

Abstract
Purpose.
We determined bioactivity of lysophospholipids generated by degradation of the low-density (LDL), very low-density (VLDL), and high-density (HDL) lipoproteins with hepatic lipase (HL), cholesterol esterase (CE), and lipoprotein-associated phospholipase A2 (Lp-PLA2).
Methods.
The LDL, VLDL, and HDL were treated with HL, CE, and Lp-PLA2 after immobilization on plates, and complement activation studies were performed with diluted human serum. Complement component 3 (C3) fixation, a marker for complement activation, was determined with a monoclonal anti-human C3d antibody. Enzymatic properties of HL and CE were assayed with triglyceride and phosphatidylcholine substrates for triglyceride hydrolase and phospholipase A activities. The ARPE-19 cells were used for viability studies.
Results.
The HL degradation of human lipoproteins LDL, VLDL, or HDL results in the formation of modified lipoproteins that can activate the complement pathway. Complement activation is dose- and time-dependent upon HL and occurs via the classical pathway. Enzymatic studies suggest that the phospholipase A1 activity of HL generates complement-activating lysophospholipids. C-reactive protein (CRP), known to simultaneously interact with complement C1 and complement factor H (CFH), further enhances HL-induced complement activation. The lysophospholipids, 1-Palmitoyl-sn-glycero-3-phosphocholine and 1-Oleoyl-sn-glycero-3-phosphocholine, can be directly cytotoxic to ARPE-19 cells.
Conclusions.
The HL degradation of lipoproteins, known to accumulate in the outer retina and in drusen, can lead to the formation of bioactive lysophospholipids that can trigger complement activation and induce RPE cellular dysfunction. Given the known risk associations for age-related macular degeneration (AMD) with HL, CRP, and CFH, this study elucidates a possible damage pathway for age-related macular degeneration (AMD) in genetically predisposed individuals, that HL activity may lead to accumulation of lysophospholipids to initiate complement activation, with CFH dysregulation exacerbating the effects of this process.
Keywords: age-related macular degeneration, hepatic lipase, lipoproteins, lysophospholipids, complement activation, C-reactive protein, RPE cytotoxicity
Hepatic lipase modifies the lipoproteins HDL, LDL, and VLDL and the resultant products are capable of complement activation via the classical pathway.
Introduction
Age-related macular degeneration (AMD) is a degenerative eye disease associated with genetic and environmental risk factors. This disease is the leading cause of vision loss in senior populations of developed countries and remains a major public health problem.1 Drusen commonly develop in the aging macula and, when present in large size or number, represent a hallmark risk factor for late stage AMD. There are two advanced forms of AMD, neovascular disease, in which new blood vessels grow under or within the retinal layers, and atrophic disease, in which there is damage to photoreceptors and RPE cells without abnormal vascularization. The neovascular form of AMD currently is treated with anti-VEGF therapy but may develop associated anatomic complications, such as hemorrhage, fibrosis, and RPE rips that limit visual outcomes. There is no known treatment for the atrophic form of AMD, though antioxidants and carotenoid supplements may slow progression of the disease. Eyes may develop both forms of AMD over the course of a lifetime.
Genetic studies have identified that the complement pathway and another locus, in chromosomal region 10q26, confer major susceptibilities to the disease.2–8 Several additional loci with smaller odds ratios also have been shown to be associated with AMD, including apolipoprotein E (apoE) and hepatic lipase.9–11 Smaller odds ratio and lower allele frequency attributable to certain genetic variants do not necessarily reflect the role of a gene and its encoded protein in the pathophysiology of disease. A single nucleotide polymorphism (SNP) can alter gene expression and/or its protein function, but the level of alteration can vary, making it difficult to clarify the importance of a single gene in a disease process by genetic studies alone.
As the presence of larger and more numerous drusen in the macula is a common risk factor for AMD, the analysis of drusen components has aided the understanding of AMD pathogenesis. Components of the complement system are present in significant quantity in drusen, further evidence that complement activation is important in the development of AMD.12,13 Drusen also contain many other proteins and lipoprotein-like particles.14,15 Lipid profiles of such deposits have shown high levels of lysophospholipids and free fatty acids, suggesting that hydrolysis of phospholipids, such as phosphatidylcholine, has occurred.16,17 The Y402H polymorphism of complement factor H (CFH) has been demonstrated to have a strong association with AMD.2–5 The CFH is a regulator protein of complement activation; it competes with factor B for binding to C3b and functions as a cofactor for Factor I–mediated C3b inactivation. The amino acid 402 of CFH is not involved with C3b binding, but it has been demonstrated that the AMD-associated 402H variant of CFH has lower binding affinity to C-reactive protein (CRP) than the 402Y variant.18,19 Patients with AMD and individuals who are homozygous for the CFH 402H haplotype have increased levels of CRP in drusen and basal deposits,20,21 but the mechanisms of recruitment of CRP in these patients remain unclear.
