

Abstract
Efflux pumps of the resistance nodulation division (RND) superfamily, such as AcrB, make a major contribution to multidrug resistance in Gram-negative bacteria. The development of inhibitors of the RND pumps would improve the efficacy of current and next-generation antibiotics. To date, however, only one inhibitor has been cocrystallized with AcrB. Thus, in silico structure-based analysis is essential for elucidating the interaction between other inhibitors and the efflux pumps. In this work, we used computer docking and molecular dynamics simulations to study the interaction between AcrB and the compound MBX2319, a novel pyranopyridine efflux pump inhibitor with potent activity against RND efflux pumps of Enterobacteriaceae species, as well as other known inhibitors (D13-9001, 1-[1-naphthylmethyl]-piperazine, and phenylalanylarginine-β-naphthylamide) and the binding of doxorubicin to the efflux-defective F610A variant of AcrB. We also analyzed the binding of a substrate, minocycline, for comparison. Our results show that MBX2319 binds very tightly to the lower part of the distal pocket in the B protomer of AcrB, strongly interacting with the phenylalanines lining the hydrophobic trap, where the hydrophobic portion of D13-9001 was found to bind by X-ray crystallography. Additionally, MBX2319 binds to AcrB in a manner that is similar to the way in which doxorubicin binds to the F610A variant of AcrB. In contrast, 1-(1-naphthylmethyl)-piperazine and phenylalanylarginine-β-naphthylamide appear to bind to somewhat different areas of the distal pocket in the B protomer of AcrB than does MBX2319. However, all inhibitors (except D13-9001) appear to distort the structure of the distal pocket, impairing the proper binding of substrates.
INTRODUCTION
In the past 3 decades, there has been a resurgence of bacterial resistance as a major problem in public health (1–3). Of particular concern is the appearance of multidrug-resistant (MDR) Gram-negative bacteria (1, 2), in which the efflux pumps of the resistance nodulation division (RND) family make a major contribution to the MDR phenotype (4–6). The substrates of the RND family pumps include antibiotics and biocides with very diverse structural and chemical features; however, they tend to have a significant lipophilic portion (6, 7). RND pumps are tripartite complexes (7–9). The major RND efflux system in Escherichia coli consists of a pump protein (AcrB), a channel that traverses the outer membrane (TolC), and a membrane fusion (adaptor) protein (AcrA). The RND pump AcrB, which is driven by proton motive force (10), determines the substrate specificity (2, 5, 6, 8, 11).
The structure of AcrB, a homotrimeric protein embedded in the bacterial inner membrane, has been determined in a 3-fold symmetric form (12) and later in an asymmetric conformation (13–15). In the asymmetric conformation, each protomer assumes a different conformation: access (loose or A), binding (tight or B), or extrusion (open or C). Furthermore, minocycline (MIN) and doxorubicin (DOX) were cocrystallized within a distal (deep) binding pocket (DP) located in the periplasmic domain of the binding conformer (13) (Fig. 1). On the basis of these findings, a functional rotation hypothesis was suggested for the mechanism of drug efflux, in which each protomer successively assumes one of the aforementioned conformations (13–15). This idea has been supported by several biochemical studies (16–18) and by molecular simulation (19, 20). Thus, the DP (Fig. 1) plays a major role in the binding and selection of substrates by AcrB. Indeed, site-directed mutagenesis and real-time efflux experiments have confirmed this hypothesis (21–24). The DP in the binding protomer is quite extensive (surrounded by >20 residues) and contains many hydrophobic residues, as well as several charged and polar ones (Fig. 1) (25). The diverse features of the DP are consistent with the extreme variety of substrates recognized by AcrB (8), ranging from basic dyes to most antibiotics, detergents, and even solvents (5, 26, 27). Recently, some antibiotics have been cocrystallized in a form bound to a more proximal binding pocket (also called the access pocket [AP]) in the access protomer (28, 29). This presumably represents an earlier stage in the drug efflux process and is consistent with earlier cocrystallization (30) and biochemical (31) studies. AP and DP are separated by a loop rich in glycine (G-loop) (also called F617 loop or switch loop), consisting of residues 614 through 621 (28, 29).
FIG 1.

(A) Reduced model of AcrB used in this work. The transmembrane domain (in transparent gray) was cut off from the protein, and only the periplasmic domain (residues 33 to 335 and 565 to 871 of the intact protein; shown in blue, red, and gray for access, binding, and extrusion protomer, respectively) was kept. The substrate MIN (PDB code 4DX5 [29]) is shown in spheres (violet), and the molecular surface of the residues free to move during the partially restrained MD is shown in transparent yellow. (B) View (perspective looking to the binding protomer) of the key regions of AcrB. The periplasmic and transmembrane domains of AcrB are shown in red and gray transparent ribbons, respectively, while the tip of the G-loop is shown in solid yellow. The residues lining the key regions (see table in panel C) are shown with colored spheres: orange, red, green, magenta, pink, and ice blue for the PC1/PC2 entrance cleft, distal pocket, access pocket, hydrophobic trap, vestibule, and exit gate, respectively. A channel leading from the cleft or the vestibule to the distal pocket is shown by small violet spheres. (C) Table identifying the residues shown in panel B. The residues common to the cleft and the access pocket are italicized, while those shared between the access and distal pockets are underlined. Residues belonging to the hydrophobic trap are in bold red type and colored magenta.
Efflux pump inhibitors (EPIs) (Fig. 2) are of extreme interest in antimicrobial research, as their use might, in principle, restore the efficacy of several antibiotics (32–40). Their biological activities, measured by decreases in antibiotic MICs (Table 1), suggest that the EPIs studied here (Fig. 2) affect the efflux of many antibiotics, but there are important differences in specificity that may be related to the modes of binding of EPI to DP, as is explained in the Discussion. Several EPIs from various research groups are currently under study, but their possible mechanisms of action remain largely unknown (32–38). They might compete with substrates for the DP or AP, bind to AcrB partners (e.g., to the access mouth of TolC, thus interfering with the translocation of substrates), or interfere with the assembly of the AcrB trimer (which should assemble after insertion and folding of the three monomers in the inner membrane [41]) or the tripartite AcrAB-TolC machinery.
FIG 2.

Chemical structures of the inhibitors MBX2319, D13-9001, PAβN, NMP, and of the AcrB substrates DOX and MIN.
TABLE 1.
Reduction in the MICs of several antibiotics and dyes due to the F610A mutation or to the administration of the inhibitors MBX2319, NMP, PAβN, and D13-9001a
| Inhibitormutation | Fold decrease in MICb |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OXA | NOV | CLY | ERY | LZD | EtBr | LVX | CIP | CHL | TET | PIP | NOR | NAX | CLO | CAR | AFV | RIF | |
| AcrBF610Ac | >4 | 16 | 32 | 8 | <4 | >2 | 8 | 8 | 4 | 4 | |||||||
| MBX2319 | 8d | 1d | 8d | 4d | 1d | 4d | 2d | 4d | 2d | 8d | 2d | 4d | 8d | 0.5d | 1d | ||
| NMP | 4e | 8e | 32e | 16e | 8e | 4e | 4e | ||||||||||
| 4f | 8f | 16f | 16f | 8f | 4f | 4f | |||||||||||
| ≥4 (37)g | ≥4 (7)g | ≥16 (53)g | ≥4 (85)g | ≥4 (67)g | ≥4 (8)g | ≥4 (35)g | ≥4 (31)g | ||||||||||
| PAβN | 4d | 1d | 32d | 2d | 1d | 2d | 0.5d | 4d | 1d | 0.5d | 0.5d | 8d | 2d | 0.25d | 1d | ||
| 2e | 32e | 8e | 4e | 8e | 1e | 64e | |||||||||||
| 2f | 128f | 4f | 8f | 4f | 1f | 64f | |||||||||||
| ≥4 (42)g | ≥16 (90)g | ≥16 (23)g | ≥4 (43)g | ≥4 (28)g | ≥4 (45)g | ≥16 (97)g | |||||||||||
| 128h | 2h | ||||||||||||||||
| D13-9001 | 4h | 4h | |||||||||||||||
All the E. coli strains (except AB1157) overexpressed the AcrAB-TolC efflux pump.
