

All seven possible single-site lysine-to-serine mutations of ubiquitin were tested for crystallization behavior and were found to yield crystallization ‘hit rates’ varying by two orders of magnitude. High-resolution structures of three mutants revealed that mutations can exert both promoting and permissive effects on crystallization.
Keywords: ubiquitin, surface-entropy reduction
Abstract
Ubiquitin has many attributes suitable for a crystallization chaperone, including high stability and ease of expression. However, ubiquitin contains a high surface density of lysine residues and the doctrine of surface-entropy reduction suggests that these lysines will resist participating in packing interactions and thereby impede crystallization. To assess the contributions of these residues to crystallization behavior, each of the seven lysines of ubiquitin was mutated to serine and the corresponding single-site mutant proteins were expressed and purified. The behavior of these seven mutants was then compared with that of the wild-type protein in a 384-condition crystallization screen. The likelihood of obtaining crystals varied by two orders of magnitude within this set of eight proteins. Some mutants crystallized much more readily than the wild type, while others crystallized less readily. X-ray crystal structures were determined for three readily crystallized variants: K11S, K33S and the K11S/K63S double mutant. These structures revealed that the mutant serine residues can directly promote crystallization by participating in favorable packing interactions; the mutations can also exert permissive effects, wherein crystallization appears to be driven by removal of the lysine rather than by addition of a serine. Presumably, such permissive effects reflect the elimination of steric and electrostatic barriers to crystallization.
1. Introduction
Crystallization chaperones can facilitate the crystallization of refractory targets, including membrane proteins, RNA and intrinsically disordered molecules (Zhang & Ferré-D’Amaré, 2014 ▶; Bukowska & Grütter, 2013 ▶; Griffin & Lawson, 2011 ▶; Koide, 2009 ▶). The term ‘crystallization chaperone’ is customarily applied to proteins or other macromolecules that noncovalently bind the target molecule to be crystallized; they aid crystallization by stabilizing a single conformation and/or providing additional surface area that can form lattice contacts. Our laboratory has recently developed a variation on this approach, in which a ligand known to bind to a crystallization target is covalently attached to the C-terminus of a carrier protein; afterwards, the ternary complex of carrier protein, ligand and target is crystallized. This approach has proven particularly useful for crystallizing large natural product antibiotics in complex with their ligands (Economou et al., 2012 ▶, 2013 ▶). As is true for classical crystallization chaperones, the carrier protein contributes additional surface for crystal contacts; it also enhances the solubility of the target-ligand complex and simplifies phasing of the diffraction data, since the carrier protein can serve as a molecular-replacement probe or be labeled with selenomethionine for anomalous dispersion experiments.
Ideally, carrier proteins should be stable, highly soluble and easily expressed and purified. Two examples that have worked in our hands are maltose-binding protein (MBP) and T4 lysozyme (Economou et al., 2012 ▶). However, because different target molecules behave differently with different carrier proteins, it would be advantageous to add more proteins to this list. One obvious carrier-protein candidate is ubiquitin. It is small, robust and easily expressed, and has proven to be useful as a solubility-enhancing fusion partner during protein expression (Baker, 1996 ▶; Catanzariti et al., 2004 ▶). In addition, because of its biological role as a signaling molecule, it has been evolutionarily selected to be covalently attached to various other molecules via its C-terminus. Since our strategy requires covalent attachment of ligands at the C-terminus of a carrier protein, ubiquitin would seem to be particularly well suited to this application. However, while we have succeeded in using ubiquitin as a carrier protein for several different ligand-target complexes (Economou et al., 2012 ▶, 2013 ▶), the crystallization has not been facile, nor has it produced extremely well ordered crystals. We reasoned that the disappointing performance of ubiquitin as a crystallization chaperone may be owing to the presence of multiple lysine residues on the surface of the protein.
It is well established that large, flexible side chains are excluded from crystal contacts because locking these side chains into packing interactions is accompanied by an unfavorable loss of entropy (Derewenda & Vekilov, 2006 ▶; Longenecker et al., 2001 ▶). Lysine side chains are large and flexible, and converting them to smaller amino acids such as alanine or serine is therefore predicted to promote crystallization. This ‘surface-entropy reduction’ approach has enjoyed considerable success in improving crystallization behavior, both in general (Derewenda, 2011 ▶) and in particular for a crystallization chaperone (Moon et al., 2010 ▶). In the case of ubiquitin, seven of the 76 amino acids in the protein are lysines (9.2%), whereas the average lysine composition of eukaryotic proteins is approximately 6% (Tourasse & Li, 2000 ▶; Tekaia & Yeramian, 2006 ▶). To assess how these lysine residues contribute to the crystallization behavior of ubiquitin, we have adopted a surface-entropy reduction strategy in which each lysine residue is mutated in turn to serine. Our first step focuses on ubiquitin itself, without adding any fused ligands or target molecules; the underlying assumption is that mutations that promote the crystallization of ubiquitin alone will also prove to be useful when ubiquitin is used as a carrier protein together with various target molecules. Here, we characterize the crystallization behavior of different mutants of ubiquitin alone, and describe X-ray crystal structures of three mutant proteins that exemplify some of the principal effects of lysine mutation.
