

The C. jejuni phosphoethanolamine transferase EptC functions in antibacterial resistance, motility and host intestinal colonization. In this study, the three-dimensional structure of the catalytic domain of EptC is reported in a covalent phosphoenzyme-intermediate state. EptC active-site mutant strains of C. jejuni demonstrate the potential for EptC enzyme inhibitors.
Keywords: EptC, phosphoethanolamine transferase, Campylobacter jejuni, polymyxin resistance, motility
Abstract
The foodborne enteric pathogen Campylobacter jejuni decorates a variety of its cell-surface structures with phosphoethanolamine (pEtN). Modifying lipid A with pEtN promotes cationic antimicrobial peptide resistance, whereas post-translationally modifying the flagellar rod protein FlgG with pEtN promotes flagellar assembly and motility, which are processes that are important for intestinal colonization. EptC, the pEtN transferase required for all known pEtN cell-surface modifications in C. jejuni, is a predicted inner-membrane metalloenzyme with a five-helix N-terminal transmembrane domain followed by a soluble sulfatase-like catalytic domain in the periplasm. The atomic structure of the catalytic domain of EptC (cEptC) was crystallized and solved to a resolution of 2.40 Å. cEptC adopts the α/β/α fold of the sulfatase protein family and harbors a zinc-binding site. A phosphorylated Thr266 residue was observed that was hypothesized to mimic a covalent pEtN–enzyme intermediate. The requirement for Thr266 as well as the nearby residues Asn308, Ser309, His358 and His440 was ascertained via in vivo activity assays on mutant strains. The results establish a basis for the design of pEtN transferase inhibitors.
1. Introduction
Bacteria maintain a complex outer surface that gives rise to a variety of biological functions. O-Linked phospho-form modifications such as phosphoethanolamine (pEtN), phosphoglycerol and phosphocholine are found on a variety of surface molecules, where they promote biosynthesis and pathogenic functions in certain organisms. For example, pEtN modifications are commonly associated with lipopolysaccharide/lipooligosaccharide (LPS/LOS) in Gram-negative bacteria and have dedicated roles in the recognition and evasion of host innate immune responses (Raetz et al., 2007 ▶; Trent et al., 2006 ▶; Needham & Trent, 2013 ▶). More recently, post-translational phospho-form modifications of the type IV pili subunits of pathogenic Neisseria species and the flagellar rod protein FlgG of Campylobacter jejuni have extended these modifications to cell-surface proteins (Cullen & Trent, 2010 ▶; Chamot-Rooke et al., 2011 ▶; Naessan et al., 2008 ▶).
Our laboratory previously identified the EptC protein, a unique pEtN transferase in C. jejuni (Cullen & Trent, 2010 ▶). C. jejuni is a Gram-negative enteric pathogen responsible, in large part, for foodborne illness and bacteria-associated diarrhea in the United States and abroad (Scallan et al., 2011 ▶; Havelaar et al., 2013 ▶). In terms of burden to health and cost, campylobacteriosis ranks highest among foodborne infections (Batz et al., 2012 ▶). Contracted by ingestion of contaminated poultry and beef products, the disease is characterized by gastrointestinal colonization followed by acute diarrhea. In rare instances, secondary neurological symptoms may persist in the form of the debilitating autoimmune disease Guillain–Barré syndrome (Yuki & Hartung, 2012 ▶; Israeli et al., 2012 ▶).
C. jejuni has evolved a single pEtN transferase, EptC, that modifies an array of cell-surface molecules, including the 1- and 4′-phosphoryl groups of lipid A (Cullen & Trent, 2010 ▶), the first heptose residue of the inner-core oligosaccharide of LOS (Cullen et al., 2013 ▶), residue Thr75 of the flagellar rod protein FlgG (Cullen et al., 2012 ▶) and the N-linked glycans of numerous glycoproteins (Scott et al., 2012 ▶) (Fig. 1 ▶). Conversely, most mucosal pathogens appear to express several pEtN transferases dedicated to the modification of one specific target. For example, pathogenic Neisseria species produce the enzymes LptA, Lpt3, Lpt6 and PptA that specifically transfer pEtN to only one of the following: lipid A phosphoryl groups (Cox et al., 2003 ▶), position 3 of LOS core heptose II (Mackinnon et al., 2002 ▶; Wenzel et al., 2010 ▶), position 6 of LOS core heptose II (Wright et al., 2004 ▶) or the major pilin protein PilE (Naessan et al., 2008 ▶), respectively. Likewise, Escherichia coli species possess at least three functional pEtN transferases (EptA, EptB and CptA) that attach pEtN groups to specific positions of LPS (Raetz et al., 2007 ▶). Owing to its uniquely promiscuous nature, EptC is linked to multiple and distinct biological functions in C. jejuni. Mutational studies demonstrate that the loss of pEtN modifications causes a drastic reduction in flagellar assembly/motility and a significantly higher sensitivity to a range of cationic antimicrobial peptides (Cullen & Trent, 2010 ▶; Cullen et al., 2012 ▶). Most importantly, EptC expression is required for proper intestinal colonization of C. jejuni in avian and mouse infection models (Cullen et al., 2013 ▶).
Figure 1.

Topology and biological functions of the C. jejuni phosphoethanolamine transferase EptC. (a) EptC is predicted to be anchored to the inner membrane by a five-helix transmembrane domain, with its sulfatase-like catalytic domain positioned in the periplasm. EptC transfers pEtN (red circles) from the head groups of phosphatidylethanolamine onto the LOS core (gray rectangles) and phosphates (white circles) of lipid A and Thr75 of FlgG proteins (blue boxes). Modifications of these C. jejuni surface structures with pEtN promote flagellar assembly, motility, cationic antimicrobial peptide resistance and host intestinal colonization (Cullen & Trent, 2010 ▶; Cullen et al., 2012 ▶, 2013 ▶). The organization of flagellar components has previously been proposed (Chevance & Hughes, 2008 ▶). (b) Chemical structures of C. jejuni LOS and phosphatidylethanolamine, with pEtN moieties in red. R 1, β-d-glucose; R 2, outer core sugars; pEtN, phosphoethanolamine; PtdEtN, phosphatidylethanolamine; DAG, diacylglycerol; LOS, lipooligosaccharide.
