondary antibody conjugated to Cy3 to identify B. thailandensis (red). Images were captured under ×40 magnification. Actin tails are seen as bright green protrusions from the red bacteria in panels A and B. Images are representative of data from three independent experiments.
FIG 6.
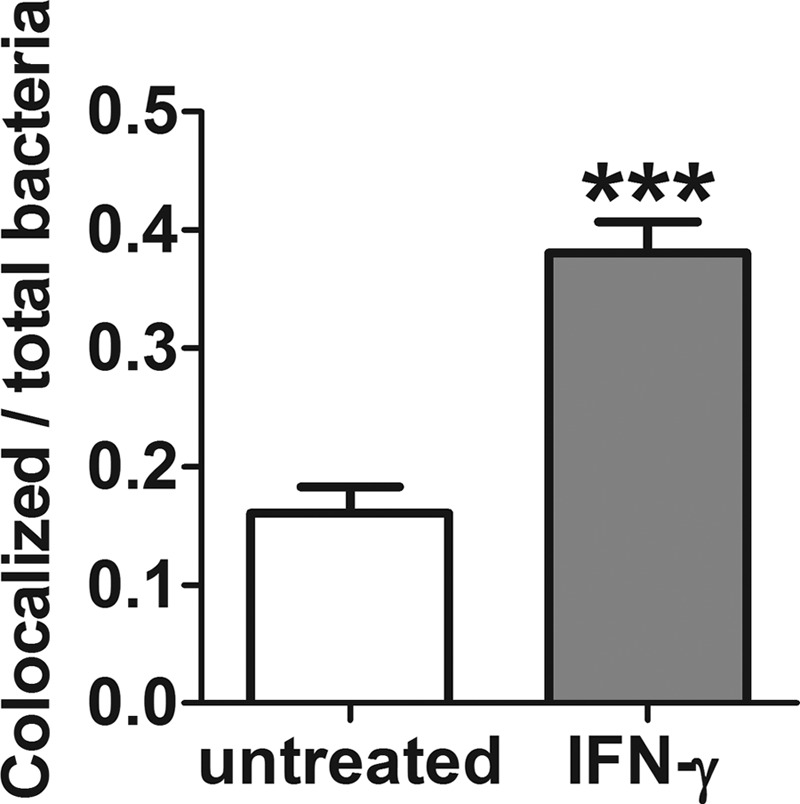
Burkholderia bacteria inside IFN-γ-treated macrophages have higher proportions of LAMP-1+ colocalization than the untreated control. Quantitation of B. thailandensis colocalization with LAMP-1 antibody after 8 h of IFN-γ treatment of infected macrophages. Data are presented as the ratio of colocalized bacteria (LAMP-1+) to total bacteria per field of view. Data represent 10 fields of view for each treatment group (***, P < 0.0001), and results are representative of data from 3 independent experiments.
We also revealed a decrease in the proportion of macrophage nuclei associated with multinucleated giant cells (MNGCs) in macrophages treated with IFN-γ compared to that of untreated controls (Fig. 5A to D). In untreated controls, an average of 83% (±7%) of nuclei were associated with MNGCs, whereas only 6% (±3%) of nuclei of IFN-γ-treated macrophages were associated with MNGCs (P < 0.0001 by Student's t test). Noting the similarities of appearance (including MNGC formation and actin tail polymerization) between ceftazidime and untreated controls, we hypothesized that the main contribution of ceftazidime to the synergism must be primarily through extracellular killing, while the role of IFN-γ may be more focused on intracellular control of bacterial burden. We next conducted experiments to determine the specific contribution of ceftazidime to the mechanism of synergy.
Ceftazidime controls primarily extracellular bacterial burden.