2. Methods
2.1. Protein production and purification
The gene encoding untagged, wild-type human ubiquitin (76 residues) was subcloned into the pRSETA expression vector between the NdeI and EcoRI restriction sites. Single-site mutations were introduced into the wild-type sequence using the QuikChange II site-directed mutagenesis kit (Agilent Technologies); the K11S/K63S double mutant was produced by engineering the K63S mutation into the K11S plasmid.
Ubiquitin expression plasmids were transformed into Escherichia coli Rosetta 2(DE3) competent cells (EMD/Millipore). A single transformed colony was picked and grown for protein production using auto-inducing ZYP-5052 medium (Studier, 2005 ▶). Bacterial cells were grown in this medium for 20 h at 30°C with shaking at 225 rev min−1. The cells were harvested at 3500g for 15 min at 277 K and the pellet was washed with phosphate-buffered saline (PBS; 12 mM sodium/potassium phosphate pH 7.4, 137 mM NaCl, 2.7 mM KCl), re-pelleted and stored at 193 K.
All protein purification steps were carried out at 277 K. The cell pellet was thawed and suspended in eight volumes of PBS supplemented with 2 µg ml−1 DNase, 2 µg ml−1 RNase, 10 mM Mg2SO4 (final concentrations). Cells were lysed by two passes through an Emulsiflex C5 cell disruptor and the cell lysate was clarified by centrifugation at 10 000g for 30 min. Perchloric acid was added to the supernatant to a final concentration of 0.5%(v/v) and the solution was stirred for 30 min, after which it was centrifuged again at 10 000g for 30 min. The supernatant was dialyzed against 50 mM sodium acetate pH 5.2 (overnight, three changes) using 3000 MWCO dialysis tubing. The sample was then passed through a 0.22 µm filter and loaded onto a 5 ml SP1 cation-exchange column (GE Life Sciences). The column was pre-equilibrated in 50 mM sodium acetate buffer pH 4.5 and samples were eluted with a 0–40% NaCl gradient in the same buffer over 20 column volumes. The flow rate was 3 ml min−1. Fractions corresponding to the ubiquitin protein peak were pooled, concentrated to 2 ml and loaded onto a Sephacryl S100 size-exclusion column (1.6 × 60 cm; GE Life Sciences) equilibrated with 50 mM MES, 150 mM NaCl pH 6. The column was eluted using a flow rate of 0.3 ml min−1. Fractions corresponding to the ubiquitin protein peak were pooled and concentrated. The buffer concentration was lowered by several rounds of concentration, dilution with water and re-concentration. The final protein concentration was 20 mg ml−1 and the final buffer composition was 1 mM MES, 3 mM NaCl pH 6.
2.2. Crystallization
All crystallization experiments were carried out at 291 K. The commercial screens chosen for crystal screening were the The Classics, Classics II, PEGs and PEGs II Suites from Qiagen. Initial screening was conducted in 96-well sitting-drop vapor-diffusion plates (Greiner). In each well, 1 µl protein sample (20 mg ml−1 in 1 mM MES, 3 mM NaCl pH 6) was mixed with 1 µl reservoir solution; the volume of the reservoir solution was 100 µl. Crystals of the K11S and K11S/K63S mutants were optimized in 24-well plates using hanging-drop vapor diffusion, with drops consisting of 1 µl protein solution plus 1 µl reservoir solution and a reservoir volume of 1 ml. Reservoir solutions for the different mutant proteins were as follows: K11S, 0.05 M calcium chloride, 0.1 M HEPES pH 7.5, 19% PEG 4000; K33S, 0.2 M calcium chloride, 0.1 M Tris pH 8.5, 25% PEG 4000; K11S/K63S, 0.2 M ammonium sulfate pH 4.5, 25% PEG 4000.
2.3. Diffraction analysis
Crystals were mounted in nylon loops, briefly dunked in a cryoprotectant solution consisting of three volumes of glycerol plus seven volumes of reservoir solution and flash-cooled by immersion into liquid nitrogen. Data were collected at 100 K in the home laboratory equipped with a MicroMax-007 rotating-anode X-ray source and an R-AXIS IV++ image-plate detector (K11S) or on beamline 17-ID of the Advanced Photon Source equipped with a PILATUS 6M pixel-array detector (K33S and K11S/K63S).
Data processing was conducted with XDS (Kabsch, 1988 ▶) and molecular replacement was carried out using the AutoMR mode of Phaser as implemented in the PHENIX suite of programs (Adams et al., 2010 ▶; Bunkóczi et al., 2013 ▶). The A chain from PDB entry 3h7p (Weeks et al., 2009 ▶) was used as the probe molecule for molecular replacement. Refinement was carried out using PHENIX v.1.8.2-1309 (Adams et al., 2010 ▶). Rounds of refinement were alternated with map inspection and manual modification using Coot (Emsley et al., 2010 ▶). For the K33S and K11S/K63S mutants, TLS refinement was incorporated in the final stages of refinement, treating each protein monomer as a TLS group (Painter & Merritt, 2006 ▶). For all three structures described here, electron density was poor at the C-terminus of the protein; such C-terminal disorder is common in ubiquitin structures and is likely to reflect the inherent flexibility of the ubiquitin linkage site. In the case of the K11S and K11S/K63S mutants, it was not possible to fit the final residue (Gly76) into density for half (K11S) or all (K11S/K63S) of the molecules in the asymmetric unit. Validation analysis of the final refined structures was carried out using MolProbity (Chen et al., 2010 ▶). Representative examples of the final refined 2F o − F c maps are shown in Supplementary Fig. S11. Refined models and diffraction data have been deposited in the Protein Data Bank with accession codes 4pig (K11S), 4pih (K33S) and 4pij (K11S/K63S). The Nearest-cell program was used to search the PDB for ubiquitin crystals having unit cells similar to those of the mutants (Ramraj et al., 2012 ▶); none were found. Analysis of the crystal-packing interfaces was performed using the European Bioinformatics Institute PISA server (https://www.ebi.ac.uk/pdbe/pisa/; Krissinel & Henrick, 2007 ▶).
