

Abstract
Fibrosis pathophysiology is critically regulated by Smad 2– and Smad 3–mediated transforming growth factor-β (TGF-β) signaling. Disintegrin metalloproteases (Adam) can manipulate the signaling environment, however, the role and regulation of ADAMs in renal fibrosis remain unclear. TGF-β stimulation of renal cells results in a significant up-regulation of Adams 10, 17, 12, and 19. The selective Smad2/3 inhibitor SB 525334 reversed these TGF-β–induced changes. In vivo, using ureteral obstruction to model renal fibrosis, we observed increased Adams gene expression that was blocked by oral administration of SB 525334. Similar increases in Adam gene expression also occurred in preclinical models of hypertension-induced renal damage and glomerulonephritis. miRNAs are a recently discovered second level of regulation of gene expression. Analysis of 3′ untranslated regions of Adam12 and Adam19 mRNAs showed multiple binding sites for miR-29a, miR-29b, and miR-29c. We show that miR-29 family expression is decreased after unilateral ureter obstruction and this significant decrease in miR-29 family expression was observed consistently in preclinical models of renal dysfunction and correlated with an increase in Adam12 and Adam19 expression. Exogenous overexpression of the miR-29 family blocked TGF-β–mediated up-regulation of Adam12 and Adam19 gene expression. This study shows that Adams are involved in renal fibrosis and are regulated by canonical TGF-β signaling and miR-29. Therefore, both Adams and the miR-29 family represent therapeutic targets for renal fibrosis.
Fibrosis of the tubulointerstitium is the common end point of most progressive renal diseases and consistently has been shown to be the best histologic predictor of progression toward end-stage renal failure.1 This point of no return involves tubular atrophy, leukocyte infiltration, myofibroblast differentiation, activation, and proliferation and the excess accumulation of extracellular matrix, resulting ultimately in scarring and loss of function.2 Transforming growth factor-β (TGF-β) initiates fibrosis pathogenesis predominantly mediated by downstream effector molecules, known as Smads.3–5 Binding of active TGF-β1, synthesized by epithelial and mesangial cells in response to injury and by activated macrophages, to TGF-β type 1 and type 2 receptors results in the initiation of a signaling cascade brought about by the phosphorylation, nuclear translocation, and regulation of gene expression by Smads 2 and 3; a phenomenon termed canonical signaling.6 Short endogenous 22-nucleotide noncoding RNAs capable of posttranscriptional gene regulation7 also have been shown to regulate renal fibrosis.8,9 Recently, we and others described the presence of a canonical TGF-β–regulated miRNA signature in renal disease.10,11 The miR-29 family, consisting of miR-29a, miR-29b, and miR-29c, has been shown to be a critical mediator of disease pathophysiology.12 Emerging evidence suggests that the miR-29 family is regulated by canonical TGF-β signaling via p-Smad 3.13 The miR-29 family is expressed differentially in healthy renal tissue and fibrosis. In the experimental unilateral ureter obstruction (UUO) model of renal fibrosis, it previously was shown that expression of the miR-29 family is reduced after injury.13 Furthermore, forced expression of miR-29b before or 4 days after UUO was protective from interstitial fibrosis induced by UUO.13 The miR-29 family is able to post-transcriptionally regulate a cohort of fibrosis-relevant genes, including collagens and various metalloproteases.14,15
Metzincin metalloproteases, which includes the classic matrix metalloproteases and their tissue inhibitors, have been shown to play important roles in regulating matrix turnover in normal nephrogenesis and in fibrotic renal disease.16 Disintegrin metalloproteases (Adams) are members of this class that can regulate key cellular and acellular processes.17 Thus, they potentially can regulate both the inflammatory and the fibrotic aspects of renal pathophysiology. Adams 10 and 17 classically have been studied in the proinflammatory tumor necrosis factor turnover in tissue,18 which in turn determines the activation status of the cellular components of renal inflammation.19,20 Adams 10 and 12 have been shown to facilitate leukocyte infiltration21,22 and cleave E-cadherin, thereby promoting invasiveness in differentiated myofibroblasts.23 Members of the epidermal growth factor signaling pathway, crucial in myofibroblast differentiation and resulting basement membrane invasion, have been shown to be cleaved selectively by Adams 10, 17, and 19.24,25 Adams are the central and rate-limiting factors in the cross-talk between epidermal growth factor and the angiotensin II pathways, a phenomenon now termed triple membrane pass signaling.26 Similar to other metzincin proteases, Adams can regulate extracellular matrix turnover in tissue.27–29
The role of Adams in fibrosis, including renal fibrosis, remains largely unknown. There is considerable evidence to associate Adams with renal disease. Melenhorst et al30–32 and Mulder et al33showed increased expression of Adam17 and Adam19 mRNA in renal transplant and non–transplant-related fibrotic disease in humans. Lautrette et al34 showed that inhibition of Adam17 with the pharmacologic inhibitor WTACE2 reduced angiotensin II–induced renal fibrotic lesions in mice and Schramme et al35 showed that Adam10-mediated shedding of CXCL16 from cultured thick ascending limb distal tubular cells induced CXCR6-expressing leukocyte and lymphocyte chemotaxis in vitro. Although regulation of matrix metalloproteases and tissue inhibitors of matrix metalloproteases at both the gene and functional levels by TGF-β has been well documented,36–38 we hypothesized that the regulation Adams also are mediated by TGF-β.
Therefore, we determined ADAMs expression in relevant in vitro and in vivo experimental models of renal fibrosis. Because TGF-β is involved in fibrosis pathophysiology, we then determined if canonical TGF-β signaling regulates Adams using a specific small molecule inhibitor. Furthermore, because the miR-29 family targets included Adam12 and Adam19, we examined whether these Adams potentially can be regulated by modulation of miR-29.
Materials and Methods
In Vitro Model of Renal Fibrosis
Rat tubular epithelial cells (NRK52E) were cultured according to ATCC (Manassas, VA) protocols. They were seeded, grown to subconfluent densities, and were subjected to a 48-hour serum starvation (0.2% fetal bovine serum vol/vol in media) to synchronize the cell cycle. Cells were stimulated with 10 ng/mL (wt/vol) recombinant TGF-β (R&D Systems, Abingdon, UK) over a 4-day time course, with TGF-β being replenished every 48 hours. 6-[2-Tert-butyl-5-(6-methyl-pyridin-2-yl)-1H-imidazol-4-yl]-quinoxaline (SB 525334; R&D Systems) is an orally bioavailable and selective small-molecule inhibitor that blocks ALK5 serine/threonine kinase activity and prevents phosphorylation of the R-Smads 2 and 3 and their subsequent nuclear translocalization and gene activation.39 SB 525334 used at 1 μmol/L wt/vol in vehicle (0.1% dimethyl sulfoxide in PBS) was added to cells 60 minutes before TGF-β stimulation and replenished every 24 hours.
Animal Models
All protocols and surgical procedures were approved by the local animal care committee. Animal experiments were in accordance with the Animals Scientific Procedures Act UK of 1986.
Unilateral Ureter Obstruction
Eight-week-old male C57/BL6 mice weighing approximately 21 to 23 g were used. Animals were anesthetized by isoflurane inhalation and underwent a midline laparotomy; the right ureter was exposed by blunt dissection. Complete ureteral obstruction was performed by tying two 4-0 silk sutures around the isolated ureter. The midline incision then was resutured. Control groups consisted of sham surgeries including laparotomy and ureter handling but not ligation. Animals were sacrificed 3, 7, and 14 days after surgery and kidneys were removed for analysis. To assess the effect of TGF-β in vivo, SB 525334 in 0.1% vol/vol dimethyl sulfoxide in PBS was administered daily at 10 mg/kg/d body weight by standard oral gavage in a final volume of 100 μL. Animals were sacrificed on day 7 and kidneys were snap frozen and stored at −80°C before analysis.
