

Abstract
Coadministration of dihydroartemisinin-piperaquine (DHA-PQ) with fat may improve bioavailability and antimalarial efficacy, but it might also increase toxicity. There have been no studies of these potential effects in the pediatric age group. The tolerability, safety, efficacy, and pharmacokinetics of DHA-PQ administered with or without 8.5 g fat were investigated in 30 Papua New Guinean children aged 5 to 10 years diagnosed with uncomplicated falciparum malaria. Three daily 2.5:11.5-mg-base/kg doses were given with water (n = 14, group A) or milk (n = 16, group B), with regular clinical/laboratory assessment and blood sampling over 42 days. Plasma PQ was assayed by high-performance liquid chromatography with UV detection, and DHA was assayed using liquid chromatography-mass spectrometry. Compartmental pharmacokinetic models for PQ and DHA were developed using a population-based approach. DHA-PQ was generally well tolerated, and initial fever and parasite clearance were prompt. There were no differences in the areas under the concentration-time curve (AUC0–∞) for PQ (median, 41,906 versus 36,752 μg · h/liter in groups A and B, respectively; P = 0.24) or DHA (4,047 versus 4,190 μg · h/liter; P = 0.67). There were also no significant between-group differences in prolongation of the corrected electrocardiographic QT interval (QTc) initially during follow-up, but the QTc tended to be higher in group B children at 24 h (mean ± standard deviation [SD], 15 ± 10 versus 6 ± 15 ms0.5 in group A, P = 0.067) and 168 h (10 ± 18 versus 1 ± 23 ms0.5, P = 0.24) when plasma PQ concentrations were relatively low. A small amount of fat does not change the bioavailability of DHA-PQ in children, but a delayed persistent effect on ventricular repolarization cannot be excluded.
INTRODUCTION
Dihydroartemisinin-piperaquine (DHA-PQ) is a widely used artemisinin combination therapy (ACT) that has proved highly effective in a range of studies of children and adults with uncomplicated falciparum or vivax malaria (1). However, in a recent large-scale multiarm randomized trial in Papua New Guinean (PNG) children (2), DHA-PQ proved inferior to artemether-lumefantrine for treatment of Plasmodium falciparum infections and was associated with a treatment failure rate above that required by WHO for the adoption of a new antimalarial therapy (3). Among possible explanations for this unexpected result was the fact that the PNG children allocated to receive DHA-PQ took their tablets with water rather than in the fed state, potentially reducing bioavailability (4). Such an effect could be especially important in young children since conventional mg/kg body weight dosing gives rise to relatively low plasma PQ concentrations in this age group (5, 6).
Evidence that PQ absorption is proportional to the amount of coadministered fat comes from a variety of studies in healthy adults and adults with uncomplicated malaria. A high-fat meal (typically containing around 50 g fat) is associated with at least a doubling of area under the PQ plasma concentration-time curve (AUC0–∞) relative to that when the drug is administered with water (7, 8). A lower fat content (17 g) increases the PQ area under the plasma concentration-time curve (AUC) by up to 41% (9, 10), while only 6 g ingested fat (such as that contained in a small carton of milk, which can usually be tolerated by a patient with acute malaria who may be anorectic and nauseated) is not associated with increased PQ bioavailability (11–13). The effect of fat on PQ absorption in children is, however, unknown.
Like other chemically related drugs (14), PQ can prolong the corrected electrocardiographic QT (QTc) interval. The product information for one approved DHA-PQ formulation (Eurartesim; Sigma-Tau Industrie Farmaceutiche Riunite S.p.A., Rome, Italy) advises that DHA-PQ should not be used in people who have, or are at risk of, QTc prolongation or cardiac arrhythmias (including those with electrolyte disturbances such as hypokalemia and hypocalcemia) and should not be taken with other drugs which prolong the QTc (8). In addition, each dose should be taken orally with water and without food, no less than 3 h before or after eating, and with electrocardiographic monitoring if there is clinical concern (8). These recommendations are based on studies showing an increased risk of QTc prolongation in healthy adults who took DHA-PQ with a high-fat meal that resulted in increased plasma PQ concentrations, together with an excess of DHA-PQ-related cardiac adverse events in clinical trials (8). However, the overall evidence for clinically significant QTc prolongation with recommended DHA-PQ doses given with water or relatively small amounts of fat for uncomplicated malaria is not compelling (1, 15–17), and it has not revealed an association with cardiac dysrhythmias and their sequelae, including sudden death (18).
Since most DHA-PQ is administered to young children in health care facilities in tropical countries with limited or no capacity for electrocardiographic and/or laboratory monitoring, and given that bioavailability should be optimized to maximize efficacy in this patient group (5), it is important that the pharmacokinetics and potential toxicity of this ACT are well characterized when it is coadministered with fat in pediatric patients. The aim of the present study was, therefore, to examine the safety, tolerability, and pharmacokinetic properties of DHA-PQ given with water or milk to PNG children with uncomplicated malaria.
MATERIALS AND METHODS
Study site, approvals, and patients.
All components of the present study were conducted at the Alexishafen Health Center, Madang Province, on the north coast of Papua New Guinea, where there is hyperendemic transmission of P. falciparum and Plasmodium vivax (19). Children aged >5 years and with a body weight of >15 kg who presented with an axillary temperature of >37.5°C or a history of fever in the previous 24 h were screened for malaria with a Giemsa-stained thick blood film read by a trained field microscopist. Those with monoinfections with either P. falciparum (>500 asexual parasites/μl whole blood) or P. vivax, Plasmodium ovale, or Plasmodium malariae (>250 parasites/μl) were eligible for recruitment provided that (i) they had no features of severe malaria, (ii) they had not received treatment with any antimalarial drug in the previous 4 weeks, (iii) there was no history of allergy to artemisinin or quinoline drugs, (iv) there was no clinical evidence of another infection as a possible cause of fever, (v) there were no signs of malnutrition or another significant comorbidity, and (vi) the child's parents or guardians gave written informed consent. Approval for the present study was obtained from the PNG Institute of Medical Research Institutional Review Board and the Medical Research Advisory Committee of the PNG Health Department.
Baseline assessment, randomization, and treatment.
At enrollment, a history and a detailed symptom questionnaire were completed with the assistance of parents/guardians, and a full physical examination was performed, including body weight, height, axillary temperature, supine and erect blood pressure and pulse rate, and respiratory rate. An intravenous cannula was inserted, and a baseline venous blood sample was collected for point-of-care hemoglobin and blood glucose measurements. The remaining blood was centrifuged, and the plasma was stored at −20°C for subsequent drug and other assays. The red cell pellet was retained for parasite genotyping. Each child had a baseline electrocardiogram with the QT interval measured manually from lead II of the 12-lead trace and corrected for the heart rate using Bazett's method (QTc = QT/√RR interval).
All participants received Eurartesim tablets (containing 20 mg DHA and 180 mg PQ phosphate) at a DHA dose of 2.5 mg/kg of body weight and a PQ phosphate dose of 20 mg/kg (equivalent to 11.5 mg of PQ base/kg) given daily for 3 days at 0 h (day 0), 24 h (day 1), and 48 h (day 2). Each subject was randomized by a computer-generated schedule to receive their tablets under direct observation with either (i) water and no fat-containing food for the following 4 h (group A; n = 14) or (ii) 250 ml full-cream flavored cow's milk (8.5 g fat) with each dose (group B; n = 16), a form and volume of fat supplementation that have proved well tolerated in previous studies in this patient group (20). The exact time of dosing was recorded. Any children who vomited within the first 30 min of administration were excluded from the study and given PNG standard treatment for uncomplicated malaria (21).
Monitoring and follow-up.
