

Abstract
Cholinergic neuronal loss in the pedunculopontine nucleus (PPN) associates with abnormal functions, including certain motor and nonmotor symptoms. This realization has led to low-frequency stimulation of the PPN for treating patients with Parkinson disease (PD) who are refractory to other treatment modalities. However, the molecular mechanisms underlying PPN neuronal loss and the therapeutic substrate for the clinical benefits following PPN stimulation remain poorly characterized, hampering progress toward designing more efficient therapies aimed at restoring the PPN's normal functions during progressive parkinsonism. Here, we investigated postmortem pathological changes in the PPN of PD cases. Our study detected a loss of neurons producing gamma-aminobutyric acid (GABA) as their output and glycinergic neurons, along with the pronounced loss of cholinergic neurons. These losses were accompanied by altered somatic cell size that affected the remaining neurons of all neuronal subtypes studied here. Because studies showed that mitochondrial dysfunction exists in sporadic PD and in PD animal models, we investigated whether altered mitochondrial composition exists in the PPN. A significant up-regulation of several mitochondrial proteins was seen in GABAergic and glycinergic neurons; however, cholinergic neurons indicated down-regulation of the same proteins. Our findings suggest an imbalance in the activity of key neuronal subgroups of the PPN in PD, potentially because of abnormal inhibitory activity and altered cholinergic outflow.
CME Accreditation Statement: This activity (“ASIP 2013 AJP CME Program in Pathogenesis”) has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint sponsorship of the American Society for Clinical Pathology (ASCP) and the American Society for Investigative Pathology (ASIP). ASCP is accredited by the ACCME to provide continuing medical education for physicians.
The ASCP designates this journal-based CME activity (“ASIP 2013 AJP CME Program in Pathogenesis”) for a maximum of 48 AMA PRA Category 1 Credit(s)™. Physicians should only claim credit commensurate with the extent of their participation in the activity.
CME Disclosures: The authors of this article and the planning committee members and staff have no relevant financial relationships with commercial interests to disclose.
Patients with Parkinson disease (PD) present with a multitude of motor-related disabilities, including progressive resting tremor, rigidity, bradykinesia/akinesia, gait disturbances, and postural instability. In addition, it is recognized increasingly that various nonmotor functions are also left impaired, including mood, cognition, sleep, autonomic nervous system functions, and sensory functions.1 A neuropathological signature of PD is the progressive deterioration of dopamine-producing neurons in the substantia nigra pars compacta (SNpc).2 Although the precise cellular and molecular mechanisms underlying this neuronal death remain unknown, several reports implicate an underlying mitochondrial dysfunction, relating to energy deficits, enhanced production of free-radical species with concomitant oxidative stress,3 proteasomal deregulation,4 and neuronal excitotoxicity.5
Evidence for a mitochondrial-related cause in PD stems from studies reporting on the use of human postmortem brains of patients with PD, which found a deficiency of complex I of the mitochondrial respiratory chain in the SNpc.6 Furthermore, outside the central nervous system a mitochondrial respiratory chain complex I deficiency has also been detected in the blood platelets of patients with PD, with some patients who also displayed defects of mitochondrial respiratory chain complexes II and III.7 In this regard, Gu et al8 found that a mitochondrial DNA (mtDNA) abnormality may underlie this mitochondrial defect in at least a proportion of patients with PD. By contrast, data reporting on mitochondrial respiratory chain defects in skeletal muscle cells of patients with PD remain somewhat more controversial. In this regard, Penn et al9 performed 31P magnetic resonance spectroscopy on the resting muscles of patients with PD, to report detecting defects in oxidative phosphorylation in the patients' musculature, compared with healthy control cases. However, a study by Taylor et al10 was unable to validate this result. It has been proposed that the conflicting results that report on skeletal mitochondrial defects in patients with PD may relate to either methodological variation for assessing this biochemical defect or may be a reflection of the heterogeneity of the disease.11
Further evidence for an association between PD and a mitochondrial defect was obtained from the use of experimental neurotoxins such as rotenone and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Use of such toxins mimic parkinsonism in animals to a remarkably accurate extent, with studies showing that the pathological substrate for this defect may be due to the ability of such toxins to inhibit complex I of the mitochondrial respiratory chain.12,13 Finally, disordered mitochondrial function, including defects in oxidative phosphorylation, are also seen in rare, young-onset genetic forms of PD, such as in patients who harbor mutations in genes such as Parkinson protein 2, E3 ubiquitin protein ligase [parkin (official symbol, PARK2)], parkinson protein 7 [DJ-1, (official symbol, PARK7)], and PTEN (phosphatase and tensin homologue) induced putative kinase 1 (PINK1), where loss of function of the respective protein products associate with deregulation of the mitochondrial quality control pathways of the cells.14
Although the principal motor features of PD stem from reduced dopaminergic innervation of the striatum because of a substantial loss of SNpc dopaminergic neurons, recognition is growing that PD symptoms could result from disruption to multiple neural regions and systems.15 Although the loss of neurons is most conspicuous in the SNpc, neuronal loss and the presence of intracytoplasmic Lewy bodies (LBs) and Lewy neurites (LNs), composed of aggregation-prone proteins such as α-synuclein (αSYN) form an additional neuropathological hallmark of PD16 and have been observed in brain regions as diverse as the dorsal motor nucleus of vagus of the medulla, the locus ceruleus in the pons, the raphe nucleus, the basal forebrain, and allocortical regions such as the hippocampus and amygdala.17 Such widespread distribution of PD pathology could correlate with the variety of motor and nonmotor symptoms observed in patients with PD.18 PD-related pathologies that affect regions other than the dopaminergic-rich SNpc suggest that, although treatments that target only the nigrostriatal dopaminergic system could substantially benefit patients with PD, they are unlikely to completely resolve the PD-related deficit.19
One particular brain region, the pedunculopontine nucleus (PPN), located within the lateral tegmental region and spanning the pontine midbrain isthmus, has been deemed critically important for regulating some of the physiological functions that fail during progressive PD. Such functions include regulating the activity of the reticular activating system for controlling rapid eye movement (REM) sleep.20 Interestingly, patients with PD frequently present with abnormal REM muscle tone and concomitant REM sleep behavior disorder (RBD),21 which may be due to a loss of PPN cells and their concomitant functions during progressive PD. Moreover, PPN axons project toward and receive input from a variety of brain regions, including the thalamus, SN (both the compacta and reticular part), cortical regions, and spinal cord, all of which are involved in regulating aspects of voluntary motor function.22–30 The PPN was assigned a role in the onset and progression of PD because of reports that the nucleus undergoes degenerative changes, principally affecting the resident cholinergic neurons.31,32 The loss of these cells is believed to provide the cellular basis for the gait and postural deficits that patients with PD experience33 and nonhuman primates rendered parkinsonian via cytotoxic lesions.34,35 In addition, LBs and LNs are seen within the remaining PPN neurons in the postmortem brains of patients with PD.36 Such findings provided the rationale for the commencement of therapeutic trials of deep brain stimulation of the PPN, with trial results reporting a reduction in gait and postural dysfunction in patients with PD after receiving PPN deep brain stimulation.37,38
Here, we used serially cut sections taken from the postmortem PPN of patients with PD and compared this with elderly, healthy control persons, who died without known neurological or psychiatric deficit. After confirming a decreased number of cholinergic neurons in PD-affected PPNs, as previously reported,31,32 we studied whether the remaining cholinergic neurons undergo somatic cell size alterations. Because it is unknown whether other, noncholinergic neurons are also lost in the PPN of patients with PD, we next determined whether glycinergic and GABAergic neurons in the PPN also degenerate as a result of PD, and whether the remaining neurons undergo structural alterations. In an attempt to explain the altered cell numbers and cellular structural changes seen in the PPN of the present study's cohort of patients with PD compared with controls, and consistent with the wide amount of literature that suggests an association between PD and mitochondrial dysfunction, we determined whether the loss of different neuronal subpopulations in the PPN is linked to mitochondrial abnormalities.
Materials and Methods
Autopsy Material and Postmortem Neuropathological Examination
Ethical approval for this study was obtained from the Newcastle and North Tyneside Local Research Ethics Committee. Seventeen brains (nine controls, eight PD cases) were selected from the Newcastle Brain Tissue Resource. Brain tissue was processed for neuropathological examination with the use of a standardized protocol as previously described,39,40 with fixation of the right cerebral hemisphere, hemi-midbrain, brainstem, and cerebellum in neutral formalin, whereas the left hemisphere (used in the present study) was coronally sectioned, snap-frozen immediately in liquid nitrogen-chilled isopentane, and then stored at −80°C until neurochemical analysis.
Control cases showed an absence of any identifiable neurological disorder-related neuropathology and only mild age-related pathology (Table 1). PD cases had documented evidence of longstanding motor impairment, and evidence of dementia was present in six of eight cases at the most recent testing before death (Table 1). PD cases all showed extensive SNpc cell loss, in addition to the detected presence of LB/LNs in the brainstem, and in some cases also in the limbic system and cortex. PD cases also showed some evidence of limbic and hippocampal Alzheimer-type pathology, but none met the diagnostic criteria for Alzheimer disease.
Table 1.