After showing that ceftazidime had no role in killing intracellular bacteria (Fig. 4B), we began to investigate the specific contribution of ceftazidime to the mechanism of synergy. We found that ceftazidime appeared to play a role in controlling extracellular bacterial burden during the macrophage infection (Fig. 7A). However, we also found that while IFN-γ had no effect on extracellular bacterial burden itself, it significantly enhanced the ceftazidime effect. This result prompted us to investigate whether IFN-γ was itself bactericidal or could possibly increase antibiotic killing. In our extracellular bacterial killing assay, we reproduced the same in vitro culture conditions, this time in a macrophage-free system (see Materials and Methods), and found that IFN-γ had no bactericidal effects alone and did not significantly increase killing due to ceftazidime (Fig. 7B). Together, these results show that ceftazidime kills primarily extracellular bacteria in our macrophage infection model, though IFN-γ activation of macrophages can contribute to reduction of extracellular numbers, presumably by limiting the return of intracellular bacteria to the extracellular compartment.
FIG 7.
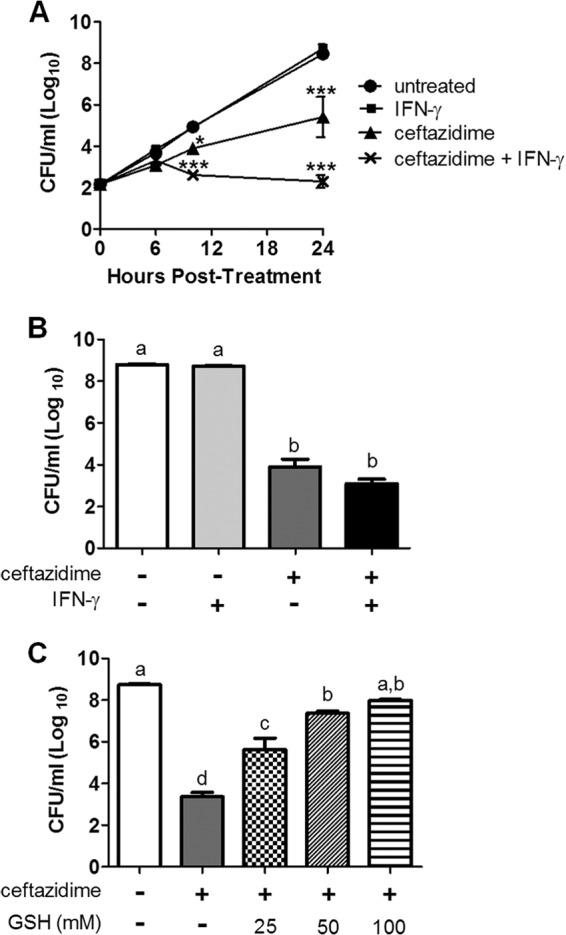
Ceftazidime controls primarily extracellular bacterial burden. (A) RAW 264.7 macrophages were infected with B. thailandensis and treated for 24 h with IFN-γ (10 ng/ml), ceftazidime (10 μg/ml), or the combination of IFN-γ and ceftazidime. Extracellular bacterial burden was assessed at 0, 6, 10, and 24 h posttreatment by plating serial dilutions of well supernatants. Significant differences were assessed at all time points by two-way repeated-measures ANOVA and compared to the untreated control (*, P < 0.05; ***, P < 0.001). (B and C) B. thailandensis was treated with ceftazidime (10 μg/ml), IFN-γ (10 ng/ml), or the combination of both treatments for 18 h in the absence of macrophages. Surviving bacteria were enumerated by plating dilutions of remaining bacteria in wells after 18 h of treatments. Significant differences were assessed by one-way ANOVA, a > b > c > d (P < 0.0001). All graphs are representative of results from two independent experiments with treatment groups run in triplicate.
After showing only a partial role for GSH to reverse IFN-γ-mediated intracellular killing (Fig. 4C), we were especially interested in whether GSH could reverse ceftazidime-mediated extracellular killing. In our macrophage-free bacterial killing assay, we showed that GSH reversed ceftazidime-mediated killing in a dose-dependent manner, with complete reversal of killing at 100 mM (Fig. 7C).