The AMD-associated SNP of hepatic lipase, rs10468017, is located in the functional promoter region of the gene; its minor T allele decreases hepatic lipase expression and has a protective effect on the development of AMD.11 Hepatic lipase, together with lipoprotein lipase and endothelial lipase, comprise the triglyceride lipase gene subfamily, which has a central role in plasma lipoprotein metabolism and homeostasis. These lipases are differentiated by their tissue-specific gene expression and substrate specificity. Lipoprotein lipase mainly hydrolyzes triglycerides, whereas endothelial lipase exerts significant phospholipase activity. Hepatic lipase (HL) appears to have equal hydrolytic activities upon triglycerides and phospholipids.22,23 The purpose of this study was to investigate the role of HL in modifying lipoproteins to become bioactive in biological processes involved in AMD development, specifically complement activation via the CRP-mediated classical pathway and direct RPE cell toxicity in vitro.
Materials and Methods
Materials
Two different recombinant human HL preparations were used. One was obtained from GeneTex (GTX48178-PRO; Irvine, CA, USA) and the other purchased from OriGene (TP315870; Rockville, MD, USA). Cholesterol esterase (CE) was obtained from MP Biomedicals (0210543950; Solon, OH, USA). The 1,2-bis(heptanoylthio)glycerophosphocholine, phosphocholine, and horseradish peroxidase (HRP)-conjugated secondary antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) was obtained from AbboMax (San Jose, CA, USA). Lipoprotein-associated phospholipase A2 (Lp-PLA2) was purchased from R&D Systems (Minneapolis, MN, USA). Human low-density lipoprotein (LDL), human high-density lipoprotein (HDL), phospholipase C, human CRP, 1-Palmitoyl-sn-glycero-3-phosphocholine (LPC [16:0]), 1-Oleoyl-sn-glycero-3-phosphocholine (LPC [18:1]), 2,3-dimercapto-1-propanol tributyrate (DMPTB), and BSA prepared by heat shock fractionation were obtained from Sigma-Aldrich (St. Louis, MO, USA). Human very low-density lipoprotein (VLDL) was from Kalen Biomedical (Montgomery Village, MD, USA). Normal human serum, C1q-depleted human serum, factor B-depleted human serum, and a monoclonal antibody to a neoepitope in the C3d domain of C3 were obtained from Quidel (San Diego, CA, USA). All cell culture products were from Life Technologies (Grand Island, NY, USA).
Human LDL, VLDL, and HDL Degradation and Complement Activation
The 96-well NUNC MaxiSorp plates were coated with 40 μL of human LDL, VLDL, or HDL at 200 μg/mL in PBS at 4°C overnight. The plates then were warmed to 37°C for 1 hour and remaining binding sites then were blocked with 3% BSA in PBS at 37°C for 1 hour. The wells were washed and then incubated with 40 μL of HL, CE, or Lp-PLA2 in PBS containing 2% BSA at 37°C for 1 or 2 hours as indicated in the Results section. The degradation reaction was stopped by washing the plate with PBS. For complement activation, the reacted plate was incubated with 20 μL of 1:1 diluted human sera at 21°C for 30 minutes. The diluent for serum dilutions was PBS containing calcium and magnesium. Human sera that were used in our study were native serum, C1q-depleted serum, factor B-depleted serum, native serum with the addition of 10 mM MgCl2 and 2 mM EGTA (Mg-EGTA), and native serum with the addition of 10 mM EDTA. After thorough washing of the plate, complement component 3 (C3) fixation was determined with anti-human C3d antibody in combination with HRP-conjugated secondary antibody. The monoclonal anti-human C3d antibody is reactive to all C3d-containing fragments of C3, but not with C3 itself, so it can detect C3 fixation on the plate and not simply C3 absorption. The final peroxidase activity was monitored at 450 nm with 3,3′,5,5′-tetramethyl-benzidine and hydrogen peroxide as substrates after 10 to 15 minutes reaction time at room temperature. Addition of EDTA to the native human serum totally blocked complement activation; thus it was used as a control for the complement activation studies and was set as the baseline reading for every experiment.
Enzymatic Activities
Triglyceride hydrolase activity of CE and HL was determined using DMPTB as a substrate according to the method of Choi et al.24 Phospholipase A activity of CE and HL was assayed similarly as for its triglyceride hydrolase activity, but with 1.5 mM of 1,2-bis(heptanoylthio) glycerophosphocholine as a substrate and by substituting 10 mM CaCl2 for 1 mM EDTA in the assay buffer.25
Cell Culture and Cell Viability Assay
Human retinal pigment epithelial cell, ARPE-19, purchased from ATCC (Manassas, VA, USA), were cultured in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 IU/mL of penicillin, and 100 μg/mL of streptomycin (all cell culture products from Invitrogen-Gibco, Rockville, MD, USA). Cells were maintained at 37°C in a 5% CO2 incubator with medium changes every 3 to 4 days. Subculture of cells was performed with a 0.05% trypsin-EDTA solution.