3. Results
3.1. Crystal screening
Seven single-site mutants of ubiquitin were constructed corresponding to the seven possible lysine-to-serine mutations. Only single-site mutations were considered, although in many instances mutating two or three adjacent residues has been shown to produce superior crystals (Longenecker et al., 2001 ▶); we reasoned that the small size of ubiquitin ensured that mutating even a single lysine would produce a meaningful change in the surface properties of the protein. Serine was chosen as the replacement residue, rather than the more commonly used alanine (Cooper et al., 2007 ▶), in an effort to maximize the solubility of the mutant proteins (Trevino et al., 2007 ▶). All seven mutants were expressed in E. coli using a pET-based vector; all seven expressed at levels comparable to that of the wild type and all seven mutant proteins were readily purified using the wild-type protocol. To assess the crystallization behavior of the various mutant proteins, the seven mutants plus wild-type protein were subjected to four commercially available crystallization screens, each containing 96 conditions, at a single temperature (291 K). Crystallization behavior was scored by examining each drop for obviously crystalline structures, including both single crystals and clusters of plates or needles; any drop containing such structures was scored as a ‘hit’. No effort was made to verify that all hits were actually protein crystals rather than salt crystals. However, given that salt and buffer concentrations were deliberately kept low in the protein solutions, and that the crystallization screening kits have been pre-optimized to minimize salt-crystal formation, we judged that the rate of false positives owing to salt crystallization was likely to be quite low. Crystallization experiments were allowed to incubate for 6 d before scoring.
The results of the crystal screening experiments are summarized in Fig. 1 ▶ and Supplementary Table S1. For the 384 experimental conditions tested, the wild-type protein gave a hit rate of 3.1%. Different mutants displayed markedly different behaviors in the crystallization screen, with some mutants showing a reduced propensity to crystallize while others crystallized much more readily than the wild type. Hit rates for the seven mutants ranged from less than 1% to over 20%. The best-performing mutant by far was the K11S variant, with a hit rate of 20.8%; two other mutants also showed improved crystallization behavior, namely K33S and K63S, with hit rates of 7.3 and 6.3%, respectively. The remaining four mutant proteins displayed lower hit rates than the wild-type protein. It should be emphasized that these hit rates reflect a single protein preparation and single crystal screen for each mutant; since vapor-diffusion experiments are known to exhibit intrinsic variability (Newman et al., 2007 ▶), small numerical differences in these hit rates are unlikely to be significant.
Figure 1.

Hit rates obtained for wild-type ubiquitin and the seven single-site lysine-to-serine mutations. The total number of crystalline hits obtained out of 384 conditions representing four 96-well commercial screens are shown. Hits are broken down on a per-screen basis in Supplementary Table S1.
To probe whether combining different mutants might lead to synergistic effects, a double mutant was prepared combining the K11S and K63S mutations. This mutant protein was also readily expressed and purified. It was tested in two of the four crystal screens (The Classics and Classics II Suites), where it behaved in an intermediate manner: for the 192 conditions in these two screens, the K11S and K63S single mutants and the K11S/K63S double mutant gave hit rates of 26, 9.4 and 17.7%, respectively. Hence, for this pair of mutations at least, no evidence of synergy was observed.
3.2. Examples of mutant protein structures
Crystals from several particularly promising conditions were selected for structural analysis. In one case (K33S) a crystal was taken directly from one of the initial screening conditions; in two other cases (K11S and the K11S/K63S double mutant) conditions that appeared encouraging in the initial screens were subjected to optimization, subsequently yielding large single crystals. Structures were determined and refined for all three mutants, using data measured at resolutions ranging from 1.95 to 1.50 Å (Table 1 ▶).