The eptC gene (locus tag Cj0256) is clustered in a family of inner-membrane metalloenzymes (COG2194) containing a five-helix transmembrane domain and a periplasmic catalytic domain that is currently grouped in the sulfatase family (Pfam00884; Marchler-Bauer et al., 2013 ▶; Fig. 1 ▶). Placement of the catalytic domain dictates that pEtN modification occurs on the periplasmic side of the inner membrane, where EptC transfers head groups from the phospholipid phosphatidylethanolamine to target molecules. Indeed, EptC activity is not observed when the enzyme is heterologously expressed in a phosphatidylethanolamine-deficient E. coli strain (Cullen & Trent, 2010 ▶). To gain insight into the structural basis of enzyme function, we crystallized and determined the atomic coordinates of the catalytic domain of EptC (cEptC) to a resolution of 2.40 Å. Our structural data along with mutational analysis of EptC allowed us to identify zinc-ligand residues, a putative nucleophile and conserved active-site residues required for in vivo activity. Moreover, we juxtaposed the structures of cEptC and the catalytic domains of the related enzymes cLtaS (lipoteichoic acid synthase from Bacillus subtilis and Staphylococcus aureus) and cLptA (lipid A pEtN transferase from N. meningitidis). The observed structural differences around the active sites of cEptC and the lipid A-specific cLptA may provide clues to the promiscuous nature of EptC.
2. Materials and methods
2.1. Strains and plasmid construction
The primers, plasmids and strains used in this study are listed in Tables 1 ▶ and 2 ▶. Plasmid pETcEptC was constructed for the overexpression of the catalytic domain of EptC (cEptC; residues 203–512) fused to a Gly6His8 tag at the C-terminus using the Polymerase Incomplete Primer Extension (PIPE) method (Klock & Lesley, 2009 ▶). Briefly, the plasmid pET-28b(+) (Novagen) was linearized using the V-PIPE primers 1 and 2 and subsequently gel-purified and treated with the restriction enzyme DpnI (New England Biolabs). The insert was colony PCR-amplified from C. jejuni 81-176 using the I-PIPE primers 3 and 4. The insert and linearized vector were mixed, transformed into XL1-Blue E. coli cells (Stratagene) and incubated overnight at 37°C on LB–agar plates containing 50 µg ml−1 kanamycin.
Table 1. Bacterial strains and plasmids.
| Strains and plasmids | Description | Source |
|---|---|---|
| Escherichia coli | ||
| XL1-Blue | General cloning strain, recA1 endA1 gyrA96thi-1 hsdR17 | Stratagene |
| BL21(DE3) | Protein-expression strain, F− ompT gal dcm lon hsdSB(rB − mB −) λ(DE3) | Novagen |
| Campylobacter jejuni | ||
| 81-176 | Serotype HS: 23, 26 | Cullen & Trent (2010 ▶) |
| 81-176 A1 | 81-176 Δcj0256 clone1, CamR | Cullen & Trent (2010 ▶) |
| 81-176 B | Complement strain, 81-176 Δcj0256 ΔastA::cj0256, CamR, KanR | Cullen & Trent (2010 ▶) |
| 81-176 eptCT266A | 81-176 Δcj0256 ΔastA::cj0256 T266A, CamR, KanR | This study |
| 81-176 eptCT266S | 81-176 Δcj0256 ΔastA::cj0256 T266S, CamR, KanR | This study |
| 81-176 eptCN308A | 81-176 Δcj0256 ΔastA::cj0256 N308, CamR, KanR | This study |
| 81-176 eptCS309A | 81-176 Δcj0256 ΔastA::cj0256 S308A, CamR, KanR | This study |
| 81-176 eptCH358A | 81-176 Δcj0256 ΔastA::cj0256 H358A, CamR, KanR | This study |
| 81-176 eptCH440A | 81-176 Δcj0256 ΔastA::cj0256 H4440A, CamR, KanR | This study |
| Plasmids | ||
| pET-28b(+) | Expression vector containing a T7 promoter, AmpR | Novagen |
| pETcEptC | pET-28b(+)-derived expression plasmid used to express cEptC, AmpR | This study |
| pAtsAKO::cj0256:KanR | pGEM-atsAKO:KanR with cj0256 inserted | Cullen & Trent (2010 ▶) |
| pETcEptCT266S | T266S mutant derivative of pETcEptC, AmpR | This study |
| pAstAEptCT266A | T266A mutant derivative of pAtsAKO::cj0256:KanR | This study |
| pAstAEptCT266S | T266S mutant derivative of pAtsAKO::cj0256:KanR | This study |
| pAstAEptCN308A | N308A mutant derivative of pAtsAKO::cj0256:KanR | This study |
| pAstAEptCS309A | S309A mutant derivative of pAtsAKO::cj0256:KanR | This study |
| pAstAEptCH358A | H358A mutant derivative of pAtsAKO::cj0256:KanR | This study |
| pAstAEptCH4440A | H440A mutant derivative of pAtsAKO::cj0256:KanR | This study |
Table 2. Primers used in this study.
Underlined regions indicate introduced mutations.
| 1. F-VPIPEcEptC | 5′-ACCCCCGCCTCCCCTGGATTTGCCTTAAGTTTAGGATTTAA-3′ |
| 2. R-VPIPEcEptC | 5′-AGGAGATATACCATGTTTAAAACTATAGCTAATGATGCTTATA-3′ |
| 3. F-IPIPEcEptC | 5′-GGGGAGGCGGGGGTGGACACCACCACCACCACCACCACCACTGAGAT-3′ |
| 4. R-IPIPEcEptC | 5′-CATGGTATATCTCCTTCTTAAAGTTAAACAAAATTATT-3′ |
| 5. F-EptCT266S | 5′-ATTTTTCATCATGTGGAACAGCAAGCGCGGTAAGTTTGCCTTGTATGTTT-3′ |
| 6. R-EptCT266S | 5′-AAACATACAAGGCAAACTTACCGCGCTTGCTGTTCCACATGATGAAAAAT-3′ |
| 7. F-EptCT266A | 5′-ATTTTTCATCATGTGGAACAGCAGCGGCGGTAAGTTTGCCTTGTATGTTT-3′ |
| 8. R-EptCT266A | 5′-AAACATACAAGGCAAACTTACCGCCGCTGCTGTTCCACATGATGAAAAAT-3′ |
| 9. F-EptCN308A | 5′-GCATGGTTTGATAATGCATCTGGTGGTTGTAAA-3′ |
| 10. R-EptCN308A | 5′-TTTACAACCACCAGATGCATTATCAAACCATGC-3′ |
| 11. F-EptCS309A | 5′-TGGTTTGATAATAATGCAGGTGGTTGTAAAGGG-3′ |
| 12. R-EptCS309A | 5′-CCCTTTACAACCACCTGCATTATTATCAAACCA-3′ |
| 13. F-EptCH358A | 5′-CATTTGCAAGGTTCTGCAGGGCCAACTTATTAT-3′ |
| 14. R-EptCH358A | 5′-ATAATAAGTTGGCCCTCGAGAACCTTGCAAATG-3′ |
| 15. F-EptCH440A | 5′-AATGGTATTTATCTTGCAGGTATGCCTTATGCT-3′ |
| 16. R-EptCH440A | 5′-AGCATAAGGCATACCTCGAAGATAAATACCATT-3′ |
EptC active-site mutants were constructed by site-directed mutagenesis of plasmid pAtsAKO::cj0256-KanR (Cullen et al., 2012 ▶) using primers 5–16 (Tables 1 ▶ and 2 ▶). The mutant alleles were introduced into the eptC mutant strain C. jejuni 81-176 Δcj0256 as described previously (Cullen & Trent, 2010 ▶).