Further 2.5-ml blood samples for drug assay were drawn from all participants at 1, 2, 3, 4, 6, 10, 18, and 24 h (the last sample immediately before the second dose) through the intravenous cannula and by venesection on days 4, 7, 14, 28, and 42. In addition, four other blood samples were taken at randomly selected times (computer-generated randomization schedule) from the following time points: 26, 28, 30, 32, 34, 40, 48 (immediately before the third dose), 50, 52, 56, 58, 60, 66, and 72 h. Daily clinical assessment, including a side effect questionnaire and blood smear, was performed at each follow-up appointment (days 1, 2, 3, 7, 14, 28, and 42). Additional monitoring comprised (i) blood glucose measurement at 4 h and on days 1, 2, 3, 7, 14, and 28; (ii) hemoglobin concentration measurement on days 2, 7, 14, 28, and 42; and (iii) electrocardiography and measurement of lying/standing blood pressures at 4 and 24 h and on day 7.
All blood smears from each patient subsequently were reexamined, and parasite densities were quantified independently by two skilled microscopists in a central laboratory, with discrepancies being adjudicated by a third microscopist. Parasite densities were calculated from the number of parasites per 200 or 500 white cells (depending on parasitemia) and an assumed total peripheral white cell count of 8,000/μl (22), with the final density taken as the geometric mean of the two values. The presence and species of Plasmodium at baseline and in follow-up samples were confirmed by PCR, but further analysis to differentiate reinfection from recrudescence was not performed.
Drug assays.
Plasma PQ concentrations were measured by previously validated high-performance liquid chromatography (23, 24). In brief, extracted plasma samples were injected onto a Chromolith Performance column (E. Merck GmbH, Darmstadt, Germany). Analytes were detected at 340 nm and quantified by using Chemstation software (version 9; Agilent Technology, Waldbronn, Germany). The intraday relative standard deviations (RSDs) were ≤10.8, and the interday RSDs were ≤11.6% at plasma PQ concentrations between 9 and 1,867 nmol/liter, respectively. The limits of quantification and detection were 3 nmol/liter and 1.3 nmol/liter, respectively.
Plasma DHA concentrations were measured by liquid chromatography-mass spectrometry as previously described (25). In brief, extracted plasma samples were injected onto a Synergy fusion-RP C18 (150-mm by 2.0-mm-inside-diameter [i.d.]) column coupled with a 4-mm by 3-mm-i.d., 5-μm-particle C18 guard column (Phenomenex, Lane Cove, Australia). Optimized mass spectra were acquired, and quantitation was performed by selected ion monitoring using the dual ionization source mode on a single quad mass spectrometer (Shimadzu, Kyoto, Japan). Interday accuracy deviations across clinically relevant concentration ranges were <15%. The limits of quantification and detection for DHA were 2 and 1 μg/liter, respectively.
Pharmacokinetic modeling.
Loge plasma concentration-time data sets for PQ and DHA were analyzed by nonlinear mixed effects modeling using NONMEM (v7.2.0; Icon Development Solutions, Ellicott City, MD, USA) with an Intel Visual FORTRAN 10.0 compiler. The Laplacian method with interaction estimation was used for DHA, as required for the M3 method of handling below-the-limit-of-quantification (BLQ) data. The first-order conditional estimation (FOCE) method with interaction estimation was used for PQ. The minimum value of the objective function (OFV) and conditional weighted residual (CWRES) plots were used to choose suitable models during the model-building process. A significance level of P < 0.01 was set for comparison of nested models. Allometric scaling was employed a priori, with volume terms multiplied by (WT/70)1.0 and clearance terms by (WT/70)0.75 (26), where WT is body weight. Residual variability (RV) was estimated as additive error for the log-transformed data. Secondary pharmacokinetic parameters, including area under the curve (AUC0–∞), maximum plasma concentration (Cmax), and elimination half-lives (t1/2) for the participants were obtained from post hoc Bayesian prediction in NONMEM using the final model parameters. Base models were parameterized using ka (absorption rate constant), VC/F (central volume of distribution), CL/F (clearance), and VP/F and Q/F (peripheral volumes of distribution[s] and their respective intercompartmental clearance[s], respectively).
For PQ, two- and three-compartment models (ADVAN 4 and 12) with first-order absorption with and without lag time were tested. Since inspection of the plasma concentration-time curves indicated that there was significant variability in the absorption phase, a transit compartment model was also tested, as used previously to model PQ disposition (12). In this model, the dose passes through a series of transit compartments before entering the absorption compartment in order to model the delay often associated with drug absorption. A single rate constant (ktr) describes the entry and exit for all transit compartments. Using a previously described implementation of the transit compartment model in NONMEM (27), the number of transit compartments (NN) and the mean transit time [MTT = (1 + NN)/ktr] were estimated as continuous variables. For the DHA data set, one- and two-compartment models (ADVAN 2 and 4) with first-order absorption with and without lag time were evaluated. A transit compartment model was also assessed but was not supported by the data. Once the structure of the models was established, exponentially modeled interindividual variability (IIV) and interoccasion variability (IOV), as well as correlations between IIV terms, were evaluated for each suitable parameter and included where supported by the data.
The potential effect on relative bioavailability and absorption kinetics with the coadministration of milk was then assessed. The inclusion of an extra parameter to account for differences in relative bioavailability was considered only if accompanied by a significant fall in the OFV (>6.63, P < 0.01) and an improvement in the CWRES plot. Differences in absorption parameters (ka, NN, and MTT) between the two groups were also assessed within NONMEM. As described below, the effect size of the difference (%) was estimated. To maintain the extra parameter estimating this difference, a significant fall in the OFV (>6.63, P < 0.01) was required.
Finally, relationships between model parameters and the covariates age, sex, log(baseline parasitemia), and fever were identified through inspection of scatter plots and box plots of individual IIV estimates versus covariates and subsequently evaluated within NONMEM. The effect size (%) of categorical data (sex and fever) was assessed, while both linear and power relationships were evaluated for continuous covariates [age, log(baseline parasitemia)]. For effect size, the individual parameter value equals population parameter value × [1 + effect parameter × covariate value (0 or 1)]. For linear relationships, the individual parameter value equals population parameter value × [1 + effect parameter × (covariate value for individual)/(average value of covariate)]. For power relationships, the individual parameter value equals population parameter value × [(covariate value for individual)/(average value of covariate)effect parameter]. A stepwise forward inclusion and backward elimination method was used with a P of <0.05 required for inclusion of a covariate relationship and a P value of <0.01 required to retain a covariate relationship.
Initially, plots of observed versus individual and population predicted values, and time versus CWRES, were assessed. A bootstrap using Perl Speaks NONMEM (PSN) with 1,000 samples was performed with stratification according to dose group, and the parameters derived from this analysis were summarized as median and 2.5th and 97.5th percentiles (95% empirical confidence interval [CI]) to facilitate evaluation of final model parameter estimates. In addition, prediction-corrected visual predictive checks (pcVPCs) (28) and numerical predictive checks (NPCs) were performed with 1,000 data sets simulated from the final models. The observed 10th, 50th, and 90th percentiles were plotted with their respective simulated 95% CIs to assess the predictive performance of the model. For DHA, this was plotted against time from last dose (rather than time from first dose) and the fraction of BLQ data observed with the simulated 95% CI was also plotted for each time point. NPCs were assessed by comparing the actual with the expected number of data points within the 20, 40, 60, 80, 90, and 95% prediction intervals (PIs). Shrinkage of population variability parameters and residual variability was incorporated to help determine whether models were overparameterized and to determine the reliability of diagnostic plots (29).
Statistical analysis.
Statistical analysis was performed using IBM SPSS Statistics version 20 (IBM Corporation, Somers, NY, US). Two-sample comparisons for normally distributed variables were performed by Student's t test, those for nonnormally distributed variables were performed by the Mann-Whitney U-test, and those for proportions were performed by Fisher's exact test. Generalized linear mixed modeling was used to compare repeated measures of continuous variables across time points. Unless otherwise stated, all P values are two-tailed and unadjusted for multiple comparisons.