OXA, oxacillin; NOV, novobiocin; CLY, clarithromycin; ERY, erythromycin; LZD, linezolid; EtBr, ethidium bromide; LVX, levofloxacin; CIP, ciprofloxacin; CHL, chloramphenicol; TET, tetracycline; PIP, piperacillin; NOR, norfloxacin; NAX, nalidixic acid; CLO, cloxacillin; CAR, carbenicillin; AFV, acriflavine; RIF, rifampin.
Compared with the strain producing unaltered AcrB in E. coli K-12 strain 3-AG100 (22).
Strain E. coli AB1157; concentration of MBX2319 and PAβN, 25 μM (44).
Strain E. coli 2-DC14PS; concentrations of NMP and PAβN, 100 μM and 25 μM, respectively (59).
Strain E. coli 3-AG100MKX; concentrations of NMP and PAβN, 100 μM and 25 μM, respectively (59).
Averages of 60 clinical isolates of E. coli; concentrations of NMP and PAβN, 100 μM and 25 μM, respectively (67). Values in parentheses are the percentages of strains showing reduction in MICs.
Strain E. coli MG1655; concentration of D13-9001 and PAβN, 32 μM (66).
The first (and so far the only) structure of an inhibitor cocrystallized with an RND efflux pump is that of D13-9001 bound to AcrB of E. coli and MexB of Pseudomonas aeruginosa, which was released very recently (42). The inhibitor binds to the DP of the two transporters, and Nakashima et al. (42) suggested that it competitively inhibits the functional rearrangements of the efflux pumps at work, sterically preventing the binding and transport of typical substrates.
As stressed in a previous investigation of the binding of a dozen compounds to AcrB (25), the availability of a single experimental structure, although constituting a milestone in the field, does not allow a comparison with other inhibitors or the identification of a general mechanism of pump inactivation. Thus, we have utilized computational methods, such as docking and all-atom molecular dynamics (MD) simulations, to predict the molecular details of the interaction between other EPIs and RND transporters (25, 43), including the binding of 1-(1-naphthylmethyl)-piperazine (NMP) and phenylalanylarginine-β-naphthylamide (PAβN) to the DP of AcrB. In this work, we extend this approach in order to study a novel synthetic inhibitor (MBX2319) (Fig. 2), which is a potent inhibitor of AcrB and some of its orthologs (44). We performed MD simulations on the complexes of AcrB with MBX2319, the substrate MIN, and the inhibitor D13-9001, for which experimental structural data are available (13, 28, 29, 42). Finally, we discovered that the predicted binding mode of MBX2319 was very similar to that of DOX in the F610A variant of AcrB (AcrBF610A) (22, 45, 46), in which DOX export is severely impaired in spite of its predicted strong binding to the DP. Our results pinpoint the similarities and differences in the binding of the four inhibitors and suggest a significant concordance of the mechanism of inhibition by MBX2319 with that suggested for D13-9001, as well as with that inducing a reduced efflux of substrates in the F610A variant of AcrB.
MATERIALS AND METHODS
The structures of the bimolecular adducts between the ligands and AcrB were taken from previous works (25, 42) or obtained by docking using AutoDock Vina (47). A grid of 30 by 30 by 30 Å centered in the middle of the DP was used. The MD simulations were performed using the program NAMD 2.9 (48), starting from the best pose obtained from docking. Further details about the protocols for docking, MD simulation, and postprocessing, as well as about the analysis of the data, are described in the supplemental material.
RESULTS
We organized the results as follows. First we describe, in brief, the main approximations and limitations of our approach. Next, we discuss the main features of the binding of MBX2319 to the DP of AcrB and compare them with the binding of the other inhibitors and the substrate MIN (13, 25, 29), as well as with that of DOX in the F610A variant of AcrB (22, 45, 46). Finally, using MIN as a probe in docking and MD simulations, we describe how the inhibitors might affect the binding of substrates to the DP. The choice of the systems made here allows for a solid comparison of the molecular mechanisms of inhibition by different molecules. Table S1 in the supplemental material contains a summary of the simulations performed in this work. Selected properties of all the compounds are reported in Table S2 in the supplemental material.
Limitations of our approach.
In comparison with docking, all-atom MD simulations have the main advantage of intrinsically describing the dynamics of the system under study and the role of solvents. However, with the inclusion of these additional degrees of freedom, the number of configurations that should be examined becomes huge, so that insufficient sampling might become an issue (49). In addition, one has to consider the inaccuracies in the model. For instance, inhibitors of AcrB contain weakly acidic or basic groups whose charge states might be different from the standard one (in aqueous solvent) in the low dielectric constant environment of the binding site; this is not taken into account in our MD simulation protocol. Similarly, a quantitative comparison among the affinities of the various compounds is compromised by the intrinsic limitations of the molecular mechanics/generalized Born surface area (MM/GBSA) methodology used to estimate binding free energies (see, e.g., 50) and by the quite large standard deviations accompanying the calculated values of ΔGb, the total free energy of binding (Table 2).
TABLE 2.
Free energy of binding, H-bonds, and surface matching coefficients for the inhibitors MBX2319, NMP, PAβN, and D13-9001, the substrate MIN, all bound to wild-type AcrB, and the substrate DOX bound to the F610A variant of AcrB
| Compound | Calculated binding free energy |
H-bond interactions and surface matchingb |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔGb (kcal/mol)a |
Contribution to ΔGsolv (%) |
||||||||||
| DPc |
G-loop | Interface | Cleft | ||||||||
| ΔGb | ΔGsolv | TΔSconf | Total | Trapf | H-bonds | SMTotd | SMLd | ||||
| MBX2319 | −12.5 ± 6.6 | −37.7 ± 2.8 | −25.2 ± 3.8 | 31 | 21 (68) | 0 | 2 | 4 | 0 (0) | 0.77 (0.77) | 0.87 (0.87) |
| NMP | −10.6 ± 7.9 | −26.9 ± 3.9 | −16.3 ± 4.0 | 26 | 18 (69) | 4 | 1 | 0 | 0 (0) | 0.82 (0.61) | 0.91 (0.67) |
| PAβN | −13.4 ± 10.8 | −40.8 ± 5.8 | −27.4 ± 5.0 | 32 | 17 (53) | 2 | 0 | 0 | 5.4 (0) | 0.71 (0.44) | 0.81 (0.63) |
| D13-9001 | −18.2 ± 12.3 | −48.8 ± 4.8 | −30.6 ± 7.5 | 49 | 30 (61) | 1 | 1 | 1 | 5.9 (0) | 0.74 (0.71) | 0.88 (0.91) |
| MIN | −7.2 ± 7.7 | −29.3 ± 4.7 | −22.1 ± 3.0 | 41 | 12 (29) | 0 | 0 | 0 | 0 (1.2) | 0.65 (0.62) | 0.82 (0.85) |
| DOXF610A | NCe | −30.0 ± 4.5 | NC | 54 | 28 (52) | 3 (NC) | 1 (NC) | 0 | 1.1 (NC) | 0.71 (NC) | 0.84 (NC) |
The total free energy of binding (ΔGb) is the sum of the contribution calculated with the MM/GBSA method (ΔGsolv) and of the conformational entropy of the solute (TΔSconf). The contributions to ΔGb from residues belonging to selected regions are also reported. In the case of the hydrophobic trap, embedded in the DP, the weight of the contribution from the trap relative to the whole pocket is reported in parentheses.