2.2. Expression, purification and crystallization of cEptC
E. coli BL21(DE3) cells expressing cEptC were cultured in LB with 50 mg l−1 kanamycin at 37°C. Upon reaching an OD600 of 0.6, the cultures were cooled to 18°C and induced with 0.5 mM IPTG. After 18 h, the cells were pelleted, resuspended in lysis buffer [500 mM NaCl, 30 mM HEPES pH 7.5, 10%(v/v) glycerol] and sonicated. The lysate was clarified by centrifugation and the soluble portion was poured over an Ni–NTA column (Expedeon) equilibrated with lysis buffer. The beads were washed with 15 mM imidazole in lysis buffer and the bound protein was eluted with 150 mM imidazole in lysis buffer. The eluate was further purified on a Superdex 200 gel-filtration column (GE Healthcare Life Sciences) equilibrated with 150 mM NaCl, 10 mM HEPES pH 7.5. Selenomethionine (SeMet)-derivative protein was purified similarly but was expressed in minimal medium with 50 mg l−1 kanamycin supplemented with amino acids for methionine-pathway inhibition (Doublié, 1997 ▶).
SeMet-derivative and native proteins were concentrated to 30 mg ml−1 in 25 mM NaCl, 10 mM HEPES pH 7.5. Crystals were grown over the course of several days by sitting-drop vapor diffusion at 22°C by combining 4 µl protein solution with 1 µl 200 mM diammonium phosphate, 15%(w/v) PEG 3350. SeMet-derived protein crystallized in space group P212121, while the native protein crystallized in space group C2221. Before flash-cooling in liquid nitrogen, the crystals were cryoprotected in 200 mM diammonium phosphate, 16.5%(w/v) PEG 3350, 10%(v/v) glycerol for 10 s.
2.3. X-ray diffraction data collection, processing, phasing and refinement
Native and SeMet-derivative data were collected on Advanced Photon Source beamline 23-ID-B and processed and scaled in HKL-2000 (Otwinowski & Minor, 1997 ▶) (Table 3 ▶). Reflection intensities were converted to amplitudes in TRUNCATE (French & Wilson, 1978 ▶) from the CCP4 software suite (Winn et al., 2011 ▶). A single-wavelength anomalous dispersion (SAD) data set was collected at the Se K edge (λ = 0.9795 Å) from one SeMet-derived cEptC crystal that diffracted to 2.80 Å resolution. Initial phases were obtained from AutoSol of the PHENIX software suite (Adams et al., 2010 ▶), which identified 46 Se sites (30 were expected for six monomers based on the sequence; figure of merit = 0.329) in a P212121 unit cell and generated six partial cEptC monomers in the asymmetric unit (R work/R free = 32.9/37.8%; map–model correlation coefficient = 0.747). One of the six monomers was subjected to several cycles of refinement in Coot (Emsley & Cowtan, 2004 ▶) and REFMAC5 (Murshudov et al., 2011 ▶) from CCP4 and was then employed as a molecular-replacement search model to phase the native data set in Phaser (McCoy et al., 2007 ▶) from CCP4. One native cEptC crystal diffracted to 2.50 Å resolution (Zn remote, λ = 1.0332 Å), with a C2221 unit cell containing three monomers. Iterative refinement was carried out in Coot and phenix.refine of PHENIX, with default NCS restraints applied in the latter. Notably, for the P212121 (SeMet-derivative) data set, phenix.xtriage from PHENIX identified an off-origin Patterson peak that was 30% of the origin peak, which strongly indicates the presence of translational pseudo-symmetry. No pseudo-symmetry was detected for the C2221 (native) data set.
Table 3. Data-collection and refinement statistics for cEptC.
Values in parentheses are for the outer shell.
| Native, Zn K edge | Native, Zn remote wavelength | SeMet derivative, Se K edge | |
|---|---|---|---|
| Data collection | |||
| Space group | C2221 | C2221 | P212121 |
| X-ray source | Advanced Light Source, 4.2.2 | Advanced Photon Source, 23-ID-B | Advanced Photon Source, 23-ID-B |
| Wavelength (Å) | 1.2680 | 1.0332 | 0.9795 |
| Temperature (K) | 100 | 100 | 100 |
| Unit-cell parameters (Å, °) | a = 121.9, b = 183.1, c = 121.5, α = β = γ = 90.0 | a = 122.5, b = 183.6, c = 121.7, α = β = γ = 90.0 | a = 119.1, b = 120.8, c = 182.7, α = β = γ = 90.0 |
| Resolution (Å) | 55–2.40 (2.46–2.40) | 50–2.50 (2.54–2.50) | 50–2.80 (2.85–2.80) |
| R merge † (%) | 11.2 (99.6) | 11.9 (95.5) | 12.1 (82.9) |
| CC1/2 ‡ (%) | 99.6 (67.4) | 99.0 (85.4) | 99.8 (75.6) |
| Average I/σ(I) | 10.6 (1.5) | 18.2 (2.8) | 14.3 (2.0) |
| No. of observed reflections | 392584 | 349675 | 729109 |
| No. of unique reflections | 53319 (3892) | 47144 (2327) | 119456 (5831) |
| Oscillation step (°) | 0.2 | 1.0 | 1.0 |
| No. of images | 900 | 180 | 360 |
| Exposure time per image (s) | 0.4 | 1.0 | 1.0 |
| Completeness (%) | 99.7 (99.6) | 99.9 (100) | 99.7 (97.6) |
| Multiplicity | 7.4 (7.4) | 7.4 (7.2) | 6.1 (4.9) |
| Monomers in asymmetric unit | 3 | 3 | 6 |
| Solvent content (%) | 61.8 | 60.6 | 59.0 |
| Matthews coefficient (Å3 Da−1) | 3.22 | 3.12 | 3.00 |
| Refinement | |||
| Resolution (Å) | 54.58–2.40 (2.43–2.40) | ||
| R work/R free § (%) | 21.5/25.6 (33.0/38.8) | ||
| No. of atoms | |||
| Overall | 7411 | ||
| Protein | 7378 | ||
| Ligand¶ | 15 | ||
| Water | 18 | ||
| Average B factors (Å2) | |||
| Overall | 49.8 | ||
| Protein | 49.8 | ||
| Ligand¶ | 44.3 | ||
| Water | 38.7 | ||
| R.m.s.d., bond lengths (Å) | 0.009 | ||
| R.m.s.d., bond angles (°) | 1.28 | ||
| Ramachandran favored (%) | 96.8 | ||
| Ramachandran allowed (%) | 3.2 | ||
| PDB code | 4tn0 | ||
R
merge = 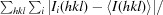
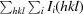 , where Ii(hkl) is the ith intensity measurement of reflection hkl and 〈I(hkl)〉 is the average intensity for all reflections.