Summary of Clinical and Neuropathological Characteristics of Patients and Controls
| Case | Sex | Age of PD symptom onset (y) | Age at death (y) | PMI (h) | Clinical history | Meds | Neuropathology diagnosis | AD Braak stage | Cortical LB score |
|---|---|---|---|---|---|---|---|---|---|
| PD1 | M | 73 | 86 | 18 | Ischemic heart disease; myocardial infarct; transient ischemic attack; acute bronchopneumonia; pulmonary thromboembolism; postural hypotension; falls | Madopar; Sinemet | PD dementia; neocortex: ubiquitin+ and αSYN+ neurons; SN: severe neuron loss; LC: severe neuron loss; remainder pale with occasional LBs | IV | 17 |
| PD2 | M | 70 | 78 | 13 | Epilepsy; bronchopneumonia; increased motor tone and cogwheeling | Sinemet; phenytoin | PD; ischemia in hippocampus CA1; SN: neuron loss, LBs; LC: LBs | II | 1 |
| PD3 | F | 75 | 83 | 19 | Bronchopneumonia; postural hypotension | Unknown | PD; neocortex: αSYN+ neurons | III | 9 |
| PD4 | F | 70 | 76 | 21 | Bronchopneumonia; septicemia; adult-onset diabetes | Unknown | PD-dementia; brainstem: αSYN+ neurons; SN: severe neuron loss; remainder pale with LBs; LC: moderate neuron loss, multiple LBs; hippocampus: LBs | IV | 11 |
| PD5 | F | 63 | 72 | 24 | Dementia | Unknown | PD; DLB; minimal ATP | II | 7 |
| PD6 | M | 60 | 68 | 11 | Dementia; visual hallucinations; delusions | Unknown | PD; DLB; neocortex: αSYN+ neurons | IV | 0 |
| PD7 | F | 55 | 65 | 24 | Pulmonary embolism; pulmonary edema; bronchopneumonia; dementia | Unknown | PD; dementia; neocortex: αSYN+ neurons | IV | 13 |
| PD8 | M | 65 | 78 | 23 | Partial vertical supranuclear gaze palsy; postural myoclonus; dementia | Donepezil; Sinemet; Madopar | PD; mixed DLB; mild ATP | III | 13 |
| Cntr1 | M | - | 70 | 18 | Cognitively normal; triple ascending aortic aneurism | Unknown | Mild amyloid angiopathology; mild degree of diffuse amyloid plaques; | 0 | 0 |
| Cntr2 | M | - | 68 | 21 | Cognitively normal; bowel cancer with pulmonary and liver cancer metastasis; no brain cancer metastasis | Unknown | Small superficial micro-infarcts in temporal and frontal cortex; trauma | 0 | 0 |
| Cntr3 | M | - | 72 | 17 | Cognitively normal; esophageal adenocarcinoma | Unknown | Mild ATP: some tangles in the neocortex | I | 0 |
| Cntr4 | F | - | 93 | 21 | Cognitively normal; pneumonia | Unknown | Mild AD-type pathology; small cerebellar infarct; striking perivascular dilation | I | 0 |
| Cntr5 | F | - | 69 | 16 | Cognitively normal; blind; hypertension; renal and cardiovascular disease; possible heart infarct | Unknown | Focal pathology: vessel wall collagenosis and axon/myelin loss in periventricle and DWM of parietal lobe; axon/myelin loss in optic tract; perivascular lacunae in GP | I | 0 |
| Cntr6 | F | - | 75 | 30 | Cognitively normal; endometrial cancer metastasis | Unknown | No evidence of neurodegenerative/ cerebrovascular disease | 0 | 0 |
| Cntr7 | M | - | 75 | 19 | Cognitively normal; MI; chest infection; skin cancer | Unknown | Mild DWM pathology: activated microglia; mild vascular changes in central DWM and cortex; age-related ubiquitin dot profiles: profusion in entorhinal cortex; pons: mild demyelination | I | 0 |
| Cntr8 | F | - | 85 | 18 | Cognitively normal; chest infection; pneumonia | Unknown | No evidence of neurodegenerative/cerebrovascular disease in spinal cord, hippocampus, multiple cortical lobes, motor cortex, and cerebellum | 0 | 0 |
| Cntr9 | M | - | 80 | 22 | Cognitively normal; MI | Unknown | No evidence of neurodegeneration in CNS | 0 | 0 |
For PD cases sex ratio (M/F) was 4:4, mean age of PD symptom onset was 66.38 ± 6.84 years, mean age at death was .75 ± 7.16, and mean PMI was 19.13 ± 4.94 hours. For control patients, sex ratio (M/F) was 5:4, mean age at death was 76.33 ± 8.31years, and mean PMI was 20.22 ± 4.18 hours.
F, female; M, male; AD, Alzheimer disease; ATP, Alzheimer type pathology (ie, plaques and tangles); CNS, central nervous system; DLB, dementia with Lewy bodies; DWM, deep white matter; GP, globus pallidus; LC, locus coeruleus; MI, myocardial infarction; SN, substantia nigra.
IHC for Anatomical Identification of the PPN
Serial frozen sections 20 μm thick were obtained from the frozen left midbrain at the pontine midbrain border with the use of a cryostat (Bright Instrument Company Ltd., Huntingdon, UK). To determine architectural boundaries, sections were air dried at room temperature for 60 minutes, followed by fixation in 4% paraformaldehyde dissolved in 0.1 mol/L PBS for 30 minutes and another 10 minutes in Carnoy fixative solution (60% EtOH, 30% chloroform, 10% glacial acetic acid) for Loyez/Cresyl fast violet (CFV) staining. The sections were hydrated through a graded series of ethanol (2 × 100%, 95%, 70%) before being rinsed in distilled H2O. Sections were then placed in mordant (4% iron alum) for 6 hours before rinsing in distilled H2O. The sections were stained overnight in hematoxylin solution. The next morning the sections were washed in distilled H2O, before incubation in CFV solution for 15 minutes at 60°C. The sections were left in the CFV solution while it was being cooled, before finally washing in distilled H2O, dehydrating, clearing, and mounting with DPX.
To identify the anatomical boundaries of the PPN from surrounding neural structures, the Loyez/CFV-stained sections were used (Figure 1, A–D). Previously published criteria for delineating the boundaries of the PPN,31,41,42 combined with knowledge of the cytoarchitectural features of the PPN, served as the guideline. The PPN was anatomically defined as a sickle-shaped zone lying within the dorsolateral mesopontine tegmentum, consisting of large neurons (presumed to be the Ch5 cholinergic population), which extended rostrocaudally from the level of the rostral locus coeruleus.31,41,42 Fibers of the superior cerebellar peduncle and peduncular decussation bordered the PPN medially, whereas its lateral border comprised the medial leminuscus. Its rostral end was seen to contact the SNpc, whereas its most caudal pole was adjacent to neurons of the locus coeruleus (Figure 1). In the present study, we were unable to distinguish between neurons inherent to the core and those locating to the diffuse aspects of the PPN, which may represent ventral parts of the nucleus subcuneiformis/cuneiformis.
Figure 1.

Loyez-CFV stained sections show the most caudal (A) and rostral (B) aspects of the series of serial sections stained with various antibody combinations for the present study. Mapped outlines (C and D) indicate the anatomical location of the PPN in relation to major surrounding structures, including the PAG, CnF, and DRN that form along the axis of the brainstem. The individually tiled images that make up A and B were captured with a Zeiss Axio ImagerZ2. The images were stitched together to make up the mosaic by using AxioVision software version 4.8 (Carl Zeiss). The insets show magnified images of the PPN region (A and B), which prominently features large, triangular-shaped neurons that characterize the PPN. A10, ventral tegmentum (paranigral nucleus) A10 cell population; CnF, nucleus cuneiformis; CP, midbrain cerebral peduncle; DRN, dorsal raphe nucleus; IC, inferior colliculus; ML, medial lemniscus; MLF, medial longitudinal fasciculus; PAG, periaqueductal gray; PN, pontine gray nuclei; PPN, pedunculopontine nucleus compactus; RRF, retrorubral fields; SNpc, substantia nigra pars compacta; SCP, superior cerebellar peduncle; III, third cranial nerve. Original magnification: ×20 (A and B); ×2.5 (insets). Scale bars: 500 μm (A and B); 50 μm (insets).
Antibodies Used
Antibodies used were as follows: mouse monoclonal anti–mitochondrial porin (VDAC1; 1:400; Mitosciences, Eugene, OR; code MSA08); mouse monoclonal anti–mitochondrial Complex-I, subunit 19 kDa (GRIM-19; 1:100; Mitosciences; code MS103); mouse monoclonal anti–mitochondrial Complex-I, subunit 20 kDa (NDUFB8; 1:100; Mitosciences; code MS105); mouse monoclonal anti–mitochondrial Complex IV, subunit 1 (COX-1; 1:100; Mitosciences; serum code MS404); goat polyclonal anti–choline acetyltransferase (1:500; Millipore, Temecula, CA; code AB144P-200UL; rabbit polyclonal anti–glycine transporter 2 (GlyT-2; 1:500; Abcam, Cambridge, UK; code ab99098); rabbit polyclonal anti–glutamic acid decarboxylase 65/67 (GAD65/67; 1:500; Sigma-Aldrich, St. Louis, MO; code G5163); and mouse monoclonal anti-αSYN (1:20; Novocastra/Leica Biosystems, Newcastle-On-Tyne, UK; code NCL-L-ASYN; clone KM51).
The secondary antibodies used were biotinylated goat anti–mouse IgG (H + L chain specific) for primary antibody 1 (1:150) and 2 to 4 (1:200; Vector Laboratories Inc., Peterborough, UK; code: BA-9200); for the immunofluorescence reaction that involved primary antibody 5, the secondary biotinylated rabbit anti–goat IgG (H + L chain specific; 1:200; Vector Laboratories; code BA-5000) and Alexa Fluor 488-conjugated streptavidin (1:200; Molecular Probes, Eugene, OR; code S-11223) were used. For detecting anti–GlyT-2 (primary antibody 6), rhodamine red (TRITC) affinity-pure goat anti–rabbit IgG (H + L chain specific; 1:200; Jackson ImmunoResearch Laboratories, West Grove, PA; code 111-025-045) was used. Anti-GAD65/67 (primary antibody 7) was detected with fluorescein-conjugated goat anti–rabbit IgM + IgG (H + L chain specific; 1:200; Southern Biotech; Birmingham, AL; code: 4010-02). Finally, anti-αSYN (primary antibody 8) detection was performed with biotinylated horse anti–mouse IgG (H + L chain specific; 1:200; Vector Laboratories; code BA-2000).