RESULTS
Patient characteristics.
The baseline characteristics of participants by randomized therapy are summarized in Table 1. The two groups were well matched for key demographic, anthropometric, clinical, and laboratory characteristics. On screening microscopy at the study site, all children had detectable baseline P. falciparum parasitemia. No children presented with a P. vivax infection. On subsequent confirmatory expert microscopy, one child in each group was found to have a negative blood slide. These two children were also negative for malaria by Plasmodium species PCR. All remaining children (13 in group A and 15 in group B) were positive for P. falciparum infection by both expert microscopy and PCR. All recruited children were included in the tolerability and safety analyses.
TABLE 1.
Admission characteristics of the children in each group
| Characteristic | Group A | Group B | P value |
|---|---|---|---|
| n | 14 | 16 | |
| Age (mo), median (interquartile range) | 88 (72 to 109) | 85 (79 to 109) | 0.55 |
| Male sex, no. (%) | 8 (57) | 11 (69) | 0.71 |
| Duration of illness (days), mean (interquartile range) | 3 (2 to 4) | 3.5 (2 to 5) | 0.71 |
| Plasmodium falciparum density (per μl), median (interquartile range) | 22,905 (11,840 to 73,264) | 16,063 (950 to 33,255) | 0.35 |
| Body wt (kg), mean ± SD | 20 ± 4.3 | 19.3 ± 3.3 | 0.52 |
| Ht (cm), mean ± SD | 117 ± 12 | 116.2 ± 9.8 | 0.86 |
| Axillary temp (°C), mean ± SD | 37.5 ± 1.4 | 37.2 ± 1.1 | 0.47 |
| Mid-upper-arm circumference (cm), mean ± SD | 15.9 ± 1.6 | 15.7 ± 1.2 | 0.65 |
| Pulse rate (per min), median (interquartile range) | |||
| Supine | 92 (84 to 100) | 92 (83 to 109) | 0.76 |
| Standing | 96 (92 to 115) | 100 (100 to 120) | 0.048 |
| Blood pressure (mm Hg), median (interquartile range) | |||
| Supine systolic | 90 (75 to 100) | 90 (80 to 90) | 0.54 |
| Supine diastolic | 52 (45 to 60) | 50 (50 to 59) | 0.98 |
| Systolic change on standing | 0 (−8 to 4) | −10 (−10 to 3) | 0.30 |
| Diastolic change on standing | 0 (−5 to 2) | −3 (−10 to 0) | 0.055 |
| Respiratory rate (breaths per min), median (interquartile range) | 22 (20 to 28) | 23 (20 to 26) | 0.86 |
| Hemoglobin (g/liter), median (interquartile range) | 99.5 (77 to 108) | 105 (93 to 115) | 0.49 |
| Blood glucose (mmol/liter), median (interquartile range) | 6.6 (6.1 to 7.4) | 6.5 (6.0 to 6.9) | 0.44 |
| QTc (ms0.5), median (interquartile range) | 431 (421 to 440) | 430 (426 to 445) | 0.51 |
Safety and tolerability. (i) Adverse events and symptoms.
No significant adverse events occurred in any of the 30 children. Most (93% and 88% for groups A and B, respectively) presented with symptoms consistent with fever, including hot skin, chills, and lethargy. Other common symptoms at presentation included headache (69% and 50%), anorexia (46% and 25%), nausea (54% and 29%), and cough/dyspnea (46% and 56%, respectively; P ≥ 0.28 for between-group comparisons in each case). Symptoms were all mild or moderate. Anorexia and vomiting were reported by some children in both groups on day 1 (4% in total), but all symptoms had resolved by day 2 apart from persistent cough/dyspnea in a small minority of cases in each group.
(ii) Hemoglobin and blood glucose.
There was no significant difference in hemoglobin concentrations in the two patient groups at baseline (Table 1). There were reductions in mean hemoglobin to minima on day 2 in both groups (88 ± 5 g/liter in group A and 88 ± 5 in group B) but no significant between-group differences over time by generalized linear modeling (P ≥ 0.32). The mean hemoglobin concentration had exceeded the level at presentation by day 14. No child developed severe anemia (hemoglobin, ≤50 g/liter). No child was found to be hypoglycemic (blood glucose, <4.0 mmol/liter) at enrollment or during follow-up.
(iii) Hemodynamic changes.
There were no significant differences in systolic blood pressure (SBP) or diastolic blood pressure, or their changes on standing (Δ), between the two groups at baseline (Table 1), and no individual child had an SBP fall of ≥20 mm Hg at this time. There were also no between-group differences in resting pulse rate (PR) at baseline, but there was a borderline significant greater increase in PR on standing in group B (P = 0.048). Generalized linear modeling did not show any significant between-group differences in ΔSBP or ΔPR at 4 h, 24 h (day 1), and 168 h (day 7) after correction for day 0 (baseline) values (P ≥ 0.40).
(iv) QTc interval changes.
There was no significant difference in QTcs in the two patient groups at baseline (Table 1). There was prolongation of the mean QTc to maxima at 4 h in group A and at 24 h in group B (Fig. 1). Ten of the 14 group A children (71%) had a QTc of ≥440 ms0.5 at 4 h, with the longest QTc at this time being 488 ms0.5. By 24 h, only five group A children (36%) had a QTc of ≥440 ms0.5. Ten group B children (63%) had a QTc of ≥440 ms0.5 at 4 h (P = 0.71 versus group A), and 12 (75%) had a QTc of ≥440 ms0.5 at 24 h (P = 0.06 versus group A), with the longest QTc at the latter time point being 476 ms0.5. Generalized linear modeling did not show any significant between-group differences in QTc at 4 h (P = 0.28), 24 h (P = 0.067), or 168 h (P = 0.24) after correction for day 0 (baseline) values.
FIG 1.
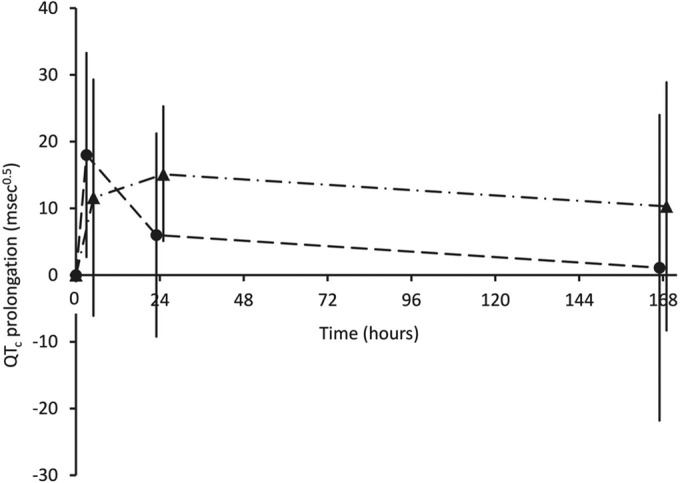
Changes in the electrocardiographic QTc interval during follow-up to day 7 in the two groups of children treated with dihydroartemisinin-piperaquine. Data are means for groups A (●) and B (▲) and SDs (vertical bars).
Antimalarial efficacy.
For the 13 children in group A who were positive by microscopy for P. falciparum at baseline, there was a rapid decline in asexual parasitemia after drug administration with a mean parasite clearance time of 1.8 ± 0.4 days and slide negativity in all cases by 48 h. Two children (14%) developed recurrent parasitemia by microscopy and PCR during the 42-day follow-up period, both cases being P. falciparum infections detected on day 42. At baseline, 31% of the group A children were positive for P. falciparum gametocytes by microscopy, with 23% and 15% remaining positive at days 7 and 14, respectively.