, where Ii(hkl) is the ith intensity measurement of reflection hkl and 〈I(hkl)〉 is the average intensity for all reflections.
CC1/2 is the correlation coefficient between two random half data sets (Karplus & Diederichs, 2012 ▶; Diederichs & Karplus, 2013 ▶).
R
work/R
free = 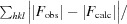
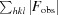 , where F
obs and F
calc are the observed and calculated structure factors, respectively.
, where F
obs and F
calc are the observed and calculated structure factors, respectively.
Includes phosphoryl group and Zn atoms.
The X-ray fluorescence of native cEptC crystals was examined on Advanced Light Source beamline 4.2.2 to identify the catalytic metal ion. A data set was collected at the Zn K edge (λ = 1.2680 Å) from one crystal to 2.40 Å resolution; this was processed in XDS (Kabsch, 2010 ▶) and scaled in SCALA (Evans, 2006 ▶) from CCP4. Data were truncated at a resolution at which a compromise was reached between the 〈I/σ(I)〉 and CC1/2 values (Karplus & Diederichs, 2012 ▶; Diederichs & Karplus, 2013 ▶). Refinement was carried out as described above. The two native data sets were merged in CAD to generate an anomalous difference map in FFT (Read & Schierbeek, 1988 ▶; both programs are from CCP4). The Zn K edge data set was selected for deposition owing to less apparent radiation damage to disulfides and zinc ions, as evident in the F o − F c density (Supplementary Fig. S11; Weik et al., 2000 ▶; Burmeister, 2000 ▶). The structure-factor amplitudes and atomic coordinates of cEptC were deposited in the RCSB Protein Data Bank as PDB entry 4tn0.
2.4. Protein mass spectrometry
LC-MS (AB Sciex 4000 QTrap hybrid triple quadrupole–linear ion trap with Shimadzu Prominence analytical LC) was employed in the collection of mass spectra. The protein sample was eluted with a 0.5 × 50 mm PLRP-S column (8 µm, 4000 Å; Michrom BioResources) with mobile phases A and B (acetonitrile:water:formic acid:trifluoroacetic acid; A = 2:98:0.1:0.01; B = 90:10:0.1:0.01). A linear gradient of 5–65% B was used to elute protein in 20 min at a flow rate of 20 µl min−1. Automated acquisition of full-scan MS spectra over the range 450–2000 Da was executed by Xcalibur (Thermo Scientific). The acquired convoluted protein spectra were deconvoluted by ProMass for Xcalibur 2.5.0 (Novatia) to yield the [M + H]+ mass-to-charge values.
2.5. Motility assays
C. jejuni strains were initially grown from freezer stocks on MH agar with 10 µg ml−1 trimethoprim for 48 h at 37°C under microaerobic conditions (5% O2, 10% CO2, 85% N2). Following the initial growth, the strains were re-streaked and grown under the same conditions. After 16 h, the strains were resuspended from agar plates in MH broth and diluted to an OD600 of 0.05, stab-inoculated into semisolid MH motility medium (containing 0.4% agar) and incubated for 24 h at 37°C under microaerobic conditions. Relative motilities were assessed from the diameter of the halos of growth. Experiments were performed in duplicate.
2.6. Polymyxin B resistance assay
Strains were grown in the same manner as described for the motility assays in §2.5 and swabbed onto MH agar plates with 10 µg ml−1 trimethoprim. Polymyxin B Etest strips (bioMérieux) were applied to the swabbed plates and incubated in microaerobic conditions for 48 h at 37°C before reading minimal inhibitory concentrations (MICs). Experiments were performed in duplicate.
3. Results and discussion
3.1. Overall structure of the catalytic domain of EptC
In order to examine the structural basis for enzyme function, we overexpressed the catalytic domain of EptC (cEptC; residues 203–512) fused to a C-terminal Gly6His8 tag and determined the X-ray crystal structure (Fig. 2 ▶). After testing many gene truncations, we obtained one that expressed sufficient soluble protein suitable for crystallography and confirmed the molecular weight of the gene product (Supplementary Fig. S2). Including the His tag, cEptC is composed of 325 residues and has a molecular weight of 36.7 kDa. The phases of cEptC were elucidated from a SAD data set collected from a SeMet-derivative cEptC crystal that diffracted to 2.80 Å resolution in space group P212121 (Table 3 ▶). Molecular replacement was carried out using one monomer from the SeMet derivative as a search model for the native data set. One native crystal diffracted to 2.40 Å resolution in space group C2221. With the exception of residues 213–215 of chains A and C and residues 212–214 of chain B, there is full coverage of the peptide backbone by the electron-density map between the initiating Met202 and Pro512 (preceding the C-terminal His tag). Elevated B factors (overall average = 49.8 Å2), particularly for the lower half of the structure as arranged in Fig. 2 ▶, indicate increased mobility in atomic positions: this may be owing to the absence of the N-terminal transmembrane region of the enzyme or to the dearth of crystal contacts in this region of each chain in the asymmetric unit.