Cell viability was determined using the colorimetric MTT metabolic activity assay. ARPE-19 cells cultured in 96-well cluster plates were incubated with 0.15 mL of serum-free medium containing 0.5 mg/mL MTT at 37°C for 2 hours in a 5% CO2 incubator. After removal of the cell culture medium, formation of formazan from MTT reduction was detected at 540 nm by adding 0.1 mL of dimethyl sulfoxide (DMSO).
Statistical Analysis
All values are presented as a mean ± SD. Where applicable, a 2-tailed Student's t-test was used to analyze the differences between groups (Excel software; Microsoft, Redmond, WA, USA). Minimum values of P less than 0.01 were considered statistically significant and are denoted by an asterisk in each figure.
Results
HL Degradation of LDL, VLDL, and HDL Generates Lipid Molecules That Can Activate the Classical Complement Pathway
The HL degradation of human lipoproteins LDL, VLDL, or HDL caused the modified lipoproteins to activate the complement system (Fig. 1). The HL degradation of LDL, VLDL, and HDL occurred through a calcium independent mechanism (data not shown). The complement activation was dose- and time-dependent upon HL degradation (Figs. 1D, 1F, respectively). To identify which complement pathway was being activated, we used known manipulations of the complement system from native human serum. The C1q depletion prevents classical pathway at initiation and the addition of Mg-EGTA chelates calcium, a necessary component for classical pathway activation. Factor B depletion halts alternative pathway activation from the beginning. When either C1q-depleted serum or Mg-EGTA–containing serum was used, complement activation did not occur, indicating that the classical pathway is involved. When factor B depleted serum was used, there was no change in the level of complement activation, indicating that the alternative pathway is not involved. We compared two commercially available HL products (see Materials) with regard to degradation of human lipoproteins, and both enzymes had similar activities for modifying lipoproteins to activate the complement system (data not shown). The HL from GeneTex was used for most of our studies.
Figure 1.
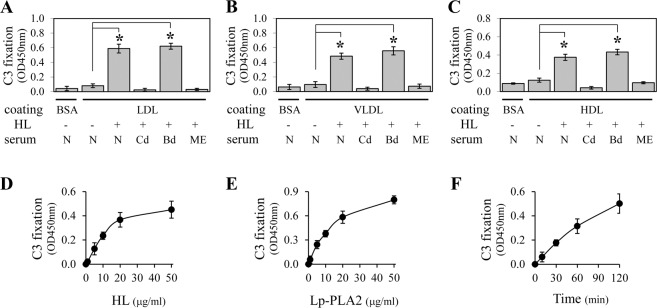
Phospholipase-digested human LDL, VLDL, and HDL can activate the classical pathway of the complement system. MaxiSorp plates (96-well) were coated with fresh human LDL (A, D–F), VLDL (B), and HDL (C). After blocking with BSA, the plate was digested with HL (10 μg/mL for [A–C, F], and 0–50 μg/mL for [D]) or Lp-PLA2 (0–50 μg/mL for [E]). The HL digestion time was 1 hour for (D), 2 hours for (A–C), and 0 to 2 hours for (F). The Lp-PLA2 digestion time was 2 hours for (E). Color development was 15 minutes for (A–C, E, F) and 10 minutes for (D). To test complement activation, the plate was incubated with dilute native human serum (N), C1q-depleted human serum (Cd), factor B-depleted human serum (Bd), or human serum containing Mg-EGTA (ME). The C3 fixation on the plate then was detected with a monoclonal anti-C3d antibody followed by HRP-conjugated secondary antibody. (A) Shows large statistically significant increases in complement activation for HL digested LDL. Since increased C3 fixation was observed with normal and factor B-depleted serum, but not with either C1q-depleted or Mg-EGTA supplemented serum, this indicates that activation is via the classical pathway (see text for further description). A similar response was observed using VLDL and HDL, as shown in (B) and (C), respectively. (D, E) Show a concentration-dependent increase in C3 fixation induced by HL and Lp-PLA2 digestion of LDL, respectively. The effect of HL digestion of LDL also was time-dependent (F).