In group B, 15 of the 16 recruited children were positive by microscopy for P. falciparum at baseline. After drug administration, a rapid decline in parasitemia was observed with a mean parasite clearance time of 1.5 ± 0.5 days (P = 0.08 versus group A). All children were slide negative by 48 h. No participant in group B had a recurrent infection during the 42-day follow-up period (P = 0.21 versus group A). At baseline, 27% of group B participants were positive by microscopy for P. falciparum gametocytes with 20% remaining positive at day 7, but all gametocytes had cleared by day 14 (P ≥ 0.21 versus group A). All 13 children with fever at baseline (6 in group A and 7 in group B) became afebrile within 48 h.
Pharmacokinetic modeling. (i) Piperaquine.
There were 477 individual plasma PQ concentrations available for analysis, none of which were BLQ during the 42-day follow-up period. A three-compartment model fitted the data better than a two-compartment model with a significant decrease in the OFV (ΔOFV = −32.365, P < 0.001) and improved CWRES plot. Although the addition of a lag time improved the model significantly (ΔOFV = −13.867, P < 0.001), the absorption phase was poorly described with first-order absorption with or without lag time. Therefore, a transit compartment model was tested where the number of transit compartments (NN) and the mean transit time (MTT) through the transit compartments were estimated as continuous variables. The transit compartment was significantly better than a model with first-order absorption and lag time, resulting in a 21.007 point reduction in the OFV (P < 0.001).
The structural model parameters were ka, NN, MTT, FPQ, VC/FPQ, VP1/FPQ, VP2/FPQ, CL/FPQ, Q1/FPQ, and Q2/FPQ. There was poor precision for the estimate of ka (% relative standard error [RSE] of >100%) as well as a high correlation between ka and MTT (r >0.95). Therefore, with the data available in this study, these two parameters could not be estimated simultaneously and ka was set to be the same as ktr, i.e., equal to (1 + NN)/MTT (30). Interoccasion variability on FPQ was estimated, and including this in the model resulted in a substantial decrease in OFV (ΔOFV = −134.338, P < 0.001) as well as RV (48% to 36%). Interindividual variability was estimable for MTT, CL/FPQ, and VC/FPQ. Correlation between IIV terms was estimated for CL/FPQ and VC/FPQ; however, as this relationship was poorly defined in the bootstrap, it was removed from the final model. Although inspection of the concentration-time curves appeared to indicate a delay in the peak with the administration of milk, when differences in NN and MTT were evaluated in NONMEM, no significant effect was identified. Likewise, the coadministration of milk did not significantly affect relative bioavailability, and none of the other tested covariates improved the model.
The final model parameter estimates and the bootstrap results for PQ pharmacokinetics in both groups are summarized in Table 2. Bias was less than 6% and 12% for all fixed and random model parameters, respectively. Figures 2 and 3 show goodness-of-fit plots and pcVPCs, respectively. The pcVPCs demonstrate wide 95% confidence intervals for the 10th, 50th, and 90th percentiles between 24 and 72 h due to a smaller number of observations than at other time points where all patients contributed. The actual 10th, 50th, and 90th percentiles fell into their respective 95% CIs for all but two time points. The stratified NPCs demonstrated good predictive performance with the expected number of points above and below the 20, 40, 60, 80, 90, and 95% PIs. The post hoc individual parameters, half-lives, and AUC0–∞ are shown in Table 3. There was a trend for a longer MTT in children who had received milk; however, as noted in the model building procedure this did not reach statistical significance (P = 0.052). There was no significant difference in the two groups for AUC0–∞ (P = 0.20). The first distribution, second distribution, and terminal elimination t1/2 for all participants had median values of 2.2, 28.0, and 432 h, respectively, and the median PQ AUC0–∞ was 38,590 μg · h/liter.
TABLE 2.
Final population pharmacokinetic variable estimates and bootstrap results for piperaquine in childrena
| Parameter | Mean | RSE, % | Bootstrap median (95% CI) |
|---|---|---|---|
| Objective function value | −325.935 | −341.177 (−412.947 to −275.879) | |
| Structural model parameters | |||
| MTT (h) | 1.09 | 16 | 1.09 (0.73 to 1.40) |
| NN | 1.16 | 72 | 1.10 (0.10 to 4.31) |
| CL/FPQ (liters/h/70 kg) | 40.4 | 8 | 40.9 (35.8 to 58.6) |
| VC/FPQ (liters/70 kg) | 2,160 | 14 | 2,189 (1,557 to 3,096) |
| Q1/FPQ (liters/h/70 kg) | 63.7 | 11 | 64.5 (52.7 to 96.7) |
| VP1/FPQ (liters/70 kg) | 18,900 | 13 | 19,052 (15,716 to 28,261) |
| Q2/FPQ (liters/h/70 kg) | 224 | 17 | 226 (158 to 351) |
| VP2/FPQ (liters/70 kg) | 3,340 | 15 | 3,325 (2,321 to 5,055) |
| Variability model parameters (shrinkage %) | |||
| IOV in FPQ (%) | 52 (4, 16, 37) | 8 | 51 (43 to 59) |
| IIV in MTT (%) | 60 (14) | 17 | 58 (42 to 79) |
| IIV in VC/FPQ (%) | 19 (31) | 26 | 18 (3 to 27) |
| IIV in CL/FPQ (%) | 46 (26) | 22 | 44 (21 to 63) |
| RV for PQ (%) | 32 (13) | 4 | 32 (29 to 34) |
Abbreviations: MTT, mean transit time; NN, number of transit compartments; CL/FPQ, clearance relative to bioavailability; VC/FPQ, central volume of distribution relative to bioavailability; Q1/FPQ, intercompartmental clearance for VP1/FPQ; VP1/FPQ, first peripheral volume of distribution relative to bioavailability; Q2/FPQ, intercompartmental clearance for VP2/FPQ; VP2/FPQ, second peripheral volume of distribution relative to bioavailability; IOV, interoccasion variability; IIV, interindividual variability; RV, residual variability. IOV and IIV are presented as .
FIG 2.

Goodness-of-fit plots for piperaquine. The observed plasma piperaquine has been plotted against population (A) and individual (B) predicted plasma concentrations, and conditional weighted residuals have been plotted against time (C) and population predicted plasma concentrations (D).
FIG 3.

Prediction-corrected visual predictive check for piperaquine (μg/liter on log10 scale) with observed 50th (solid line) and 10th and 90th (dashed lines) percentiles within their simulated 95% CI (gray shaded areas) with overlying data points (○) for the whole time period of the study (A). Given the overlap of the 95% CI, the 10th (B), 50th (C), and 90th (D) percentiles with their respective 95% CIs are shown separately for the first 96 h.
TABLE 3.