Genetic Rat Model of Hypertension-Induced Renal Damage
Inbred colonies of stroke-prone, spontaneously hypertensive and Wistar-Kyoto rats have been developed and maintained at the University of Glasgow since 1991, as described previously.40 All animals were sacrificed at 21 weeks. After sacrifice, kidneys were removed, snap frozen, and stored at −80°C before analysis.
Induction of Glomerulonephritis
Male Wistar-Kyoto rats (Harlan, Wyton, UK) (age, 7 to 12 weeks) were used throughout the study. All protocols and surgical procedures were approved by the local animal care committee. Animals were age-matched at sacrifice to minimize age-related differences in miRNA expression and gene expression. For the induction of glomerulonephritis, rats were infused intravenously with 2 mg/kg anti-Thy1 antibody (ER4) three times, 1 week apart. After sacrifice at 7 days, kidneys were removed and portions were fixed in 10% formalin or snap frozen and stored at −80°C before analysis.
RNA Extraction and Gene Expression Analysis
RNA was extracted lysis using the miRNeasy kit (Qiagen, Manchester, UK) following the manufacturer's instructions, and quantified using a NanoDrop ND-1000 Spectrophotometer (ThermoFisher Scientific, Waltham, MA). For cDNA synthesis, 1 μg of total RNA (cell or whole tissue) was reverse-transcribed using the manufacturer's instructions (Life Technologies, Paisley, UK) and quantitative real-time PCR (qPCR) was performed using specific inventoried gene expression or stem-loop miRNA primer/probes (Life Technologies) on the Applied Biosystems 7900 HT real-time PCR system. Mouse glyceraldehyde-3-phosphate dehydrogenase or U87 (rat)/U6 (mouse) primer/probes were used as endogenous controls for gene and miRNA expression, respectively.
Immunocytochemistry
Immunocytochemistry was performed on cells grown on sterile 18-mm coverslips (Menzel-Glazer, Braunschweig, Germany) and fixed with 4% (wt/vol) paraformaldehyde and permeabilized by using 0.1% (vol/vol) Triton X-100 (Sigma-Aldrich, Gillingham, UK). These cells were probed using rabbit polyclonal anti-Adam (all 1:100; Abcam, Cambridge, UK) and p-Smad2/3 specific (1:100; Santa Cruz, Santa Cruz, CA) antibodies followed by Alexa-Flour 488 tagged secondary antibody (Invitrogen, Paisley, UK). Coverslips were mounted onto slides using DAPI Prolong-Gold (Molecular Probes, Paisley, UK) mountant and analyzed by fluorescence microscopy with a Zeiss LSM510 (Carl-Zeiss AG, Oberkochen, Germany).
Tissue sections (3 μm) were deparaffinized, hydrated, and endogenous peroxidases were quenched using 0.3% H2O2 in methanol. Heat-mediated antigen retrieval was performed using citrate buffer (pH 6.6). Sections were serum- and avidin/biotin-blocked (Vector Laboratories, Peterborough, UK) and probed using 1:100 dilution of rabbit polyclonal primary antibody (Abcam) overnight at 4°C. They then were washed and probed with 1:400 dilution of biotinylated anti-rabbit secondary antibody (Vector Laboratories) and horseradish-peroxidase–conjugated streptavidin, and detected using diaminobenzidine substrate (Vector Laboratories). The sections were visualized and imaged using a Zeiss Axiovert microscope (Carl-Zeiss AG).
Immunofluorescence
Sections of mouse kidneys (3 μm) were prepared as described earlier and heat-mediated antigen retrieval was performed using citrate buffer (pH 6.6). Sections were blocked in buffer containing 15% (vol/vol) goat serum and 0.05% (vol/vol) Tween 20 in Tris-buffered saline and probed using p-smad 2/3 (1:100) overnight at 4°C, washed, and probed with a 1:400 dilution of biotinylated anti-rabbit secondary antibody (Vector Laboratories). Slides were treated with 0.1% vol/vol Sudan Black in methanol (Sigma) for 10 minutes at room temperature in the dark to minimize background fluorescence, followed by treatment with 100 μg/mL DAPI for 10 minutes. ProLong Gold with DAPI (Invitrogen) was placed on clean plain glass coverslips and slides were placed onto coverslips, tissue-side down, over the drop, and cured overnight on the bench top. The sections were imaged and imaged using a Zeiss LSM510 confocal microscope. Green fluorescent nuclei were counted in six nonoverlapping images per kidney section per animal and are represented as a percentage ratio to the number of DAPI-stained total nuclei. At least 100 nuclei were counted per image.
Adam10/17 Activity Assay
An Adam17 activity assay was performed based on cleavage of fluorescent substrate. Briefly, an enzyme versus substrate reaction was set up at a final volume of 100 μL, using half the dilution range from 1.6 to 0.2 ng/μL of recombinant murine Adam17 peptide (R&D Systems) or 20 μg whole-tissue protein lysate. The reaction was initiated with the addition of 10 μmol/L Adam10/17-specific substrate Mca-P-L-A-Q-A-V-Dpa-R-S-S-S-R-NH2 Fluorogenic Peptide Substrate III (R&D Systems) and performed at 37°C. Fluorescence intensity was determined with the fluorescence plate reader SpectraMax 2.0 Station (Molecular Devices) using an excitation wavelength at 320 nm and emission at 405 nm every 5 minutes for 20 minutes. Fluorescence kinetics curves were constructed by plotting fluorescence intensity versus enzyme concentration. A further Vmax versus enzyme concentration curve was constructed from log-phase values from the kinetics curve. The Adam10/17 concentration in tissue lysates was extrapolated from the Vmax standard curve.
Manipulation of miR-29 Levels
NRK52E cells were plated in 6-well plates at 8 × 104 cells per well. Cells were transfected with mirVana miR-29a, miR-29b, or miR-29c mimics (Ambion, Paisley, UK) individually or in combination at a final concentration of 50 nmol/L complexed with Lipofectamine (Invitrogen). The Lipofectamine:miRNA complex was left on cells for 5 hours with intermittent shaking, then supplemented with 10% serum containing media. Twelve hours later the cells were washed with PBS and cells were replenished with fresh serum-free media. After 48 hours of serum starving, cells were stimulated with 10 ng/mL TGF-β and left for 72 hours. Total RNA was extracted and miRNA and gene levels were quantified.
Statistical Analysis
All in vitro work was performed in triplicate and repeated on at least three independent occasions. In vivo experiments were conducted in groups of six animals. Analysis of significance across two groups was performed by a standard unpaired 2-tailed Student’s t-test and analysis across multiple groups was performed by one-way analysis of variance followed by the Tukey post hoc correction. Significance was set at a P value less than 0.05.
Results
TGF-β Stimulation Induces Adams Expression in Cultured Renal Cells
We and others previously have shown that NRK52E in response to 10 ng/mL of TGF-β treatment acquire a profibrotic, secretory, and invasive mesenchymal-like phenotype.10,41 We determined changes in Adams gene expression in response to TGF-β stimulation in NRK52E over a 4-day time course. TGF-β stimulation induced Adams gene expression in a time-dependent manner. Significant increases were seen in Adam10, Adam17, Adam12, and Adam19 by 72 hours compared with time-matched untreated controls (Figure 1). Similar time-dependent TGF-β–induced up-regulation of Adams gene expression also was seen in mesangial cells in culture (Supplemental Figure S1). Adam 9, 15, and 33, the other remaining members of the proteolytic group of Adams with renal expression, however, remained unresponsive to TGF-β treatment of renal cells and therefore were omitted from further analysis (Supplemental Figure S2).
Figure 1.

Effect of TGF-β on Adam expression in tubular epithelial cells. Rat tubular epithelial NRK52E cells were stimulated with 10 ng/mL TGF-β over a 4-day time course. Gene expression was measured by qPCR using specific TaqMan probes (Life Technologies) for Adam10, Adam17, Adam12, and Adam19 and normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh). RQ ± RQmax. ∗P < 0.05, ∗∗P < 0.01 versus unstimulated time-matched controls. Results are representative of three independent experiments. RQ, relative quantification.