Lysophospholipids in Phospholipase-Digested Lipoproteins Activate the Complement System
To identify the lipid molecules that can activate the classical complement pathway in our system, we tested three lipases – HL, CE, and Lp-PLA2 on generation of the lipid molecules. The Lp-PLA2 can degrade phospholipids in lipoproteins into lysophospholipids and fatty acids. As shown in Figure 1E, Lp-PLA2-digested LDL could activate complement system, implying that either lysophospholipids or fatty acids are the lipid molecules that can activate the classical complement pathway in our system. The CE has a broad spectrum of substrates that include triglycerides, phospholipids, cholesterol esters. The CE-digested LDL already is known to activate the complement system via the classical pathway.26 Although HL and CE have phospholipase A1 and triglyceride hydrolase activities, their proportional enzymatic activities vary. Specifically, when equivalent phospholipase A1 activity is present for CE and HL, CE has significantly greater triglyceride hydrolase activity than HL. As shown in Figure 2, we observed similar levels of complement activation by using equivalent phospholipase A1 activity, 8 μUnits, of HL and CE, while their triglyceride hydrolase activity displayed a >2700-times difference. This suggests that phospholipase A1 activity is the primary enzymatic activity that generates a complement-activating lipid species in the setting of these enzymes. Triglyceride hydrolase is known to digest LDL into fatty acids, monoglycerides, and diglycerides, while phospholipase A1 is known to digest LDL into fatty acids and lysophospholipids. Thus we can infer that phospholipase activity is responsible for complement activation in this case and that the lysophospholipids likely mediate this effect (i.e., are the complement activating lipid species in these settings).
Figure 2.
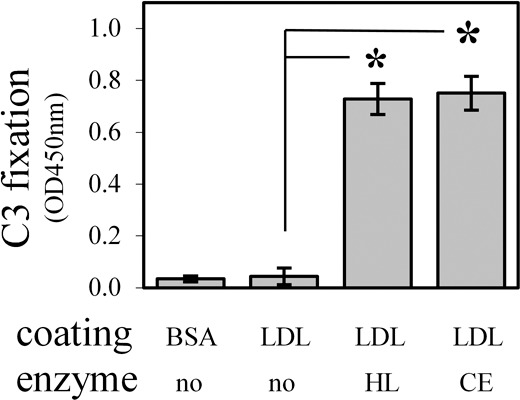
Phospholipase A1 activity of HL and CE is responsible for the generation of complement-activating lipid molecules. Concentrations of HL and CE were normalized based on equivalent phospholipase A1 activity (8 μUnits each, data not shown). Immobilized LDL was digested by each enzyme for 2 hours followed by the complement activation assay using dilute native human serum (as in Fig. 1) and HRP color development for 15 minutes. When normalized based on phospholipase A1 activity, HL and CE have equivalent complement activation activity, indicating that phospholipase A1 activity is responsible for complement activation. In other words, if triglyceride hydrolase activity were responsible for complement activation, a much higher response from CE would have been expected since at this concentration, the triglyceride hydrolase activity of each enzyme differ markedly (3 μUnits for HL and 8125 μUnits for CE)
Inhibition of HL-Mediated Complement Activation
Phosphatidylcholine is the most abundant phospholipid in cell membranes and lipoproteins, helping to maintain the structure of the membrane. It then might be expected that the major lysophospholipid generated from the phospholipase A1 activity of HL upon lipoproteins is lysophosphatidylcholine (Fig. 3), a well-studied ligand for CRP in membrane structures.27 As it is known that immobilization of CRP can activate the classical complement pathway by interaction with C1, we next focused upon ways of blocking the complement activation due to lysophosphatidylcholine. When phosphocholine was added into native human serum as a competitive binding inhibitor with lysophosphatidylcholine for CRP,28 it dose-dependently decreased complement activation and approached a maximum inhibitory effect at 10 μM (Fig. 4A). In addition, as shown in Figure 4B, phospholipase C, which specifically cleaves phospholipids at the phosphate group, was shown to reduce complement activation in a concentration dependent manner. This latter finding further supports the notion that the lipid species responsible for complement activation are lysophospholipids.
Figure 3.
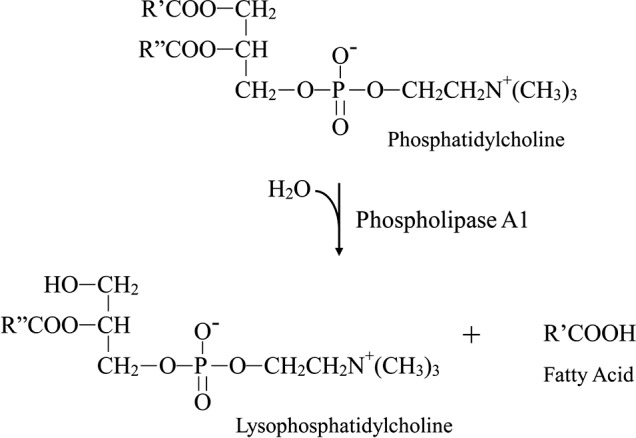
Hydrolysis of phosphatidylcholine occurs via the phospholipase A1 activity of the HL enzyme.
Figure 4.