Figure 2.

Structure of the C-terminal catalytic domain of the pEtN transferase EptC (cEptC) from C. jejuni. (a) Secondary-structural features of a monomer. α-Helices (green), β-strands (red) and loops (gray) are labeled as in Supplementary Fig. S3. Disulfides (yellow) and the phosphothreonine residue are displayed in ball-and-stick representation, Zn2+ (purple) is displayed as a sphere and the N- and C-termini are labeled. The dotted line indicates residues 213–215, for which there is no observable electron density in monomer A. (b) Zinc-binding residues of the phosphoenzyme intermediate. 2F o − F c map density (blue mesh) is contoured at 2.0 r.m.s.d. The metal ion was identified as Zn2+ by anomalous diffraction (see Supplementary Fig. S7). Coordination bonds between side chains and Zn2+ are indicated by dashed lines. All molecular images were generated in PyMOL (Schrödinger).
The cEptC catalytic domain assumes an α/β/α fold with a six-stranded, central β-sheet that is enveloped by a series of 11 α-helices (Fig. 2 ▶), imparting an overall hemispherical shape. β-Strands and α-helices are labeled β1–β8 and α1–α11, respectively (Supplementary Fig. S3). The α/β/α fold is common to proteins in the alkaline phosphatase superfamily (pfam00245), which includes sulfatases. Enzymes of this family cleave ester bonds such as sulfoester and phosphoester linkages, a reaction that is likely to be required for EptC function. A zinc-binding site, identified by anomalous diffraction, is located on the flat surface of the hemisphere and is fairly exposed to solvent (Supplementary Fig. S4a). As the putative nucleophile Thr266 resides at the N-terminus of α2, a helical dipole may help to stabilize a nucleophilic alkoxide form of its side chain. A phosphoryl group was found attached to Thr266 to form phosphothreonine (Supplementary Fig. S5), which we speculate mimics a pEtN–enzyme intermediate. Consistent with the location of cEptC in the oxidizing environment of the periplasm, three disulfide bonds are present (Fig. 2 ▶): between residues Cys262 and Cys272, Cys312 and Cys316, and Cys377 and Cys385. Cys272 is located on the nucleophilic helix, while Cys262 is on the loop extending from the N-terminus of that helix. Cys312 resides on a loop near the nucleophilic Thr266 and, with Cys316, may play a role in properly positioning an active-site loop, as discussed below. In contrast, the Cys377–Cys385 pair is more distant from the active site.
3.2. Comparison of cEptC with known phospho-form transferase structures
Phospho-form transferase enzymes transfer phospho-form structures such as pEtN and phosphoglycerol to the hydroxyl and phosphoryl groups of target molecules to promote their biological functions. In addition to the cEptC structure presented here, the structures of two other phospho-form transferase proteins have previously been reported: the catalytic domain of S. aureus (Lu et al., 2009 ▶) and B. subtilis (Schirner et al., 2009 ▶) lipoteichoic acid synthase LtaS (cLtaS; PDB entries 2w5s and 2w8d), and most recently the catalytic domain of the lipid A pEtN transferase LptA (cLptA; PDB entry 4kay) of N. meningitidis (Wanty et al., 2013 ▶). LtaS, a phosphoglycerol transferase that is distantly related to EptC, synthesizes the polyphosphoglycerol backbone structure of lipoteichoic acid in Gram-positive bacteria. LptA is functionally similar to EptC in that it adds pEtN to the phosphoryl groups of lipid A; however, LptA displays specificity for lipid A alone, while EptC has multiple molecular targets. Like cEptC, the C-terminal domains cLptA and cLtaS adopt the α/β/α alkaline phosphatase-like fold (Fig. 3 ▶), which is fused to a predicted N-terminal five-helix transmembrane domain. Together, these three proteins represent a class of bacterial membrane-associated, biosynthetic enzymes with similar activities (phospho-form transfer), although the products and biological outcomes are widely varied.
Figure 3.

Overall structures of the pEtN transferases cEptC and cLptA and the B. subtilis lipoteichoic acid synthase cLtaS. (a) cEptC (green), with phosphothreonine and disulfides shown in ball-and-stick representation and the Zn2+ ion as a purple sphere. (b) cLptA (PDB entry 4kay; cyan), with phosphothreonine and disulfides shown in ball-and-stick representation and Zn2+ ions as purple spheres. Structural insertions found in cLptA, but not cEptC, are colored yellow. cLptA and cEptC superpose with a Cα r.m.s.d. of 0.59 Å over 211 residues. (c) B. subtilis cLtaS (PDB entry 2w8d; orange), with phosphothreonine shown in ball-and-stick representation and the Mg2+ ion as a green sphere. Structural features unique to cLtaS are colored blue. cLtaS and cEptC superpose with a Cα r.m.s.d. of 3.74 Å over 216 residues.
The cEptC and cLtaS structures share a number of common core β-strands and α-helices that align with a Cα r.m.s.d. of 3.74 Å over 216 residues (Figs. 3 ▶ a and 3 ▶ c; Supplementary Figs. S4a and S4c). The main differences between the two structures are the positioning of the loop regions and the presence of insertions that are only observed in cLtaS. For instance, cLtaS has an extra 79-residue C-terminal region composed of a five-stranded β-sheet and a long α-helix. Additionally, a helix–loop–helix structure spanning residues 477–500 corresponds to a loop region between β6 and β7 of cEptC. Interestingly, this loop in the cEptC structure is oriented towards the active site and positions His440 N∊2 within hydrogen-bonding distance (3.0 Å) of a phosphoryl O atom of phospho-Thr266 (Figs. 3 ▶ a and 4 ▶ a). In contrast, the helix–loop–helix structure of cLtaS is oriented in the opposite direction, causing this region of the active site to be more open in cLtaS (Figs. 3 ▶ c and 4 ▶ c). Finally, LtaS contains an additional helix positioned in a loop region between the corresponding β2 and β3 of cEptC (Figs. 4 ▶ a and 4 ▶ c). Side chains on the N-terminal end of this helix and the preceding loop have been shown to bind phosphoglycerol in S. aureus cLtaS (PDB entry 2w5s; Lu et al., 2009 ▶). The major structural differences observed between cEptC and cLtaS presumably contribute to their distinct enzyme–substrate interactions.
Figure 4.