Increased ADAMs Expression Is Observed in Obstructed Mouse Kidneys
To investigate whether Adams expression was altered in vivo, we used the well-established UUO model in which TGF-β is known to be the primary profibrotic mediator42,43 and quantified gene expression of Adam at days 3, 7, and 14 from whole-tissue lysates of obstructed kidneys. A similar time-dependent increase was observed in vivo, with all Adams studied showing significantly increased expression profiles compared with time-matched sham kidneys at all time points after obstruction (Figure 2A). Renal protein lysates from obstructed kidneys contained significantly higher twofold active Adams 10 and 17 levels on day 3 after injury (Figure 2B). Immunohistochemical analysis on renal cortices for Adams 12 and 19 showed up-regulation in UUO (Figure 2, C and D). Adam12 expression appeared to be highest in the proximal tubules with only marginal glomerular, distal tubular, or interstitial staining. Adam19 expression was greatest in dilating tubules by day 3, with little glomerular mesangial and no interstitial staining. However, by day 7 the expressions appeared pan-corticular.
Figure 2.

Adam expression in kidneys after UUO. RNA was extracted from whole kidneys after UUO surgery. A: Gene expression was measured by qPCR using specific TaqMan probes for Adam10, Adam17, Adam12, and Adam19 and normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh). RQ ± RQmax. B: Adam10/17 enzyme activity assay on whole-kidney protein. Fluorescent substrate generation versus time curves. Active enzyme concentrations in whole lysates (ng of enzyme/μg whole protein). Means ± SEM. ∗∗P < 0.01, ∗∗∗P < 0.001 versus sham kidneys. C: Adam12 and Adam19 (D) expression in renal cortex analyzed by immunohistochemistry using specific antibodies. Inset: Isotype (ISO) stained sections. Arrowheads denote proximal tubules, and arrows denote dilating tubules. Scale bars: 100 μm. N = 6 mice per group. RQ, relative quantification.
These results suggest that a specific cohort of Adams (ie, Adams 10, 12, 17, and 19) is up-regulated in an experimental model of TGF-β–driven renal fibrosis.
SB 525334 Abolishes TGF-β–Induced Adam Expression in Vitro and in Vivo
We next sought to determine whether TGF-β–induced ADAMs expression was mediated by Smad 2/3. We used a selective Smad phosphorylation inhibitor SB 525334 concomitantly with TGF-β stimulation. SB 525334 treatment blocked TGF-β–induced profibrotic phenotypic changes, including up-regulation of collagen, α-smooth muscle actin, and loss of E-cadherin, and diminished p-Smad 2/3 nuclear staining compared with vehicle-treated cells stimulated with TGF-β alone (Supplemental Figure S3).
Abrogation of canonical TGF-β signaling by SB 525334 substantially affected growth factor–induced Adams gene expression in vitro (Figure 3A). Similar changes in ADAMs were observed at the protein expression level as analyzed by immunofluorescent imaging using specific ADAMs antibodies in which the readily detectable up-regulation resulting from TGF-β stimulation was abrogated by SB 525334 treatment (Figure 3B).
Figure 3.

SB 525334 abrogates TGF-β–induced Adam expression in vitro. A: Gene expression measured by qPCR using specific TaqMan probes for Adam10, Adam17, Adam12, and Adam19 and normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh). RQ ± RQmax. ∗∗P < 0.01, versus unstimulated time-matched controls. †P < 0.05 versus +TGF-β. B: Immunofluorescent analysis on rat tubular epithelial NRK52E cells after 72 hours TGF-β ± SB 525334 using Adam-specific antibodies. Scale bars: 20 μm. Results are representative of three independent experiments. RQ, relative quantification; Un, unstimulated time matched control cells.
SB 525334 previously was shown to be orally bioavailable and daily administration at 10 mg/kg body weight ameliorated collagen I a, III a, and plasminogen activator inhibitor-1 expression, and decreased proteinuria in a model of puromycin aminonucleoside nephritis.44 We therefore used SB 525334 in vivo and examined kidneys at day 7 after obstruction to reflect the transition phase from acute to chronic injury.
Inhibition of canonical TGF-β signaling decreased Adam10, Adam17, and Adam19 gene expression by 50%, 60%, and 70%, respectively, compared with control animals undergoing UUO (Figure 4A). The increase in Adam12 gene expression in UUO was completely abolished in the group administered SB-525334. Enzyme activity assays showed that UUO animals administered SB-525334 expressed lower levels of active ADAMs 10 and 17 in kidneys compared with the vehicle UUO group, although values remained significantly greater than those in sham controls (Figure 4B). Proximal tubular ADAM12 expression observed in control UUO kidneys largely was absent in the cortices of obstructed kidneys of animals receiving SB-525334 (Figure 4C). A similar loss of ADAM19 was observed but some dilating tubules still showed ADAM19 expression (Figure 4D).
Figure 4.

SB 525334 decreases ADAM expression in UUO. A: Gene expression measured on whole-kidney RNA by qPCR using specific TaqMan probes for Adam10, Adam17, Adam12, and Adam19 and normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh). RQ ± RQmax. B: Adam10/17 enzyme activity assay on whole-kidney protein. Fluorescent substrate generation versus time curves. Active enzyme concentrations in whole lysates (ng of enzyme/μg whole protein). Means ± SEM. ∗∗P < 0.01, ∗∗∗P < 0.001 versus vehicle sham kidneys. †P < 0.05 versus vehicle UUO. ADAM12 (C) and ADAM19 (D) expression in renal cortex analyzed by immunohistochemistry using specific antibodies. Inset: Isotype (ISO) stained sections. Arrowheads denote proximal tubules, and arrows denote dilating tubules. Scale bars: 100 μm. N = 6 mice per group. RQ, relative quantification.
Taken together, these results show that increased expression of ADAMs 10, 17, 12, and 19 in renal fibrosis is mediated by canonical Smad 2/3 signaling.
miR-29 Can Regulate Adam12 and Adam19 Expression
Members of the miR-29 family are emerging as key regulatory switches in gene expression in fibrotic injury and development through their ability to negatively regulate gene expression by binding to 3′-untranslated regions of target mRNAs that include various metzincin metalloproteases and prevent expression.45,46 The 3′- untranslated regions of both Adam12 and Adam19 mRNA contain multiple conserved putative 7 and 8mer miR-29 binding sites (Figure 5A) that are conserved across species. We therefore assessed whether Adam expression in this model can be regulated by miR-29 modulation. Consistent with previously published reports,13,15,47 TGF-β stimulation of NRK52E resulted in progressive loss of all three members of the miR-29 family (Figure 5B) and this loss was rescued by SB-525334 treatment of TGF-β–stimulated cells (Figure 5C).
Figure 5.

miR-29 expression in rat tubular epithelial NRK52E cells. A: Predicted miR-29 target sites in the Adam12 and Adam19 3′-untranslated regions (UTRs), based on TargetScan Release 5.1 (http://www.targetscan.org, last accessed October 15, 2013). Nucleotides are highlighted in the 3′-UTRs that show exact matches to residues 2 to 8 of the mature miR-29a, miR-29b, and miR-29c. miRNA expression was measured by qPCR using specific TaqMan probes for miR-29a, miR-29b, and miR-29c, and normalized to U87 after TGF-β stimulation at the indicated time points (B), and TGF-β stimulation and SB-525334 inhibitor treatment at 72 hours (C). RQ ± RQmax. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 versus unstimulated time-matched controls. †P < 0.05 versus CTL+ TGF-β. Results are representative of three independent experiments. RQ, relative quantification; Un, unstimulated time matched controls.