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Ubiquitin mutant | K11S | K33S | K11S/K63S |
|---|---|---|---|
| Data collection | |||
| X-ray source | Rotating anode | APS beamline 17-ID | APS beamline 17-ID |
| Wavelength () | 1.5418 | 1.000 | 1.000 |
| Space group | P212121 | P1 | P212121 |
| Unit-cell parameters (, ) | a = 47.70, b = 61.82, c = 91.50 | a = 27.35, b = 32.74, c = 40.34, = 69.77, = 72.55, = 73.12 | a = 31.97, b = 48.85, c = 80.32 |
| Resolution range () | 20.01.95 (2.001.95) | 23.21.50 (1.541.50) | 25.01.50 (1.541.50) |
| No. of observations | 179401 (8164) | 82699 (3672) | 121636 (4830) |
| No. of unique reflections | 20225 (1388) | 19305 (3672) | 20416 (1254) |
| Completeness (%) | 99.4 (94.2) | 98.1 (93.7) | 98.0 (82.2) |
| Mean multiplicity | 8.9 (5.9) | 4.3 (2.7) | 6.0 (3.8) |
| Mean I/(I) | 22.0 (9.1) | 8.7 (5.6) | 21.8 (4.3) |
| R merge † | 0.087 (0.212) | 0.115 (0.090) | 0.044 (0.252) |
| R meas ‡ | 0.092 (0.233) | 0.129 (0.108) | 0.049 (0.291) |
| CC1/2 § | 99.8 (97.5) | 99.0 (98.3) | 99.9 (92.6) |
| Refinement | |||
| No. of molecules in asymmetric unit | 4 | 2 | 2 |
| Solvent content¶ (%) | 37.9 | 33.7 | 33.1 |
| Resolution range () | 19.81.95 | 23.21.50 | 24.41.50 |
| No. of reflections used | |||
| Working set | 19139 | 17374 | 18414 |
| Test set | 1037 | 1931 | 2000 |
| No. of protein atoms | 2396 | 1198 | 1193 |
| No. of solvent atoms | |||
| Glycerol + ions | 13 | 5 | 31 |
| Water | 221 | 134 | 81 |
| Estimated Wilson B value (2) | 10.0 | 10.8 | 13.4 |
| Mean B values (2) | |||
| Protein | 11.2 | 14.0 | 17.1 |
| Ions | 23.4 | 19.6 | 24.2 |
| Water | 15.1 | 22.8 | 28.7 |
| R.m.s. deviations from ideal geometry | |||
| Bond distances () | 0.0075 | 0.010 | 0.0072 |
| Bond angles () | 1.10 | 1.32 | 1.15 |
| Ramachandran statistics | |||
| Most favored (%) | 100.0 | 100.0 | 100.0 |
| Additionally allowed (%) | 0.0 | 0.0 | 0.0 |
| R cryst/R free | 0.171/0.217 | 0.165/0.190 | 0.176/0.198 |
| Clashscore | 1.02 | 0.00 | 1.62 |
R
merge is calculated by the equation R
merge = 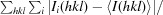
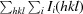 , where I
i(hkl) is the ith measurement.
, where I
i(hkl) is the ith measurement.
R
meas (or the redundancy-independent R
merge) is calculated by the equation 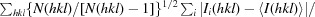 , where I
i(hkl) is the ith measurement and N(hkl) is the redundancy of each unique reflection hkl.
, where I
i(hkl) is the ith measurement and N(hkl) is the redundancy of each unique reflection hkl.
CC1/2 is the correlation coefficient between two randomly chosen half data sets (Diederichs Karplus, 1997 ▶).
Calculated using the Matthews Probability Calculator (http://www.ruppweb.org/mattprob/default.html), considering the mass of the protein chains only (Kantardjieff Rupp, 2003 ▶).
3.3. Comparison of overall folds
For all three mutants, the overall fold of the protein was compared with that of wild-type ubiquitin. In order to make this comparison, we first assessed the range of normal structural variation expected for the unmutated protein by superposing nine independent structures of wild-type ubiquitin (Supplementary Table S2) using the SALIGN server (Braberg et al., 2012 ▶). Fig. 2 ▶ shows how the root-mean-square (r.m.s.) deviation from the average position varies with residue number for these nine superposed structures. The most conformationally variable positions in the wild-type protein are seen to be the N- and C-termini and three surface-exposed turns located near residues 8, 33 and 46, respectively.
Figure 2.

Comparison of deviations in backbone position for wild-type ubiquitin and three mutants. In all three panels, the solid black line represents the r.m.s. deviation from the average Cα position for nine independent wild-type ubiquitin structures (Supplementary Table S2). (a) K11S mutant. Differences in Cα position are shown for the mutant versus the average wild-type structure after superposition; the four independent chains in the K11S asymmetric unit are shown as open circles, filled circles, filled squares and inverted triangles for chains A, B, C and D, respectively. (b) K33S mutant. Differences in Cα position are shown for the mutant versus the average wild-type structure after superposition; the mutant A and B chains are represented by filled circles and inverted triangles, respectively. (c) K11S/K63S double mutant. Differences in Cα position are shown for the mutant versus the average wild-type structure after superposition; the mutant A and B chains are represented by filled circles and inverted triangles, respectively.
Once we had established the degree of conformational variability for the wild-type protein, we then determined whether each mutant differed significantly from the wild-type conformation by superposing all independent copies of each mutant structure upon the average wild-type structure (Kabsch, 1976 ▶, 1978 ▶) and calculating the distance of each mutant Cα position from the average wild-type position (Fig. 2 ▶). This comparison revealed the folds of the mutant and wild-type proteins to be essentially similar. As seen for the wild type, the positions of greatest variability in the mutants are centered around residues 8, 33 and 46; this holds true regardless of whether or not a given mutation lies near one of these variable regions. We therefore conclude that the mutations do not elicit any significant perturbations in the fold of the protein.