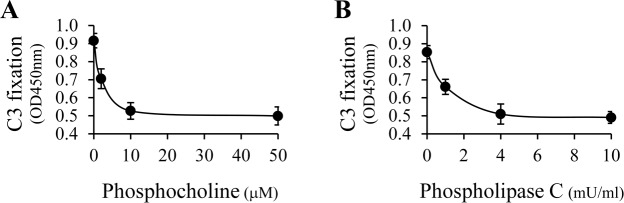
Inhibition of HL-mediated complement activation. Plate coating with human LDL and 2 hours of HL digestion were performed as described in Figure 1. (A) Phosphocholine (0–50 μM), a competitive binding inhibitor for CRP, was added into dilute native human serum and the mixture was used for complement activation. (B) HL-digested LDL was further reacted with phospholipase C (0–10 mU/mL), an enzyme that specifically cleaves phosphocholine groups, followed by complement activation. For both experiments, the HRP-catalyzed color development was measured after a 15-minute incubation. As shown, competitive inhibition of CRP binding by phosphocholine (A) as well as elimination of the CRP binding ligand via phosphocholine cleavage (B) are shown to reduce HL-mediated complement activation in a concentration dependent manner.
CRP Enhances Complement Activation From HL-Digested LDL
The CRP has multiple ligand binding sites and can bind lysophosphatidylcholine, C1, and factor H to form a complex structure. In further experiments, we studied the role of CRP in lipoprotein-induced complement activation. As shown in Figure 5, BSA alone, BSA with HL digestion, or native undigested LDL did not activate the complement system. As well, increasing the CRP level did not alter the complement activation activity of these molecules. In contrast, HL degradation of LDL resulted in C3 fixation as well as a CRP dose-dependent increase in complement activation. It should be noted that an individual's serum CRP level will vary in different situations and can rise up dramatically in acute inflammation, such as during an infection. Basal levels of CRP also can be elevated in people with a variety of illnesses, including AMD.29 Thus, elevated CRP levels may escalate complement activation induced via a HL-mediated pathway.
Figure 5.
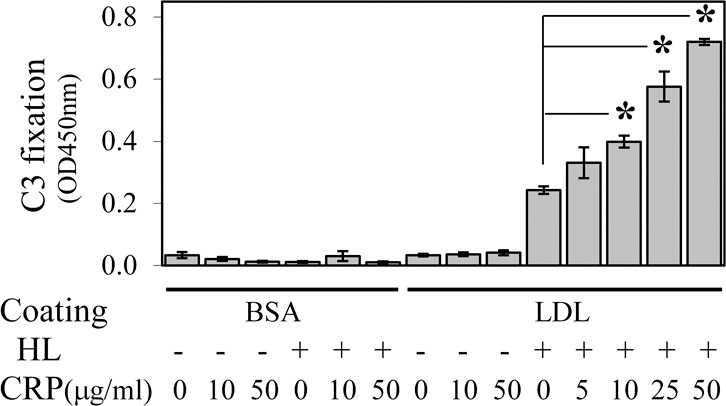
The CRP enhances complement activation from HL digested LDL. Human LDL plate coating with a 1-hour HL digestion was performed as described in Figure 1. Complement activation by HL-digested LDL was performed with dilute native human serum supplemented with purified human CRP. The HRP-catalyzed color development time was 10 minutes. The Figure shows that very little C3 fixation occurred in control samples that lacked either LDL coating or HL digestion. In contrast, in samples containing both LDL and HL digestion, the addition of CRP increased complement activation in a concentration-dependent manner.
Lysophosphatidylcholine is Cytotoxic to RPE Cells
We assessed the effects of lysophosphatidylcholine with regard to cytotoxic activity against RPE cells. The saturated lysophospholipid 1-palmitoyl-sn-glycero-3-phosphocholine (LPC [16:0]) is one of the most predominant lysophospholipid products resulting from the hydrolysis of biological membranes and is a molecule with known cytotoxic activity toward many different cell types, including endothelial cells, vascular smooth muscle cells, and red blood cells.30–32 However, cytotoxicity to RPE cells has not been reported previously to our knowledge. As shown in Figure 6, LPC (16:0) displayed cytotoxicity to ARPE-19 cells, a human RPE cell line, when it was added to the cell culture medium at 20 μM or greater. Nearly all the cells were killed when greater than 100 μM LPC (16:0) was used. ARPE-19 cells displayed typical dead cell morphology after incubation with 100 μM LPC (16:0) for 22 hours. As control, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) did not show any cytotoxicity on ARPE-19 cells when the same concentrations of POPC was used in cell culture medium. The unsaturated lysophospholipid, 1-Oleoyl-sn-glycero-3-phosphocholine (LPC [18:1]), also was tested in its effect upon ARPE-19 cells, and it displayed less toxicity though still substantial (Fig. 6).
Figure 6.