Post hoc Bayesian estimates of pharmacokinetic parameters and derived secondary parameters for piperaquine in childrena
| Parameter | Median (IQR) |
P value | |
|---|---|---|---|
| Group A (n = 14) | Group B (n = 16) | ||
| MTT (h) | 0.942 (0.602–1.318) | 1.559 (1.049–1.802) | 0.052 |
| NN | 1.16 | 1.16 | |
| CL/FPQ (liters/h) | 15.4 (13.3–16.7) | 15.3 (14–16.6) | 0.95 |
| VC/FPQ (liters) | 436 (380–826) | 604 (452–750) | 0.22 |
| Q1/FPQ (liters/h) | 23.2 (21.3–27.6) | 23.7 (22.7–25.1) | 0.82 |
| VP1/FPQ (liters) | 4,930 (4,390–6,213) | 5,065 (4,795–5,470) | 0.82 |
| Q2/FPQ (liters/h) | 81.6 (74.8–97) | 83.3 (79.9–88.2) | 0.82 |
| VP2/FPQ (liters) | 870 (775–1,097) | 894 (846–965) | 0.82 |
| t1/2α (h) | 2.06 (1.88–2.93) | 2.38 (2.12–2.98) | 0.18 |
| t1/2β (h) | 26.4 (25.9–31) | 28.2 (26.6–30.1) | 0.38 |
| t1/2γ (h) | 439 (399–455) | 422 (406–446) | 0.61 |
| Cmax (first dose; μg/liter) | 267 (211–390) | 146 (99–342) | 0.27 |
| AUC0–∞ (μg · h/liter) | 41,906 (33,101–49,175) | 36,752 (33,051–43,031) | 0.24 |
Abbreviations: MTT, mean transit time; NN, number of transit compartments; CL/FPQ, clearance; VC/FPQ, central volume of distribution; Q1/FPQ, intercompartmental clearance for VP1/FPQ; VP1/FPQ, first peripheral volume of distribution; Q2/FPQ, intercompartmental clearance for VP2/FPQ; VP2/FPQ, second peripheral volume of distribution; t1/2α, t1/2β, and t1/2γ (first and second distribution and elimination half-lives, respectively); Cmax, maximum plasma concentration; AUC0–∞, area under the plasma concentration-time curve.
(ii) Dihydroartemisinin.
There were 237 individual plasma DHA concentrations available for analysis, of which 43 (18.1%) were BLQ. As these represented a significant proportion of the data, a method previously described, namely, M3 (31), and successfully utilized in the PK analysis of antimalarial drugs (25, 32) was employed. Initial modeling of the DHA data set demonstrated that a two-compartment model was not significantly better than a one-compartment model (ΔOFV = −6.171, P = 0.023, degrees of freedom [df] = 2) and that the absorption phase was best described by a first-order absorption with a lag time. Therefore, the structural model parameters were ka, lag, V/FDHA, and CL/FDHA. The IIV of both V/FDHA and CL/FDHA was estimable, as was the IOV on FDHA. There was no significant relationship between the coadministration of milk and relative bioavailability or absorption parameters. No other significant covariate relationships were identified.
The final model parameter estimates and the bootstrap results for DHA are summarized in Table 4. Bias was <5% for all fixed and random parameters. Figures 4 and 5 show goodness-of-fit plots and pcVPCs, respectively. Although there appeared to be bias in the CWRES plots, this can be attributed to the data points that are BLQ. These points would be expected to have negative residuals, given their low values, and would therefore balance the bias seen in the plots. The pcVPCs showed that all observed 50th and 90th percentiles were within their simulated 95% CIs, with the first two 10th percentile points just outside the 95% CI. The observed fraction BLQ was within the 95% CI at all points. The NPC demonstrated good predictive performance with the expected number of points above and below the 20, 40, 60, 80, 90, and 95% PIs. The post hoc individual parameters, t1/2 and AUC0–∞, for the study participants are summarized in Table 5. There were no significant differences between the two groups. The median t1/2 and AUC0–∞ for all participants were 0.77 h and 4.13 μg · h/liter, respectively.
TABLE 4.
Final population pharmacokinetic estimates and bootstrap results for dihydroartemisinin in childrena
| Parameter | Mean | RSE % | Bootstrap median (95% CI) |
|---|---|---|---|
| Objective function value | 173.607 | 165.806 (112.984–205.681) | |
| Structural model parameters | |||
| Lag (h) | 0.611 | 4 | 0.615 (0.461–0.846) |
| ka (per h) | 1.88 | 16 | 1.95 (1.39–3.21) |
| CL/FDHA (liters/h/70 kg) | 82.4 | 10 | 81.4 (68.1–97.9) |
| V/FDHA (liters/70 kg) | 146 | 42 | 144 (112–180) |
| Variability model parameters (shrinkage %) | |||
| IOV in FDHA (%) | 31 (39, 58, 78) | 33 | 31 (6–49) |
| IIV in CL/FDHA (%) | 19 (12) | 12 | 18 (3–33) |
| IIV in V/FDHA (%) | 21 (18) | 18 | 21 (2–37) |
| RV for DHA (%) | 73 (12) | 8 | 71 (58–81) |
Abbreviations: lag, lag time; ka, absorption rate constant; CL/FDHA, clearance relative to bioavailability; V/FDHA, central volume of distribution relative to bioavailability; IOV, interoccasion variability; IIV, interindividual variability; RV, residual variability. IOV and IIV are presented as .
FIG 4.

Goodness-of-fit plots for dihydroartemisinin. The observed plasma dihydroartemisinin has been plotted against population (A) and individual (B) predicted plasma concentrations, and conditional weighted residuals have been plotted against time (C) and population predicted plasma concentrations (D).
FIG 5.
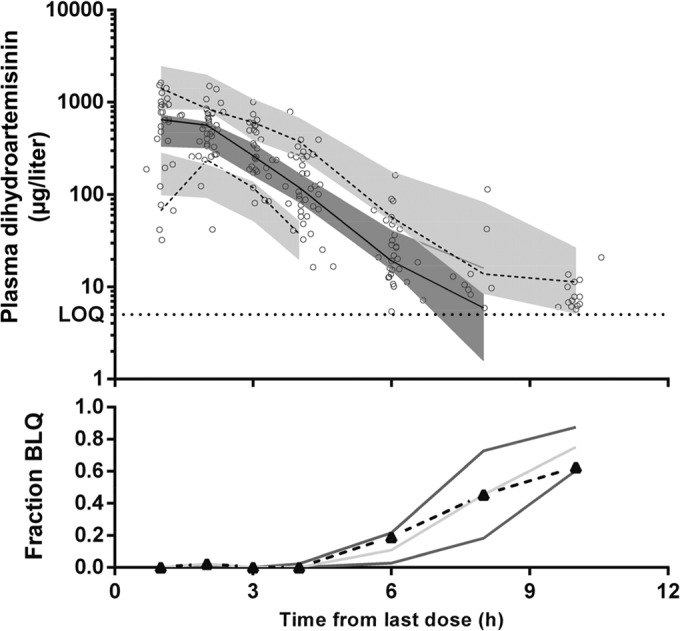
Prediction-corrected visual predictive check for dihydroartemisinin (μg/liter on log10 scale) with observed 50th (solid line) and 10th and 90th (dashed lines) percentiles within their simulated 95% CIs (gray shaded areas) with overlying data points (○).The fraction of observations below the limit of quantification (BLQ) from the data (▲ connected with a dashed black line) with the simulated 95% prediction interval is also shown.
TABLE 5.
Post hoc Bayesian estimates of pharmacokinetic parameters and derived secondary parameters for dihydroartemisinin in childrena
| Parameter | Water (n = 14) | Milk (n = 16) | P value |
|---|---|---|---|
| Lag (h) | 0.611 | 0.611 | |
| ka (per h) | 1.88 | 1.88 | |
| CL/FDHA (liters/h), median (IQR) | 32.2 (30.2–36.4) | 30.2 (27.2–33.6) | 0.52 |
| V/FDHA (liters), median (IQR) | 37.2 (32.5–46) | 41.1 (35.4–45.3) | 0.58 |
| t1/2 (h), median (IQR) | 0.851 (0.616–0.947) | 0.757 (0.647–0.952) | 0.79 |
| AUC0–∞ (μg · h/liter), median (IQR) | 4,047 (3,672-4,528) | 4,190 (3,853-4,464) | 0.67 |
Abbreviations: lag, lag time; ka, absorption rate constant; CL/FDHA, clearance relative to bioavailability; V/FDHA, central volume of distribution relative to bioavailability; t1/2, elimination half-life; AUC0–∞, area under the plasma concentration-time curve; IQR, interquartile range.
Pharmacokinetic/pharmacodynamic relationships.