Juxtaposed active sites of (a) cEptC, (b) cLptA (PDB entry 4kay) and (c) S. aureus cLtaS (PDB entry 2w5s). Active-site architectures and zinc-binding residues are conserved between cEptC and cLptA (Supplementary Fig. S6), although only one Zn2+ ion associates with cEptC in crystallo. His358/His383 and metal-ligand residues of cEptC and cLptA are also conserved in cLtaS. Side chains on an active-site loop/helix of cLtaS, corresponding to a loop between β2 and β3 of cEptC, bind phosphoglycerol (PG, white C atoms; Lu et al., 2009 ▶). In cEptC and cLptA, this loop contains Asn308/Asp324 and Ser309/Ser325, which may contribute to substrate binding. Notably, Thr266, His358, His440, Asn308 and Ser309 mutant strains of eptC in C. jejuni displayed polymyxin B sensitivity and a severe loss of motility (Figs. 5 ▶ and 6 ▶).
The cEptC and cLptA structures are strikingly similar, as noted by their 44% amino-acid sequence identity and observed Cα r.m.s.d. of 0.59 Å over 211 residues (Figs. 3 ▶ a and 3 ▶ b; Supplementary Figs. S4a and S4b). The disulfide-bonding pattern of cLptA is identical to that of cEptC, with the exception of two additional disulfides between residues Cys348–Cys353 and Cys499–Cys540 in cLptA. Interestingly, these extra disulfides occur in the only regions that differ significantly from cEptC. Cys499 is part of an α-helix that corresponds to α10 of cEptC. Also, cLptA contains an extra eight-residue α-helix between the corresponding β8 and α10 of cEptC that, together with the Cys499–Cys540 disulfide, causes α10 to take on a different pitch than observed in cEptC. The second additional disulfide between Cys348 and Cys353 of cLptA is in a loop spanning residues 339–354 adjacent to the active site. Curiously, this loop is significantly shorter in the cEptC structure (residues 325–329), despite the similarity in sequence and structure to cLptA around this region. It is possible that this extended loop plays a part in substrate specificity in cLptA, and its absence from the cEptC structure may allow the entry of more diverse molecular targets.
3.3. Active-site architecture of cEptC
The cEptC active site contains a tetrahedrally coordinated zinc ion and a putative nucleophilic threonine, Thr266, covalently bound to a phosphoryl group to form phosphothreonine (Fig. 4 ▶ a). We reason that phospho-Thr266 mimics an enzyme reaction intermediate representing the first step in a two-step transferase reaction. Phosphothreonine was also observed in the cLptA (Wanty et al., 2013 ▶) and B. subtilis cLtaS (Schirner et al., 2009 ▶) structures (Figs. 3 ▶ b and 3 ▶c).
Coordinated divalent metals are important cofactors for enzymes in the alkaline phosphatase protein superfamily, as they are implicated in substrate binding and catalysis (Kim & Wyckoff, 1991 ▶; O’Brien & Herschlag, 2001 ▶; Stec et al., 2000 ▶). The cEptC zinc ion is tetrahedrally coordinated by residues Glu227, phospho-Thr266, Asp427 and His428. By sequence alignment and structural comparison, these residues are highly conserved in the characterized phospho-form transferase members of COG2194 (includes pEtN transferases) and COG1368 (includes phosphoglycerol transferases) (Supplementary Fig. S6). The orientation of these residues is well conserved between cEptC, cLptA and cLtaS, with r.m.s.d. values of 0.26 Å over 30 atoms and 0.18 Å over 21 atoms when comparing cEptC with cLptA and B. subtilis cLtaS, respectively (Supplementary Fig. S7). Of the two cLtaS structures solved, one was shown to bind a magnesium ion (Schirner et al., 2009 ▶) and the other a manganese ion (Lu et al., 2009 ▶) (compare Supplementary Fig. S7c and Fig. 4 ▶ c). The cLptA structure, on the other hand, contains two zinc ions (Fig. 4 ▶ b), reminiscent of the E. coli alkaline phosphatase PhoA (PDB entry 1ed8; Wanty et al., 2013 ▶; Stec et al., 2000 ▶). It is worth noting that only one zinc ion, corresponding to the zinc and magnesium/manganese ions of cEptC and cLtaS, was observed unless the cLptA crystals were soaked with 1 mM zinc sulfate. Nevertheless, the zinc-coordinating residues of cLptA superimpose well onto corresponding residues of alkaline phosphatase (Wanty et al., 2013 ▶) and, importantly, most of these residues are conserved in cEptC, cLtaS and several diverse pEtN transferases (Supplementary Fig. S6). More specifically, the first-zinc ligand residues Glu227/240, Thr266/280, Asp427/452 and His428/453 of cEptC/cLptA directly correspond to Asp51, Ser102, Asp369 and His370 of alkaline phosphatase, and the second-zinc ligand residues His358/383 and His440/465 correspond to Asp327 and His412 of alkaline phosphatase. Another second-zinc ligand residue, His331, is found in alkaline phosphatase but not in cLptA or cEptC. Additional studies will be necessary to determine the extent of this similarity, as well as the capacity of cEptC to bind multiple zinc ions.
3.4. Mutational characterization of conserved cEptC active-site residues
Based on a sequence alignment of COG2194 members (Supplementary Fig. S6), we identified conserved amino-acid residues that correspond to the cEptC active-site residues Thr266, Asn308, Ser309, His358 and His440 (Fig. 4 ▶ a). In order to assess the functional requirement of these active-site residues, we prepared mutant strains of C. jejuni 81-176 carrying point mutations for each residue, as described in §2.1. We previously established that a functional eptC allele is required for pEtN modification of the flagellar rod protein FlgG and lipid A in C. jejuni (Cullen & Trent, 2010 ▶; Cullen et al., 2012 ▶). These modifications are unequivocally responsible for proper flagellar assembly, motility and resistance to polymyxin B. To this end, we monitored the in vivo activity of mutant EptC enzymes by comparing the minimal inhibitory concentration (MIC) of polymyxin B and the halo-of-growth diameter in motility assays. All of the active-site mutants were significantly less resistant to polymyxin B when compared with the parent strain 81-176 (Fig. 5 ▶). The S309A mutant displayed a sixfold decrease in polymyxin B resistance, while the remaining active-site mutants and the ΔeptC null mutant displayed a 15.8-fold decrease. All of the mutants were also significantly impaired with respect to motility (Fig. 6 ▶). Interestingly, the N308A, S309A, H440A, H358A and T266S mutants displayed the most severe motility defects, while the T266A mutant was comparable to the ΔeptC null strain after 24 h of growth. It was previously determined that roughly 10% of the ΔeptC mutant population produces visible flagella (Cullen & Trent, 2010 ▶), giving rise to partial recovery of motility as demonstrated in Fig. 6 ▶ (compare 81-176, ΔeptC and the nonmotile strain H358A as a reference). The more pronounced motility defects observed for most of the active-site mutants presented here may reflect the fact that these strains express inactive protein that precludes partial recovery of motility via unknown mechanisms.