Calculated on the conformation of the complex with the lower RMSD from the average extracted from the unbiased MD simulations. SMTot and SML refer to the total and lipophilic surface matching coefficients, respectively. See the supplemental material for further details.
The residues within the various regions are listed in Fig. 1.
Values in parentheses are those for the starting structures.
NC, not calculated.
Values in parentheses are the percentages of the binding energy contributed by the hydrophobic trap in relation to that contributed by the entire DP.
Finally, our system may be incomplete because other accessory proteins, such as AcrA (8) and AcrZ (51, 52), may be needed for full activity and thus for the active conformation of AcrB. However, it is likely that the conformation of the complexes of AcrB with substrates and inhibitors is only marginally affected by the presence of accessory proteins.
Binding of MBX2319 to the DP of AcrB.
The docking of MBX2319 to AcrB was performed using the AutoDock Vina (47) package, as described previously (25). The binding of MBX2319 to AcrB was further characterized through extensive MD simulations of >300 ns in length (see Table S1 in the supplemental material), starting from the best docking pose and using a reduced model of the protein lacking the transmembrane domain (Fig. 1). This model of AcrB was previously validated (25).
The root mean square deviation (RMSD) of the complex, calculated with respect to the initial conformation obtained by docking, became stable after ∼90 ns of MD simulation (see Fig. S1 in the supplemental material). Moreover, after a few nanoseconds, the inhibitor moved away from the initial pose toward the AP (Fig. 3) (the displacement of the center of mass was ∼8 Å) and bound stably in a new position for the last 250 ns of the simulation (see Fig. S1). In this position, MBX2319 binds with high affinity at a site that is close to the interface between DP and AP (Table 2) and appears to be in contact with four out of the five phenylalanine residues lining the hydrophobic trap (Fig. 4A) (F136, F178, F610, F615, and F628; Fig. 1). Note that this trap, defined by Nakashima et al. (42) for the inhibitor D13-9001 bound to the B protomer of AcrB (Fig. 3), is actually a lower part of the DP identified previously (13, 31). The pyridine ring of MBX2319 stacks with the ring of F628 on one side and interacts with F136 on the other, while the morpholine ring is close to Y327, and the phenylethylthio group is loosely located in a hydrophobic pocket surrounded by F610, F628, P326, and Y327 (Fig. 4C). Finally, the upper part of the DP, closer to the exit gate, shrinks instead of making a deep cleft as in the ligand-free or MIN-bound B protomer, and this phenomenon is discussed below. The high affinity of MBX2319 for AcrB (calculated ΔGb, −12.5 kcal/mol; Table 2) is supported by the large value of the matching between the hydrophobic surfaces of inhibitor and protein (53) (Table 2, surface matching). This is consistent with the nature of key residues stabilizing the inhibitor in its pocket, which are almost exclusively aromatic or aliphatic (Fig. 5; see also Table S3 in the supplemental material). In accordance with the results reported by Vargiu and Nikaido (25) and confirmed here for NMP, PAβN, and MBX2319, the residues of the DP outside the hydrophobic trap do not contribute significantly to the solvation free energy (ΔGsolv) (Table 2). Indeed, in the case of MBX2319, almost 70% of the ΔGsolv comes from residues belonging to the hydrophobic trap (see Fig. 4A). According to Nakashima et al. (42), this trap is not in the main part of the substrate extrusion channel leading from the PC1/PC2 cleft (or from the vestibule [15, 54]) to the gate of the TolC docking domain (Fig. 1B). Interestingly, the binding of MBX2319 to AcrB does not cause a significant perturbation in the overall morphology of the substrate extrusion channel (Fig. 4A, F, and H; see also Fig. S3 in the supplemental material), except for a reduction of its radius at the AP with respect to the value in the transporter free of ligands (Fig. 4H).
FIG 3.

(A) Representative position of MBX2319 (in thick sticks colored according to the atom type: red, oxygen; yellow, sulfur; dark blue, nitrogen; cyan, carbon; nonpolar hydrogens are removed) in the equilibrium phase of the MD simulation, compared to the initial pose from docking (thinner gray sticks). The DP (transparent red surface) and AP (green surface), the cleft (orange surface), and the tip of the G-loop (gray cartoon) are also shown. The residues that are within 3.5 Å from the ligand are shown as beads (red, green, orange, and yellow for those of distal and access pockets, cleft, and G-loop, respectively). Those shared by the two pockets are colored blue, while the bigger magenta beads represent residues F136, F178, F610, and F628 of the hydrophobic trap. The residues of the exit gate (far from the ligand) are shown as gray beads. (B) Closer view of interactions realized by the compound with residues within 3.5 Å in the docking geometry. (C) Closer view of interactions realized by the compound with residues within 3.5 Å in the final phase of the MD (also, the initial docking pose and conformation of the G-loop are shown in gray sticks and cartoon, respectively).
FIG 4.

(A to E) Positions of the various ligands used in this study with respect to the hydrophobic trap in protomer B of AcrB, as found in representative average structures of the complexes from MD simulations. The ligands are shown in thick sticks colored according to the atom type (red, oxygen; yellow, sulfur; dark blue, nitrogen; cyan, carbon; white, hydrogen), and the side chains of the residues constituting the hydrophobic trap are shown with sticks (thick if the residue is within 3.5 Å of the ligand, thinner otherwise). The rest of the protein is shown with the molecular surface colored in orange, yellow, and ice blue at the PC1/PC2 cleft, the G-loop tip, and the exit gate, respectively, and white elsewhere. The channels leading to the proximity of the exit gate (see Fig. 1) and passing through the residues of the DP are also shown in the presence and absence of the ligand, with blue and green transparent surfaces, respectively, while the centers of gravity of the points defining them are shown with points. The centers of mass of the AP and DP are shown with green and red transparent spheres, respectively. No contiguous substrate translocation channel was found in the AcrB-PAβN complex (and also in AcrBF610A-DOX [see Fig. S2 in the supplemental material]). (A to E) MBX2319 (A), D13-9001 (B), NMP (C), PAβN (D), and MIN(E). (F) Channel found in AcrB free of ligands, again as found in the representative average structure of the transporter. (G) Superposition among the channels in panels A (black), B (red), C (green), E (magenta), and F (yellow). C-α atoms of the cleft, exit gate, and distal and access pockets are shown with orange, ice blue, red, and green beads, respectively. (H) Profile radii of the channels calculated in the presence of the ligands and shown in panel G. The positions of DP, AP, cleft, and gate on the x axis are approximate.
FIG 5.