Figure 6 shows the relationship between the mean ± SD plasma PQ concentration and mean ± SD QTc prolongation from baseline at 4 h, 24 h (day 1), and 168 h (day 7) in the two groups. While there was a linear relationship between these two variables in the case of group A, there was persistent mean QTc prolongation at later time points in group B children in the presence of declining plasma PQ concentrations.
FIG 6.
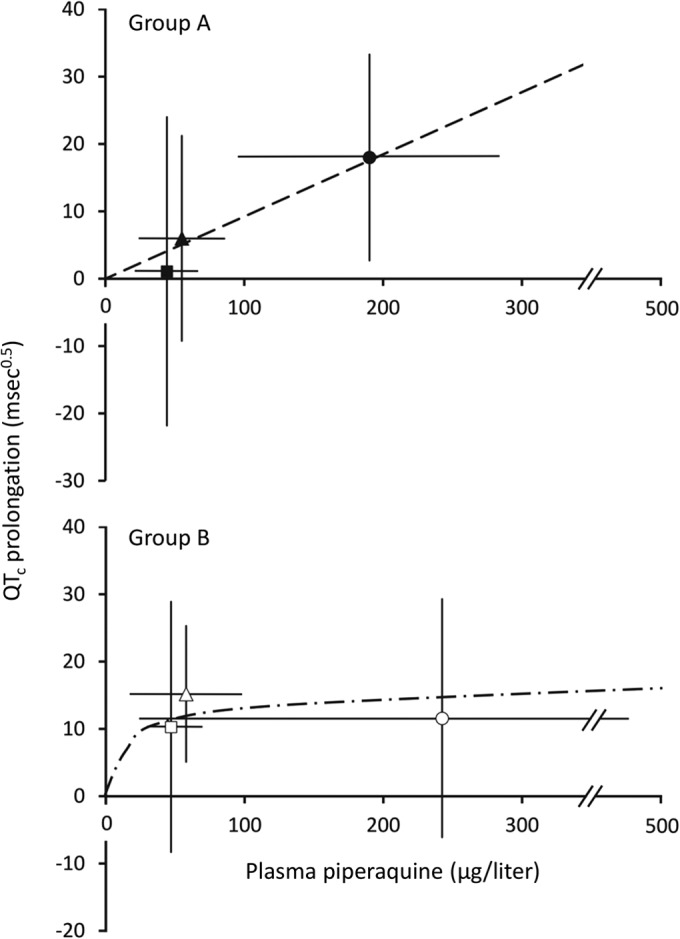
Plot of mean plasma piperaquine ± SD against mean QTc prolongation ± SD. Data are for group A (top; solid symbols) and group B (bottom; open symbols) at 4 h (●), 24 h (▲), and 168 h (■). The least-squares line of best fit through the origin for group A is shown. For group B, an illustrative polynomial curvilinear line of best fit through the origin is also shown.
DISCUSSION
The present study is the first to assess the effect of coadministered fat on the pharmacokinetics and pharmacodynamics of the widely used ACT DHA-PQ in children. Since most of our children were anorectic and/or nauseated at presentation because of the infection, and since PQ is a drug which is itself associated with nausea and vomiting (1, 33), we selected a tolerable volume of milk as the most suitable way of ensuring fat ingestion. None of our group B subjects vomited the doses of DHA-PQ administered with 8.5 g fat. Adult studies have used a similar approach and, as in the present study, have shown no effect of a small amount of fat on the bioavailability of PQ (11–13). Our data extend these findings to include an assessment of the effects of coadministered fat on the most important safety concern associated with PQ-based treatment regimens, specifically QTc prolongation (8). Consistent with the pharmacokinetic data, there was no statistically significant difference in QTc prolongation between the two groups posttreatment, but we cannot exclude a delayed and persistent effect of fat coadministration on ventricular repolarization.
Conventional doses of DHA-PQ were generally well tolerated by the children in both groups, and there were no severe adverse events, including progression to severe malarial illness. Initial parasite clearance was prompt, and consistent with our large intervention trial (2), a relatively small minority of subjects (7%) developed recurrent P. falciparum parasitemia during the 42-day follow-up period. Gametocyte carriage also declined during follow-up. There was a mild early fall in hemoglobin, but mean concentrations had exceeded those at presentation by day 14. The expected PQ-associated posttreatment QTc prolongation (34) occurred in both groups, but there was no other evidence of cardiotoxicity or other adverse effects. Overall, coadministration of fat was associated with neither benefit nor harm. In particular, the present data provide no convincing evidence that, as has been suggested previously (4), the DHA-treated children who participated in our previous intervention trial (2) should have been given milk rather than water with each dose to improve outcome.
It is unlikely that patients with malaria-associated symptoms, including nausea and anorexia, would tolerate the sort of high-fat meal that can increase the bioavailability and potential toxicity of DHA-PQ (7, 8), even if it were inadvertently available. However, since all studies of PQ bioavailability to date involving higher fat supplementation than that in the present study have been conducted in healthy adult volunteers (7–10), it is difficult to assess tolerability in the field. On the basis of the present and other studies (11, 12), we suggest that small amounts of fat (<10 g) can be given safely to patients treated with conventional DHA-PQ doses but that caution be exercised with higher fat loads or where the amount of fat to be consumed cannot be reliably estimated (8).
The QTc prolongation observed in the present patients was consistent with that in previous studies of DHA-PQ in children with uncomplicated malaria (15, 16, 34). Our primary focus was on the QTc interval at around the time of the maximum concentration after the first dose (4 h) (23). This was because a recent U.S. study of children and adolescents presenting to an emergency department showed that one-third had QTc prolongation of >440 ms0.5 which did not have an obvious underlying cause but which then reverted to normal within days in most cases (35). This suggests that factors with the potential to prolong the QTc, such as fever (36) and serum electrolyte disturbances such as hypocalcemia (37), may be influential in affecting ventricular repolarization early in the course of infections such as malaria in children, but without leading to serious sequelae (35), even when a drug such as PQ is given (18).
Although only hypothesis generating, there were some nonsignificant trends in the data in the present study which may be worthy of further consideration. First, there appeared to be persistent mean QTc prolongation with fat coadministration that was evident at 24 h and 168 h despite relatively low mean plasma PQ concentrations in group B subjects at these times. This contrasted with the apparently linear relationship between these two variables in group A and seems paradoxical since there is evidence that feeding shortens the QTc interval (38). Second, clearance of both asexual and sexual parasite forms tended to be more rapid in group B patients, who were also less likely to develop late recurrent parasitemia. This finding is consistent with a similar study of DHA-PQ in adults in which reinfection occurred earlier and more frequently in patients in the fasting group than in the fed group during prolonged (126-day) follow-up (12).
Since PQ is lipophilic and predominantly bound to lipoproteins in the blood (33), these various observations might indicate that the lipidemia resulting from fat supplementation in some way enhances tissue PQ uptake from the circulation with possible implications for improved efficacy but potentially greater toxicity. It may be of relevance to this hypothesis that the in vitro antimalarial potency of the phenanthrene drug halofantrine (an ACT partner drug that has been withdrawn because of cardiotoxicity) is increased when cultured P. falciparum is incubated with postprandial serum (39). In addition, there is evidence of greater vulnerability to halofantrine-induced QTc prolongation in the hyperlipidemic state in animal models (40).
The pharmacokinetic properties of PQ in the present children were consistent with those reported in previous pediatric patient samples (6, 23, 30, 41), including a median terminal elimination t1/2 of 18 days versus 14 to 23 days in other studies. There are few published pharmacokinetic data from children given oral DHA per se rather than other artemisinin drugs that are metabolized to DHA (42). Nevertheless, its relatively short elimination t1/2 in our patients (0.8 h) is in accord with available data (42), and the lack of effect of a small amount of fat is also consistent with similarly designed studies of other artemisinin drugs in PNG children (20).