DISCUSSION
We previously described how IFN-γ synergistically enhanced killing of intracellular Burkholderia bacteria by ceftazidime and suggested that such an approach might be used to improve the effectiveness of antibiotic therapy for melioidosis (23). Here, we have shown that the mechanism of synergy between IFN-γ and ceftazidime is mediated primarily by IFN-γ-induced ROS responses to control intracellular spread and replication of Burkholderia bacteria, combined with ceftazidime control of extracellular bacterial burden. Initially, ROS involvement was supported by experiments in which ROS inhibitors reversed the synergistic killing elicited by the combination of IFN-γ and ceftazidime. Moreover, a chemical inducer of ROS (BSO) essentially recapitulated the IFN-γ effect in terms of potentiating bacterial killing by ceftazidime. We further showed evidence that IFN-γ-induced ROS could directly kill intracellular bacteria and that both prooxidant drugs, IFN-γ and BSO, prevented bacteria from polymerizing host cell actin, which led to our discovery that IFN-γ prevents bacterial escape from the phagolysosome. Previous reports indicate a role for IFN-γ, and IFN-γ-induced ROS, in preventing vacuolar escape of other intracellular bacterial pathogens (45–47). Given that vacuolar escape and actin polymerization are thought to be essential for intracellular replication and cell-to-cell spread of Burkholderia infection (5, 9, 10, 48), our results shed new light on the importance of IFN-γ to limit the spread of infection by controlling intracellular bacterial burden. We also found that macrophages activated by IFN-γ contributed to reducing the extracellular bacterial burden when combined with ceftazidime (Fig. 7A). This suggests that by controlling the intracellular bacterial burden, IFN-γ indirectly controls the extracellular bacterial burden as well, presumably by limiting actin tail protrusions and escape into the extracellular space.
We next showed that the contribution of ceftazidime to the synergistic killing was mediated entirely by killing and blocking replication of extracellular bacteria. When applied extracellularly at 10 μg/ml, the intracellular concentration of ceftazidime inside macrophages was still approximately 50-fold below the MIC and was unable to kill or inhibit the growth of intracellular bacteria. It is possible that at much higher concentrations, ceftazidime may better penetrate into the macrophages and therefore have a small role in killing intracellular bacteria or at least inhibiting their growth. Future experiments will determine if the role of ceftazidime would change with much higher doses. We also observed a complete reversal of antibiotic killing when GSH was added to our macrophage-free bacterial killing assay. While this effect might indicate a role for ROS in the mechanism of ceftazidime killing, newer evidence suggests that GSH protects bacteria against antibiotics through a mechanism independent of ROS scavenging, which may involve blocking or inactivating antibiotic activity (49). Thus, we now show that GSH can block bacterial killing by scavenging IFN-γ-induced ROS in the intracellular compartment and secondly by interference with ceftazidime killing by some yet-unknown mechanism.
In summary, we propose that the in vitro macrophage infection model is a dynamic system involving both extracellular and intracellular bacteria, in which the bacterial burden of one compartment may influence the burden of the other compartment (Fig. 8). In the absence of an antibiotic, the extracellular bacteria replicate to large quantities and continue to invade into macrophages, leading to high intracellular bacterial burdens as well. In the absence of IFN-γ, the bacteria that invade the macrophages are capable of rapid intracellular replication, leading to high intracellular bacterial burdens. Actin polymerization and protrusions from the cell membrane contribute to spread of the infection but likely increase extracellular bacterial burden, too. Therefore, it seems that IFN-γ-induced ROS and ceftazidime are necessary but insufficient by themselves to defend against Burkholderia infection in vitro. It is only with the combination of intracellular control by IFN-γ and extracellular control by ceftazidime that the bacterial burden is synergistically reduced in both compartments and the system as a whole.
FIG 8.