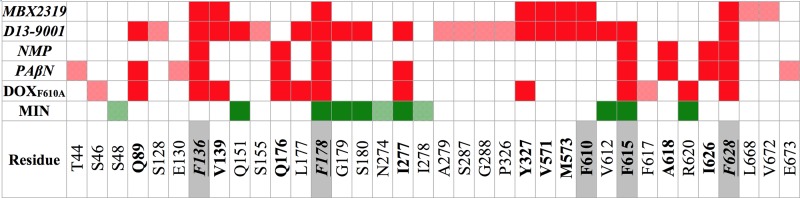
Comparison of per-residue contributions to ΔGb for the inhibitors MBX2319, NMP, PAβN, and D13-9001, the substrate MIN, and the substrate DOX in the F610A variant of AcrB. Residues contributing more than kT (where k is the Boltzmann constant and T is the absolute temperature) at room temperature (0.593 kcal/mol) for a compound are indicated by colored cells (red, inhibitors and DOX; green, MIN). Cells with semitransparent colors and gray labels identify residues contributing to ΔGb for one compound only, while cells with solid colors identify residues contributing to ΔGb for at least two molecules. The residues contributing to the binding of two or more inhibitors are in bold type, and those contributing to binding of all inhibitors are also italicized. The residues belonging to the hydrophobic trap, as defined by Nakashima et al. (42), are underlined, and the corresponding row is shaded gray.
Comparison of the binding of MBX2319 with that of NMP, PAβN, D13-9001, MIN, and DOX in AcrBF610A.
In order to consistently compare the behavior of MBX2319 with that of NMP and PAβN (25), we extended the MD simulations of those compounds within the DP of AcrB to >300 ns (see Table S1 in the supplemental material). In addition, we performed an equally long simulation of the complex between AcrB and the inhibitor D13-9001 (42). Finally, we prolonged the simulation of the AcrB-MIN complex of Vargiu and Nikaido (25) to 300 ns. As a reference, we used both the available high-resolution X-ray structures of AcrB (15, 29) and a previous 100-ns MD trajectory of the protein free of ligands (25). Before discussing these results further, we would like to point out that our computational protocol is able to reproduce very well the binding of MIN and D13-9001 to AcrB, as seen in the available crystallographic structures (RMSD, 2.7 ± 1.0 Å and 2.8 ± 0.4 Å from the compounds in the X-ray structures 4DX5 [29] and 3W9H [42], respectively (see Fig. S1 in the supplemental material).
First, we compared the binding position of MBX2319 with that of the other inhibitors, as well as with that of DOX in AcrBF610A (Fig. 6). The largest overlap is seen between MBX2319 and the region of D13-9001 containing the pyridopyrimidine rings and the t-butylthiazole moiety (Fig. 6A) that occupy the hydrophobic trap in the X-ray structure (42) and in our simulation. According to our data, the conformation of the G-loop is very similar in these two systems, as well as for the AcrBF610A-DOX complex (Fig. 6D). The binding sites of NMP and PAβN partially overlap with that of MBX2319 within the pocket of protomer B, but to a lesser extent than with D13-9001. Note that only NMP and PAβN straddle the tip of the G-loop (Fig. 4B and C).
FIG 6.
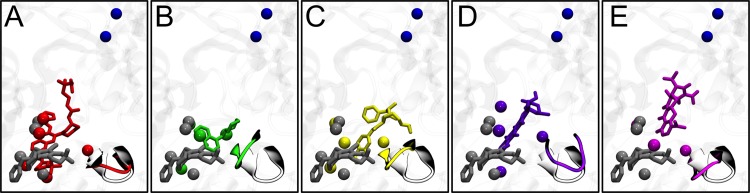
Superposition among representative structures of AcrB in complex with MBX2319 (gray sticks in all panels), D13-9001 (A) (red), NMP (B) (green), PAβN (C) (yellow), DOX in complex with AcrBF610A (D) (violet), and MIN (E) (magenta). The G-loop tip is shown in ribbons (gray in free AcrB and colored like the ligands in complexes), while the residues of the hydrophobic trap within 3.5 Å of the ligands are shown by solid spheres. The two residues of the gate to the TolC docking domain (Q124 and Y758) are shown by blue spheres.
While MBX2319 is almost totally packed by the residues lining the hydrophobic trap, a large portion of D13-9001 extends into the channel (42) that in the absence of ligands leads from the PC1/PC2 cleft to the gate of the TolC docking domain passing through the AP and DP (Fig. 4A and B). This behavior of D13-9001 is similar to that of DOX in AcrBF610A (see Fig. S2 in the supplemental material), as evidenced by the good superposition of these two molecules in the complexes they form with AcrB (see Fig. S4 in the supplemental material). However, in AcrBF610A, DOX extends out of the trap much closer to the cleft than does D13-9001 (the occluding arm of which is in the upper region of the DP), and no functional channel was found at all. Namely, no contiguous substrate translocation channels starting from the PC1/PC2 cleft and leading to the gate through the AP and DP were found. Several other nonfunctional channels, leading to other regions of the protein or to the cleft through other routes, were found in all systems.
NMP and PAβN affect to a larger extent the morphology of the substrate extrusion channel, with PAβN completely occluding the passageway (Fig. 4G and H). The position of the bottleneck in AcrB-NMP, which is closer to the AP than the other inhibitors, and the total occlusion caused by PAβN might be explained by considering that: (i) these compounds bind to the hydrophobic trap more peripherally than does MBX2319 (see also in Table 2 the lower contributions to ΔGb from this region for these two inhibitors compared to the others), and (ii) they significantly drag (and thus might prevent the movement of) the tip of the G-loop (Fig. 6), which is suggested to act as a gate for the passage of substrates from the AP to the DP along the A-to-B transition of the functional rotation (28, 29). MBX2319 is unique in this respect among the inhibitors considered here, as it does not significantly affect the morphology of the substrate extrusion channel when bound to AcrB (Fig. 4H). Importantly, the features of the channels described above were retained over the MD trajectories of all the systems studied here (see Fig. S3 in the supplemental material).
To compare the binding affinities of the EPIs and MIN for AcrB, we estimated their free energies of binding (ΔGb) from our MD simulations using the MM/GBSA approach (25, 55–58) (Table 2). It is striking to notice (despite the limitations outlined in “Limitations of our approach” above; note that we did not estimate the entropy contribution to ΔGb for the AcrBF610A-DOX system, as we have not extended the simulation of Vargiu et al. [46] to an acceptable length [25]) that all the inhibitors have binding free energies in the range of −10 to −18 kcal/mol, with only the noninhibitor MIN featuring a smaller value of about −7 kcal/mol. Based on the ΔGb values, D13-9001 is predicted to bind with the highest affinity; however, MBX2319 and DOX in AcrBF610A are also predicted to bind with high affinity. NMP and PAβN exhibited slightly lower ΔGb values. The finding is consistent with the differences described above regarding the structure and dynamics of the inhibitors versus those of MIN.
The contributions from the residues of the hydrophobic trap, PC1/PC2 cleft, AP/DP interface, and G-loop were all negligible only for MIN (Table 2). Previously, we highlighted how the contributions to ΔGb from the residues of DP were approximately >40% for the substrates of AcrB only, while the inhibitors NMP and PAβN had values of <30% (25). This result is confirmed here for MBX2319 but not for D13-9001, which features a contribution to ΔGb from residues of the DP that are much higher than the value of ∼30% found for the other inhibitors and even higher than that found for MIN (Table 2). It should be noted that since this inhibitor is significantly bigger than all the others considered here (see Table S1 in the supplemental material), it is perhaps not surprising that the surface contact with the DP is the largest. Moreover, if one focuses on the contributions from the residues of the hydrophobic trap, it clearly appears that only for the inhibitors and DOX in the F610A variant are the values >50% of the total contribution from the DP, and they drop to <30% for MIN. As expected, NMP and PAβN feature the highest contributions from the residues of the G-loop.