All primary and secondary antibody dilutions were made in Tris-buffered saline (TBS; pH 7.4).
IHC for Detecting Intraneuronal αSYN Aggregates
For detecting intraneuronal αSYN aggregates within the PPN, standard immunohistochemistry (IHC) methods were applied to a single frozen section per patients with PD of control case (Table 2) were air dried for 60 minutes at room temperature and fixed in 4% paraformaldehyde in PBS for 30 minutes. Endogenous peroxidase activity was quenched by incubating the sections in 3% hydrogen peroxide (H2O2) for 20 minutes at room temperature. Because frozen sections allow for better antigen preservation than the excessive fixation and processing regime associated with formalin-fixed, paraffin-embedded brain tissue,43 leaving antigens in a more native state, no antigen-retrieval step was required. The sections were rinsed in TBS before incubation with blocking buffer (3% normal horse serum in PBS; Vector Laboratories) for 60 minutes at room temperature. Anti-αSYN was then applied for 90 minutes at room temperature. After washing in PBS, an appropriate secondary antibody was applied for 30 minutes at room temperature, followed by incubation in the avidin-biotin-peroxidase complex (ABC), prepared as directed (Vectastain Elite ABC kit; Vector Laboratories). The slides were washed several times in TBS and then rinsed in TBS that contained 1% Tween-20 before the antibody binding sites were visualized with a 3,3′-diaminobenzidine tetrahydrochloride (DAB) substrate (Peroxidase Substrate Kit; Vector Laboratories), prepared as directed and applied to the sections for 10 minutes at room temperature. Slides were rinsed in distilled H2O for 10 minutes and then counterstained for 10 minutes with Mayer's hematoxylin (Histolab Products AB, Göteborg, Sweden) to view the nuclei. Finally, the sections were dehydrated, cleared, and mounted with DPX mounting media (Leica Biosystems, Wetzlar, Germany). For negative control slides, anti-αSYN antibody was omitted (blocking buffer only).
Table 2.
IHC Staining Scheme Design for Immunodetection of Antigens
| Section no. | Stained for |
|---|---|
| 1 (most caudal) | Loyez-CFV (high-contrasting histological stain, to confirm presence of PPN using anatomical identifiers) |
| 2 | Porin + cholinergic neuron identifier + nuclear marker counterstain |
| 3 | Porin + GABAergic neuron identifier + nuclear marker counterstain |
| 4 | Porin + glycinergic neuron identifier + nuclear marker counterstain |
| 5 | CO-1 (19 kDA) + cholinergic neuron identifier + nuclear marker counterstain |
| 6 | CO-1 (19 kDA) + GABAergic neuron identifier + nuclear marker counterstain |
| 7 | CO-1 (19 kDA) + glycinergic neuron identifier + nuclear marker counterstain |
| 8 | COX/SDH immunoassay |
| 9 | IHC for detecting αSYN, including LBs and LNs |
| 10 | CO-1 (20 kDA) + Cholinergic neuron identifier + nuclear marker counterstain |
| 11 | CO-1 (20 kDA) + GABAergic neuron identifier + nuclear marker counterstain |
| 12 | CO-1 (20 kDA) + glycinergic neuron identifier + nuclear marker counterstain |
| 13 | COX1 + cholinergic neuron identifier + nuclear marker counterstain |
| 14 | COX1 + GABAergic neuron identifier + nuclear marker counterstain |
| 15 | COX1 + glycinergic neuron identifier + nuclear marker counterstain |
| 16 (most rostral) | Loyez-CFV high-contrasting histological stain (to confirm presence of PPN) + nuclear marker counterstain |
Staining was performed on serial 20-μm thick fresh/frozen sections that followed and contained the PPN. Sections containing the PPN were identified with combined Loyez-CFV IHC, performed at the start of the most caudally available aspect of the serial sections and at the most rostral end, but still within the PPN nucleus. An example of a Loyez-CFV stained section can be seen in Figure 1. In each case where a mitochondrial marker is co-stained with a marker identifying a particular neuronal phenotype (cholinergic, GABAergic, and glycinergic neurons), the mitochondrial protein was detected by using a chromogen (DAB), whereas the neurochemical phenotype was detected by use of a fluorescence-coupled secondary antibody.
IHC for Detecting Mitochondrial Mass and Mitochondrial Respiratory Chain Protein Subunits
Table 2 provides an overview of the staining protocols followed for the various immunodetection combinations used here. Sections were stained with primary antibodies directed against specific subunits of respiratory chain complexes I, II, and IV and detection of porin, a voltage-dependent anion-selective channel, located to the outer mitochondrial membrane, regarded as a reliable marker of mitochondrial density.40 Specifically, the mitochondrial respiratory chain proteins assessed here comprised mtDNA encoded subunit-I of complex IV (COX-I) and the two nuclear encoded subunits, Co-I 19 kDa and Co-I 20 kDa.
Briefly, frozen sections were air-dried, fixed in 4% paraformaldehyde in 0.1 mol/L PBS for 30 minutes, after which they were thoroughly washed in TBS. Endogenous peroxidase activity was quenched with 3% H2O2 for 30 minutes followed by washing in TBS. Nonspecific binding was then blocked in TBS that contained 3% normal goat serum and 2% bovine serum albumin for 30 minutes. The blocking solution was then tipped off each section, before they were incubated for 90 minutes at room temperature in the respective primary antibody solution used for detecting the mitochondrial proteins of interest here (for the concentrations used, see above). Sections were washed in TBS (3 × 3 minutes), before the biotinylated secondary antibody diluted in TBS was applied for 30 minutes at room temperature. This was followed by horseradish peroxidase–conjugated ABC reagent from the Vector Elite kit (Vector Laboratories) prepared according to kit protocol. Bound peroxidase was visualized by incubation with DAB substrate chromogen, using a commercial kit (Vector Laboratories; SK-4100) for 8 minutes. Care was taken to ensure that each section was exposed to DAB for this length of time only, after which time the reaction was stopped by rinsing the sections in cold PBS solution. Moreover, to eliminate variation in intensity of immunostaining, staining with the use of one antibody combination (Table 2) was performed for all patients with PD and control subjects on the same day.
Immunofluorescence Histochemistry for Detecting PPN Neuronal Subtypes
A dual IHC assay was designed, combining staining for the mitochondrial markers of interest (see above) with the detection of neuron-specific markers in the PPN (Table 2). To correlate mitochondrial staining with subtype-specific PPN neurons, we performed a sequential staining protocol, by first applying a chromogen detection strategy for the various mitochondrial proteins of interest (see above). Immediately after this step, we performed immunofluorescence labeling for either GAD 65 or GAD 67 for identifying GABAergic cells; GlyT-2, a neuronal marker of glycinergic neurons, or anti–choline acetyltransferase that serves as a marker of cholinergic cells. We ensured that sections did not dry out after the immunohistochemical staining protocols for the various mitochondrial proteins. Immediately after the chromogen staining protocol, sections were reblocked in 5% normal goat serum, 2% bovine serum albumin, and 0.5% Triton X-100, before applying the primary antibody for detecting GABAergic, glycinergic, or cholinergic neurons and then incubating the sections overnight at 4°C. The sections were then washed in TBS (3 × 3 minutes), before applying the fluorochrome-conjugated secondary antibodies (see above). The sections were incubated in the secondary antibody solution for 60 minutes at 37°C. Nuclei were labeled with TBS-diluted Bisbenzimide Hoechst 33342 nucleic acid stain (1:200, no. B2883; Sigma-Aldrich) for 30 minutes at room temperature. The sections were given a final wash in TBS, before being covered with ProLong Gold anti-fade mounting media (Invitrogen, Paisley, UK) and protected with a coverslip. In each case, no signal was observed in control sections where the primary antibody to the various neuronal-type (GABAergic, glycinergic, and cholinergic) marker protein was omitted.
Quantification of Neurons
Quantification of the numbers of GABAergic, cholinergic, and glycinergic neurons in the PPN was achieved by a semistereological method. Cell counts for these neurons were performed on the same images that had been captured for densitometry and somatic cell size analyses (hence, serially cut PPN sections).
GABAergic, cholinergic, or glycinergic neurons were considered as such, if they met the staining criteria deemed for the respective neuronal subtype (see Neurochemical Identification of PPN Neurons). The counts were performed with the cell counter function in ImageJ image analysis software version 1.4 (National Institutes of Health, Bethesda, MD), which allowed for labeling of neurons. In all instances, a GABAergic, cholinergic, or glycinergic neuron was marked (once only) for counting, if it displayed large, spherical DAPI-stained neuronal nuclei.44 In addition, a neuron was considered immunopositive if the nucleus was surrounded by cytoplasm filled by colored precipitate, respective to the specific neuronal type being immubolabeled (GABAergic and cholinergic, green; glycinergic, red). In addition, a neuron was only flagged for inclusion in the overall cell count of its respective PPN cell population if it was deemed to be clearly in focus at a specific focal plane. Overlapping cells were omitted from the analysis.
Each of the four PPN sections used for calculating neuronal density for each neuronal subtype covered an area of 1.949 mm2, representing 15 adjacent PPN regions, which had been stitched together with AxioVision software version 4.6 (Carl Zeiss, Jena, Germany). The counts are presented as number of neurons/mm2 ± SD of the means.
Neuronal Cell Body Areas
The same images captured for densitometric analyses were used for determining area size (μm2) of the somas of cells identified fluorescently as GABAergic, glycinergic, and cholinergic and was calculated by using ImageJ software version 1.4 (National Institutes of Health). Only well-defined individual cells were used, whereas overlapping cells were excluded from the analysis.