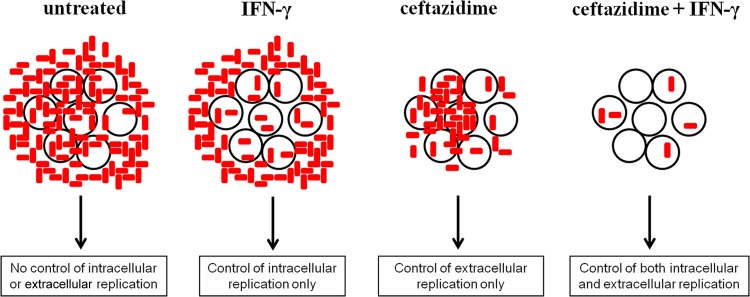
Compartmentalized killing describes the mechanism of synergy between IFN-γ and ceftazidime. We propose that during the macrophage infection assay, there is a dynamic exchange between bacteria in the intracellular and extracellular compartments. Therefore, it is only with IFN-γ control of intracellular spread and replication, combined with ceftazidime control of extracellular bacterial burden, that synergy is achieved with low bacterial burden in the system as a whole.
There are multiple reports that IFN-γ treatment of macrophages can greatly increase killing of intracellular B. pseudomallei (20, 22, 50–52). However, in this report, we first show that IFN-γ by itself has a negligible role in controlling intracellular B. thailandensis infection (Fig. 1). We believe this discrepancy may be explained by differences in the macrophage infection models, in particular the relative order and timing of IFN-γ stimulation prior to macrophage infection. We (Fig. 4) and others have shown that when macrophages are preactivated with IFN-γ prior to infection, or when IFN-γ is coadministered during the infection, there is significant antimicrobial activity and killing of intracellular bacteria (20, 22, 50–53). However, when macrophages are activated with IFN-γ after infection (Fig. 1), then the activity of IFN-γ is considerably diminished. One explanation for the decreased activity of IFN-γ in the postactivation model is that by the time ROS is maximally induced by IFN-γ stimulation, the macrophage is already overwhelmed by the infection and the ROS response is too little too late in eliminating the pathogen. Another explanation is that Burkholderia bacteria have already induced expression of suppressor of cytokine signaling 3 (SOCS3) and cytokine-inducible Src homology 2-containing protein (CIS) in the macrophage, both of which interfere with IFN-γ signaling and can lead to decreased elimination of Burkholderia bacteria in infected macrophages (53). Either way, it is apparent that preactivation and postactivation of infected macrophages elicit very different antimicrobial responses. While both models (preactivation and postactivation of infected macrophages) have their advantages and disadvantages, we argue that the postactivation model may more closely mimic the events in a natural infection, wherein the host cell is infected first and an immune response is mounted secondarily.
In the current study, we have provided unambiguous evidence of a key role for IFN-γ-induced ROS responses to synergize with antibiotics to control B. pseudomallei infection in macrophages. There is already a well-documented role for IFN-γ-induced reactive nitrogen species (RNS) in the killing of B. pseudomallei, typically through induction of inducible nitric oxide synthase (iNOS) (18, 19, 50–52). However, there are far fewer studies investigating a role for IFN-γ-induced ROS responses. For example, in vivo knockout studies by Brietbach and colleagues showed that NADPH oxidase, but not iNOS, was important for controlling mortality during acute-phase infection with B. pseudomallei (16). Furthermore, two studies have shown a role for IFN-γ-induced ROS response control of bacterial burden during acute-phase infection with B. pseudomallei (16, 18). Finally, melioidosis susceptibility is reported to be higher in patients with chronic granulomatous disease, who lack a functional NADPH phagocyte oxidase, suggesting a role for NADPH oxidase-generated ROS responses in melioidosis protection (54, 55). While IFN-γ-induced antimicrobial mechanisms are complex, we believe that there are important roles for both IFN-γ-induced RNS and ROS responses and that the relative importance of these roles depends in part on the current activation state of the host immune cells at the time of initial infection.
Finally, based on our results, we suggest caution when using the standard kanamycin protection assay to define IFN-γ effects on regulation of intracellular bacterial infections. We propose that in most macrophage infection models, any addition of extracellular antibiotics during the treatment period is likely enhancing the overall bacterial killing in the system as a whole, by reducing the number of viable bacteria that can reinfect healthy cells. By this logic, the kanamycin protection assay is likely another example of immuno-antimicrobial synergy, and the observed results are due to the effects of both the antibiotic and IFN-γ.