These results are consistent with the binding positions of MBX2319 and D13-9001, which are as tightly packed by the hydrophobic trap as is DOX in the F610A variant of AcrB (Fig. 4A to E; see also Fig. S2 and S4 in the supplemental material). As discussed before, NMP and PAβN are only partially bound within the hydrophobic trap and basically are found between this subpocket and the tip of the G-loop, which they straddle significantly compared to MBX2319 and D13-9001 (Fig. 6). Concerning MIN, this substrate binds to the upper portion of the DP, almost totally outside the hydrophobic trap (Fig. 4E), and it does not contact the tip of the G-loop (Fig. 6E). Consistently, the contributions to the ΔGb from the residues of the trap and the G-loop are the lowest for MIN (Table 2).
The similarity between the binding of MBX2319 and that of D13-9001 and DOX in AcrBF610A is strengthened by the observation of the large overlap between the sets of residues contributing to the stability of the complexes (Fig. 5). Out of 10 residues contributing to the binding of MBX2319, eight and six residues also contribute to the stabilization of D13-9001 and DOX, respectively (Fig. 5; see also Table S3 in the supplemental material). This number is reduced to three for NMP and PAβN and to only one (F178) for MIN. Furthermore, half of the residues shared with D13-9001 and DOX and all those shared with NMP and PAβN belong to the hydrophobic trap. More in detail, four out of the five residues lining the hydrophobic trap contribute significantly to the binding of each inhibitor and DOX, with F136, F178, and F628 contributing to the binding of all of them (Fig. 5; see also Table S3).
Docking of MIN to AcrB-inhibitor complexes.
To investigate how the structural deformations of DP induced by inhibitors might affect the binding of substrates, MIN was docked to the DP of the B protomer using the representative average structures of AcrB-inhibitor complexes obtained from the final equilibrium phases of the MD simulations. As a test, we first docked MIN to the AcrB-MIN complex after removal of the ligand; MIN was found to bind to virtually the same position previously occupied in the MD-simulation complex as well as in the cocrystal structure (Fig. 7A). However, when MIN was docked on the AcrB-inhibitor complexes, in all cases, it bound to an area outside the DP proper (Fig. 7B to E) in a manner that was similar to the manner in which the nonsubstrates glucose and kanamycin A were predicted to bind to AcrB in our previous MD simulation study (25). These results were confirmed by docking MIN on the representatives of the top 5 structural clusters extracted from the equilibrium trajectories of each of the AcrB-inhibitor systems. Moreover, the conformations of the complexes were stable after four consecutive rounds of structural optimization (see supplemental material for details). This failure to bind to DP might explain, in principle, how the inhibitor might work. In examining the structure of the DP in various complexes (Fig. 7), one is struck by the observation that MBX2319, NMP, and PAβN cause a conformational change that closes the upper part of the DP, i.e., the portion that appears as a deep crevice that binds many substrates, including MIN (13, 20, 25, 29, 43). The distances between C-γ of F178 on the left wall of this crevice and C-β of I277 on the right wall, which was 8.5 ± 0.5 Å in the MIN-AcrB complex, were found to decrease to 6.6 ± 0.5 Å for MBX, 6.6 ± 0.5 Å for NMP, and 6.6 ± 0.4 Å for PAβN in the inhibitor complexes. This reduction is enough to close the crevice in the DP and possibly prevents the DP binding of MIN (Fig. 7). This distance does not decrease in the D13-9001 complex (10.5 ± 0.5 Å), but MIN presumably cannot bind because the crevice is occluded by the long tail of the inhibitor containing both positive and negative charges. Similarly, in the DOXF610A mutant complex, the distance does not change (8.5 ± 0.3 Å), but DOX again prevents the binding of a second substrate by steric hindrance.
FIG 7.

Top docking poses of MIN on representative structures AcrB extracted from the AcrB-MIN MD trajectory (A) and AcrB-inhibitor complex extracted from the corresponding MD trajectories (B to E). The docked MIN and the inhibitors are shown as sticks colored according to the atom type (white carbons, MIN; yellow carbons, inhibitors). The green sticks represent the molecule of MIN as found in the highest resolution X-ray structure available to date (29). The residues of the hydrophobic trap, distal pocket, and PC1/PC2 entrance cleft are shown with magenta, red, and orange spheres, respectively. Residues F136, V139, F178, I277, A279, E280, P285, Y327, F610, V612, F615, F617, I626, and F628 are rendered with semitransparent surfaces colored according to the atom type (white carbons). The rest of the protein is shown in gray ribbons.
DISCUSSION
In this work, we compared the binding features of four different inhibitors, including the novel potent inhibitor MBX2319, as well as two substrates of the RND efflux pump AcrB of E. coli, by means of computational modeling. The inhibition of this pump and its homologs is highly desirable in view of combined therapies with antibiotics (6, 32–35, 44, 59–62). Since AcrB is able to expel perhaps the largest variety of substrates among the transporters of the RND superfamily, the importance of understanding the molecular mechanism behind the inhibition and the differences between inhibitors and substrates with atomic-level detail cannot be overemphasized (3, 5, 6, 63). In recent years, we have performed several investigations addressing the functional mechanisms of AcrB and MexB, the homologue of AcrB in P. aeruginosa, as well as the reasons for impaired efflux by selected mutations and inhibitors (see references 20, 25, 46, 58, 64, and 65; reviewed in reference 6), and in 2013, the first X-ray structure of AcrB in complex with the inhibitor D13-9001 was released (42). However, to our knowledge, a thorough and consistent comparison of the binding features of several inhibitors has never been published. The importance of such a study can be understood simply by examining the molecular properties of the various inhibitors considered here, which are diverse in terms of molecular weight, flexibility, charge, H-bond patterns, and octanol-water partition coefficients (see Table S1 in the supplemental material).
The first notable result of our study is that all inhibitors competed very efficiently with MIN (and likely with other substrates) in binding to AcrB. Indeed, although the standard deviations accompanying the ΔGb calculations preclude a quantitative discussion of affinity, all inhibitors were found to have higher affinities for the DP of protomer B than did MIN (Table 2), corresponding to dissociation constants well below the μM range. Moreover, all but D13-9001 caused a closure of the crevice where MIN has been found to bind (by crystallography and by MD simulations [13, 25, 29, 43]). Thus, this (competitive) binding of the inhibitors might alter the properties of the MIN binding site, reducing the affinity of this and other substrates to the DP of AcrB. D13-9001 extends from the hydrophobic trap to the MIN binding crevice, which is thus open in the AcrB-D13-9001 complex. Nonetheless, the much higher affinity of this inhibitor than that of MIN to the DP points to a very efficient competitive inhibition mechanism. Irrespective of the details, the net effect is that the binding of MIN to the DP becomes impossible (Fig. 7). In the simplest interpretation, this might explain how all these EPIs work. However, the above hypothesis is likely an oversimplification. This is suggested by the close analysis of the biological activities of these compounds. Table 1 shows that out of 9 compounds for which the reduction in the MIC was evaluated for MBX2319 and in the F610A variant of AcrB, 7 (oxacillin [OXA], erythromycin [ERY], linezolid [LZD], ethidium bromide [EtBr], levofloxacin [LVX], chloramphenicol [CHL], and tetracycline [TET]) showed comparable values. Similarly, ERY and ciprofloxacin (CIP), the only two compounds for which data are available for both MBX2319 and D13-9001 (44, 66), show approximately the same decreases in MIC. Although these comparisons might be flawed by the fact that different E. coli strains were used in other studies (22, 44, 66), the correlation between the MICs and the structural features of MBX2319, D13-9001, and DOX binding seems remarkable. Thus, it seems possible that these inhibitors and the F610A mutation might also block or retard further conformational changes in the pump, as was proposed earlier for the mutant (46). In this respect, it is interesting to notice that MBX2319 is almost completely “hidden” within the hydrophobic trap of protomer B. As this region undergoes large conformational changes during the functional rotation of AcrB, the binding of MBX2319 with high affinity might hinder these rearrangements (or those triggering the translocation of protons from the periplasm to the cytoplasm upon substrate binding). Indeed, the binding of MBX2319 resembles that of DOX in AcrBF610A (Fig. 6; see also Fig. S2 and S4 and Table S3 in the supplemental material).