3.4. Differences in crystal packing
The three mutants studied display marked differences in crystal packing. Each of the mutants crystallizes in a different crystal form, and these three crystal forms are distinct from those of any monoubiquitin or polyubiquitin structures previously found in the Protein Data Bank. The specifics of packing for each of the three mutants are described below.
3.4.1. K11S
The asymmetric unit of the K11S crystals contains four ubiquitin monomers arranged as two dimers (Fig. 3 ▶ a). The two dimers are highly similar to one another, and can be superposed with an r.m.s.d. of 0.8 Å for Cα positions. Within each dimer, the two monomers are related by a twofold rotational axis of symmetry. The dimers themselves are related by a pseudo-translational symmetry, which is reflected by a strong peak in the native Patterson function at approximately (0.0, 0.5, 0.0).
Figure 3.

Crystal packing in the K11S crystals. (a) The asymmetric unit of the K11S crystals, containing two dimers (blue/red and cyan/magenta). The Ser11 residues are shown as gold spheres. The orientation of the unit-cell axes is also shown. (b) Close-up view of the interaction between the two monomers making up one dimer. The hydroxyl groups of Thr9 and Ser11 on one molecule (cyan) form hydrogen bonds to the backbone atoms of Leu71 and Leu73 on the facing molecule (magenta). (c) Superposition of the K11S dimer (green) upon the Lys11-linked diubiquitin structure (purple; chains A and G from PDB entry 2xew are shown).
The interfaces within each dimer appear to be the most significant packing interactions in the crystal. Each such interface buries approximately 840 Å2 of surface area, while the next largest packing interaction (which corresponds to one dimer packing against a neighboring dimer) buries less than 40% of this area. Interestingly, the K11S mutation lies within the dimeric interface. In one dimer, both Ser11 side chains form hydrogen bonds to the Leu73 backbone amides on the facing monomers, while in the other dimer both copies of Ser11 form water-mediated hydrogen bonds to the corresponding Leu73 backbone amides (Fig. 3 ▶ b). Thus, all four copies of the mutant residue in the asymmetric unit are engaged in packing interactions. This particular packing interaction would not be compatible with the wild-type protein, as a free lysine side chain at position 11 would be placed into close proximity with Arg72 on the facing monomer, giving rise to electrostatic repulsion.
Dimeric structures equivalent to those described here can be found in other crystal structures of ubiquitin or ubiquitin-like proteins, notably those of Lys11-linked ubiquitin dimers (Bremm et al., 2010 ▶; Matsumoto et al., 2010 ▶), an insertion mutant of ubiquitin (Ferraro et al., 2006 ▶) and the ubiquitin-like protein NEDD8 (Whitby et al., 1998 ▶; Fig. 3 ▶ c). Importantly, in all of these structures the electrostatic repulsion between Lys11 on one chain and Arg72 on the facing chain is alleviated either by neutralizing the charge of Lys11 via covalent modification (as in the Lys11-linked dimers), by moving Lys11 to a different position (which is what occurs in the insertion mutant) or by removing Arg72 entirely (as in the case of NEDD8, which contains no Arg72 equivalent).
3.4.2. K33S
The asymmetric unit of the K33S crystal structure contains two molecules (chains A and B). These molecules are arranged in the triclinic cell so as to form stacked layers of protein molecules (Fig. 4 ▶ a). The interactions between the layers are modest, but within each layer the A and B chains interact through two distinct and extensive interfaces (Fig. 4 ▶ b). The larger of these interfaces represents a novel dimeric packing interaction for ubiquitin (i.e. no similar packing interface was found in a PISA search of the PDB). The larger interface buries 720 Å2 of surface, much of it hydrophobic; among the side chains that are at least partially buried in this interface are those of Leu8, Ile44, Val70, Leu71 and Leu73 from the A and/or B chains. No direct polar contacts are observed between the two proteins in this interface; however, two water molecules and one chloride ion are found in the interface and bridge the two protein chains.
Figure 4.

Crystal packing in the K33S crystals. (a) The crystal lattice contains stacked layers of protein molecules. The A and B chains are colored yellow and blue, respectively; the Ser33 residues are shown as red spheres, emphasizing their positioning at the junction between adjacent layers in the lattice. The outline of the triclinic unit cell is shown. (b) An orthogonal view of the lattice packing, showing a single layer. The larger and smaller interfaces are shown as green and red dashed lines. (c, d) Detailed views showing neighboring basic residues in the environment of the Ser33 side chain in the A chain (c) and B chain (d); a lysine residue at position 33 would cause electrostatic repulsion in each case.
The smaller interface (area = 435 Å2) is much more open and polar in character than the larger interface. A number of hydrogen bonds connect the A and B chains across this interface, including between the side chains of Lys6 and Gln62, between the side chain of His68 and the backbone carbonyl of Glu64 and between the carbonyl O atom of Ala46 on one chain and the backbone amide of the same residue on the opposing chain. In addition, over a dozen water molecules and chloride ions populate this interface.