In conclusion, we have shown that IFN-γ-induced ROS controls intracellular bacterial burden while ceftazidime controls primarily extracellular bacterial burden of Burkholderia-infected macrophages. Together, they synergize to reduce the bacterial burden of the system as a whole. The ability of IFN-γ-induced ROS to contain bacteria within the phagolysosome and prevent their escape into the cytoplasm suggests that IFN-γ may help control cell-to-cell spread of this intracellular pathogen. Our results also suggest that certain antioxidants may prevent bacterial killing by scavenging ROS and by interference with the action of antibiotics. On the other hand, prooxidant drugs that increase intracellular ROS responses may be used to nonspecifically enhance antibiotic activity against Burkholderia and other intracellular bacterial pathogens.
Supplementary Material
Footnotes
Published ahead of print 28 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02781-14.
REFERENCES
- 1.Wuthiekanun V, Peacock SJ. 2006. Management of melioidosis. Expert Rev. Anti Infect. Ther. 4:445–455. 10.1586/14787210.4.3.445 [DOI] [PubMed] [Google Scholar]
- 2.Limmathurotsakul D, Peacock SJ. 2011. Melioidosis: a clinical overview. Br. Med. Bull. 99:125–139. 10.1093/bmb/ldr007 [DOI] [PubMed] [Google Scholar]
- 3.Fisher DA, Harris PN. 2013. Melioidosis: refining management of a tropical time bomb. Lancet 383:762–764. 10.1016/S0140-6736(13)62143-1 [DOI] [PubMed] [Google Scholar]
- 4.Jones AL, Beveridge TJ, Woods DE. 1996. Intracellular survival of Burkholderia pseudomallei. Infect. Immun. 64:782–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kespichayawattana W, Rattanachetkul S, Wanun T, Utaisincharoen P, Sirisinha S. 2000. Burkholderia pseudomallei induces cell fusion and actin-associated membrane protrusion: a possible mechanism for cell-to-cell spreading. Infect. Immun. 68:5377–5384. 10.1128/IAI.68.9.5377-5384.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stevens MP, Galyov EE. 2004. Exploitation of host cells by Burkholderia pseudomallei. Int. J. Med. Microbiol. 293:549–555. 10.1078/1438-4221-00292 [DOI] [PubMed] [Google Scholar]
- 7.Chieng S, Carreto L, Nathan S. 2012. Burkholderia pseudomallei transcriptional adaptation in macrophages. BMC Genomics 13:328. 10.1186/1471-2164-13-328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.French CT, Toesca IJ, Wu TH, Teslaa T, Beaty SM, Wong W, Liu M, Schroder I, Chiou PY, Teitell MA, Miller JF. 2011. Dissection of the Burkholderia intracellular life cycle using a photothermal nanoblade. Proc. Natl. Acad. Sci. U. S. A. 108:12095–12100. 10.1073/pnas.1107183108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allwood EM, Devenish RJ, Prescott M, Adler B, Boyce JD. 2011. Strategies for intracellular survival of Burkholderia pseudomallei. Front. Microbiol. 2:170. 10.3389/fmicb.2011.00170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burtnick MN, DeShazer D, Nair V, Gherardini FC, Brett PJ. 2010. Burkholderia mallei cluster 1 type VI secretion mutants exhibit growth and actin polymerization defects in RAW 264.7 murine macrophages. Infect. Immun. 78:88–99. 10.1128/IAI.00985-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burtnick MN, Brett PJ, Nair V, Warawa JM, Woods DE, Gherardini FC. 2008. Burkholderia pseudomallei type III secretion system mutants exhibit delayed vacuolar escape phenotypes in RAW 264.7 murine macrophages. Infect. Immun. 76:2991–3000. 10.1128/IAI.00263-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galyov EE, Brett PJ, DeShazer D. 2010. Molecular insights into Burkholderia pseudomallei and Burkholderia mallei pathogenesis. Annu. Rev. Microbiol. 64:495–517. 10.1146/annurev.micro.112408.134030 [DOI] [PubMed] [Google Scholar]