In vivo, we initially checked that miR-29 expression after UUO was decreased as previously reported.13 Using the same RNA extracted from the tissue we observed the increase in Adam10, Adam12, Adam17, and Adam19, after UUO we examined the expression of miR-29a, miR-29b, and miR-29c. We found significant decreases in the miR-29 family 7 days after UUO surgery (P < 0.05), which remained significantly reduced at 14 days (Figure 6). To examine whether the link between miR-29 family loss and an increase in Adam12 and Adam19 was more broadly relevant in models of renal pathology we used two additional preclinical models that resulted in renal damage induced by divergent pathologic stimuli; the stroke-prone, spontaneously hypertensive rat10,40 and the anti-Thy1.1 model of glomerulonephritis.10,48 We observed that the miR-29 family was reduced significantly in both models (Figure 7, A and C). Examining ADAM expression we observed that Adam10, Adam12, Adam17, and Adam19 were increased in the stroke-prone, spontaneously hypertensive compared with the normotensive reference strain (Figure 7B), whereas in the anti-Thy1.1 model (a mild renal damage was observed) there were significant changes in Adam12 and Adam19 only (Figure 7D).
Figure 6.
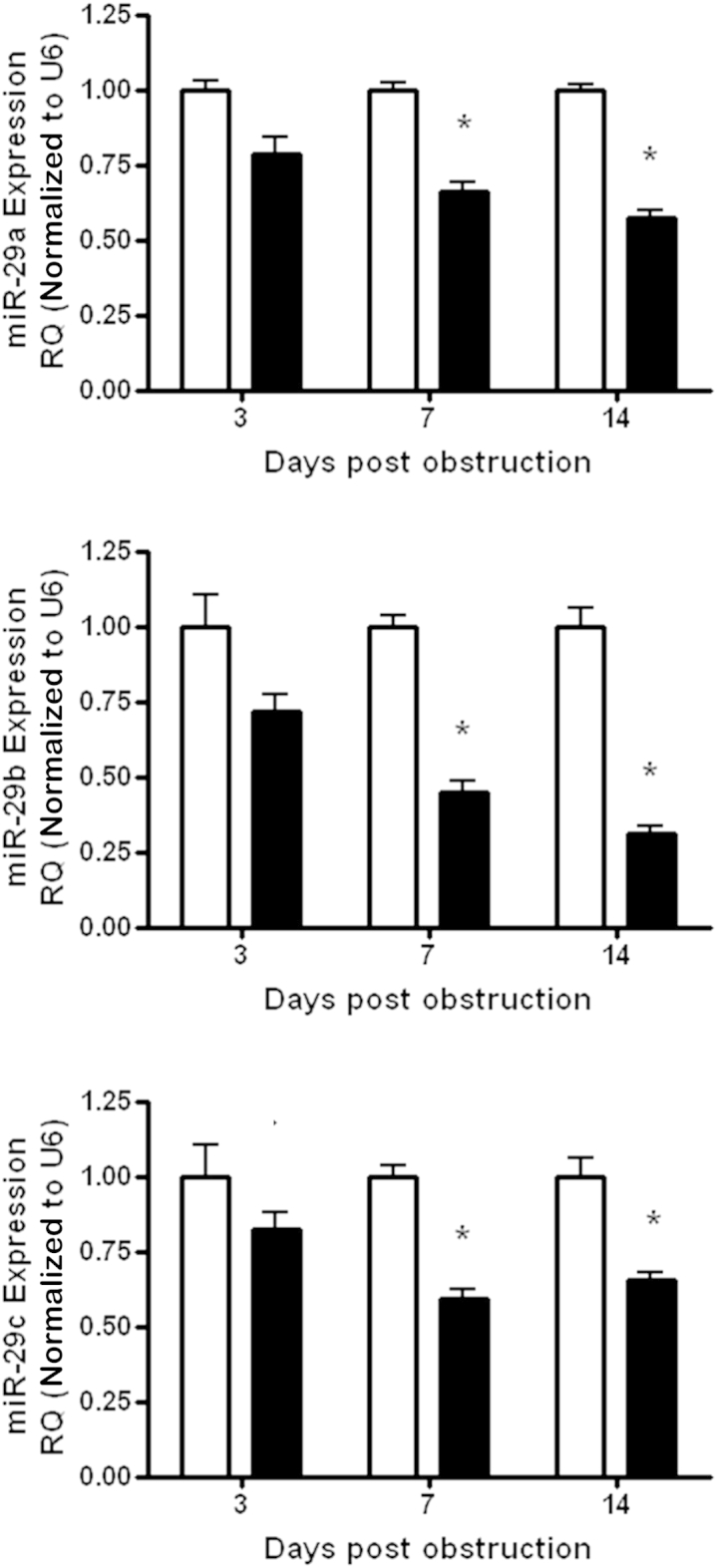
Adam expression in kidneys after UUO. RNA was extracted from whole kidney after UUO surgery. miRNA expression was measured by quantitative RT-PCR using a specific TaqMan probes for miR-29a, miR-29b, and miR-29c, and normalized to U87 after RQ ± RQmax. ∗P < 0.05. n = 6 per group. RQ, relative quantification. White bars indicate sham operated animals; black bars, animals who have undergone UUO surgery.
Figure 7.

The miR-29 family and Adam expression correlate in preclinical models of renal damage. A and B: RNA was extracted from whole kidneys from animals at 21 weeks of age (n = 3 to 4 per group). A: The miR-29 family expression was measured by qPCR using specific TaqMan probes for miR-29a, miR-29b, and miR-29c, and normalized to U87. B: Gene expression was measured by qPCR using specific primers and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). ∗P < 0.05, ∗∗P < 0.01 versus Wistar-Kyoto (WKY). C and D: RNA was extracted from whole kidneys from animals with induced glomerulonephritis and the animals were sacrificed at 7 days. C: The miR-29 family expression was measured by qPCR using specific TaqMan probes for miR-29a, miR-29b, and miR-29c, and normalized to U87. D: Gene expression was measured by qPCR using specific primers and normalized to Gapdh. ∗P < 0.05 versus control. RQ, relative quantification.
We next sought to determine whether exogenous overexpression of the three individual members of the miR-29 family or an equimolar mix of miR-29a, miR-29b, and miR-29c could modulate Adam12 and Adam19 expression in response to TGF-β treatment. To achieve this, miR-mimics containing mature sequences of the miR-29 family members were transfected into NRK52E cells. Successful overexpression of these miRNA was confirmed by qPCR at 48 hours after transfection (Figure 8A).
Figure 8.

miR-29 overexpression in rat tubular epithelial NRK52E cells. A: Exogenous overexpression of the miR-29 family measured by qPCR using specific TaqMan probes for miR-29a, miR-29b, and miR-29c, and normalized to U87. Gene expression of miR-29 targets (B) and nontargets (C) measured by qPCR in NRK52E overexpressing miR-29. RQ ± RQmax. ∗P < 0.05, ∗∗∗P < 0.001 versus unstimulated controls. ††P < 0.01 versus stimulated CTL miR-mimic. ‡‡P < 0.01 versus unstimulated CTL miR-mimic. α-SMA, α-smooth muscle actin; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; TIMP, tissue inhibitor of matrix metalloprotease. RQ, relative quantification.
Induction of differentiation from the epithelial to fibroblast phenotype in NRK52E by TGF-β resulted in increased Col1a and Col3a expression, and this effect was rescued by overexpression of the miR-29 family (Figure 8B). However, the increase in α-smooth muscle actin and tissue inhibitor of matrix metalloprotease 1 expression was not altered under these conditions (Figure 8C). This concurs with published data that showed that Col1a and 3a are direct targets of the miR29 family,15,49 whereas α-smooth muscle actin and tissue inhibitor of matrix metalloprotease 1 are not direct targets.