Ser33 is not found in either of these two major packing interfaces. Instead, both copies of this residue lie at the junction between adjacent layers in the crystal (Fig. 4 ▶ a). They do not, however, form any interactions linking the adjacent layers; rather, one copy of the Ser33 side chain participates in an internal hydrogen bond to the side chain of Glu34, while the other copy participates in no interactions whatsoever, even with water molecules. Although they do not form any direct packing interactions, both copies of the Ser33 side chain lie close to several basic residues on neighboring molecules. In the A chain Ser33 is close to the Arg42 and Arg72 residues on a neighboring molecule in an adjacent layer, and in the B chain Ser33 lies close to Arg54 on a molecule in an adjacent layer (Figs. 4 ▶ c and 4 ▶ d). The side chains of these neighboring residues are not well ordered, judging from the electron density; however, their backbone positions are well defined, meaning that their side chains cannot stray far from the position occupied by the side chain of residue 33. Therefore, a lysine at position 33 would lead to electrostatic repulsion between adjacent layers of molecules in the crystal and would be incompatible with this particular crystal packing.
3.4.3. K11S/K63S double mutant
Crystals of the double mutant contain two molecules in the asymmetric unit (again labeled A and B). There are two major packing interfaces through which the A and B chains interact (Fig. 5 ▶ a), as well as additional small points of contact. The larger interface, with an area of 502 Å2, is a rather open interface with multiple bridging solvent molecules and ions; this interface also partially buries the so-called Ile44 patch, a hydrophobic patch on the ubiquitin surface that is frequently exploited for recognition. Packing interfaces similar to this one can be found in many different ubiquitin crystal structures (see, for example, PDB entries 4jqw, 1f9j, 3alb, 3m3j, 1aar and 2o6v). Neither mutation site is near this packing interface.
Figure 5.

Crystal packing in crystals of the K11S/K63S double mutant. (a) The two principal interfaces through which A and B chains interact. The larger interface relates the blue B chain and the green A chain; the smaller interface relates the same blue B chain to the violet A chain. The Ser11 residues are shown as yellow spheres and the Ser63 residues are shown as red spheres. (b) A detailed view of the smaller interface, showing the hydrogen bonds formed between Ser11 on the violet A chain and Thr9 on the blue B chain. A sulfate ion (shown in ball-and-stick representation) also helps to bridge these two protein molecules and interacts with threonine residues on both chains. Ser11 on the blue B chain forms a hydrogen bond to Gln62 on a different symmetry mate of the A chain (shown in cyan).
The second major packing interface has an area of 464 Å2, and no equivalent interfaces were identified in a PISA search. This interface relies on polar interactions, many of which utilize bridging water molecules and sulfate ions. Notably, Ser11 from the A chain participates in an interaction stabilizing this interface, forming a hydrogen bond to Thr9 on the facing B chain. Ser11 from the B chain also forms a hydrogen bond to Gln62 on a symmetry-related copy of the A chain. Thus, both copies of Ser11 in the asymmetric unit are engaged in interactions that promote crystal packing. Lysine residues would not be capable of making these specific interactions; furthermore, the tight packing around these positions would not accommodate the larger lysine side chain.
Neither copy of Ser63 lies in either of the two major packing interfaces described above, but both copies are nonetheless engaged in packing interactions. Ser63 of the A chain forms hydrogen bonds to water molecules that bridge this molecule to a neighboring copy of the B chain; Ser63 of the B chain forms a hydrogen bond to the backbone carbonyl O atom of Thr9 on another copy of the B chain. Again, steric constraints would prevent a lysine side chain from occupying either of these positions.
4. Discussion
Surface-entropy reduction and related protein-engineering approaches to crystallization are based on the notion that small, localized changes on the surface of a protein can profoundly affect its crystallization behavior (Ruggiero et al., 2012 ▶). The experiments described in this paper support this idea by showing that single-site mutations can alter the ‘hit rates’ of ubiquitin crystallization by two orders of magnitude.
An algorithm has been developed to predict mutations that are likely to enhance crystallization behavior (Goldschmidt et al., 2007 ▶). To compare this prediction scheme with our results, we submitted the ubiquitin sequence to the Surface Entropy Reduction prediction (SERp) server (http://services.mbi.ucla.edu/SER/). The prediction server suggested three positions as good candidates for mutation, Lys33, Lys63 and Lys11, the same three sites for which we find that lysine-to-serine mutations improve the crystallization behavior. We must note, however, that the SERp recommendations included mutating additional residues adjacent to both Lys33 and Lys63, whereas we chose to mutate only the lysines. This difference may explain why the rank order of the efficacy of these mutations differs between the SERp prediction (Lys33 > Lys63 ≃ Lys11) and our actual results (Lys11 > Lys33 ≃ Lys63). Thus, because single-site mutants can behave very differently from double or triple mutants, caution should be used when comparing the SERp predictions and our results for the Lys33 and Lys63 positions.
How do local alterations of surface structure enhance crystallization? A simple model is that mutations can be promoting or permissive. Promoting mutations contribute favorable interactions at crystal contact sites; permissive mutations remove an impediment that might otherwise prevent the formation of a crystal contact. The three structures discussed in this paper provide examples of both effects.