- 13.Wiersinga WJ, van der Poll T, White NJ, Day NP, Peacock SJ. 2006. Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat. Rev. Microbiol. 4:272–282. 10.1038/nrmicro1385 [DOI] [PubMed] [Google Scholar]
- 14.Schweizer HP. 2012. Mechanisms of antibiotic resistance in Burkholderia pseudomallei: implications for treatment of melioidosis. Future Microbiol. 7:1389–1399. 10.2217/fmb.12.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santanirand P, Harley VS, Dance DA, Drasar BS, Bancroft GJ. 1999. Obligatory role of gamma interferon for host survival in a murine model of infection with Burkholderia pseudomallei. Infect. Immun. 67:3593–3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Breitbach K, Klocke S, Tschernig T, van Rooijen N, Baumann U, Steinmetz I. 2006. Role of inducible nitric oxide synthase and NADPH oxidase in early control of Burkholderia pseudomallei infection in mice. Infect. Immun. 74:6300–6309. 10.1128/IAI.00966-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haque A, Easton A, Smith D, O'Garra A, Van Rooijen N, Lertmemongkolchai G, Titball RW, Bancroft GJ. 2006. Role of T cells in innate and adaptive immunity against murine Burkholderia pseudomallei infection. J. Infect. Dis. 193:370–379. 10.1086/498983 [DOI] [PubMed] [Google Scholar]
- 18.Miyagi K, Kawakami K, Saito A. 1997. Role of reactive nitrogen and oxygen intermediates in gamma interferon-stimulated murine macrophage bactericidal activity against Burkholderia pseudomallei. Infect. Immun. 65:4108–4113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tangsudjai S, Pudla M, Limposuwan K, Woods DE, Sirisinha S, Utaisincharoen P. 2010. Involvement of the MyD88-independent pathway in controlling the intracellular fate of Burkholderia pseudomallei infection in the mouse macrophage cell line RAW 264.7. Microbiol. Immunol. 54:282–290. 10.1111/j.1348-0421.2010.00205.x [DOI] [PubMed] [Google Scholar]
- 20.Charoensap J, Utaisincharoen P, Engering A, Sirisinha S. 2009. Differential intracellular fate of Burkholderia pseudomallei 844 and Burkholderia thailandensis UE5 in human monocyte-derived dendritic cells and macrophages. BMC Immunol. 10:20. 10.1186/1471-2172-10-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bast A, Schmidt IH, Brauner P, Brix B, Breitbach K, Steinmetz I. 2011. Defense mechanisms of hepatocytes against Burkholderia pseudomallei. Front. Microbiol. 2:277. 10.3389/fmicb.2011.00277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Breitbach K, Sun GW, Kohler J, Eske K, Wongprompitak P, Tan G, Liu Y, Gan YH, Steinmetz I. 2009. Caspase-1 mediates resistance in murine melioidosis. Infect. Immun. 77:1589–1595. 10.1128/IAI.01257-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Propst KL, Troyer RM, Kellihan LM, Schweizer HP, Dow SW. 2010. Immunotherapy markedly increases the effectiveness of antimicrobial therapy for treatment of Burkholderia pseudomallei infection. Antimicrob. Agents Chemother. 54:1785–1792. 10.1128/AAC.01513-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brett PJ, Deshazer D, Woods DE. 1997. Characterization of Burkholderia pseudomallei and Burkholderia pseudomallei-like strains. Epidemiol. Infect. 118:137–148. 10.1017/S095026889600739X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeShazer D, Brett PJ, Carlyon R, Woods DE. 1997. Mutagenesis of Burkholderia pseudomallei with Tn5-OT182: isolation of motility mutants and molecular characterization of the flagellin structural gene. J. Bacteriol. 179:2116–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bosio CM, Dow SW. 2005. Francisella tularensis induces aberrant activation of pulmonary dendritic cells. J. Immunol. 175:6792–6801. 10.4049/jimmunol.175.10.6792 [DOI] [PubMed] [Google Scholar]