The present study had limitations. First, especially given the high cure rate, the present sample size is well below that required to assess the comparative efficacy of DHA-PQ given with or without fat. Second, although measured plasma PQ concentrations 4 h after the first dose (median [interquartile range], 185 [107 to 264] μg/liter) were not significantly different from model-derived Cmax values (257 [126 to 356] μg/liter) at a time to Cmax (Tmax) of 3.1 (2.2 to 4.4) h (P = 0.19 by Wilcoxon signed-rank test), we did not draw blood samples or measure the QTc around the Tmax after the other two doses (at 28 and 52 h) in all participants. This was because, as discussed above, there is greatest clinical concern early in treatment but also because the sparse sampling and electrocardiographic monitoring procedures were designed to minimize the burden on children and their parents/guardians. In any case, in 5 of the children (17%), the estimated Cmax after the third dose was less than the Cmax after the first dose, consistent with significant between-dose variability in PQ disposition.
In summary, we found that coadministration of 8.5 g of fat did not change the bioavailability of either component of DHA-PQ given as treatment for uncomplicated malaria in PNG children. There was no evidence of an effect of fat supplementation on the QTc when plasma PQ concentrations were at their maximum after the first dose, but there was a trend to persistent QTc prolongation subsequently in the children who took DHA-PQ with milk. The latter observation and possibly enhanced effects of coadministered fat on parasitological response in the present and other studies may warrant further investigation, but the present data indicate that DHA-PQ can be safely given with a small amount of fat (<10 g) in uncomplicated malaria in the pediatric age group.
ACKNOWLEDGMENTS
We thank the children and their parents/guardians for their participation. We are also most grateful to Sister Valsi Kurian and the staff of Alexishafen Health Centre for their kind cooperation during the study and to staff of the Papua New Guinea Institute of Medical Research for clinical, laboratory, and logistic assistance.
The study was funded by the National Health and Medical Research Council (NHMRC) of Australia (project grant 634343). B.R.M. was supported by an NHMRC Early Career Fellowship (1036951), L.J.R. was supported by an NHMRC Early Career Fellowship (1016443), I.M. was supported by an NHMRC Senior Research Fellowship (1043345), and T.M.E.D. was supported by an NHMRC Practitioner Fellowship (572561 and 1058260).
No conflicts of interest are declared.
Footnotes
Published ahead of print 21 July 2014
REFERENCES
- 1.Zani B, Gathu M, Donegan S, Olliaro PL, Sinclair D. 2014. Dihydroartemisinin-piperaquine for treating uncomplicated Plasmodium falciparum malaria. Cochrane Database Syst. Rev. 1:CD010927. 10.1002/14651858.CD010927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karunajeewa HA, Mueller I, Senn M, Lin E, Law I, Gomorrai PS, Oa O, Griffin S, Kotab K, Suano P, Tarongka N, Ura A, Lautu D, Page-Sharp M, Wong R, Salman S, Siba P, Ilett KF, Davis TM. 2008. A trial of combination antimalarial therapies in children from Papua New Guinea. N. Engl. J. Med. 359:2545–2557. 10.1056/NEJMoa0804915 [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization. 2010. WHO Global Malaria Programme. Guidelines for the treatment of malaria. World Health Organization, Geneva, Switzerland [Google Scholar]
- 4.Price RN, Dorsey G, Nosten F. 2009. Antimalarial therapies in children from Papua New Guinea. N. Engl. J. Med. 360:1254–1255. 10.1056/NEJMc090023 [DOI] [PubMed] [Google Scholar]
- 5.WorldWide Antimalarial Resistance Network (WWARN) DP Study Group. 2013. The effect of dosing regimens on the antimalarial efficacy of dihydroartemisinin-piperaquine: a pooled analysis of individual patient data. PLoS Med. 10:e1001564. 10.1371/journal.pmed.1001564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tarning J, Zongo I, Some FA, Rouamba N, Parikh S, Rosenthal PJ, Hanpithakpong W, Jongrak N, Day NJP, White NJ, Nosten F, Ouedraogo J-B, Lindegardh N. 2012. Population pharmacokinetics and pharmacodynamics of piperaquine in children with uncomplicated falciparum malaria. Clin. Pharmacol. Ther. 91:497–505. 10.1038/clpt.2011.254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sim I-K, Davis TME, Ilett KF. 2005. Effects of a high-fat meal on the relative oral bioavailability of piperaquine. Antimicrob. Agents Chemother. 49:2407–2411. 10.1128/AAC.49.6.2407-2411.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.European Medicines Agency. 28 November 2011. Eurartesim: Annex I. Summary of product characteristics. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/001199/human_med_001450.jsp&mid=WC0b01ac058001d124 Accessed May 2014
- 9.Hai TN, Hietala SF, van Huong N, Ashton M. 2008. The influence of food on the pharmacokinetics of piperaquine in healthy Vietnamese volunteers. Acta Trop. 107:145–149. 10.1016/j.actatropica.2008.05.013 [DOI] [PubMed] [Google Scholar]
- 10.Nguyen TC, Nguyen NQ, Nguyen XT, Bui D, Travers T, Edstein MD. 2008. Pharmacokinetics of the antimalarial drug piperaquine in healthy Vietnamese subjects. Am. J. Trop. Med. Hyg. 79:620–623 [PubMed] [Google Scholar]
- 11.Annerberg A, Lwin KM, Lindegardh N, Khrutsawadchai S, Ashley E, Day NJP, Singhasivanon P, Tarning J, White NJ, Nosten F. 2011. A small amount of fat does not affect piperaquine exposure in patients with malaria. Antimicrob. Agents Chemother. 55:3971–3976. 10.1128/AAC.00279-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tarning J, Lindegardh N, Lwin KM, Annerberg A, Kiricharoen L, Ashley E, White NJ, Nosten F, Day NPJ. 2014. Population pharmacokinetic assessment of the effect of food on piperaquine bioavailability in patients with uncomplicated malaria. Antimicrob. Agents Chemother. 58:2052–2058. 10.1128/AAC.02318-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lwin KM, Phyo AP, Tarning J, Hanpithakpong W, Ashley EA, Lee SJ, Cheah P, Singhasivanon P, White NJ, Lindegardh N, Nosten F. 2012. Randomized, double-blind, placebo-controlled trial of monthly versus bimonthly dihydroartemisinin-piperaquine chemoprevention in adults at high risk of malaria. Antimicrob. Agents Chemother. 56:1571–1577. 10.1128/AAC.05877-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White NJ. 2007. Cardiotoxicity of antimalarial drugs. Lancet Infect. Dis. 7:549–558. 10.1016/S1473-3099(07)70187-1 [DOI] [PubMed] [Google Scholar]