Relative Densitometric Measurements
In each case the PPN boundary was manually outlined with a camera lucida attached to the microscope. The section boundary was then used as a guide to locate the PPN with the use of an epifluorescent microscope (Axioplan 2; Carl Zeiss) equipped with a motorized stage used to systematically cover the entire PPN region. For capturing images, a high-resolution objective lens (Plan-Neofluar, 40×/0.75) and an Axiocam HRc digital camera (Carol Zeiss) were used. Identical exposure times were used throughout. Images were captured as separate or unmixed fluorescent images by using the multichannel acquisition function of the image capturing software (AxioVision; Carl Zeiss). Hoechst 33342 signal was captured with a DAPI filter cube, green-fluorescent fluorescein with a fluorescein isothiocyanate cube, and Rhodamine with a TRITC cube (Carl Zeiss). All images were collected under the same microscope settings with the use of previously described procedures. We used a protocol for analysis of bright field images, which, by being automatic and nonsubjective, has been shown to facilitate accurate measurement of regional variations in optical density.40,45–49 The light microscopy images were converted into an eight-bit gray scale (monochromatic) digital image, for processing binary (black and white) color information, representative of the DAB content. Because the DAB stain intensity represents a unitless value that ranges between 0 (black) to 255 (white), values were inverted by subtracting them from 255. Therefore, more intense staining gave a higher value, before applying a weighting calculation for estimating the amount of DAB (antigen) as a dimensionless index. An identical threshold was applied to all images to eliminate background.
Contiguous, slightly overlapping individual images were stitched together with ImageJ software version 1.4 (National Institutes of Health) to create a mosaic from which global computerized densitometric values for porin, COX-I, Co-I 19 kDa, and Co-I 20 kDa expressions were obtained. On the basis of morphology and the overlap of the nuclear marker with neurochemical subtype-specific fluorescent staining, single GABAergic, glycinergic, and cholinergic neuronal cells were outlined and copied onto the unmixed monochrome images of complex I and IV activity. The densitometric parameter computed was absorbance (integrated optical density; IOD), as a proxy marker of protein levels, reported as arbitrary values of mean pixel density per selection area. The investigator (I.S.P.) were blinded to clinical diagnosis.
COX-SDH Histochemistry
The presence of COX-deficient neurons is regarded as a pathological hallmark of mtDNA mutations.50 Succinate dehydrogenase (SDH) is entirely encoded by nuclear DNA, whereas COX contains subunits encoded for by both the mtDNA and nuclear genome, thereby allowing the COX-SDH assay to detect mitochondrial genomic deficiency that affects COX. We performed dual histochemistry to identify and quantify deficiency of COX alongside SDH-positive cells within the PPN, using a previously described method.51
Statistical Analysis
Comparison of neuronal densities, cell size, mitochondrial respiratory chain subunit immunoreactivity between cases and controls are reported as mean values ± SD. For the analyses of cell size and protein expression, the mean value for each participant, along with the SD and number of observations was used in significance testing. All these data sets were analyzed by using an unpaired, two-tailed Student's t-test. Spearman's rank correlation coefficient was used to assess whether a correlation between mean protein expression and either LB or Braak stage could be detected. Spearman's rank correlation coefficient was used to assess whether a correlation between mitochondrial protein expression levels and postmortem delay could be detected. Statistical analysis was performed with GraphPad Prism software version 4.0 (GraphPad Inc., San Diego, CA).
Results
Clinical Details and Neuropathological Features of Study Cases
The clinical and postmortem neuropatholological characteristics of the control and PD cases used in the present study are summarized in Table 1. PD cases showed the presence of αSYN-positive LBs and LNs in SNpc and also in some PD cases the presence of cortical αSYN pathology. Investigation of the PPN showed the presence of LNs (Figure 2A) and LBs (Figure 2B) within PPN neurons in six of eight PD cases but not in any of the neurologically intact control cases. This accumulation of αSYN aggregates, a protein known to localize presynaptically, indicates that the PPN is pathologically affected in PD, consistent with previous observations.52
Figure 2.
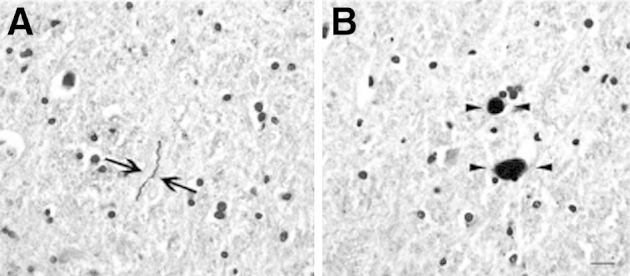
IHC with the use of a peroxidase-DAB detection method for indicating the accumulation of αSYN deposits within neurons of the PPN region in postmortem brains of patients with PD. Shown is magnification of the thread-like LNs (arrows, A) and dot-like LBs (arrowheads, B), both of which are visible as a dark brown colored precipitate. Lewy pathology in the PPN was not detected in any of the case controls studied here. Original magnification: ×20 (A and B). Scale bars: 20 μm (A and B).
Neurochemical Identification of PPN Neurons
GABAergic neurons were identified by labeling with an antibody that recognized both GAD65 and GAD67, the enzymes involved in GABA synthesis, thus serving as a GABAergic cell-specific marker.53 GAD65/67-immunopositive neurons were small (<20 μm in diameter) and round (Figure 3, A and B), while morphologically appearing very dissimilar to neurons labeled for either glycinergic or cholinergic neurons. With the use of an antibody specific for GlyT-2 as a marker of glycinergic neurons,54 immunoreactivity was observed in glycinergic neurons (<15 μm in diameter) both in the soma and neurites, with a higher concentration of expression noted at the polar end of the cell body, similar to previous descriptions (Figure 3, C and D). The PPN Ch5 cholinergic neurons were identified (Figure 3, E and F) as a cluster of relatively large neurons (approximately 30 μm diameter), principally of pyramidal shape. Our labeling scheme identified three specific types of neurons within the PPN, but it is plausible that other neuronal subtypes exist (eg, glutamatergic neurons), although these were not assessed in the present study.
Figure 3.

Fluorescence IHC with the use of an anti-GAD65/67 antibody indicates the presence of small, round GABAergic neurons in the PPN of postmortem brain specimens (in green, control case, arrows, A; PD-affected case, arrows, B). Immunofluorescence detection of an antibody that targets GlyT-2 was used for labeling glycinergic neurons in the PPN (in red, control case, arrows, C; PD-affected case, arrows, D). Choline acetyltransferase fluorescence immunoreactivity indicated the presence of cholinergic neurons in the PPN (in green, control case, arrows, E; PD-affected case, arrows, F). For each neuronal subtype investigated here, the sections were counterstained with a nuclear marker, Bisbenzimide Hoechst 33342, indicated in blue. Right panels show the merged images. Images were processed after capture in Adobe Photoshop software version 7.0 (Adobe Systems, Mountain View, CA), adjusting for levels of brightness and contrast and merged automatically with the built-in functions of the software program. Original magnification, ×64/1.4 NA oil-based. Scale bars: 50 μm (A, B, E, and F); 20 μm (C and D).
Quantification of Neurons
We estimated the numerical density of different neurochemically defined neurons in the PPN, expressed as the average number of neurons per square millimeter. Confirming previous reports,31,32,36 a reduced density of the PPN cholinergic neurons, by approximately 50%, was seen in PD cases (30.07 ± 0.82 neurons/mm2) compared with controls (62.70 ± 1.37 neurons/mm2; P < 0.0001; four sections were analyzed for each of the PD and control cases) (Figure 4). Our study reports, for the first time, that GABAergic cell counts were also significantly decreased in the PD-affected PPN compared with control PPN (152.0 ± 2.76 GABAergic neurons/mm2 for PD; 184.3 ± 3.2 for controls; P < 0.0001; four N sections analyzed/control or PD case), equating to a neuronal loss of 18% in PD cases (Figure 4). At the same time, another novel observation was that a reduction was observed for PPN glycinergic neuronal densities (98.64 ± 1.28 neurons/mm2 in PD cases, 113.7 ± 1.1 neurons/mm2 in controls; P < 0.0001; four sections analyzed per neuronal marker per control or PD case) (Figure 4), equating to a loss of just >13% in glycinergic cell density in the PPN of PD cases.
Figure 4.
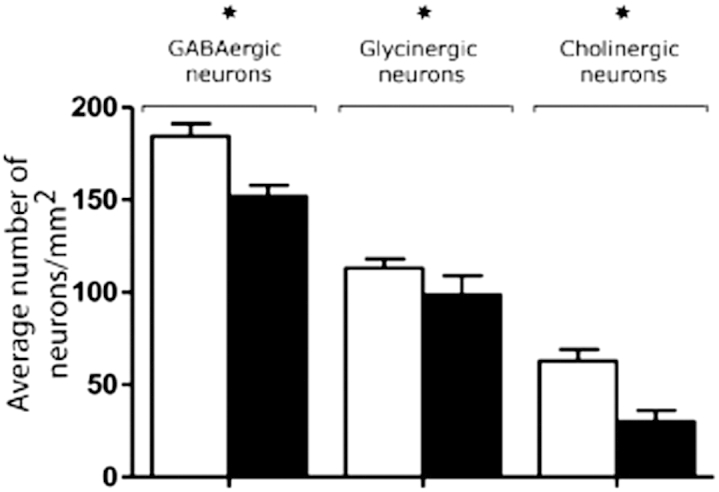
The neuronal density for GABAergic, glycinergic, and cholinergic neurons (± SD) within the PPN of PD-affected cases compared with controls. Each neuronal type was counted in four serially cut sections of the PPN region for each of the control and PD cases, the PPN counting region comprised an area of 1.949 mm2. Analyses found a decreased number of GABAergic, glycinergic, and cholinergic cells in PD cases compared with controls, reaching a statistically highly significant extent. ∗P < 0.001, unpaired Student's t-test. The measurements indicated that the loss of cholinergic cells in the PD-affected cases was most pronounced (48%) compared with GABAergic and glycinergic cell loss.