In contrast to D13-9001 and DOX in AcrBF610A, the binding features of PAβN and NMP overlap to a lesser extent with those of MBX2319. In particular, these compounds feature a weaker interaction with the hydrophobic trap (Fig. 4 and 5; see also Table S3 in the supplemental material), partly compensated by a small stabilization from the tip of the G-loop (Table 2), which PAβN and NMP straddle significantly with respect to the conformation in AcrB free of ligands (Fig. 6). Perhaps these different binding positions of MBX2319 and PAβN are consistent with the different spectra of compounds potentiated by the two inhibitors (44). Indeed, ERY, piperacillin (PIP), norfloxacin (NOR), and cloxacillin (CLO) behaved in a markedly different manner in a study carried out in the same laboratory with the same strain of E. coli (44) (Table 1). However, as pointed out by Opperman et al. (44), additional factors, such as the increase of the outer membrane permeability by PAβN (61), might also be the reason for this difference. This argument is weakened in a comparison of the activities of MBX2319 and NMP, as these compounds are not suspected to alter the permeability of the outer membrane. In this case, two drugs (LZD and LVX) behaved very differently among the five compounds that can be compared (Table 1).
In summary, the observations above suggest that the hypothesis based on the closure of the DP can be only the first approximation, and there are probably subtle differences in the ways various EPIs inhibit AcrB function. Ideally, our conclusions based on simulations should be confirmed by experimental results, such as the isolation of inhibitor-insensitive mutants, or the chemical modification of residues suspected in inhibitor binding. Unfortunately, efforts to isolate mutants resistant to MBX2319 have not been successful.
Although we have considered only MIN among the substrates of AcrB, our main findings are confirmed by the analysis of the complex between the wild-type AcrB and DOX, the only other substrate of the pump for which binding to the DP of the B protomer has been established by X-ray crystallography (29). Like MIN, DOX is also not buried within the hydrophobic trap, being in contact (within 4 Å) with residues F178 and F615 only (see Fig. S5 in the supplemental material) and thus susceptible to the same impairment in binding by the inhibitors considered here.
In summary, all the inhibitors appear to hinder the binding of substrates to the upper part of the DP, where MIN, a good substrate of AcrB, is bound, by reducing the space available or by blocking access to this region (Fig. 6 and 7), although there are likely to be small differences in the way each EPI acts. Obviously, it is worth pointing out that our discussion has been focused on the binding of substrates and inhibitors to the DP of AcrB, while other binding spots not considered here (for instance, the AP in protomer A) or other factors (such as binding to protein partners) might play a relevant role in the inhibition of the transporter.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported in part by grants (AI-009644 and AI-100332) from the Public Health Service.
We thank the staff of the Lawrencium cluster at the Lawrence Berkeley National Laboratory for their help in running simulations, as well as for computer time from CINECA (ISCRA-A grant HP10ARSZ26).
We declare no conflicts of interest.
Footnotes
Published ahead of print 11 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03283-14.
REFERENCES
- 1.Tam VH, Rogers CA, Chang KT, Weston JS, Caeiro JP, Garey KW. 2010. Impact of multidrug-resistant Pseudomonas aeruginosa bacteremia on patient outcomes. Antimicrob. Agents Chemother. 54:3717–3722. 10.1128/AAC.00207-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nikaido H. 2009. Multidrug resistance in bacteria. Annu. Rev. Biochem. 78:119–146. 10.1146/annurev.biochem.78.082907.145923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wong K, Ma J, Rothnie A, Biggin PC, Kerr ID. 2014. Towards understanding promiscuity in multidrug efflux pumps. Trends Biochem. Sci. 39:8–16. 10.1016/j.tibs.2013.11.002 [DOI] [PubMed] [Google Scholar]
- 4.Li XZ, Nikaido H. 2009. Efflux-mediated drug resistance in bacteria: an update. Drugs 69:1555–1623. 10.2165/11317030-000000000-00000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nikaido H, Pagès JM. 2012. Broad-specificity efflux pumps and their role in multidrug resistance of Gram-negative bacteria. FEMS Microbiol. Rev. 36:340–363. 10.1111/j.1574-6976.2011.00290.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruggerone P, Murakami S, Pos KM, Vargiu AV. 2013. RND efflux pumps: structural information translated into function and inhibition mechanisms. Curr. Top. Med. Chem. 13:3079–3100. 10.2174/15680266113136660220 [DOI] [PubMed] [Google Scholar]
- 7.Nikaido H. 1996. Multidrug efflux pumps of Gram-negative bacteria. J. Bacteriol. 178:5853–5859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nikaido H. 2011. Structure and mechanism of RND-type multidrug efflux pumps. Adv. Enzymol. Relat. Areas Mol. Biol. 77:1–60. 10.1002/9780470920541.ch1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blair JM, Piddock LJ. 2009. Structure, function and inhibition of RND efflux pumps in Gram-negative bacteria: an update. Curr. Opin. Microbiol. 12:512–519. 10.1016/j.mib.2009.07.003 [DOI] [PubMed] [Google Scholar]
- 10.Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. 1993. Molecular cloning and characterization of acrA and acrE genes of Escherichia coli. J. Bacteriol. 175:6299–6313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nikaido H, Takatsuka Y. 2009. Mechanisms of RND multidrug efflux pumps. Biochim. Biophys. Acta 1794:769–781. 10.1016/j.bbapap.2008.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murakami S, Nakashima R, Yamashita E, Yamaguchi A. 2002. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 419:587–593. 10.1038/nature01050 [DOI] [PubMed] [Google Scholar]
- 13.Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. 2006. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 443:173–179. 10.1038/nature05076 [DOI] [PubMed] [Google Scholar]
- 14.Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Pos KM. 2006. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science 313:1295–1298. 10.1126/science.1131542 [DOI] [PubMed] [Google Scholar]