We then subjected miR-29 mimic transfected cells to 72 hours of TGF-β stimulation and determined the effect on Adams gene expression. Expression of Adam12 and Adam19 were both down-regulated after miR-29 overexpression (Figure 9, A and B). Overexpression of miR-29b or equimolar doses of miR29-a, miR-29b, and miR-29c together significantly blocked TGF-β–induced Adam12 expression, whereas miR-29a or miR-29c had no effect on gene expression (Figure 9A). Adam19 expression was decreased significantly with overexpression of miR-29b or miR-29c and in cells receiving equimolar miR-29a, miR-29b, and miR-29c (Figure 9B).
Figure 9.

miR-29 regulates Adam12 and Adam19 in vitro. Gene expression of Adam12 (A) and Adam19 (B) after miR-29 family overexpression. ∗P < 0.05, ∗∗P < 0.01 versus unstimulated controls. ††P < 0.01 versus stimulated CTL miR-mimic. ‡P < 0.05, ‡‡P < 0.01 versus unstimulated CTL miR-mimic. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. CTL, control time matched cells; RQ, relative quantification.
Collectively, this suggests that the beneficial effect observed with manipulation of the miR-29 family likely is mediated in part through the regulation of collagen overexpression and Adams 12 and 19, which are induced by TGF-β.
Discussion
In this article we report that Adams 10, 12, 17, and 19 are up-regulated in TGF-β–driven renal fibrosis in vitro and in vivo and that canonical Smad 2/3 signaling regulates Adams gene expression. We show that the loss of the miR-29 family is a significant feature in renal damage, occurs regardless of the injury, and this loss correlates with an increase in the expression of ADAM family members. In addition, we show that Adam12 and Adam19 are regulated by miR-29 manipulation in vitro.
Adam10 and Adam17 are perhaps the most widely studied members of the ADAMs family.50,51 Recently, Lu et al52 showed that TGF-β stimulation up-regulated Adam17 expression and increased the invasiveness of T98G glioma cells, whereas preconditioning with a pan ALK inhibitor SB431542 reversed these changes. This is in accordance with changes we have observed in renal tubular epithelial and mesangial cells by administering the specific ALK5 inhibitor SB-525334. Treatment with SB-525334 resulted in a 50% to 60% reduction in Adam10 and Adam17 gene expression, respectively. Similar time-dependent up-regulation and inhibition in response to SB-525334 administration also was observed in vivo. Promoter analysis of Adam10 and Adam17 showed that these Adams are devoid of cis-regulatory elements such as the TATA or CAAT sequences in their promoters; a category of genes that also includes the classic TGF-β–regulated matrix metalloprotease 2 and matrix metalloprotease 14.53 Work by Prinzen et al54 in characterizing the Adam10 promoter showed the presence of multiple CpG islands with potential TGF-β–related transcription factors Sp1 and Smad 3 binding sites in the region lying between −508 and the transcriptional start site. Similar sequences were found between -290 bp and the transcriptional start site of the Adam17 promoter.55 Taken together with data reported here, this suggests that Adam10 and Adam17 are directly responsive to TGF-β transcriptional regulation. Consistent with previously published reports, immunofluorescent staining showed perinuclear localization of Adams in unstimulated NRK52E.56
Adam12 and Adam19 belong to the meltrin subfamily of ADAM and share significant sequence homology and partial substrate specificity.57 There is evidence to suggest that TGF-β–mediated up-regulation of Adam is owing to inhibition of repressors such as ski-related novel protein N in a Smad2/3-dependent manner.58 In this study, Adam did not show early responses to TGF-β stimulation but increased rapidly after 48 hours, and SB 525334 treatment completely blocked Adam12 expression. Adam12 also is regulated via NF-κB p65 as a response to Notch1 activation in breast cancer models.59,60 Although Adam17 is thought to act in the counter-regulatory axis on TGF-β signaling,61 Adam12 has been shown to enhance TGF-β signaling via stabilization of the TGF-βR1 and TGF-βR2 dimer and to enhance receptor Smad 2/3 phosphorylation.62 Taken together, this suggests that TGF-β signaling inhibition potentially could affect a feed-forward mechanism in Adam12 gene expression and that it can be regulated by TGF-β at multiple levels.
Transcriptional regulation of Adam19 is not very well understood. It previously was reported via microarray analysis that TGF-β stimulation of A549 alveolar epithelial cells led to a significant increase in Adam19 gene expression as early as 4 hours after stimulation.63 Here, Adam19 expression was seen to change only after 48 hours of stimulation. Chan et al64 reported that Adam19 expression in ovarian cancer cells was regulated by histone deacylation mediated by TGF-β1 via Smad 4. Early sequence analysis studies showed potential upstream and downstream regulatory elements in murine Adam19 gene. The positive regulatory sequence upstream of the transcriptional start site contained, similar to the case of Adam10 and Adam17, multiple CpG islands with potential Sp1 binding sites.
miRNA offer an additional post-transcriptional checkpoint in gene regulation. miRNA previously have been shown to regulate Adam gene regulation in experimental models of disease and development. Computational analysis and Adam10 3′-untranslated region luciferase assays showed significant inhibition via miR-1306, miR-103, and miR-107.65 miR-126 overexpression was able to inhibit Adam9-mediated EMT-like processes in pancreatic cancer cells66 and Adam17 has been implicated as a target of miR-14567 and miR-222.67 miR-29 has conserved binding sites at the 3′ untranslated regions of Adam12.68 Our study shows miR-29 regulation in canonical TGF-β–driven models of renal fibrosis, confirming previous reports in similar models.69 In addition, we provide evidence that miR-29 modulation regulates Adam19 expression. Although triple transfections repress Adam12 and Adam19 expression to baseline, the individual isoforms miR-29b and miR-29c are sufficient to significantly inhibit Adam12 and Adam19 expression, respectively. The physiologic relevance of such isoform-specific inhibition needs to be investigated further. It previously has been shown that miR-29b localizes to the tubules.13 In our study, we show that Adam12 and Adam19 expression also is localized within the tubules (Figure 2). This similarity in localization may be important because we hypothesized that there is interplay between miR-29 expression and regulation of Adam expression. However, because we show correlation between the miR-29 family and Adam expression in vitro, coupled with the observation from others that forced expression of miR29b is therapeutic in renal fibrosis,13 this suggests that the antifibrotic effect observed could be mediated, at least in part, through miR-29 family repression of Adams 12 and 19. Further detailed experiments are warranted to address this.
In this study we show changes and regulation at the gene expression level of Adams by canonical TGF-β signaling (Figure 10). Adams could well be the conduits for TGF-β cross-talks with other key signaling pathways such as angiotensin II, epidermal growth factor, and Notch, whose activity precipitates fibrosis in the tubulointerstitium. Adam-specific intervention strategies, either pharmacologically or genetically, are needed to further elucidate their individual roles in pathogenesis or resolution of renal fibrosis, making them potential candidates for targeted fibrosis therapy in renal disease.
Figure 10.

Mechanism of ADAM expression in fibrosis and the Smad2/3-miR-29-ADAM axis. TGF-β binding induces receptor kinase–mediated phosphorylation of Smads 2 and 3. Phosphorylated Smads then translocate to the nucleus and initiate Adam gene expression. p-Smad 2/3 negatively regulates miR-29 expression, the presence of which represses Adam12 and Adam19 expression post-transcriptionally. Treatment with SB-525334 inhibits Smad 2/3 phosphorylation and hence its downstream effects on gene and miRNA expression. These mechanisms combine to result in decreased Adam gene expression.
Acknowledgment
We thank Dr. Christian Delles for critical appraisal of the manuscript.
Footnotes
Supported by the Medical Research Council UK, the British Heart Foundation Chair of Translational Cardiovascular Sciences (A.H.B.), and by a personal Kidney Research UK Fellowship (PDF6/2012 to L.D.).