Neuronal Somatic Size
A statistically significant difference was observed in terms of the somatic area of GABAergic neurons, between the PPN of normal controls and those of PD affected cases (P = 0.0001) (Figure 5), with the PD-affected neurons appearing generally larger than those analyzed in control cases. Specifically, the mean somatic size for the GABAergic neurons in the PD-affected PPN sections was 32.25 ± 0.21 μm2 (667 individual neurons analyzed, across four PPN sections per PD case). In the PPNs of control brains the neuronal size of GABAergic neurons was calculated to 28.70 ± 0.17 μm2 (1428 individual neurons analyzed, across four PPN sections per control case). Similarly, the area size of glycinergic neurons in PD cases were larger (P < 0.0001) (Figure 5) than those in control sections, with a mean cell area of 16.72 ± 0.18 μm2 (496 individual neurons analyzed, across four PPN sections per PD case), compared with an average of 8.93 ± 0.08 μm2 in the controls (608 individual neurons analyzed, across four PPN sections per control case). In contrast, the area size of cholinergic neurons in the PPN was dramatically reduced in size in PD compared with controls (P = 0.0008) (Figure 5).
Figure 5.

The means ± SD of area covered by the neuronal cell body (μm2) of GABAergic, glycinergic, and cholinergic neurons were calculated by identifying such neuronal populations in the PPN region of control and PD cases, using fluorescent IHC applied to post-mortem brain sections. Hypertrophy was seen in the neuronal cell bodies of GABAergic and glycinergic neurons in PD, whereas a noted shrunken appearance was observed in cholinergic neurons in PD. Statistical analyses with an unpaired Student's t-test found a significant increase in somatic area for both GABAergic and glycinergic (∗P < 0.0001) PPN neurons in PD cases compared with controls. However, the reverse was seen for the PPN cholinergic neurons, to indicate a statistically significant decrease in the cholinergic neuronal cell area in PD cases compared with controls (P = 0.008). The numerical summary data for both patients and controls can be viewed in Supplemental Table S2.
The mean somatic area of cholinergic neurons in the PD-affected PPN was 75.78 ± 0.92 μm2 (313 individual neurons analyzed, across four PPN sections per PD case), compared with a mean of 94.59 ± 1.4 μm2 (501 individual neurons analyzed, in 4387 PPN sections per control case). Supplemental Table S1 shows the number of observations made for each of the PD cases and controls when totaled across the four sections examined for each participant. The same table also shows the means ± SD of cell size for each of the participants; again as an average of the neurons analyzed over the four sections examined per participant, for each of the neuronal subtypes studied here. Examination of the data for each study participant indicated that the significance values observed were not the result of outliers in either cohort, and thus likely reflect an important physiological difference between the two groups. The use of four PPN sections per participant, per neuronal type, is likely to have been important in the clear demarcation seen in the cell size values in the two cohorts.
Measurement of Mitochondrial-Related Protein Content in Neurochemically Distinct PPN Neurons
PPN GABAergic neurons showed a significantly increased mean expression level (measured as the IOD score; see Materials and Methods) of porin in PD cases compared with controls (P = 0.0007) (Figure 6A and Supplemental Table S2). A similar pattern was seen after IHC detection for the mitochondrial respiratory chain subunits COX-I, Co-I 19 kDa, and Co-I 20 kDa in PD cases compared with controls in the same PPN neuronal subtype (P < 0.0001) (Figure 6A and Supplemental Table S2). Analysis of porin expression levels in glycinergic PPN neurons indicated a highly significant up-regulation in PD cases versus controls (P < 0.0001) (Figure 6B and Supplemental Table S3). Similarly, expression levels of COX-I, Co-I 19 kDa, and 20 kDa showed a highly significant increase in the same neurons analyzed in PD-affected cases compared with controls (P < 0.0001) (Figure 6B and Supplemental Table S3). In contrast, analyses of PPN cholinergic neurons for porin indicated that this protein's expression level was significantly lower in patients with PD than in controls (P < 0.0001) (Figure 6C and Supplemental Table S4). This pattern repeated itself in analyses performed for COX-I, Co-I 19, and Co-I 20 kDa expression (P < 0.0001) (Figure 6C and Supplemental Table S4). Plots showing the individual data points collected for this analysis are given in Figure 7, A–H, and Figure 8, A–D; the clouds of data points are a stark illustration of the differential protein expression in the cases and controls. Examination of the tabulated data, which gives participant means, SD, and the number of observations, indicates clearly that the differences in expression, as found in Figures 7 and 8, are not the result of a few unusual cases in either cohort that have been overrepresented in the data collection.
Figure 6.

IOD values of porin, COX-I, Co-I 19 kDa, and Co-I 20 kDa from densitometric analyses with the use of ImageJ software version 1.4 (National Institutes of Health), obtained for distinct PPN neuronal populations indicated highly significant (∗P < 0.001) or extremely significant (∗∗P < 0.0001) differences between controls and patients with PD in GABAergic (A), glycinergic (B), and cholinergic (C) PPN neurons. In particular, porin IHC indicated a mitochondrial density increase in the PPN GABAergic and glycinergic neurons of PD specimens relative to controls. However, this expression pattern was reversed for cholinergic neurons, with lower porin expression seen in the PD cases compared with controls. A similar effect was seen for the mitochondrial electron transport chain proteins COX-I, Co-I 19 kDa, and Co-I 20 kDa, indicating up-regulated expression in GABAergic neurons and glycinergic neurons of PD cases. However, a reversed trend was seen in PPN cholinergic neurons of PD cases, which rather showed down-regulated expression in PD cases relative to controls. Numerical summary data for both patients and controls can be viewed in Supplemental Tables S3 to S5
Figure 7.

Dot-plot graphs show individual immunoreactivity measures taken for porin (A), COX-I (B), Co-I 19 kDa (C), and 20 kDa (D) localizing to GABAergic PPN neurons. All measures taken for the nine control cases (black) and eight patients with PD (red) are shown. A comparison of patient and control data for porin in the GABAergic PPN neurons yielded a significant difference (P = 0.0007). In addition, a highly significant difference in expression was seen for COX, Co-I 19 kDa, and Co-I 20 kDa, when analyzing the GABAergic PPN neurons of patients with PD versus neurological control cases (P < 0.0001). E–H: Dot-plots of individual immunoreactivity measures recorded in PPN glycinergic neurons for controls (black) and patients (red) for porin (E), COX-I (F), Co-I 19 kDa (G), and Co-I 20 kDa (H) expression. Statistical analysis indicated a highly significant difference in the expression levels for all proteins measured (P < 0.0001). Pts, patients.
Figure 8.

A–D: Dot-plots show individual immunoreactivity values for porin (A), COX-I (B), Co-I 19 kDa (C), and Co-I 20 kDa (D) expression in PPN cholinergic neurons. As with GABAergic and glycinergic neuronal populations, a highly significant difference (P < 0.0001) in the expression levels of these proteins was seen in cholinergic PPN neurons in PD. However, in contrast to the PPN GABAergic and glycinergic neurons, the expression of these mitochondria-related proteins in cholinergic neurons was decreased in the PD cases. Pts, patients.
The presence of prolonged agonal factors, such as respiratory arrest, multiorgan failure, or coma preceding death, resulting in decreased pH of the postmortem brain tissue, has been shown to adversely affect the quality of RNA by some.55 However, others have reported that, although brain pH decline was related to agonal state severity, this was seen to be independent of postmortem interval (PMI) and the histological presence of hypoxic changes.56,57 Moreover, it was shown that the PMI exerted no effect on either mRNA yields or the protein products, up to 48 hours after death.56 Protein level fluctuations are generally regarded as more resistant to degradation than fluctuations of RNA levels.58 None of the patients with PD or control subjects from whom postmortem brain tissue was collected for analyses in the present study indicated what could be regarded as the excessive duration or a severe agonal state preceding death (Table 1). Moreover, the collection and processing of postmortem material from cases and controls was done within the parameters set by the BrainNet Europe consortium.57 This included minimizing the time for dissection, rapid removal of the brain tissue, followed by immediate and appropriate freezing and storage of the tissue. However, because determining the effects of PMI on brain tissue samples is a complex issue, we performed a Pearson's correlation between the mean mitochondrial protein IOD levels and PMI, for both PD cases and control subjects for PPN neuronal subtypes studied here. The results of the 24 correlations conducted affirm that no relation exists between PMI and the IOD values, providing confidence that the observed IOD changes were not because of the variable PMIs among the PD cases and controls (Supplemental Table S5).
No Correlation Between Mitochondrial Protein Expression Levels and the Related Braak and LB Stage of PD Cases
Further analyses were performed to address whether a correlation existed between the mitochondrial protein expression (IOD) values and the respective Braak stage and LB score of the patients with PD included in this study. The relevant Braak and LB stage clinical information for all patients are given in Table 1. No significant correlation was detected between the mean IOD values in the patients with PD in terms of either the Braak stage or the LB stage, for any of the four proteins examined in any of the three neuronal types (P > 0.05, Spearman's rank correlation test).
Limited Mitochondrial Dysfunction (COX-deficiency) Detected in PPN Neurons
COX-SDH histochemistry of sections (one section per participant) of PD and control cases was used to look for indications of mitochondrial respiratory chain dysfunction within the PPN. All of the neurons inspected in control cases indicated normal respiration, identified as neurons and neuronal fibers that stained brown (Figure 9, A and B). In contrast, respiratory-deficient cells (identified as those that stained blue) were seen in all eight PD cases included in this study cohort (Figure 9, C and D), but to a limited extent. Overtly, COX-negative neurons that were entirely blue were not seen in any of the PD cases; however, many neurons that appeared to be COX intermediate could be viewed.