- 27.Webb C, Bedwell C, Guth A, Avery P, Dow S. 2006. Use of flow cytometry and monochlorobimane to quantitate intracellular glutathione concentrations in feline leukocytes. Vet. Immunol. Immunopathol. 112:129–140. 10.1016/j.vetimm.2006.02.009 [DOI] [PubMed] [Google Scholar]
- 28.Slinker BK. 1998. The statistics of synergism. J. Mol. Cell. Cardiol. 30:723–731. 10.1006/jmcc.1998.0655 [DOI] [PubMed] [Google Scholar]
- 29.Meister A. 1995. Glutathione metabolism. Methods Enzymol. 251:3–7. 10.1016/0076-6879(95)51106-7 [DOI] [PubMed] [Google Scholar]
- 30.Samuni Y, Goldstein S, Dean OM, Berk M. 2013. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta 1830:4117–4129. 10.1016/j.bbagen.2013.04.016 [DOI] [PubMed] [Google Scholar]
- 31.Kerksick C, Willoughby D. 2005. The antioxidant role of glutathione and N-acetyl-cysteine supplements and exercise-induced oxidative stress. J. Int. Soc. Sports Nutr. 2:38–44. 10.1186/1550-2783-2-2-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meister A. 1988. Glutathione metabolism and its selective modification. J. Biol. Chem. 263:17205–17208 [PubMed] [Google Scholar]
- 33.Kaplowitz N, Aw TY, Ookhtens M. 1985. The regulation of hepatic glutathione. Annu. Rev. Pharmacol. Toxicol. 25:715–744. 10.1146/annurev.pa.25.040185.003435 [DOI] [PubMed] [Google Scholar]
- 34.Noctor G, Queval G, Mhamdi A, Chaouch S, Foyer CH. 2011. Glutathione. Arabidopsis Book 9:e0142. 10.1199/tab.0142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foyer CH, Noctor G. 2011. Ascorbate and glutathione: the heart of the redox hub. Plant Physiol. 155:2–18. 10.1104/pp.110.167569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forman HJ, Zhang H, Rinna A. 2009. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 30:1–12. 10.1016/j.mam.2008.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu SC. 2009. Regulation of glutathione synthesis. Mol. Aspects Med. 30:42–59. 10.1016/j.mam.2008.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang H, Forman HJ. 2012. Glutathione synthesis and its role in redox signaling. Semin. Cell Dev. Biol. 23:722–728. 10.1016/j.semcdb.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Griffith OW. 1981. Depletion of glutathione by inhibition of biosynthesis. Methods Enzymol. 77:59–63. 10.1016/S0076-6879(81)77011-3 [DOI] [PubMed] [Google Scholar]
- 40.Meister A. 1995. Glutathione biosynthesis and its inhibition. Methods Enzymol. 252:26–30. 10.1016/0076-6879(95)52005-8 [DOI] [PubMed] [Google Scholar]
- 41.Armstrong JS, Steinauer KK, Hornung B, Irish JM, Lecane P, Birrell GW, Peehl DM, Knox SJ. 2002. Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ. 9:252–263. 10.1038/sj.cdd.4400959 [DOI] [PubMed] [Google Scholar]
- 42.Ault JG, Lawrence DA. 2003. Glutathione distribution in normal and oxidatively stressed cells. Exp. Cell Res. 285:9–14. 10.1016/S0014-4827(03)00012-0 [DOI] [PubMed] [Google Scholar]
- 43.Brodie AE, Reed DJ. 1985. Buthionine sulfoximine inhibition of cystine uptake and glutathione biosynthesis in human lung carcinoma cells. Toxicol. Appl. Pharmacol. 77:381–387. 10.1016/0041-008X(85)90177-2 [DOI] [PubMed] [Google Scholar]