- 15.Bassat Q, Mulenga M, Tinto H, Piola P, Borrmann S, Menendez C, Nambozi M, Valea I, Nabasumba C, Sasi P, Bacchieri A, Corsi M, Ubben D, Talisuna A, D'Alessandro U. 2009. Dihydroartemisinin-piperaquine and artemether-lumefantrine for treating uncomplicated malaria in African children: a randomised, non-inferiority trial. PLoS One 4:e7871. 10.1371/journal.pone.0007871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valecha N, Phyo AP, Mayxay M, Newton PN, Krudsood S, Keomany S, Khanthavong M, Pongvongsa T, Ruangveerayuth R, Uthaisil C, Ubben D, Duparc S, Bacchieri A, Corsi M, Rao BHK, Bhattacharya PC, Dubhashi N, Ghosh SK, Dev V, Kumar A, Pukittayakamee S. 2010. An open-label, randomised study of dihydroartemisinin-piperaquine versus artesunate-mefloquine for falciparum malaria in Asia. PLoS One 5:e11880. 10.1371/journal.pone.0011880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borsini F, Crumb W, Pace S, Ubben D, Wible B, Yan GX, Funck-Brentano C. 2012. In vitro cardiovascular effects of dihydroartemisin-piperaquine combination compared with other antimalarials. Antimicrob. Agents Chemother. 56:3261–3270. 10.1128/AAC.05688-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keating GM. 2012. Dihydroartemisinin/piperaquine: a review of its use in the treatment of uncomplicated Plasmodium falciparum malaria. Drugs 72:937–961. 10.2165/11203910-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 19.Michon P, Cole-Tobian JL, Dabod E, Schoepflin S, Igu J, Susapu M, Tarongka N, Zimmerman PA, Reeder JC, Beeson JG, Schofield L, King CL, Mueller I. 2007. The risk of malarial infections and disease in Papua New Guinean children. Am. J. Trop. Med. Hyg. 76:997–1008 [PMC free article] [PubMed] [Google Scholar]
- 20.Batty KT, Salman S, Moore BR, Benjamin J, Lee ST, Page-Sharp M, Pitus N, Ilett KF, Mueller I, Hombhanje FW, Siba P, Davis TM. 2012. Artemisinin-naphthoquine combination therapy for uncomplicated pediatric malaria: a pharmacokinetic study. Antimicrob. Agents Chemother. 56:2472–2484. 10.1128/AAC.06250-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Papua New Guinea Department of Health. 2000. Standard treatment of common illnesses of children in Papua New Guinea, 7th ed. Government of Papua New Guinea, Port Moresby, Papua New Guinea [Google Scholar]
- 22.Laman M, Moore BR, Benjamin J, Padapu N, Tarongka N, Siba P, Betuela I, Mueller I, Robinson LJ, Davis TM. 2014. Comparison of an assumed versus measured leucocyte count in parasite density calculations in Papua New Guinean children with uncomplicated malaria. Malar. J. 13:145. 10.1186/1475-2875-13-145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karunajeewa HA, Ilett KF, Mueller I, Siba P, Law I, Page-Sharp M, Lin E, Lammey J, Batty KT, Davis TM. 2008. Pharmacokinetics and efficacy of piperaquine and chloroquine in Melanesian children with uncomplicated malaria. Antimicrob. Agents Chemother. 52:237–243. 10.1128/AAC.00555-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hung T-Y, Davis TME, Ilett K. 2003. Measurement of piperaquine in plasma by liquid chromatography with ultraviolet absorbance detection. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 791:93–101. 10.1016/S1570-0232(03)00209-5 [DOI] [PubMed] [Google Scholar]
- 25.Salman S, Page-Sharp M, Griffin S, Kose K, Siba PM, Ilett KF, Mueller I, Davis TM. 2011. Population pharmacokinetics of artemether, lumefantrine, and their respective metabolites in Papua New Guinean children with uncomplicated malaria. Antimicrob. Agents Chemother. 55:5306–5313. 10.1128/AAC.05136-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson BJ, Holford NH. 2009. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab. Pharmacokinet. 24:25–36. 10.2133/dmpk.24.25 [DOI] [PubMed] [Google Scholar]
- 27.Savic RM, Jonker DM, Kerbusch T, Karlsson MO. 2007. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J. Pharmacokinet. Pharmacodyn. 34:711–726. 10.1007/s10928-007-9066-0 [DOI] [PubMed] [Google Scholar]
- 28.Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. 2011. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 13:143–151. 10.1208/s12248-011-9255-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Savic RM, Karlsson MO. 2009. Importance of shrinkage in empirical Bayes estimates for diagnostics: problems and solutions. AAPS J. 11:558–569. 10.1208/s12248-009-9133-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salman S, Page-Sharp M, Batty KT, Kose K, Griffin S, Siba P, Ilett KF, Mueller I, Davis TME. 2012. A pharmacokinetic comparison of two piperaquine-containing artemisinin combination therapies in Papua New Guinean children with uncomplicated malaria. Antimicrob. Agents Chemother. 56:3288–3297. 10.1128/AAC.06232-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahn JE, Karlsson MO, Dunne A, Ludden TM. 2008. Likelihood based approaches to handling data below the quantification limit using NONMEM VI. J. Pharmacokinet. Pharmacodyn. 35:401–421. 10.1007/s10928-008-9094-4 [DOI] [PubMed] [Google Scholar]
- 32.Manning L, Laman M, Page-Sharp M, Salman S, Hwaiwhanje I, Morep N, Siba P, Mueller I, Karunajeewa HA, Davis TM. 2011. Meningeal inflammation increases artemether concentrations in cerebrospinal fluid in Papua New Guinean children treated with intramuscular artemether. Antimicrob. Agents Chemother. 55:5027–5033. 10.1128/AAC.00375-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis TM, Hung TY, Sim IK, Karunajeewa HA, Ilett KF. 2005. Piperaquine: a resurgent antimalarial drug. Drugs 65:75–87. 10.2165/00003495-200565010-00004 [DOI] [PubMed] [Google Scholar]
- 34.Karunajeewa H, Lim C, Hung TY, Ilett KF, Denis MB, Socheat D, Davis TM. 2004. Safety evaluation of fixed combination piperaquine plus dihydroartemisinin (Artekin) in Cambodian children and adults with malaria. Br. J. Clin. Pharmacol. 57:93–99. 10.1046/j.1365-2125.2003.01962.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Dorn CS, Johnson JN, Taggart NW, Thorkelson L, Ackerman MJ. 2011. QTc values among children and adolescents presenting to the emergency department. Pediatrics 128:e1395–1401. 10.1542/peds.2010-1513 [DOI] [PubMed] [Google Scholar]
- 36.Lim SM, Pak HN, Lee MH, Kim SS, Joung B. 2011. Fever-induced QTc prolongation and ventricular fibrillation in a healthy young man. Yonsei Med. J. 52:1025–1027. 10.3349/ymj.2011.52.6.1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis TM, Li GQ, Guo XB, Spencer JL, St. John A. 1993. Serum ionized calcium, serum and intracellular phosphate, and serum parathormone concentrations in acute malaria. Trans. R. Soc. Trop. Med. Hyg. 87:49–53 [DOI] [PubMed] [Google Scholar]
- 38.Taubel J, Wong AH, Naseem A, Ferber G, Camm AJ. 2012. Shortening of the QT interval after food can be used to demonstrate assay sensitivity in thorough QT studies. J. Clin. Pharmacol. 52:1558–1565. 10.1177/0091270011419851 [DOI] [PubMed] [Google Scholar]
- 39.Humberstone AJ, Cowman AF, Horton J, Charman WN. 1998. Effect of altered serum lipid concentrations on the IC50 of halofantrine against Plasmodium falciparum. J. Pharm. Sci. 87:256–258. 10.1021/js970279q [DOI] [PubMed] [Google Scholar]
- 40.Patel JP, Brocks DR. 2010. Effect of experimental hyperlipidaemia on the electrocardiographic effects of repeated doses of halofantrine in rats. Br. J. Pharmacol. 161:1427–1440. 10.1111/j.1476-5381.2010.00983.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hung TY, Davis TME, Ilett K, Karunajeewa H, Hewitt S, Denis MB, Lim C, Socheat D. 2004. Population pharmacokinetics of piperaquine in adults and children with uncomplicated falciparum or vivax malaria. Br. J. Clin. Pharmacol. 57:253–262. 10.1046/j.1365-2125.2003.02004.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pawluk SA, Wilby KJ, Ensom MH. 2013. Pharmacokinetic profile of artemisinin derivatives and companion drugs used in artemisinin-based combination therapies for the treatment of Plasmodium falciparum malaria in children. Clin. Pharmacokinet. 52:153–167. 10.1007/s40262-012-0026-5 [DOI] [PubMed] [Google Scholar]