Figure 9.

A representative section of PPN tissue stained with COX/SDH histochemistry shows respiratory-normal activity (brown staining) in the neurons (arrowheads, A) of a control patient, as opposed to the COX-deficiency (blue staining) seen in the PPN tissue of PD cases (arrow, C). B: Shown is the apparent lack of COX-deficiency/SDH positivity throughout the neuronal projection fibers and particularly for neuronal somata (arrowhead) within the PPN region of a representative control case, in contrast to the blue staining seen for neurons in PD cases (arrow, D). However, in PD cases, COX deficiency was only seen in a limited number of PPN neurons, most of them appearing with intact respiratory activity. Original magnification: ×40 (A and C); ×10 (B and D). Scale bars: 10 μm (A and C); 20 μm (B and D).
Discussion
Our study aimed to characterize the PPN of patients with PD in terms of neurochemically distinct neuronal population loss, altered neuronal cell size, and differential mitochondrial protein expression within the remaining PPN neurons. Although the PPN is traditionally defined as a major cholinergic nucleus, several findings support the view of the PPN being neurochemically a highly heterogeneous nucleus. Noncholinergic PPN neuronal subgroups described so far include GABAergic,59 glutamatergic,60 and glycinergic61–63 neurons. In the present study, we quantified GABAergic neurons within the PPN and found a significant loss in PD cases compared with controls (P < 0.001) (Figure 4). This implies that the GABAergic innervation from the PPN to the SNpc and other areas of the central nervous system22,64 may be significantly diminished in PD, potentially contributing to clinical symptoms. Although no study before the present one has directly measured the number of GABAergic neurons, previous work reported a loss (>40%) of substance-P–containing (presumably GABAergic) neurons in the PPN in PD cases compared with controls,65 indicating that GABAergic neuronal loss is a feature of the PPN in PD. Our results suggest a more modest reduction of GABAergic neurons of 15% to 20% (Figure 4) compared with the extensive PPN cholinergic neuronal loss we observed in PD-affected brains (approximately 50%) (Figure 4). This potential imbalance may underlie some of the clinical observations seen in PD. In this regard, a role has been assigned to GABA disequilibrium in RBD,66 a frequent co-diagnosis in patients with PD. Moreover, drugs such as clonazepam and melatonin, which modulate GABAergic function, have been shown to provide an effective treatment option for alleviating RBD.67,68 Under normal circumstances, GABA agonist administration to the PPN results in reduced motor activity, whereas GABA antagonists are without major effects when administered to non-human primates. However, under conditions of SNpc damage, GABA antagonists resulted in improved motor function.69 Although it has been assumed that this effect of GABAergic antagonists in experimental PD models may be due to a blockade of major inhibitory inputs to the PPN from the internal part of the globus pallidus and the substantia nigra pars reticulata,26,60,70 it is possible that local inhibitory activity onto cholinergic and glutamatergic neurons may also play a role. Our finding of higher than normal levels of mitochondrial protein expression within PPN GABAergic neurons may indicate that local inhibitory neurotransmission within the PPN may be elevated despite the modest cell loss, requiring increased mitochondrial function to sustain. Future studies should aim to identify the GABAergic receptor subtypes within the PPN to determine whether selective targeting of the PPN could alleviate aspects of parkinsonism.
We also observed a reduction (approximately 13%) of PPN glycinergic cell density in PD cases (P < 0.001) (Figure 4). Glycinergic inhibition is involved in major spinal motor circuits, and glycinergic receptors have been shown to complement other inhibitory receptors involved in modulating cholinergic, dopaminergic, and GABAergic neuronal pathways in the basal ganglia.71 PPN neurons receive inhibitory glycinergic fast synaptic inputs,63 whereas postsynaptic inhibition mediated by glycine is also critical for suppressing motor neuron discharge during the tonic and phasic periods of REM sleep.72 In addition, Krenzer et al73 provided evidence that in the mouse brainstem glycinergic neurons serve to regulate REM sleep, while fulfilling a role in atonia, a frequent feature of PD.21 Our result, of reduced glycinergic neuronal density in the PPN in advanced PD, therefore suggests that potential RBD symptoms may at least in part stem from a loss of glycinergic PPN neurons. Further investigative work is required to be performed on cases with prospective assessment of RBD symptoms, with such case material that were not available with the present cases.
As with GABAergic neurons, glycinergic neurons had elevated mitochondrial protein expression, potentially translating to increased mitochondrial function and respiration. Should future work prove that this feature also translates into enhanced glycinergic tone, REM sleep disturbance and loss of atonia may be the consequences of enhanced local glycinergic inhibition. Dysfunction of other parts of the circuits that control muscle tone, including a reciprocal substantia nigra pars reticulate-to-PPN projection, may also underlie the postural dysfunction seen in PD.74 Although not investigated here, excitatory glutamatergic neurons form an additional PPN subgroup.26,75,76 Given that the SNpc innervation by the PPN is also mediated by glutamatergic afferents, along with glycinergic modulation, may provide suitable therapeutic candidates for treating specific PD symptoms.
Quantification of PPN cholinergic neuron content found extensive (approximately 50%) cell loss in patients with PD (Figure 4), to support previous findings.31,32,36 In this respect the ascending cholinergic projection from the PPN primarily innervates the SNpc, with up to 25% of cholinergic neurons in the PPN providing this function.26 Cholinergic neuronal loss in the PPN would clearly relate to the findings of gait and postural disturbances and would further exacerbate SNpc neuronal loss, with emergence of axial involvement, balance, and gait problems. In relation to REM sleep disorder, the cholinergic projection from the PPN to the thalamus may play a role because the projection is known to regulate REM sleep and the presence of atrophic PPN cholinergic neurons would clearly exacerbate this problem in PD.36
In patients with PD, changes in the density of the cholinergic neuron population of the PPN were accompanied by cell shrinkage. Both physiological neuronal activity and brain disorders can lead to cell swelling and shrinkage.77,78 Hirsch et al31 described the PPN of patients with PD as appearing shrunken, while displaying a loss of its characteristic sickle shape. In light of the suggestion that neuronal shrinkage rather than cell death may be the main phenomenon in neurodegenerative diseases,79 our present findings support the concept that these cells could be targeted for potential therapeutic relief, because atrophied cells can be activated. The cholinergic cell shrinkage seen in PD may be due to loss of target innervation and loss of trophic support from the SNpc and also the thalamus that shows structural and pathological changes, particularly in cases with concomitant dementia.80,81 It would be of interest to determine what trophic factors govern support of PPN neurons or whether activation of the SNpc provides neurotrophic support. Because PPN stimulation can ameliorate sleep disturbances accompanying advanced PD,82 improving the function of cholinergic projections to the thalamus may be beneficial.
The present report is the first to describe selective changes in mitochondrial proteins in specified neuronal types of the PPN, suggesting that widespread mitochondrial abnormalities occur during PD, to not just affect the SNpc. Increasing evidence implicates mitochondria in the pathogenesis of PD.5 Here, we analyzed the expression patterns of mitochondrial respiratory chain subunits of PPN neurons, finding a significant decrease in expression of mitochondrial proteins in cholinergic neurons, suggestive of mitochondrial injury. A similar decrease in expression was seen for voltage dependent anion-selective channel protein 1 (VDAC1/porin), a major mitochondrial outer membrane protein, to indicate a loss of mitochondrial mass in PPN cholinergics. Loss of mitochondrial proteins and mass may relate to our finding of atrophy of cholinergic neurons, although it is difficult to ascribe a causal role for the mitochondrial changes. A logical question that emerges from this finding is whether an underlying mtDNA deficit accompanies the altered expression of mitochondrial respiratory chain subcomponents. mtDNA anomalies can be effectively indicated by a dual COX/SDH histochemical assay.83 Here, PD cases indicated COX deficiency, albeit limited, in scattered neuronal soma (Figure 9, A and C) and in neuronal projection fibers (Figure 9, B and D), but with an atypical structure, including patches of soma appearing COX-deficient in a generally COX-positive cytoplasm. It is possible that this represents mitochondrial heteroplasmy with certain mitochondria within a cell showing COX deficiency and others being normal, with entirely COX-deficient cells only appearing as such when mtDNA mutations reach critical levels.84,85 Previous studies have shown that mtDNA deletions accumulate to high levels with age in SNpc neurons,50,86 with a close relationship seen between high levels of mtDNA deletions and a respiratory chain defect. In aged healthy persons, COX-deficient skeletal muscle cells also show mtDNA mutations, including clonal expansion of the individual mtDNA.87,88 All these studies concluded that COX-negative cells, with concomitantly high levels of mtDNA mutation, appear similar to those seen in patients with inherited mtDNA mutations with similar biochemical and pathological consequences.
Our study also provides evidence for GABAergic and glycinergic neuronal losses in the PPN of patients with PD accompanied by altered somatic structure, similar to cholinergic neuronal loss seen here and in previous reports.31,32,36 Moreover, we report the presence of mitochondrial changes within neurochemically identified neuronal populations of the PPN in patients with PD, by finding a reduction in subunits of complexes I and IV of the mitochondrial respiratory chain in cholinergic neurons and an up-regulated mitochondrial subunit expression in GABAergic and glycinergic neurons.
Overall, the strategic location of the PPN in the mesencephalic locomotor region of the brain, where it reciprocally connects with a range of subcortical and thalamic nuclei as well as the SN, underscores its critical modulatory role in locomotion and sleep. This suggests that a correlation may exist between PPN neuronal cell loss and neuronal structure, altered homeostasis of mitochondrial-related events in the PPN, and the degree of motor and nonmotor disability seen in patients with PD.