Figure 5.

Minimal inhibitory concentrations (MICs) of polymyxin B for C. jejuni EptC active-site mutants. Parent strain (81-176), eptC null mutant (ΔeptC), complemented ΔeptC strain carrying a copy of the native eptC allele (ΔeptC/eptC +) and mutant strains carrying eptC alleles with single amino-acid mutations (T266A, T266S, N308A, S309A, H358A and H440A) were swabbed onto MH agar plates with 10 µg ml−1 trimethoprim and subjected to the polymyxin B Etest strip assay. MICs were recorded after 48 h. The numbers above each bar in the graph indicate polymyxin B MICs in µg ml−1.
Figure 6.

EptC active-site mutant motility assays. Parent strain (81-176), eptC null mutant (ΔeptC), complemented ΔeptC strain (ΔeptC/eptC +) and strains carrying eptC active-site mutant alleles (T266A, T266S, N308A, S309A, H358A and H440A) were stab-inoculated into semisolid MH motility medium and incubated under microaerobic conditions. To assess relative motility, bacterial growth diameters were visually compared 24 h post-inoculation.
Taken together, the loss of motility and polymyxin B resistance suggest a crucial role for Thr266, Asn308, Ser309, His358 and His440 in EptC function. Notably, Asn308 Oδ1, Asn308 Nδ2 and Ser309 Oγ are 5.5, 6.7 and 6.4 Å from phospho-Thr266 O3P (chain A), respectively, which places them within hydrogen-bonding distance of a pEtN moiety covalently linked to Thr266.
To further explore the phosphorylation of Thr266, we expressed and purified soluble cEptC protein that contained a T266S mutation and investigated the presence of phosphoserine. The molecular weights of native and mutant cEptC proteins were determined by mass spectrometry in the positive mode with an accuracy of ±5 u. The deconvoluted cEptC spectrum showed a major peak at 36 750 u, consistent with the calculated [M + H]+ of native cEptC (Supplementary Fig. S2a). As expected, another major peak was observed with a mass shift of +80 u, demonstrating the presence of phosphothreonine as observed in the cEptC crystal structure. Corresponding peaks with masses of 36 477 and 36 556 u suggest the loss of two histidine residues (−273 u), likely owing to proteolysis of the disordered His tag during expression and purification. In contrast to native cEptC, the T266S mutant spectrum showed one major peak at 36 740 u, consistent with unmodified protein, and a corresponding minor peak indicating the loss of two histidine residues (−272 u; Supplementary Fig. S2c). Taken together, these results suggest that serine cannot replace threonine in EptC catalysis and that the phosphothreonine observed in the cEptC crystal structure and mass spectrum is indeed most likely owing to the catalytic activity of cEptC.
4. Conclusion
We present the structure of the catalytic domain of EptC, the only known phosphoethanolamine transferase from the Gram-negative pathogen C. jejuni. Despite the wide array of molecules modified by EptC, our structure displays extensive similarities to the lipid A-specific cLptA of N. meningitis and fewer similarities to the distantly related phosphoglycerol-specific cLtaS of B. subtilis and S. aureus. Like cLptA and cLtaS, cEptC assumes an α/β/α fold that coordinates a divalent metal ion and possesses a nucleophilic threonine modified with a phosphoryl group. We suggest that this residue mimics a covalent phosphoethanolamine–enzyme intermediate. Given the similarities among the active sites of these phospho-form transferases and alkaline phosphatase, the enzymes are expected to share mechanistic features. C. jejuni strains with point mutations at EptC residues Thr266, Asn308, Ser309, His358 and His440 suffered polymyxin B sensitivity and severe loss of motility, indicating that the active sites of EptC and other lipid A phosphoethanolamine transferases may be effective targets for drug design. C. jejuni has increasingly been recognized as a major causative agent of foodborne illness worldwide. Our analysis of the catalytic domain of EptC augments the structural characterization of phosphoethanolamine transferases and sets the foundation for the development of inhibitory drugs to combat infections by C. jejuni and related bacteria.
5. Related literature
The following references are cited in the Supporting Information for this article: Laskowski (2009 ▶) and Sievers & Higgins (2014 ▶).
Supplementary Material
PDB reference: EptC, 4tn0
Supporting Information . DOI: 10.1107/S1399004714017623/mh5132sup1.pdf
Acknowledgments
This work was supported by the National Institutes of Health (grants AI064184 and AI076322 to MST and grant GM106112 to ATK) and the Army Research Office (grant W911NF-12-1-0390 to MST). Instrumentation and technical assistance for this work were provided by the Macromolecular Crystallography Facility, with financial support from the College of Natural Sciences, the Office of the Executive Vice President and Provost, and the Institute for Cellular and Molecular Biology at the University of Texas at Austin. Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the US DOE under Contract No. DE-AC02-06CH11357. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences of the US Department of Energy under Contract No. DE-AC02-05CH11231. We thank Maria Person and the Proteomics Facility at the University of Texas at Austin and funding sources ES007784 (CRED) and RP110782 (CPRIT) for producing protein mass-spectrometry data. We also thank Jay Nix for assistance with anomalous data collection and processing.
Footnotes
Supporting information has been deposited in the IUCr electronic archive (Reference: MH5132).