- 15.Sennhauser G, Amstutz P, Briand C, Storchenegger O, Grütter MG. 2007. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol. 5:e7. 10.1371/journal.pbio.0050007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takatsuka Y, Nikaido H. 2007. Site-directed disulfide cross-linking shows that cleft flexibility in the periplasmic domain is needed for the multidrug efflux pump AcrB of Eschetichia coli. J. Bacteriol. 189:8677–8684. 10.1128/JB.01127-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takatsuka Y, Nikaido H. 2009. Covalently linked trimer of the AcrB multidrug efflux pump provides support for the functional rotating mechanism. J. Bacteriol. 191:1729–1737. 10.1128/JB.01441-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seeger MA, von Ballmoos C, Eicher T, Brandstätter L, Verrey F, Diederichs K, Pos KM. 2008. Engineered disulfide bonds support the functional rotation mechanism of multidrug efflux pump AcrB. Nat. Struct. Mol. Biol. 15:199–205. 10.1038/nsmb.1379 [DOI] [PubMed] [Google Scholar]
- 19.Yao XQ, Kenzaki H, Murakami S, Takada S. 2010. Drug export and allosteric coupling in a multidrug transporter revealed by molecular simulations. Nat. Commun. 1:117. 10.1038/ncomms1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schulz R, Vargiu AV, Collu F, Kleinekathöfer U, Ruggerone P. 2010. Functional rotation of the transporter AcrB: insights into drug extrusion from simulations. PLoS Comput. Biol. 6:e1000806. 10.1371/journal.pcbi.1000806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bohnert JA, Schuster S, Fähnrich E, Trittler R, Kern WV. 2007. Altered spectrum of multidrug resistance associated with a single point mutation in the Escherichia coli RND-type MDR efflux pump YhiV (MdtF). J. Antimicrob. Chemother. 59:1216–1222. 10.1093/jac/dkl426 [DOI] [PubMed] [Google Scholar]
- 22.Bohnert JA, Schuster S, Seeger MA, Fähnrich E, Pos KM, Kern WV. 2008. Site-directed mutagenesis reveals putative substrate binding residues in the Escherichia coli RND efflux pump AcrB. J. Bacteriol. 190:8225–8229. 10.1128/JB.00912-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bohnert JA, Karamian B, Nikaido H. 2010. Optimized Nile Red efflux assay of AcrAB-TolC multidrug efflux system shows competition between substrates. Antimicrob. Agents Chemother. 54:3770–3775. 10.1128/AAC.00620-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wehmeier C, Schuster S, Fähnrich E, Kern WV, Bohnert JA. 2009. Site-directed mutagenesis reveals amino acid residues in the Escherichia coli RND efflux pump AcrB that confer macrolide resistance. Antimicrob. Agents Chemother. 53:329–330. 10.1128/AAC.00921-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vargiu AV, Nikaido H. 2012. Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proc. Natl. Acad. Sci. U. S. A. 109:20637–20642. 10.1073/pnas.1218348109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsukagoshi N, Aono R. 2000. Entry into and release of solvents by Escherichia coli in an organic-aqueous two-liquid-phase system and substrate specificity of the AcrAB-TolC solvent-extruding pump. J. Bacteriol. 182:4803–4810. 10.1128/JB.182.17.4803-4810.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.White DG, Goldman JD, Demple B, Levy SB. 1997. Role of the acrAB locus in organic solvent tolerance mediated by expression of marA, soxS, or robA in Escherichia coli. J. Bacteriol. 179:6122–6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakashima R, Sakurai K, Yamasaki S, Nishino K, Yamaguchi A. 2011. Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature 480:565–569. 10.1038/nature10641 [DOI] [PubMed] [Google Scholar]
- 29.Eicher T, Cha HJ, Seeger MA, Brandstätter L, El-Delik J, Bohnert JA, Kern WV, Verrey F, Grütter MG, Diederichs K, Pos KM. 2012. Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc. Natl. Acad. Sci. U. S. A. 109:5687–5692. 10.1073/pnas.1114944109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu EW, Aires JR, McDermott G, Nikaido H. 2005. A periplasmic drug-binding site of the AcrB multidrug efflux pump: a crystallographic and site-directed mutagenesis study. J. Bacteriol. 187:6804–6815. 10.1128/JB.187.19.6804-6815.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Husain F, Nikaido H. 2010. Substrate path in the AcrB multidrug efflux pump of Escherichia coli. Mol. Microbiol. 78:320–330. 10.1111/j.1365-2958.2010.07330.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Bambeke F, Pagès JM, Lee VJ. 2010. Inhibitors of bacterial efflux pumps as adjuvants in antibacterial therapy and diagnostic tools for detection of resistance by efflux, p 138–175 In Atta-ur-Rahman, Choudhary MI. (ed), Frontiers in anti-infective drug discovery, vol 1 Bentham Science Publishers, Sharjah, United Arab Emirates [Google Scholar]
- 33.Zechini B, Versace I. 2009. Inhibitors of multidrug resistant efflux systems in bacteria. Recent Pat. Antiinfect. Drug Discov. 4:37–50. 10.2174/157489109787236256 [DOI] [PubMed] [Google Scholar]
- 34.Kourtesi C, Ball AR, Huang YY, Jachak SM, Vera DM, Khondkar P, Gibbons S, Hamblin MR, Tegos GP. 2013. Microbial efflux systems and inhibitors: approaches to drug discovery and the challenge of clinical implementation. Open Microbiol. J. 7:34–52. 10.2174/1874285801307010034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tegos GP, Haynes M, Strouse J, Khan MM, Bologa CG, Oprea TI, Sklar LA. 2011. Microbial efflux pump inhibition: tactics and strategies. Curr. Pharm. Des. 17:1291–1302. 10.2174/138161211795703726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mahamoud A, Chevalier J, Alibert-Franco S, Kern WV, Pagès J-M. 2007. Antibiotic efflux pumps in Gram-negative bacteria: the inhibitor response strategy. J. Antimicrob. Chemother. 59:1223–1229. 10.1093/jac/dkl493 [DOI] [PubMed] [Google Scholar]
- 37.Bush K, Courvalin P, Dantas G, Davies J, Eisenstein B, Huovinen P, Jacoby GA, Kishony R, Kreiswirth BN, Kutter E, Lerner SA, Levy S, Lewis K, Lomovskaya O, Miller JH, Mobashery S, Piddock LJ, Projan S, Thomas CM, Tomasz A, Tulkens PM, Walsh TR, Watson JD, Witkowski J, Witte W, Wright G, Yeh P, Zgurskaya HI. 2011. Tackling antibiotic resistance. Nat. Rev. Microbiol. 9:894–896. 10.1038/nrmicro2693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lomovskaya O, Bostian KA. 2006. Practical applications and feasibility of efflux pump inhibitors in the clinic–a vision for applied use. Biochem. Pharmacol. 71:910–918. 10.1016/j.bcp.2005.12.008 [DOI] [PubMed] [Google Scholar]
- 39.Piddock LJ, Garvey MI, Rahman MM, Gibbons S. 2010. Natural and synthetic compounds such as trimethoprim behave as inhibitors of efflux in Gram-negative bacteria. Clin. Microbiol. Rev. 65:1215–1223. 10.1093/jac/dkq079 [DOI] [PubMed] [Google Scholar]
- 40.Stavri M, Piddock LJ, Gibbons S. 2007. Bacterial efflux pump inhibitors from natural sources. J. Antimicrob. Chemother. 59:1247–1260. 10.1093/jac/dkl460 [DOI] [PubMed] [Google Scholar]
- 41.Lu W, Zhong M, Wei YN. 2011. Folding of AcrB subunit precedes trimerization. J. Mol. Biol. 411:264–274. 10.1016/j.jmb.2011.05.042 [DOI] [PubMed] [Google Scholar]