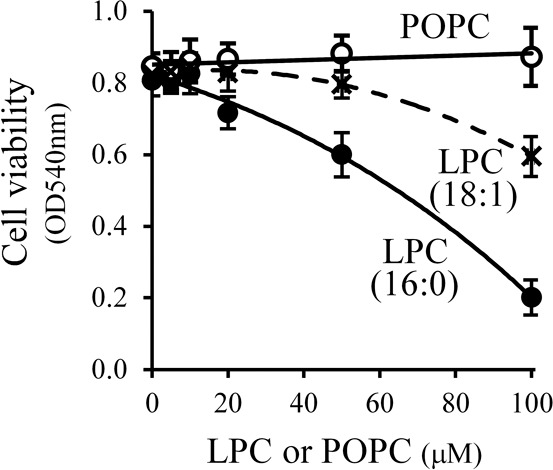
Cytotoxicity of lysophospholipids on ARPE-19 cells. The ARPE-19 cells were cultured to 40% to 50% confluence in 96-well plates, followed by a 24-hour incubation with serum-free medium. The resultant cells were incubated with 0.1 mL of serum-free medium containing 0 to 100 μM LPC (16:0) or LPC (18:1), or POPC for 22 hours. Cell viability was determined using a standard MTT assay and cell morphology was evaluated using light microscopy. The LPC (16:0) and LPC (18:1) induced a concentration-dependent decrease in ARPE-19 viability, while POPC did not demonstrate cytotoxicity to ARPE-19 cells at these concentrations.
Discussion
In this study, we identified a molecular link between known drusen components and several well-defined biological processes in the development of AMD, including complement activation and RPE cell death. Our experimental data indicated that the enzyme product of one AMD susceptibility gene, hepatic lipase, can trigger the generation of bioactive lysophospholipids, leading to complement activation via the classical pathway. This complement activation can be escalated by CRP binding. One of the best known genetic contributors for AMD, CFH can bind with CRP and has a central role in inhibiting complement activation at the C3b level. The 402H variant of CFH has weaker binding to CRP, resulting in the loss of its regulatory function and formation of membrane attack complexes. We hypothesized that bioactive lysophospholipids, in the setting of impaired CFH function, could lead to unmitigated complement activation, with generation of inflammatory activity at the outer retinal layers. Thus, the RPE cells that are in close proximity to drusen and basal deposits could be one unintended target of this physiologic process. As we have further demonstrated, lysophospholipid in sufficient quantity can be cytotoxic to RPE cells and lead to RPE cell death in vitro even in the absence of complement activation.
A common feature early in the pathogenesis of AMD is deposit formation in the region of the RPE and Bruch's membrane interface. Accumulation of deposits in Bruch's membrane results in progressive Bruch's membrane thickening and the appearance of drusen. Lipoproteins are present in all drusen, and many are in the form of lipoprotein-like particles that contain apoA-I, apoB-100, apoE, apoC-I, and apoC-II.33,34 Furthermore, transmission electron micrographs of the lipoprotein-like particles accumulating underneath RPE cells in AMD eyes show a morphology that is consistent with a model of surface degradation and particle fusion.35 Such retained lipoprotein-like particles may be subject to oxidation and degradation by local enzymes released from RPE cells (such as HL, lipoprotein lipase, and secretory phospholipase A2), or enzymes carried over via lipoproteins from the systemic circulation (such as lipoprotein-associated phospholipase A2). There are several lysophospholipid products of these enzymatic processes, such as lysophosphatidic acid, lysophosphatidylcholine, sphingosylphosphorylcholine, and sphingosine-1-phosphate (S1P). Of note, anti-S1P therapy is being investigated as a new therapy for safety and efficacy in exudative AMD.36
A human RPE cell culture model that mimics early stages of AMD displays an accumulation of sub-RPE deposits. Such deposits consist of two morphologically distinct forms, membrane-bound multivesicular material and nonmembrane-bound particle conglomerates.37 When exposed to human serum, such deposits can trigger complement activation that appears to be mediated via the classical pathway by binding of C1q to ligands in the membrane-bound multivesicular material. Such material contains apoE-rich deposits. In that cell culture model system, IgG depletion had no detectable effect on complement activation, suggesting that activation of the classical pathway occurs via an antibody-independent mechanism. The exact C1q binding partners were not identified in that study. Based upon our studies, it could be postulated that phospholipases released from human RPE cells degrade apoE-containing membrane deposits to generate lysophospholipids capable of initiating complement activation via an antibody-independent classical pathway.
A collaborative genome-wide association study, including >17,100 advanced AMD cases and >60,000 controls of European and Asian ancestry, identified 19 loci associated with AMD at P < 5 × 10−8.38 To identify biological relationships among these genetic association signals, the genes within 100 kb of the variants in each association peak were analyzed, and a total of 90 genes was obtained when the correlation was set at r2 > 0.8. Further analysis highlighted several biological pathways, particularly the complement system, atherosclerotic signaling, and angiogenesis, that were enriched in the resulting set of 90 genes. Phospholipid degradation was also identified in the top pathways with nominal P value at 0.0058. Three genes were identified in the phospholipid pathway: PLA2G12A, LIPC (hepatic lipase), and PLA2G6. The PLA2G12A is a secretory phospholipase A2, while PLA2G6 is a cytosolic calcium-independent phospholipase A2. Compared to single-gene or single-SNP analyses, such gene set/pathway association analyses can potentially reduce false positives and uncover a significant biological effect distributed over multiple loci even if changes in any individual locus have a small effect. As an example relevant to our study, each of the three genes in the phospholipid degradation pathway has a weak association with AMD, but together they form a pathway that has a strong association with AMD.