The K11S mutant promotes the formation of a dimeric packing interaction by providing hydrogen-bonding interactions that help to stabilize this interface (Fig. 3 ▶). However, this dimer interface is substantial, and so while the hydrogen bonds contributed by Ser11 are likely to assist in its formation, they are not crucial; indeed, similar dimeric structures are observed in crystals of ubiquitin variants that lack a serine at position 11. Notably, in all instances where these dimeric structures form, a potential electrostatic repulsion between Lys11 and a neighboring Arg72 must first be removed, either by covalent modification or movement of Lys11 or by removal of Arg72. This implies that the presence of Lys11 prevents formation of this particular dimer interface, and therefore the K11S mutation is playing a permissive role in crystallization as well as a promoting role. As an aside, we note that the biological implication of this is clear: this dimeric interface has evolved to provide a structural motif that is unique to Lys11-linked chains, providing a structural means by which the cellular machinery can distinguish such chains from other polyubiquitin species and from unlinked ubiquitin monomers.
In contrast to the K11S mutant, the K33S mutant appears to be playing a purely permissive role in the structure presented here; the Ser33 side chain of the mutant protein fails to participate in any specific interactions either within the same molecule or with neighboring molecules. However, these mutant side chains find themselves in environments within the crystal lattice that could not accommodate the larger, positively charged lysine side chain; hence, the only contribution of Ser33 to the formation of these crystals is to remove a barrier presented by the lysine.
Finally, the K11S/K63S double mutant shows both promoting and permissive effects. Both Ser11 and Ser63 make favorable hydrogen-bonding contacts with neighboring molecules in the lattice, thereby promoting crystal formation. In addition, the mutations also play permissive roles, since the environments of both mutant side chains are sufficiently cramped that steric effects militate against the inclusion of lysines at either position.
Thus, the crystal-enhancing mutations observed in the structures reported here can be readily rationalized in terms of promoting and/or permissive effects. However, how might one explain the mutants that are less likely to crystallize than the wild-type protein? At least three such mutants were identified in our screens: K6S, K27S and K29S (a fourth mutant, K48S, gave a slightly lower hit rate than the wild type, but the significance of this small difference is unclear).
Two of these mutations are likely to have deleterious effects on the stability of the protein, namely K27S and K29S. The Lys27 side chain is largely buried and is engaged in a salt bridge and a hydrogen bond to the side chain of Asp52 and the backbone carbonyl of Gln41, respectively. The Lys29 side chain is not buried, but lies flat along the surface of the protein, and interacts with the backbone carbonyl O atom of Glu16, as well as with the side chain of Asp21. Hence, it is reasonable to speculate that mutating either Lys27 or Lys29 would destabilize the protein structure, with a concomitant negative impact on crystallization behavior.
The K6S mutant also displays a poorer crystallization success rate than the wild type, but this is harder to explain than the K27S or K29S mutants. Lys6 does not make internal stabilizing interactions within the molecule; rather, in most ubiquitin crystal structures the Lys6 side chain projects outwards from the protein surface and is frequently poorly ordered. This residue would therefore seem to be a good candidate for surface-entropy reduction. The highest conformational variability in the backbone of ubiquitin occurs around residue 6 (Fig. 2 ▶), but it is not clear how this would cause the K6S mutant to resist crystallization. We conjecture (without supporting evidence) that the surface region around Lys6 may be ‘sticky’ and that the charged lysine group serves to prevent nonspecific aggregation that might otherwise be mediated by this spot.
In conclusion, we have shown that single lysine-to-serine mutations in ubiquitin greatly alter the propensity of the protein to crystallize, consistent with the predictions of the surface-entropy reduction theory. Three structural examples of such altered proteins reveal mutant residues engaging in favorable lattice contacts, but also present evidence for permissive effects, in which crystallization is enhanced by removing the lysine rather than by adding favorable new interactions.
Supplementary Material
PDB reference: ubiquitin, K33S mutant, 4pih
PDB reference: K11S mutant, 4pig
PDB reference: K11S/K63S double mutant, 4pij
Details of crystallization hits; structures used for r.m.s.d. calculations; electron-density maps for the structures reported.. DOI: 10.1107/S2053230X14019244/be5271sup1.pdf
Acknowledgments
This research was supported in part by grant R01GM079508 (NIH/NIGMS). We gratefully acknowledge Kim Grasty for assistance with cloning and protein purification. Partial support for our laboratory home source was provided by the Department of Biochemistry and Molecular Biology at the Drexel University College of Medicine. We gratefully acknowledge the assistance of the staff of IMCA-CAT at the Advanced Photon Source. Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman–Woodward Medical Research Institute. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-06CH11357.
Footnotes
Supporting information has been deposited in the IUCr electronic archive (Reference: BE5271).