Acknowledgments
We thank Debbie Lett, Philippa Hepplewhite, Debbie Jones, and Dr. Trevor Booth (Newcastle University) for providing excellent assistance throughout the course of this project.
Footnotes
Supported by the Newcastle University Centre for Brain Ageing and Vitality. The Centre receives grant support from the Biotechnology and Biological Sciences Research Council (BBSRC), the Engineering and Physical Sciences Research Council (EPSRC), the Economic and Social Research Council (ESRC), and the UK's Medical Research Council (MRC), as part of the cross council Lifelong Health and Wellbeing Initiative (G0700718), the Wellcome Trust Centre for Mitochondrial Research (906919), and the UK's National Institute for Health Research (NIHR) Biomedical Research Centre for Ageing and Age-related disease award to the Newcastle-upon-Tyne Hospitals NHS Foundation Trust. I.S.P. is supported by a research grant from the Rosetrees Trust and a Faculty Fellowship from the Imperial College London. J.L.E. is supported by the Research Council UK (RCUK). C.M.M. is supported by the UK Department of Health and the UK MRC. Tissue made available for this study was provided by the Newcastle Brain Tissue Resource, which is supported by the UK MRC, the Alzheimer's Research Trust and Alzheimer's Association, through the Brains for Dementia Research Initiative and through an NIHR Biomedical Research Centre Grant in Ageing and Health.
Supplemental Data
References
- 1.Jankovic J. Parkinson's disease: clinical features and diagnosis. J Neurol Neurosurg Psych. 2008;79:368–376. doi: 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- 2.Dickson D.W., Braak H., Duda J.E., Duyckaerts C., Gasser T., Halliday G.M., Hardy J., Leverenz J.B., Del Tredici K., Wszolek Z.K., Litvan I. Neuropathological assessment of Parkinson's disease: refining the diagnostic criteria. Lancet Neurol. 2009;8:1150–1157. doi: 10.1016/S1474-4422(09)70238-8. [DOI] [PubMed] [Google Scholar]
- 3.Lin M.T., Beal M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 4.McNaught K.S., Shashidharan P., Perl D.P., Jenner P., Oleo C.W. Aggresome related biogenesis of Lewy bodies. Eur J Neurosci. 2002;16:2136–2148. doi: 10.1046/j.1460-9568.2002.02301.x. [DOI] [PubMed] [Google Scholar]
- 5.Keane P.C., Kurzawa M., Blain P.G., Morris C.M. Mitochondrial dysfunction in Parkinson's disease. Parkinsons Dis. 2011;2011:716871. doi: 10.4061/2011/716871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schapira A.H., Cooper J.M., Dexter D., Jenner P., Clark J.B., Marsden C.D. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- 7.Haas R.H., Nasirian F., Nakano K., Ward D., Pay M., Hill R., Shults C.W. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson's disease. Ann Neurol. 1995;37:714–722. doi: 10.1002/ana.410370604. [DOI] [PubMed] [Google Scholar]
- 8.Gu M., Cooper J.M., Taanman J.W., Schapira A.H. Mitochondrial DNA transmission of the mitochondrial defect in Parkinson's disease. Ann Neurol. 1998;44:177–186. doi: 10.1002/ana.410440207. [DOI] [PubMed] [Google Scholar]
- 9.Penn A.M., Roberts T., Hodder J., Allen P.S., Zhu G., Martin W.R. Generalized mitochondrial dysfunction in Parkinson's disease detected by magnetic resonance spectroscopy of muscle. Neurology. 1995;45:2097–2099. doi: 10.1212/wnl.45.11.2097. [DOI] [PubMed] [Google Scholar]
- 10.Taylor D.J., Krige D., Barnes P.R., Kemp G.J., Carroll M.T., Mann V.M., Cooper J.M., Marsden C.D., Schapira A.H. A 31P magnetic resonance spectroscopy study of mitochondrial function in skeletal muscle of patients with Parkinson's disease. J Neurol Sci. 1994;125:77–81. doi: 10.1016/0022-510x(94)90245-3. [DOI] [PubMed] [Google Scholar]
- 11.Schapira A.H. Mitochondrial involvement in Parkinson's disease. Huntington's disease, hereditary spastic paraplegia and Friecheich's ataxia. Biochim Biophys Acta. 1999;1410:159–170. doi: 10.1016/s0005-2728(98)00164-9. [DOI] [PubMed] [Google Scholar]
- 12.Panov A., Dikalov S., Shalbuyeva N., Taylor G., Sherer T., Greenamyre J.T. Rotenone model of Parkinson disease: multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J Biol Chem. 2005;280:42026–42035. doi: 10.1074/jbc.M508628200. [DOI] [PubMed] [Google Scholar]
- 13.Przedborski S., Tieu K., Perier C., Vila M. MPTP as a mitochondrial neurotoxic model of Parkinson's disease. J Bioenerg Biomembr. 2004;36:375–379. doi: 10.1023/B:JOBB.0000041771.66775.d5. [DOI] [PubMed] [Google Scholar]
- 14.Narendra D.P., Youle R.J. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid Redox Signal. 2011;14:1929–1938. doi: 10.1089/ars.2010.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braak H., Del Tredici K., Rüb U., de Vos R.A.I., Jansen Steur E.N., Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 16.Spillantini M.G., Crowther R.A., Jakes R., Hasegawa M., Goedert M. Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jellinger K.A. Neuropathology of sporadic Parkinson's disease: evaluation and changes of concepts. Mov Disord. 2012;27:8–30. doi: 10.1002/mds.23795. [DOI] [PubMed] [Google Scholar]
- 18.Kim H.J. Alpha-synuclein expression in patients with Parkinson's disease: a clinician's perspective. Exp Neurobiol. 2013;22:77–83. doi: 10.5607/en.2013.22.2.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braak H., Braak E. Pathoanatomy of Parkinson's disease. J Neurol. 2000;247 doi: 10.1007/PL00007758. 113–110. [DOI] [PubMed] [Google Scholar]
- 20.Datta S., Siwek D.F. Single cell activity patterns of pedunculopontine tegmentum neurons across the sleep-wake cycle in the freely moving rats. J Neurosci Res. 2002;70:611–621. doi: 10.1002/jnr.10405. [DOI] [PubMed] [Google Scholar]
- 21.Iranzo A., Molinuevo J.L., Santamaria J., Serradell M., Marti M.J., Valldeoiola F., Tolosa E. Rapid-eye movement sleep behaviour disorder as an early marker for neurodegenerative disorder: a descriptive study. Lancet Neurol. 2006;5:572–577. doi: 10.1016/S1474-4422(06)70476-8. [DOI] [PubMed] [Google Scholar]
- 22.Granata A.R., Kitai S.T. Inhibitory substantia nigra inputs to the pedunculopontine neurons. Exp Brain Res. 1991;86:459–466. doi: 10.1007/BF00230520. [DOI] [PubMed] [Google Scholar]
- 23.Hazrati L.N., Parent A. Projection from the deep cerebellar nuclei to the pedunculopontine nucleus in the squirrel monkey. Brain Res. 1992;585:267–271. doi: 10.1016/0006-8993(92)91216-2. [DOI] [PubMed] [Google Scholar]
- 24.Inglis W.L., Winn P. The pedunculopontine tegmental nucleus: where the striatum meets the reticular formation. Prog Neurobiol. 1995;47:1–29. doi: 10.1016/0301-0082(95)00013-l. [DOI] [PubMed] [Google Scholar]
- 25.Jenkinson N., Nandi D., Muthusamy K., Ray N.J., Gregory R., Stein J.F., Aziz T.Z. Anatomy, physiology and pathophysiology of the pedunculopontine nucleus. Mov Disord. 2009;24:319–328. doi: 10.1002/mds.22189. [DOI] [PubMed] [Google Scholar]
- 26.Lavoie B., Parent A. Pedunculopontine nucleus in the squirrel monkey: cholinergic and glutamatergic projections to the substantia nigra. J Comp Neurol. 1994;344:232–241. doi: 10.1002/cne.903440205. [DOI] [PubMed] [Google Scholar]
- 27.Rye D.B., Lee H.J., Saper C.B., Wainer B.H. Medullary and spinal efferents of the pedunculopontine tegmental nucleus and adjacent mesopontine tegmentum in the rat. J Comp Neurol. 1988;269:315–341. doi: 10.1002/cne.902690302. [DOI] [PubMed] [Google Scholar]
- 28.Spann B.M., Grofova I. Origin of ascending and spinal pathways from the nucleus tegmenti pedunculopontinus in the rat. J Comp Neurol. 1989;283:13–27. doi: 10.1002/cne.902830103. [DOI] [PubMed] [Google Scholar]
- 29.Steininger T.L., Rye D.B., Wainer B.H. Afferent projections to the cholinergic pedunculopontine tegmental nucleus and adjacent midbrain extrapyramidal area in the albino rat. I. Retrograde tracing studies. J Comp Neurol. 1992;321:515–543. doi: 10.1002/cne.903210403. [DOI] [PubMed] [Google Scholar]
- 30.Winn P., Brown V.J., Inglis W.L. On the relationships between the striatum and the pedunculopontine tegmental nucleus. Crit Rev Neurobiol. 1997;11:241–261. doi: 10.1615/critrevneurobiol.v11.i4.10. [DOI] [PubMed] [Google Scholar]