Lysophospholipids, at 10 to 100 μM, have well-known cytotoxicity to many different cell types and are studied extensively in the pathogenesis of atherosclerosis.39,40 Our study demonstrated lysophospholipids also are cytotoxic to RPE cells when similar levels of lysophosphatidylcholine were present in medium. While we did not investigate multiple chemical moieties, our findings suggested that saturated lysophospholipids may be more cytotoxic than unsaturated species to ARPE-19 cells. The specific bioactive lysophospholipids that may be relevant to the pathogenesis of retinal disease, like AMD, are not yet known. The viability studies were performed with one cell line in our study, and other retinal cells may have different responses to different molecular species of lysophospholipids.
We end by summarizing a proposed damage mechanism based upon the findings of this study. Subretinal lipoproteins, such as HDL, VLDL, LDL, and lipoprotein-like particles, have an important role in the normal lipid transport mechanisms of the outer retina. These lipoproteins accumulate in the outer retina with age where they may serve as substrates for enzymatic modifications. The HL, and likely other enzymes in the outer retina with phospholipase activity, can digest these lipoproteins to form products that can initiate activation of the classical pathway of the complement system. Based upon genetic studies, there is likely to be phenotypic variation in phospholipase activity and, thus, the modification of phospholipids. Some eyes, such as those in individuals with a CFH deficiency and other polymorphisms, have a lessened ability to regulate or inhibit local complement activation, making them particularly susceptible to the deleterious effects of bioactive lipids. In such cases, bioactive lipids may serve as triggers for sustained complement activation and/or RPE cellular dysfunction, ultimately contributing to the development of AMD in its various forms.
Acknowledgments
Supported by the Eye Surgery Fund, Glaubinger Foundation, Hearst Foundation, Research to Prevent Blindness, and National Institutes of Health/National Education Institute (NIH/NEI, Bethesda, MD, USA) Core Grant P30 EY019007.
Disclosure: W. Ma, None; D.C. Paik, None; G.R. Barile, None
References
- 1. Friedman DS, O'Colmain BJ, Munoz B, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004; 122: 564–572 [DOI] [PubMed] [Google Scholar]
- 2. Edwards AO, Ritter R 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005; 308: 421–424 [DOI] [PubMed] [Google Scholar]
- 3. Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005; 102: 7227–7232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005; 308: 419–421 [DOI] [PubMed] [Google Scholar]
- 5. Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005; 308: 385–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006; 38: 458–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yates JR, Sepp T, Matharu BK, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007; 357: 553–561 [DOI] [PubMed] [Google Scholar]
- 8. Rivera A, Fisher SA, Fritsche LG, et al. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet. 2005; 14: 3227–3236 [DOI] [PubMed] [Google Scholar]
- 9. Klaver CC, Kliffen M, van Duijn CM, et al. Genetic association of apolipoprotein E with age-related macular degeneration. Am J Hum Genet. 1998; 63: 200–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Souied EH, Benlian P, Amouyel P, et al. The epsilon4 allele of the apolipoprotein E gene as a potential protective factor for exudative age-related macular degeneration. Am J Ophthalmol. 1998; 125: 353–359 [DOI] [PubMed] [Google Scholar]
- 11. Neale BM, Fagerness J, Reynolds R, et al. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc Natl Acad Sci U S A. 2010; 107: 7395–7400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hageman GS, Luthert PJ. Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch's membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001; 20: 705–732 [DOI] [PubMed] [Google Scholar]
- 13. Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002; 134: 411–431 [DOI] [PubMed] [Google Scholar]
- 14. Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000; 14: 835–846 [PubMed] [Google Scholar]