Supplemental Data
Effect of TGF-β on ADAM expression in mesangial cells. Rat mesangial cells (CRL2573) were stimulated with 10 ng/mL rTGF-β over a 4-day time course. Gene expression was measured by qPCR using specific TaqMan probes for ADAM10, ADAM12, ADAM17, and ADAM19 and normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh). RQ ± RQmax. ∗P < 0.05, ∗∗P < 0.01 versus unstimulated time-matched controls. Results are representative of three independent experiments. RQ, relative quantification.
Effect of TGF-β on ADAM expression in tubular epithelial cells. Rat tubular epithelial cells (NRK52E) were stimulated with 10 ng/mL rTGF-β over a 4day time course. Gene expression was measured by qPCR using specific TaqMan probes for ADAM9, ADAM15, and ADAM33 normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh). RQ ± RQmax. Results are representative of three independent experiments. RQ, relative quantification.
SB525334 abrogates TGF-β–induced profibrotic effects in vitro. A: Phase-contrast image of NRK52E ± TGF-β and SB525334. B: Immunofluorescent staining for p-smad 2/3 in NRK52E ± TGF-β and SB525334. Scale bars: 100 μm. C: Gene expression was measured by qPCR using specific TaqMan probes for Collagen I a, Collagen III a, α-SMA, and E-cadherin and normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh). RQ ± RQmax. ∗∗P < 0.01, versus unstimulated time-matched controls. †P < 0.05 versus + TGF-β. Representative of three independent experiments. RQ, relative quantification.
Supplemental Data
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2013.08.027.
References
- 1.Bohle A., Mackensen-Haen S., von Gise H. Significance of tubulointerstitial changes in the renal cortex for the excretory function and concentration ability of the kidney: a morphometric contribution. Am J Nephrol. 1987;7:421–433. doi: 10.1159/000167514. [DOI] [PubMed] [Google Scholar]
- 2.Wynn T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bottinger E.P. TGF-beta in renal injury and disease. Semin Nephrol. 2007;27:309–320. doi: 10.1016/j.semnephrol.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Eddy A.A., Neilson E.G. Chronic kidney disease progression. J Am Soc Nephrol. 2006;17:2964–2966. doi: 10.1681/ASN.2006070704. [DOI] [PubMed] [Google Scholar]
- 5.Schnaper H.W., Jandeska S., Runyan C.E., Hubchak S.C., Basu R.K., Curley J.F., Smith R.D., Hayashida T. TGF-beta signal transduction in chronic kidney disease. Front Biosci. 2009;14:2448–2465. doi: 10.2741/3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts A.B. Molecular and cell biology of TGF-beta. Miner Electrolyte Metab. 1998;24:111–119. doi: 10.1159/000057358. [DOI] [PubMed] [Google Scholar]
- 7.He L., Hannon G.J. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 8.Chandrasekaran K., Karolina D.S., Sepramaniam S., Armugam A., Wintour E.M., Bertram J.F., Jeyaseelan K. Role of microRNAs in kidney homeostasis and disease. Kidney Int. 2012;81:617–627. doi: 10.1038/ki.2011.448. [DOI] [PubMed] [Google Scholar]
- 9.Lorenzen J.M., Haller H., Thum T. MicroRNAs as mediators and therapeutic targets in chronic kidney disease. Nat Rev Nephrol. 2011;7:286–294. doi: 10.1038/nrneph.2011.26. [DOI] [PubMed] [Google Scholar]
- 10.Denby L., Ramdas V., McBride M.W., Wang J., Robinson H., McClure J., Crawford W., Lu R., Hillyard D.Z., Khanin R., Agami R., Dominiczak A.F., Sharpe C.C., Baker A.H. miR-21 and miR-214 are consistently modulated during renal injury in rodent models. Am J Pathol. 2011;179:661–672. doi: 10.1016/j.ajpath.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zarjou A., Yang S., Abraham E., Agarwal A., Liu G. Identification of a microRNA signature in renal fibrosis: role of miR-21. Am J Physiol Renal Physiol. 2011;301:F793–F801. doi: 10.1152/ajprenal.00273.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kriegel A.J., Liu Y., Fang Y., Ding X., Liang M. The miR-29 family: genomics, cell biology, and relevance to renal and cardiovascular injury. Physiol Genomics. 2012;44:237–244. doi: 10.1152/physiolgenomics.00141.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qin W., Chung A.C., Huang X.R., Meng X.M., Hui D.S., Yu C.M., Sung J.J., Lan H.Y. TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J Am Soc Nephrol. 2011;22:1462–1474. doi: 10.1681/ASN.2010121308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y., Taylor N.E., Lu L., Usa K., Cowley A.W., Jr., Ferreri N.R., Yeo N.C., Liang M. Renal medullary microRNAs in Dahl salt-sensitive rats: miR-29b regulates several collagens and related genes. Hypertension. 2010;55:974–982. doi: 10.1161/HYPERTENSIONAHA.109.144428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang B., Komers R., Carew R., Winbanks C.E., Xu B., Herman-Edelstein M., Koh P., Thomas M., Jandeleit-Dahm K., Gregorevic P., Cooper M.E., Kantharidis P. Suppression of microRNA-29 expression by TGF-β1 promotes collagen expression and renal fibrosis. J Am Soc Nephrol. 2012;23:252–265. doi: 10.1681/ASN.2011010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Catania J.M., Chen G., Parrish A.R. Role of matrix metalloproteinases in renal pathophysiologies. Am J Physiol Renal Physiol. 2007;292:F905–F911. doi: 10.1152/ajprenal.00421.2006. [DOI] [PubMed] [Google Scholar]
- 17.Blobel C.P. Remarkable roles of proteolysis on and beyond the cell surface. Curr Opin Cell Biol. 2000;12:606–612. doi: 10.1016/s0955-0674(00)00139-3. [DOI] [PubMed] [Google Scholar]
- 18.Black R.A. Tumor necrosis factor-alpha converting enzyme. Int J Biochem Cell Biol. 2002;34:1–5. doi: 10.1016/s1357-2725(01)00097-8. [DOI] [PubMed] [Google Scholar]
- 19.Baud L., Ardaillou R. Tumor necrosis factor in renal injury. Miner Electrolyte Metab. 1995;21:336–341. [PubMed] [Google Scholar]
- 20.Kluth D.C., Rees A.J. Inhibiting inflammatory cytokines. Semin Nephrol. 1996;16:576–582. [PubMed] [Google Scholar]
- 21.Estrella C., Rocks N., Paulissen G., Quesada-Calvo F., Noel A., Vilain E., Lassalle P., Tillie-Leblond I., Cataldo D., Gosset P. Role of A disintegrin and metalloprotease-12 in neutrophil recruitment induced by airway epithelium. Am J Respir Cell Mol Biol. 2009;41:449–458. doi: 10.1165/rcmb.2008-0124OC. [DOI] [PubMed] [Google Scholar]
- 22.Schulz B., Pruessmeyer J., Maretzky T., Ludwig A., Blobel C.P., Saftig P., Reiss K. ADAM10 regulates endothelial permeability and T-cell transmigration by proteolysis of vascular endothelial cadherin. Circ Res. 2008;102:1192–1201. doi: 10.1161/CIRCRESAHA.107.169805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maretzky T., Reiss K., Ludwig A., Buchholz J., Scholz F., Proksch E., de Strooper B., Hartmann D., Saftig P. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc Natl Acad Sci U S A. 2005;102:9182–9187. doi: 10.1073/pnas.0500918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Overall C.M., Blobel C.P. In search of partners: linking extracellular proteases to substrates. Nat Rev Mol Cell Biol. 2007;8:245–257. doi: 10.1038/nrm2120. [DOI] [PubMed] [Google Scholar]