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Batz, M. B., Hoffmann, S. & Morris, J. G. Jr (2012). J. Food Prot. 75, 1278–1291. [DOI] [PubMed]
- Burmeister, W. P. (2000). Acta Cryst. D56, 328–341. [DOI] [PubMed]
- Chamot-Rooke, J., Mikaty, G., Malosse, C., Soyer, M., Dumont, A., Gault, J., Imhaus, A. F., Martin, P., Trellet, M., Clary, G., Chafey, P., Camoin, L., Nilges, M., Nassif, X. & Duménil, G. (2011). Science, 331, 778–782. [DOI] [PubMed]
- Chevance, F. F. & Hughes, K. T. (2008). Nature Rev. Microbiol. 6, 455–465. [DOI] [PMC free article] [PubMed]
- Cox, A. D., Wright, J. C., Li, J., Hood, D. W., Moxon, E. R. & Richards, J. C. (2003). J. Bacteriol. 185, 3270–3277. [DOI] [PMC free article] [PubMed]
- Cullen, T. W., Madsen, J. A., Ivanov, P. L., Brodbelt, J. S. & Trent, M. S. (2012). J. Biol. Chem. 287, 3326–3336. [DOI] [PMC free article] [PubMed]
- Cullen, T. W., O’Brien, J. P., Hendrixson, D. R., Giles, D. K., Hobb, R. I., Thompson, S. A., Brodbelt, J. S. & Trent, M. S. (2013). Infect. Immun. 81, 430–440. [DOI] [PMC free article] [PubMed]
- Cullen, T. W. & Trent, M. S. (2010). Proc. Natl Acad. Sci. USA, 107, 5160–5165. [DOI] [PMC free article] [PubMed]
- Diederichs, K. & Karplus, P. A. (2013). Acta Cryst. D69, 1215–1222. [DOI] [PMC free article] [PubMed]
- Doublié, S. (1997). Methods Enzymol. 276, 523–530. [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- French, S. & Wilson, K. (1978). Acta Cryst. A34, 517–525.
- Havelaar, A. H., Ivarsson, S., Löfdahl, M. & Nauta, M. J. (2013). Epidemiol. Infect. 141, 293–302. [DOI] [PMC free article] [PubMed]
- Israeli, E., Agmon-Levin, N., Blank, M., Chapman, J. & Shoenfeld, Y. (2012). Clin. Rev. Allergy Immunol. 42, 121–130. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Karplus, P. A. & Diederichs, K. (2012). Science, 336, 1030–1033. [DOI] [PMC free article] [PubMed]
- Kim, E. E. & Wyckoff, H. W. (1991). J. Mol. Biol. 218, 449–464. [DOI] [PubMed]
- Klock, H. E. & Lesley, S. A. (2009). Methods Mol. Biol. 498, 91–103. [DOI] [PubMed]
- Laskowski, R. A. (2009). Nucleic Acids Res. 37, D355–D359. [DOI] [PMC free article] [PubMed]
- Lu, D., Wörmann, M. E., Zhang, X., Schneewind, O., Gründling, A. & Freemont, P. S. (2009). Proc. Natl Acad. Sci. USA, 106, 1584–1589. [DOI] [PMC free article] [PubMed]
- Mackinnon, F. G., Cox, A. D., Plested, J. S., Tang, C. M., Makepeace, K., Coull, P. A., Wright, J. C., Chalmers, R., Hood, D. W., Richards, J. C. & Moxon, E. R. (2002). Mol. Microbiol. 43, 931–943. [DOI] [PubMed]
- Marchler-Bauer, A. et al. (2013). Nucleic Acids Res. 41, D348–D352. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Naessan, C. L., Egge-Jacobsen, W., Heiniger, R. W., Wolfgang, M. C., Aas, F. E., Røhr, A., Winther-Larsen, H. C. & Koomey, M. (2008). J. Bacteriol. 190, 387–400. [DOI] [PMC free article] [PubMed]
- Needham, B. D. & Trent, M. S. (2013). Nature Rev. Microbiol. 11, 467–481. [DOI] [PMC free article] [PubMed]
- O’Brien, P. J. & Herschlag, D. (2001). Biochemistry, 40, 5691–5699. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Raetz, C. R. H., Reynolds, C. M., Trent, M. S. & Bishop, R. E. (2007). Annu. Rev. Biochem. 76, 295–329. [DOI] [PMC free article] [PubMed]
- Read, R. J. & Schierbeek, A. J. (1988). J. Appl. Cryst. 21, 490–495.
- Scallan, E., Hoekstra, R. M., Angulo, F. J., Tauxe, R. V., Widdowson, M. A., Roy, S. L., Jones, J. L. & Griffin, P. M. (2011). Emerg. Infect. Dis. 17, 7–15. [DOI] [PMC free article] [PubMed]
- Schirner, K., Marles-Wright, J., Lewis, R. J. & Errington, J. (2009). EMBO J. 28, 830–842. [DOI] [PMC free article] [PubMed]
- Scott, N. E., Nothaft, H., Edwards, A. V. G., Labbate, M., Djordjevic, S. P., Larsen, M. R., Szymanski, C. M. & Cordwell, S. J. (2012). J. Biol. Chem. 287, 29384–29396. [DOI] [PMC free article] [PubMed]
- Sievers, F. & Higgins, D. G. (2014). Methods Mol. Biol. 1079, 105–116. [DOI] [PubMed]
- Stec, B., Holtz, K. M. & Kantrowitz, E. R. (2000). J. Mol. Biol. 299, 1303–1311. [DOI] [PubMed]
- Trent, M. S., Stead, C. M., Tran, A. X. & Hankins, J. V. (2006). J. Endotoxin Res. 12, 205–223. [DOI] [PubMed]
- Wanty, C., Anandan, A., Piek, S., Walshe, J., Ganguly, J., Carlson, R. W., Stubbs, K. A., Kahler, C. M. & Vrielink, A. (2013). J. Mol. Biol. 425, 3389–3402. [DOI] [PubMed]
- Weik, M., Ravelli, R. B. G., Kryger, G., McSweeney, S., Raves, M. L., Harel, M., Gros, P., Silman, I., Kroon, J. & Sussman, J. L. (2000). Proc. Natl Acad. Sci. USA, 97, 623–628. [DOI] [PMC free article] [PubMed]
- Wenzel, C. Q., St Michael, F., Stupak, J., Li, J., Cox, A. D. & Richards, J. C. (2010). J. Bacteriol. 192, 208–216. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wright, J. C., Hood, D. W., Randle, G. A., Makepeace, K., Cox, A. D., Li, J., Chalmers, R., Richards, J. C. & Moxon, E. R. (2004). J. Bacteriol. 186, 6970–6982. [DOI] [PMC free article] [PubMed]
- Yuki, N. & Hartung, H. P. (2012). N. Engl. J. Med. 366, 2294–2304. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: EptC, 4tn0
Supporting Information . DOI: 10.1107/S1399004714017623/mh5132sup1.pdf