- 44.Stevenson D, Wokosin D, Girkin J, Grant MH. 2002. Measurement of the intracellular distribution of reduced glutathione in cultured rat hepatocytes using monochlorobimane and confocal laser scanning microscopy. Toxicol. In Vitro 16:609–619. 10.1016/S0887-2333(02)00042-5 [DOI] [PubMed] [Google Scholar]
- 45.Portnoy DA, Schreiber RD, Connelly P, Tilney LG. 1989. Gamma interferon limits access of Listeria monocytogenes to the macrophage cytoplasm. J. Exp. Med. 170:2141–2146. 10.1084/jem.170.6.2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lindgren H, Golovliov I, Baranov V, Ernst RK, Telepnev M, Sjostedt A. 2004. Factors affecting the escape of Francisella tularensis from the phagolysosome. J. Med. Microbiol. 53:953–958. 10.1099/jmm.0.45685-0 [DOI] [PubMed] [Google Scholar]
- 47.Myers JT, Tsang AW, Swanson JA. 2003. Localized reactive oxygen and nitrogen intermediates inhibit escape of Listeria monocytogenes from vacuoles in activated macrophages. J. Immunol. 171:5447–5453. 10.4049/jimmunol.171.10.5447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Breitbach K, Rottner K, Klocke S, Rohde M, Jenzora A, Wehland J, Steinmetz I. 2003. Actin-based motility of Burkholderia pseudomallei involves the Arp 2/3 complex, but not N-WASP and Ena/VASP proteins. Cell Microbiol. 5:385–393. 10.1046/j.1462-5822.2003.00277.x [DOI] [PubMed] [Google Scholar]
- 49.Dhamdhere G, Krishnamoorthy G, Zgurskaya HI. 2010. Interplay between drug efflux and antioxidants in Escherichia coli resistance to antibiotics. Antimicrob. Agents Chemother. 54:5366–5368. 10.1128/AAC.00719-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Breitbach K, Wongprompitak P, Steinmetz I. 2011. Distinct roles for nitric oxide in resistant C57BL/6 and susceptible BALB/c mice to control Burkholderia pseudomallei infection. BMC Immunol. 12:20. 10.1186/1471-2172-12-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Utaisincharoen P, Tangthawornchaikul N, Kespichayawattana W, Chaisuriya P, Sirisinha S. 2001. Burkholderia pseudomallei interferes with inducible nitric oxide synthase (iNOS) production: a possible mechanism of evading macrophage killing. Microbiol. Immunol. 45:307–313. 10.1111/j.1348-0421.2001.tb02623.x [DOI] [PubMed] [Google Scholar]
- 52.Utaisincharoen P, Anuntagool N, Arjcharoen S, Limposuwan K, Chaisuriya P, Sirisinha S. 2004. Induction of iNOS expression and antimicrobial activity by interferon (IFN)-beta is distinct from IFN-gamma in Burkholderia pseudomallei-infected mouse macrophages. Clin. Exp. Immunol. 136:277–283. 10.1111/j.1365-2249.2004.02445.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ekchariyawat P, Pudla S, Limposuwan K, Arjcharoen S, Sirisinha S, Utaisincharoen P. 2005. Burkholderia pseudomallei-induced expression of suppressor of cytokine signaling 3 and cytokine-inducible Src homology 2-containing protein in mouse macrophages: a possible mechanism for suppression of the response to gamma interferon stimulation. Infect. Immun. 73:7332–7339. 10.1128/IAI.73.11.7332-7339.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Renella R, Perez JM, Chollet-Martin S, Sarnacki S, Fischer A, Blanche S, Casanova JL, Picard C. 2006. Burkholderia pseudomallei infection in chronic granulomatous disease. Eur. J. Pediatr. 165:175–177. 10.1007/s00431-005-0022-y [DOI] [PubMed] [Google Scholar]
- 55.Tarlow MJ, Lloyd J. 1971. Melioidosis and chronic granulomatous disease. Proc. R. Soc. Med. 64:19–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.