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Baker, R. T. (1996). Curr. Opin. Biotechnol. 7, 541–546. [DOI] [PubMed]
- Braberg, H., Webb, B. M., Tjioe, E., Pieper, U., Sali, A. & Madhusudhan, M. S. (2012). Bioinformatics, 28, 2072–2073. [DOI] [PMC free article] [PubMed]
- Bremm, A., Freund, S. M. & Komander, D. (2010). Nature Struct. Mol. Biol. 17, 939–947. [DOI] [PMC free article] [PubMed]
- Bukowska, M. A. & Grütter, M. G. (2013). Curr. Opin. Struct. Biol. 23, 409–416. [DOI] [PubMed]
- Bunkóczi, G., Echols, N., McCoy, A. J., Oeffner, R. D., Adams, P. D. & Read, R. J. (2013). Acta Cryst. D69, 2276–2286. [DOI] [PMC free article] [PubMed]
- Catanzariti, A.-M., Soboleva, T. A., Jans, D. A., Board, P. G. & Baker, R. T. (2004). Protein Sci. 13, 1331–1339. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cooper, D. R., Boczek, T., Grelewska, K., Pinkowska, M., Sikorska, M., Zawadzki, M. & Derewenda, Z. (2007). Acta Cryst. D63, 636–645. [DOI] [PubMed]
- Derewenda, Z. S. (2011). Acta Cryst. D67, 243–248. [DOI] [PMC free article] [PubMed]
- Derewenda, Z. S. & Vekilov, P. G. (2006). Acta Cryst. D62, 116–124. [DOI] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol. 4, 269–275. [DOI] [PubMed]
- Economou, N. J., Nahoum, V., Weeks, S. D., Grasty, K. C., Zentner, I. J., Townsend, T. M., Bhuiya, M. W., Cocklin, S. & Loll, P. J. (2012). J. Am. Chem. Soc. 134, 4637–4645. [DOI] [PMC free article] [PubMed]
- Economou, N. J., Zentner, I. J., Lazo, E., Jakoncic, J., Stojanoff, V., Weeks, S. D., Grasty, K. C., Cocklin, S. & Loll, P. J. (2013). Acta Cryst. D69, 520–533. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Ferraro, D. M., Ferraro, D. J., Ramaswamy, S. & Robertson, A. D. (2006). J. Mol. Biol. 359, 390–402. [DOI] [PubMed]
- Goldschmidt, L., Cooper, D. R., Derewenda, Z. S. & Eisenberg, D. (2007). Protein Sci. 16, 1569–1576. [DOI] [PMC free article] [PubMed]
- Griffin, L. & Lawson, A. (2011). Clin. Exp. Immunol. 165, 285–291. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (1976). Acta Cryst. A32, 922–923.
- Kabsch, W. (1978). Acta Cryst. A34, 827–828.
- Kabsch, W. (1988). J. Appl. Cryst. 21, 916–924.
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci. 12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Koide, S. (2009). Curr. Opin. Struct. Biol. 19, 449–457. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Longenecker, K. L., Garrard, S. M., Sheffield, P. J. & Derewenda, Z. S. (2001). Acta Cryst. D57, 679–688. [DOI] [PubMed]
- Matsumoto, M. L., Wickliffe, K. E., Dong, K. C., Yu, C., Bosanac, I., Bustos, D., Phu, L., Kirkpatrick, D. S., Hymowitz, S. G., Rape, M., Kelley, R. F. & Dixit, V. M. (2010). Mol. Cell, 39, 477–484. [DOI] [PubMed]
- Moon, A. F., Mueller, G. A., Zhong, X. & Pedersen, L. C. (2010). Protein Sci. 19, 901–913. [DOI] [PMC free article] [PubMed]
- Newman, J., Xu, J. & Willis, M. C. (2007). Acta Cryst. D63, 826–832. [DOI] [PubMed]
- Painter, J. & Merritt, E. A. (2006). Acta Cryst. D62, 439–450. [DOI] [PubMed]
- Ramraj, V., Evans, G., Diprose, J. M. & Esnouf, R. M. (2012). Acta Cryst. D68, 1697–1700. [DOI] [PMC free article] [PubMed]
- Ruggiero, A., Smaldone, G., Squeglia, F. & Berisio, R. (2012). Protein Pept. Lett. 19, 732–742. [DOI] [PubMed]
- Studier, F. W. (2005). Protein Expr. Purif. 41, 207–234. [DOI] [PubMed]
- Tekaia, F. & Yeramian, E. (2006). BMC Genomics, 7, 307. [DOI] [PMC free article] [PubMed]
- Tourasse, N. J. & Li, W.-H. (2000). Mol. Biol. Evol. 17, 656–664. [DOI] [PubMed]
- Trevino, S. R., Scholtz, J. M. & Pace, C. N. (2007). J. Mol. Biol. 366, 449–460. [DOI] [PMC free article] [PubMed]
- Weeks, S. D., Grasty, K. C., Hernandez-Cuebas, L. & Loll, P. J. (2009). Proteins, 77, 753–759. [DOI] [PMC free article] [PubMed]
- Whitby, F. G., Xia, G., Pickart, C. M. & Hill, C. P. (1998). J. Biol. Chem. 273, 34983–34991. [DOI] [PubMed]
- Zhang, J. & Ferré-D’Amaré, A. R. (2014). Curr. Opin. Struct. Biol. 26C, 9–15. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: ubiquitin, K33S mutant, 4pih
PDB reference: K11S mutant, 4pig
PDB reference: K11S/K63S double mutant, 4pij
Details of crystallization hits; structures used for r.m.s.d. calculations; electron-density maps for the structures reported.. DOI: 10.1107/S2053230X14019244/be5271sup1.pdf