- 15. Ebrahimi KB, Handa JT. Lipids, lipoproteins, and age-related macular degeneration. J Lipids. 2011; 2011: 802059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang L, Li CM, Rudolf M, et al. Lipoprotein particles of intraocular origin in human Bruch membrane: an unusual lipid profile. Invest Ophthalmol Vis Sci. 2009; 50: 870–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Curcio CA, Johnson M, Huang JD, Rudolf M. Apolipoprotein B-containing lipoproteins in retinal aging and age-related macular degeneration. J Lipid Res. 2010; 51: 451–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Laine M, Jarva H, Seitsonen S, et al. Y402H polymorphism of complement factor H affects binding affinity to C-reactive protein. J Immunol. 2007; 178: 3831–3836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Okemefuna AI, Nan R, Miller A, Gor J, Perkins SJ. Complement factor H binds at two independent sites to C-reactive protein in acute phase concentrations. J Biol Chem. 2010; 285: 1053–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson PT, Betts KE, Radeke MJ, Hageman GS, Anderson DH, Johnson LV. Individuals homozygous for the age-related macular degeneration risk-conferring variant of complement factor H have elevated levels of CRP in the choroid. Proc Natl Acad Sci U S A. 2006; 103: 17456–17461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bhutto IA, Baba T, Merges C, Juriasinghani V, McLeod DS, Lutty GA. C-reactive protein and complement factor H in aged human eyes and eyes with age-related macular degeneration. Br J Ophthalmol. 2011; 95: 1323–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fan J, Wang J, Bensadoun A, et al. Overexpression of hepatic lipase in transgenic rabbits leads to a marked reduction of plasma high density lipoproteins and intermediate density lipoproteins. Proc Natl Acad Sci U S A. 1994; 91: 8724–8728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Santamarina-Fojo S, Haudenschild C, Amar M. The role of hepatic lipase in lipoprotein metabolism and atherosclerosis. Curr Opin Lipidol. 1998; 9: 211–219 [DOI] [PubMed] [Google Scholar]
- 24. Choi SJ, Hwang JM, Kim SI. A colorimetric microplate assay method for high throughput analysis of lipase activity. J Biochem Mol Biol. 2003; 36: 417–420 [DOI] [PubMed] [Google Scholar]
- 25. Reynolds LJ, Hughes LL, Dennis EA. Analysis of human synovial fluid phospholipase A2 on short chain phosphatidylcholine-mixed micelles: development of a spectrophotometric assay suitable for a microtiterplate reader. Anal Biochem. 1992; 204: 190–197 [DOI] [PubMed] [Google Scholar]
- 26. Biro A, Thielens NM, Cervenak L, Prohaszka Z, Fust G, Arlaud GJ. Modified low density lipoproteins differentially bind and activate the C1 complex of complement. Mol Immunol. 2007; 44: 1169–1177 [DOI] [PubMed] [Google Scholar]
- 27. Volanakis JE, Wirtz KW. Interaction of C-reactive protein with artificial phosphatidylcholine bilayers. Nature. 1979; 281: 155–157 [DOI] [PubMed] [Google Scholar]
- 28. Volanakis JE, Narkates AJ. Interaction of C-reactive protein with artificial phosphatidylcholine bilayers and complement. J Immunol. 1981; 126: 1820–1825 [PubMed] [Google Scholar]
- 29. Mitta VP, Christen WG, Glynn RJ, et al. C-reactive protein and the incidence of macular degeneration: pooled analysis of 5 cohorts. JAMA Ophthalmol. 2013; 131: 507–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Matsubara M, Hasegawa K. Benidipine, a dihydropyridine-calcium channel blocker, prevents lysophosphatidylcholine-induced injury and reactive oxygen species production in human aortic endothelial cells. Atherosclerosis. 2005; 178: 57–66 [DOI] [PubMed] [Google Scholar]
- 31. Hsieh CC, Yen MH, Liu HW, Lau YT. Lysophosphatidylcholine induces apoptotic and non-apoptotic death in vascular smooth muscle cells: in comparison with oxidized LDL. Atherosclerosis. 2000; 151: 481–491 [DOI] [PubMed] [Google Scholar]
- 32. Martin GP, el-Hariri LM, Marriott C. Bile salt- and lysophosphatidylcholine-induced membrane damage in human erythrocytes. J Pharm Pharmacol. 1992; 44: 646–650 [DOI] [PubMed] [Google Scholar]
- 33. Wang L, Clark ME, Crossman DK, et al. Abundant lipid and protein components of drusen. PLoS One. 2010; 5: e10329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li CM, Clark ME, Chimento MF, Curcio CA. Apolipoprotein localization in isolated drusen and retinal apolipoprotein gene expression. Invest Ophthalmol Vis Sci. 2006; 47: 3119–3128 [DOI] [PubMed] [Google Scholar]
- 35. Curcio CA, Johnson M, Rudolf M, Huang JD. The oil spill in ageing Bruch membrane. Br J Ophthalmol. 2011; 95: 1638–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sabbadini RA. Sphingosine-1-phosphate antibodies as potential agents in the treatment of cancer and age-related macular degeneration. Br J Pharmacol. 2011; 162: 1225–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Johnson LV, Forest DL, Banna CD, et al. Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc Natl Acad Sci U S A. 2011; 108: 18277–18282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fritsche LG, Chen W, Schu M, et al. Seven new loci associated with age-related macular degeneration. Nat Genet. 2013; 45: 433–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Park S, Kim JA, Choi S, Suh SH. Superoxide is a potential culprit of caspase-3 dependent endothelial cell death induced by lysophosphatidylcholine. J Phys Pharmacol. 2010; 61: 375–381 [PubMed] [Google Scholar]
- 40. Murugesan G, Fox PL. Role of lysophosphatidylcholine in the inhibition of endothelial cell motility by oxidized low density lipoprotein. J Clin Invest. 1996; 97: 2736–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]