- 25.van Tetering G., van Diest P., Verlaan I., van der Wall E., Kopan R., Vooijs M. Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J Biol Chem. 2009;284:31018–31027. doi: 10.1074/jbc.M109.006775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shah B.H., Catt K.J. TACE-dependent EGF receptor activation in angiotensin-II-induced kidney disease. Trends Pharmacol Sci. 2006;27:235–237. doi: 10.1016/j.tips.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 27.Franzke C.W., Tasanen K., Schacke H., Zhou Z., Tryggvason K., Mauch C., Zigrino P., Sunnarborg S., Lee D.C., Fahrenholz F., Bruckner-Tuderman L. Transmembrane collagen XVII, an epithelial adhesion protein, is shed from the cell surface by ADAMs. EMBO J. 2002;21:5026–5035. doi: 10.1093/emboj/cdf532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Millichip M.I., Dallas D.J., Wu E., Dale S., McKie N. The metallo-disintegrin ADAM10 (MADM) from bovine kidney has type IV collagenase activity in vitro. Biochem Biophys Res Commun. 1998;245:594–598. doi: 10.1006/bbrc.1998.8485. [DOI] [PubMed] [Google Scholar]
- 29.Roy R., Wewer U.M., Zurakowski D., Pories S.E., Moses M.A. ADAM 12 cleaves extracellular matrix proteins and correlates with cancer status and stage. J Biol Chem. 2004;279:51323–51330. doi: 10.1074/jbc.M409565200. [DOI] [PubMed] [Google Scholar]
- 30.Melenhorst W.B., van den Heuvel M.C., Stegeman C.A., van der Leij J., Huitema S., van den Berg A., van Goor H. Upregulation of ADAM19 in chronic allograft nephropathy. Am J Transplant. 2006;6:1673–1681. doi: 10.1111/j.1600-6143.2006.01384.x. [DOI] [PubMed] [Google Scholar]
- 31.Melenhorst W.B., van den Heuvel M.C., Timmer A., Huitema S., Bulthuis M., Timens W., van Goor H. ADAM19 expression in human nephrogenesis and renal disease: associations with clinical and structural deterioration. Kidney Int. 2006;70:1269–1278. doi: 10.1038/sj.ki.5001753. [DOI] [PubMed] [Google Scholar]
- 32.Melenhorst W.B., Visser L., Timmer A., van den Heuvel M.C., Stegeman C.A., van Goor H. ADAM17 upregulation in human renal disease: a role in modulating TGF-alpha availability? Am J Physiol Renal Physiol. 2009;297:F781–F790. doi: 10.1152/ajprenal.90610.2008. [DOI] [PubMed] [Google Scholar]
- 33.Mulder G.M., Melenhorst W.B., Celie J.W., Kloosterhuis N.J., Hillebrands J.L., Ploeg R.J., Seelen M.A., Visser L., van Dijk M.C., van Goor H. ADAM17 up-regulation in renal transplant dysfunction and non-transplant-related renal fibrosis. Nephrol Dial Transplant. 2012;27:2114–2122. doi: 10.1093/ndt/gfr583. [DOI] [PubMed] [Google Scholar]
- 34.Lautrette A., Li S., Alili R., Sunnarborg S.W., Burtin M., Lee D.C., Friedlander G., Terzi F. Angiotensin II and EGF receptor cross-talk in chronic kidney diseases: a new therapeutic approach. Nat Med. 2005;11:867–874. doi: 10.1038/nm1275. [DOI] [PubMed] [Google Scholar]
- 35.Schramme A., Abdel-Bakky M.S., Gutwein P., Obermuller N., Baer P.C., Hauser I.A., Ludwig A., Gauer S., Schafer L., Sobkowiak E., Altevogt P., Koziolek M., Kiss E., Grone H.J., Tikkanen R., Goren I., Radeke H., Pfeilschifter J. Characterization of CXCL16 and ADAM10 in the normal and transplanted kidney. Kidney Int. 2008;74:328–338. doi: 10.1038/ki.2008.181. [DOI] [PubMed] [Google Scholar]
- 36.Berthier C.C., Lods N., Joosten S.A., van Kooten C., Leppert D., Lindberg R.L., Kappeler A., Raulf F., Sterchi E.E., Lottaz D., Marti H.P. Differential regulation of metzincins in experimental chronic renal allograft rejection: potential markers and novel therapeutic targets. Kidney Int. 2006;69:358–368. doi: 10.1038/sj.ki.5000049. [DOI] [PubMed] [Google Scholar]
- 37.Mozes M.M., Bottinger E.P., Jacot T.A., Kopp J.B. Renal expression of fibrotic matrix proteins and of transforming growth factor-beta (TGF-beta) isoforms in TGF-beta transgenic mice. J Am Soc Nephrol. 1999;10:271–280. doi: 10.1681/ASN.V102271. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki S., Ebihara I., Tomino Y., Koide H. Transcriptional activation of matrix genes by transforming growth factor beta 1 in mesangial cells. Exp Nephrol. 1993;1:229–237. [PubMed] [Google Scholar]
- 39.Laping N.J. Therapeutic uses of smad protein inhibitors: selective inhibition of specific TGF-beta activities. IDrugs. 1999;2:907–914. [PubMed] [Google Scholar]
- 40.Davidson A.O., Schork N., Jaques B.C., Kelman A.W., Sutcliffe R.G., Reid J.L., Dominiczak A.F. Blood pressure in genetically hypertensive rats. Influence of the Y chromosome. Hypertension. 1995;26:452–459. doi: 10.1161/01.hyp.26.3.452. [DOI] [PubMed] [Google Scholar]
- 41.Fan J.M., Ng Y.Y., Hill P.A., Nikolic-Paterson D.J., Mu W., Atkins R.C., Lan H.Y. Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 1999;56:1455–1467. doi: 10.1046/j.1523-1755.1999.00656.x. [DOI] [PubMed] [Google Scholar]
- 42.Chevalier R.L., Forbes M.S., Thornhill B.A. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009;75:1145–1152. doi: 10.1038/ki.2009.86. [DOI] [PubMed] [Google Scholar]
- 43.Kaneto H., Morrissey J., Klahr S. Increased expression of TGF-beta 1 mRNA in the obstructed kidney of rats with unilateral ureteral ligation. Kidney Int. 1993;44:313–321. doi: 10.1038/ki.1993.246. [DOI] [PubMed] [Google Scholar]
- 44.Grygielko E.T., Martin W.M., Tweed C., Thornton P., Harling J., Brooks D.P., Laping N.J. Inhibition of gene markers of fibrosis with a novel inhibitor of transforming growth factor-beta type I receptor kinase in puromycin-induced nephritis. J Pharmacol Exp Ther. 2005;313:943–951. doi: 10.1124/jpet.104.082099. [DOI] [PubMed] [Google Scholar]
- 45.Fang J.H., Zhou H.C., Zeng C., Yang J., Liu Y., Huang X., Zhang J.P., Guan X.Y., Zhuang S.M. MicroRNA-29b suppresses tumor angiogenesis, invasion, and metastasis by regulating matrix metalloproteinase 2 expression. Hepatology. 2011;54:1729–1740. doi: 10.1002/hep.24577. [DOI] [PubMed] [Google Scholar]
- 46.Chen K.C., Wang Y.S., Hu C.Y., Chang W.C., Liao Y.C., Dai C.Y., Juo S.H. OxLDL up-regulates microRNA-29b, leading to epigenetic modifications of MMP-2/MMP-9 genes: a novel mechanism for cardiovascular diseases. FASEB J. 2011;25:1718–1728. doi: 10.1096/fj.10-174904. [DOI] [PubMed] [Google Scholar]
- 47.Zhou L., Wang L., Lu L., Jiang P., Sun H., Wang H. Inhibition of miR-29 by TGF-beta-Smad3 signaling through dual mechanisms promotes transdifferentiation of mouse myoblasts into myofibroblasts. PLoS One. 2012;7:e33766. doi: 10.1371/journal.pone.0033766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bagchus W.M., Hoedemaeker P.J., Rozing J., Bakker W.W. Glomerulonephritis induced by monoclonal anti-Thy 1.1 antibodies. A sequential histological and ultrastructural study in the rat. Lab Invest. 1986;55:680–687. [PubMed] [Google Scholar]