- 42.Nakashima R, Sakurai K, Yamasaki S, Hayashi K, Nagata C, Hoshino K, Onodera Y, Nishino K, Yamaguchi A. 2013. Structural basis for the inhibition of bacterial multidrug exporters. Nature 500:102–106. 10.1038/nature12300 [DOI] [PubMed] [Google Scholar]
- 43.Takatsuka Y, Chen C, Nikaido H. 2010. Mechanism of recognition of compounds of diverse structures by the multidrug efflux pump AcrB of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 107:6559–6565. 10.1073/pnas.1001460107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Opperman TJ, Kwasny SM, Kim HS, Nguyen ST, Houseweart C, D'Souza S, Walker GC, Peet NP, Nikaido H, Bowlin TL. 2014. Characterization of a novel pyranopyridine inhibitor of the AcrAB efflux pump of Escherichia coli. Antimicrob. Agents Chemother. 58:722–733. 10.1128/AAC.01866-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bohnert JA, Schuster S, Szymaniak-Vits M, Kern WV. 2011. Determination of real-time efflux phenotypes in Escherichia coli AcrB binding pocket phenylalanine mutants using a 1,2′-dinaphthylamine efflux assay. PLoS One 6:e21196. 10.1371/journal.pone.0021196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vargiu AV, Collu F, Schulz R, Pos KM, Zacharias M, Kleinekathöfer U, Ruggerone P. 2011. Effect of the F610A mutation on substrate extrusion in the AcrB transporter: explanation and rationale by molecular dynamics simulations. J. Am. Chem. Soc. 133:10704–10707. 10.1021/ja202666x [DOI] [PubMed] [Google Scholar]
- 47.Trott O, Olson AJ. 2010. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31:455–461. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802. 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grossfield A, Zuckerman DM. 2009. Quantifying uncertainty and sampling quality in biomolecular simulations. Annu. Rep. Comput. Chem. 5:23–48. 10.1016/S1574-1400(09)00502-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guimarães CW. 2012. MM-GB/SA rescoring of docking poses, p 255–268 In Baron R. (ed), Computational drug discovery and design, vol 819 Springer, New York, NY [Google Scholar]
- 51.Hobbs EC, Yin X, Paul BJ, Astarita JL, Storz G. 2012. Conserved small protein associates with the multidrug efflux pump AcrB and differentially affects antibiotic resistance. Proc. Natl. Acad. Sci. U. S. A. 109:16696–16701. 10.1073/pnas.1210093109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Du D, Wang Z, James NR, Voss JE, Klimont E, Ohene-Agyei T, Venter H, Chiu W, Luisi BF. 2014. Structure of the AcrAB-TolC multidrug efflux pump. Nature 509:512–516. 10.1038/nature13205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Efremov RG, Chugunov AO, Pyrkov TV, Priestle JP, Arseniev AS, Jacoby E. 2007. Molecular lipophilicity in protein modeling and drug design. Curr. Med. Chem. 14:393–415. 10.2174/092986707779941050 [DOI] [PubMed] [Google Scholar]
- 54.Husain F, Bikhchandani M, Nikaido H. 2011. Vestibules are part of the substrate path in the multidrug efflux transporter AcrB of Escherichia coli. J. Bacteriol. 193:5847–5849. 10.1128/JB.05759-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fogolari F, Brigo A, Molinari H. 2002. The Poisson-Boltzmann equation for biomolecular electrostatics: a tool for structural biology. J. Mol. Recognit. 15:377–392. 10.1002/jmr.577 [DOI] [PubMed] [Google Scholar]
- 56.Kollman PA, Massova I, Reyes C, Kuhn B, Huo SH, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE., III 2000. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 33:889–897. 10.1021/ar000033j [DOI] [PubMed] [Google Scholar]
- 57.Asthana S, Shukla S, Vargiu AV, Ceccarelli M, Ruggerone P, Paglietti G, Marongiu ME, Blois S, Giliberti G, La Colla P. 2013. Different molecular mechanisms of inhibition of bovine viral diarrhea virus and hepatitis C virus RNA-dependent RNA polymerases by a novel benzimidazole. Biochemistry 52:3752–3764. 10.1021/bi400107h [DOI] [PubMed] [Google Scholar]
- 58.Kinana AD, Vargiu AV, Nikaido H. 2013. Some ligands enhance the efflux of other ligands by the Escherichia coli multidrug pump AcrB. Biochemistry 52:8342–8351. 10.1021/bi401303v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bohnert JA, Kern WV. 2005. Selected arylpiperazines are capable of reversing multidrug resistance in Escherichia coli overexpressing RND efflux pumps. Antimicrob. Agents Chemother. 49:849–852. 10.1128/AAC.49.2.849-852.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoshida K, Nakayama K, Ohtsuka M, Kuru N, Yokomizo Y, Sakamoto A, Takemura M, Hoshino K, Kanda H, Nitanai H, Namba K, Yoshida K, Imamura Y, Zhang JZ, Lee VJ, Watkins WJ. 2007. MexAB-OprM specific efflux pump inhibitors in Pseudomonas aeruginosa. Part 7: highly soluble and in vivo active quaternary ammonium analogue D13-9001, a potential preclinical candidate. Bioorg. Med. Chem. 15:7087–7097. 10.1016/j.bmc.2007.07.039 [DOI] [PubMed] [Google Scholar]
- 61.Lomovskaya O, Warren MS, Lee A, Galazzo J, Fronko R, Lee M, Blais J, Cho D, Chamberland S, Renau T, Leger R, Hecker S, Watkins W, Hoshino K, Ishida H, Lee VJ. 2001. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrob. Agents Chemother. 45:105–116. 10.1128/AAC.45.1.105-116.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhardwaj AK, Mohanty P. 2012. Bacterial efflux pumps involved in multidrug resistance and their inhibitors: rejuvinating [sic] the antimicrobial chemotherapy. Recent Pat. Antiinfect. Drug Discov. 7:73–89. 10.2174/157489112799829710 [DOI] [PubMed] [Google Scholar]
- 63.Ruggerone P, Vargiu AV, Collu F, Fischer N, Kandt C. 2013. Molecular dynamics computer simulations of multidrug RND efflux pumps. Comput. Struct. Biotech. J. 5:e201302008. 10.5936/csbj.201302008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schulz R, Vargiu AV, Ruggerone P, Kleinekathöfer U. 2011. Role of water during the extrusion of substrates by the efflux transporter AcrB. J. Phys. Chem. B 115:8278–8287. 10.1021/jp200996x [DOI] [PubMed] [Google Scholar]
- 65.Collu F, Vargiu AV, Dreier J, Cascella M, Ruggerone P. 2012. Recognition of imipenem and meropenem by the RND-transporter MexB studied by computer simulations. J. Am. Chem. Soc. 134:19146–19158. 10.1021/ja307803m [DOI] [PubMed] [Google Scholar]
- 66.Matsumoto Y, Hayama K, Sakakihara S, Nishino K, Noji H, Iino R, Yamaguchi A. 2011. Evaluation of multidrug efflux pump inhibitors by a new method using microfluidic channels. PLoS One 6:e18547. 10.1371/journal.pone.0018547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kern WV, Steinke P, Schumacher A, Schuster S, von Baum H, Bohnert JA. 2006. Effect of 1-(1-naphthylmethyl)-piperazine, a novel putative efflux pump inhibitor, on antimicrobial drug susceptibility in clinical isolates of Escherichia coli. J. Antimicrob. Chemother. 57:339–343. 10.1093/jac/dki445 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.