- 49.Maurer B., Stanczyk J., Jüngel A., Akhmetshina A., Trenkmann M., Brock M., Kowal-Bielecka O., Gay R.E., Michel B.A., Distler J.H.W., Gay S., Distler O. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum. 2010;62:1733–1743. doi: 10.1002/art.27443. [DOI] [PubMed] [Google Scholar]
- 50.Crawford H.C., Dempsey P.J., Brown G., Adam L., Moss M.L. ADAM10 as a therapeutic target for cancer and inflammation. Curr Pharm Des. 2009;15:2288–2299. doi: 10.2174/138161209788682442. [DOI] [PubMed] [Google Scholar]
- 51.Gooz M. ADAM-17: the enzyme that does it all. Crit Rev Biochem Mol Biol. 2010;45:146–169. doi: 10.3109/10409231003628015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu Y., Jiang F., Zheng X., Katakowski M., Buller B., To S.S., Chopp M. TGF-beta1 promotes motility and invasiveness of glioma cells through activation of ADAM17. Oncol Rep. 2011;25:1329–1335. doi: 10.3892/or.2011.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yan C., Boyd D.D. Regulation of matrix metalloproteinase gene expression. J Cell Physiol. 2007;211:19–26. doi: 10.1002/jcp.20948. [DOI] [PubMed] [Google Scholar]
- 54.Prinzen C., Muller U., Endres K., Fahrenholz F., Postina R. Genomic structure and functional characterization of the human ADAM10 promoter. FASEB J. 2005;19:1522–1524. doi: 10.1096/fj.04-3619fje. [DOI] [PubMed] [Google Scholar]
- 55.Mizui Y., Yamazaki K., Sagane K., Tanaka I. cDNA cloning of mouse tumor necrosis factor-alpha converting enzyme (TACE) and partial analysis of its promoter. Gene. 1999;233:67–74. doi: 10.1016/s0378-1119(99)00155-9. [DOI] [PubMed] [Google Scholar]
- 56.Murphy G. The ADAMs: signalling scissors in the tumour microenvironment. Nat Rev Cancer. 2008;8:929–941. doi: 10.1038/nrc2459. [DOI] [PubMed] [Google Scholar]
- 57.Gunn T.M., Azarani A., Kim P.H., Hyman R.W., Davis R.W., Barsh G.S. Identification and preliminary characterization of mouse Adam33. BMC Genet. 2002;3:2. doi: 10.1186/1471-2156-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Solomon E., Li H., Duhachek Muggy S., Syta E., Zolkiewska A. The role of SnoN in transforming growth factor beta1-induced expression of metalloprotease-disintegrin ADAM12. J Biol Chem. 2010;285:21969–21977. doi: 10.1074/jbc.M110.133314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ray A., Dhar S., Ray B.K. Transforming growth factor-beta1-mediated activation of NF-kappaB contributes to enhanced ADAM-12 expression in mammary carcinoma cells. Mol Cancer Res. 2010;8:1261–1270. doi: 10.1158/1541-7786.MCR-10-0212. [DOI] [PubMed] [Google Scholar]
- 60.Ray B.K., Dhar S., Shakya A., Ray A. Z-DNA-forming silencer in the first exon regulates human ADAM-12 gene expression. Proc Natl Acad Sci U S A. 2011;108:103–108. doi: 10.1073/pnas.1008831108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu C., Xu P., Lamouille S., Xu J., Derynck R. TACE-mediated ectodomain shedding of the type I TGF-beta receptor downregulates TGF-beta signaling. Mol Cell. 2009;35:26–36. doi: 10.1016/j.molcel.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Atfi A., Dumont E., Colland F., Bonnier D., L‘Helgoualc’h A., Prunier C., Ferrand N., Clement B., Wewer U.M., Theret N. The disintegrin and metalloproteinase ADAM12 contributes to TGF-beta signaling through interaction with the type II receptor. J Cell Biol. 2007;178:201–208. doi: 10.1083/jcb.200612046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Keating D.T., Sadlier D.M., Patricelli A., Smith S.M., Walls D., Egan J.J., Doran P.P. Microarray identifies ADAM family members as key responders to TGF-beta1 in alveolar epithelial cells. Respir Res. 2006;7:114. doi: 10.1186/1465-9921-7-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chan M.W., Huang Y.W., Hartman-Frey C., Kuo C.T., Deatherage D., Qin H., Cheng A.S., Yan P.S., Davuluri R.V., Huang T.H., Nephew K.P., Lin H.J. Aberrant transforming growth factor beta1 signaling and SMAD4 nuclear translocation confer epigenetic repression of ADAM19 in ovarian cancer. Neoplasia. 2008;10:908–919. doi: 10.1593/neo.08540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Augustin R., Endres K., Reinhardt S., Kuhn P.H., Lichtenthaler S.F., Hansen J., Wurst W., Trumbach D. Computational identification and experimental validation of microRNAs binding to the Alzheimer-related gene ADAM10. BMC Med Genet. 2012;13:35. doi: 10.1186/1471-2350-13-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hamada S., Satoh K., Fujibuchi W., Hirota M., Kanno A., Unno J., Masamune A., Kikuta K., Kume K., Shimosegawa T. MiR-126 acts as a tumor suppressor in pancreatic cancer cells via the regulation of ADAM9. Mol Cancer Res. 2012;10:3–10. doi: 10.1158/1541-7786.MCR-11-0272. [DOI] [PubMed] [Google Scholar]
- 67.Gregersen L.H., Jacobsen A.B., Frankel L.B., Wen J., Krogh A., Lund A.H. MicroRNA-145 targets YES and STAT1 in colon cancer cells. PLoS One. 2010;5:e8836. doi: 10.1371/journal.pone.0008836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Luna C., Li G., Qiu J., Epstein D.L., Gonzalez P. Role of miR-29b on the regulation of the extracellular matrix in human trabecular meshwork cells under chronic oxidative stress. Mol Vis. 2009;15:2488–2497. [PMC free article] [PubMed] [Google Scholar]
- 69.Li H., Solomon E., Duhachek Muggy S., Sun D., Zolkiewska A. Metalloprotease-disintegrin ADAM12 expression is regulated by Notch signaling via microRNA-29. J Biol Chem. 2011;286:21500–21510. doi: 10.1074/jbc.M110.207951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of TGF-β on ADAM expression in mesangial cells. Rat mesangial cells (CRL2573) were stimulated with 10 ng/mL rTGF-β over a 4-day time course. Gene expression was measured by qPCR using specific TaqMan probes for ADAM10, ADAM12, ADAM17, and ADAM19 and normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh). RQ ± RQmax. ∗P < 0.05, ∗∗P < 0.01 versus unstimulated time-matched controls. Results are representative of three independent experiments. RQ, relative quantification.
Effect of TGF-β on ADAM expression in tubular epithelial cells. Rat tubular epithelial cells (NRK52E) were stimulated with 10 ng/mL rTGF-β over a 4day time course. Gene expression was measured by qPCR using specific TaqMan probes for ADAM9, ADAM15, and ADAM33 normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh). RQ ± RQmax. Results are representative of three independent experiments. RQ, relative quantification.
SB525334 abrogates TGF-β–induced profibrotic effects in vitro. A: Phase-contrast image of NRK52E ± TGF-β and SB525334. B: Immunofluorescent staining for p-smad 2/3 in NRK52E ± TGF-β and SB525334. Scale bars: 100 μm. C: Gene expression was measured by qPCR using specific TaqMan probes for Collagen I a, Collagen III a, α-SMA, and E-cadherin and normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh). RQ ± RQmax. ∗∗P < 0.01, versus unstimulated time-matched controls. †P < 0.05 versus + TGF-β. Representative of three independent experiments. RQ, relative quantification.