- 31.Hirsch E.C., Graybiel A.M., Duyckaerts C., Javoy-Agid F. Neuronal loss in the pedunculopontine tegmental nucleus in Parkinson disease and in progressive supranuclear palsy. Proc Natl Acad Sci U S A. 1987;84:5976–5980. doi: 10.1073/pnas.84.16.5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rinne J.O., Ma S.Y., Lee M.S., Collan Y., Röyttä M. Loss of cholinergic neurons in the pedunculopontine nucleus in Parkinson's disease is related to disability of the patients. Parkinsonism Relat Disord. 2008;14:553–557. doi: 10.1016/j.parkreldis.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 33.Rochester L., Yarnall A.J., Baker M.R., David R.V., Lord S., Galna B., Burn D.J. Cholinergic dysfunction contributes to gait disturbance in early Parkinson's disease. Brain. 2012;135:2779–2788. doi: 10.1093/brain/aws207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karachi C., Grabli D., Bernard F.A., Tandé D., Wattiez N., Belaid H., Bardinet E., Prigent A., Nothacker H.P., Hunot S., Hartmann A., Lehéricy S., Hirsch E.C., François C. Cholinergic mesencephalic neurons are involved in gait and postural disorders in Parkinson disease. J Clin Invest. 2010;120:1026–1033. doi: 10.1172/JCI42642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kojima J., Yamaji Y., Matsumura M., Nambu A., Inase M., Tokuno H., Takada M., Imai H. Excitotoxic lesions of the pedunculopontine tegmental nucleus produce contralateral hemiparkinsonism in the monkey. Neurosci Lett. 1997;226:111–114. doi: 10.1016/s0304-3940(97)00254-1. [DOI] [PubMed] [Google Scholar]
- 36.Dugger B.N., Murray M.E., Boeve B.F., Parisi J.E., Benarroch E.E., Ferman T.J., Dickson D.W. Neuropathological analysis of brainstem cholinergic and catecholaminergic nuclei in relation to REM sleep behaviour disorder. Neuropathol Appl Neurobiol. 2012;38:142–152. doi: 10.1111/j.1365-2990.2011.01203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plaha P., Gill S.S. Bilateral deep brain stimulation of the pedunculopontine nucleus for Parkinson's disease. Neuroreport. 2005;16:1883–1887. doi: 10.1097/01.wnr.0000187637.20771.a0. [DOI] [PubMed] [Google Scholar]
- 38.Stefani A., Lozano A.M., Peppe A., Stanzione P., Galati S., Tropepi D., Pierantozzi M., Brusa L., Scarnati E., Mazzone P. Bilateral deep brain stimulation of the pedunculopontine and subthalamic nuclei in severe Parkinson's disease. Brain. 2007;130:1596–15607. doi: 10.1093/brain/awl346. [DOI] [PubMed] [Google Scholar]
- 39.Lax N.Z., Hepplewhite P.D., Reeve A.K., Nesbitt V., McFarland R., Jaros E., Taylor R.W., Turnbull D.M. Cerebellar ataxia in patients with mitochondrial DNA disease: a molecular clinicopathological study. J Neuropathol Exp Neurol. 2012;71:148–161. doi: 10.1097/NEN.0b013e318244477d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reeve A.K., Park T.K., Jaros E., Campbell G.R., Lax N.Z., Hepplewhite P.D., Krishnan K.J., Elson J.L., Morris C.M., McKeith I.G., Turnbull D.M. Relationship between mitochondria and α-synuclein: a study of single substantia nigra neurons. Arch Neurol. 2012;69:385–393. doi: 10.1001/archneurol.2011.2675. [DOI] [PubMed] [Google Scholar]
- 41.Mizukawa K., McGeer P.L., Tago H., Peng J.H., McGeer E.G., Kimura H. The cholinergic system of the human hindbrain studied by choline acetyltransferase immunohistochemistry and acetylcholinesterase histochemistry. Brain Res. 1986;379:39–55. doi: 10.1016/0006-8993(86)90253-2. [DOI] [PubMed] [Google Scholar]
- 42.Alam M., Schwabe K., Krauss J.K. The pedunculopontine nucleus area: critical evaluation of interspecies differences relevant for its use as a target for deep brain stimulation. Brain. 2010;134:11–23. doi: 10.1093/brain/awq322. [DOI] [PubMed] [Google Scholar]
- 43.Renshaw S. Scion Publishing Ltd; Cambridge: 2007. Immunohistochemistry: Methods Express; p. 46. [Google Scholar]
- 44.Pienaar I.S., Van de Berg W. A non-cholinergic neuronal loss in the pedunculopontine nucleus of toxin-evoked parkinsonian rats. Exp Neurol. 2013;248:213–223. doi: 10.1016/j.expneurol.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 45.Altar C., Walter J., Neve A., Marshall J.F. Computer-assisted video analysis of ['H]spiroperidol binding in autoradiographs. J Neurosci Methods. 1984;10:173–188. doi: 10.1016/0165-0270(84)90054-2. [DOI] [PubMed] [Google Scholar]
- 46.Porro C., Fonda S., Baraldi P., Bird G.P., Cavazzuti M. Computer-assisted analyses of [I4C]2-DG autoradiographs employing a general purpose image processing system. J Neurosci Methods. 1984;11:243–250. doi: 10.1016/0165-0270(84)90086-4. [DOI] [PubMed] [Google Scholar]
- 47.Brey E.M., Lalani Z., Johnston C., Wong M., McIntyre L.V., Duke P.J., Patrick C.W., Jr. Automated selection of DAB-labeled tissue for immunohistochemical quantification. J Histochem Cytochem. 2003;51:575–584. doi: 10.1177/002215540305100503. [DOI] [PubMed] [Google Scholar]
- 48.Leal S., Diniz C., Sá C., Goncalves J., Soares A.S., Rocha-Pereira C., Fresco P. Semiautomated computer-assisted image analysis to quantify 3,3′-diaminobenzidine tetrahydrochloride-immunostained small tissues. Anal Biochem. 2006;357:137–143. doi: 10.1016/j.ab.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 49.Helps S.C., Thornton E., Kleinig T.J., Manavis J., Vink R. Automatic non-subjective estimation of antigen content visualized by immunohistochemistry using color deconvolution. Appl Immunohistochem Mol Morphol. 2012;20:89–90. doi: 10.1097/PAI.0b013e31821fc8cd. [DOI] [PubMed] [Google Scholar]
- 50.Bender A., Krishnan K.J., Morris C.M., Taylor G.A., Reeve A.K., Perry R.H., Jaros E., Hersheson J.S., Betts J., Klopstock T., Taylor R.W., Turnbull D.M. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson's disease. Nat Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 51.Krishnan K.J., Ratnaike T.E., De Gruyter H.L., Jaros E., Turnbull D.M. Mitochondrial DNA deletions cause the biochemical defect observed in Alzheimer disease. Neurobiol Aging. 2012;33:2210–2214. doi: 10.1016/j.neurobiolaging.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 52.Pienaar I.S., Burn D., Morris C.M., Dexter D. Synaptic protein alterations in Parkinson's disease. Mol Neurobiol. 2012;45:126–143. doi: 10.1007/s12035-011-8226-9. [DOI] [PubMed] [Google Scholar]
- 53.Baizabal J.M., Valencia C., Guerrero-Flores G., Covarrubias L. Telencephalic neural precursor cells show transient competence to interpret the dopaminergic niche of the embryonic midbrain. Dev Biol. 2011;349:192–203. doi: 10.1016/j.ydbio.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 54.Poyatos I., Ponce J., Aragón C., Giménez C., Zafra F. The glycine transporter GLYT2 is a reliable marker for glycine-immunoreactive neurons. Mol Brain Res. 1997;49:63–70. doi: 10.1016/s0169-328x(97)00124-1. [DOI] [PubMed] [Google Scholar]
- 55.Li J.Z., Vawter M.P., Walsh D.M., Tomita H., Evans S.J., Choudary P.V., Lopez J.F., Avelar A., Shokoohi V., Chung T., Mesarwi O., Jones E.G., Watson S.J., Akil H., Bunney W.E., Jr., Myers R.M. Systematic changes in gene expression in post-mortem human brains associated with tissue pH and terminal medical conditions. Hum Mol Genet. 2004;13:609–616. doi: 10.1093/hmg/ddh065. [DOI] [PubMed] [Google Scholar]
- 56.Harrison P.J., Heath P.R., Eastwood S.L., Burnet P.W., McDonald B., Pearson R.C. The relative importance of premortem acidosis and post-mortem interval for human brain gene expression studies: selective mRNA vulnerability and comparison with their encoded proteins. Neurosci Lett. 1995;200:151–154. doi: 10.1016/0304-3940(95)12102-a. [DOI] [PubMed] [Google Scholar]
- 57.Durrenberger P.F., Fernando S., Kashefi S., Ferrer I., Hauw J.J., Seilhean D., Smith C., Walker R., Al-Sarraj S., Troakes C., Palkovits M., Kasztner M., Huitinga I., Arzberger T., Dexter D.T., Kretzschmar H., Reynolds R. Effects of antemortem and postmortem variables on human brain mRNA quality: a BrainNet Europe study. J Neuropathol Exp Neurol. 2010;69:70–81. doi: 10.1097/NEN.0b013e3181c7e32f. [DOI] [PubMed] [Google Scholar]
- 58.Ferrer I., Martinez A., Boluda S., Parchi P., Barrachina M. Brain banks: benefits, limitations and cautions concerning the use of post-mortem brain tissue for molecular studies. Cell Tissue Bank. 2008;9:181–194. doi: 10.1007/s10561-008-9077-0. [DOI] [PubMed] [Google Scholar]
- 59.Ford B., Holmes C.J., Mainville L., Jones B.E. GABAergic neurons in the rat pontomesencephalic tegmentum: co-distribution with cholinergic and other tegmental neurons projecting to the posterior lateral hypothalamus. J Comp Neurol. 1995;363:177–196. doi: 10.1002/cne.903630203. [DOI] [PubMed] [Google Scholar]
- 60.Charara A., Smith Y., Parent A. Glutamatergic inputs from the pedunculopontine nucleus to midbrain dopaminergic neurons in primates: phaseolus vulgaris-leucoagglutinin anterograde labeling combined with post-embedding glutamate and GABA immunohistochemistry. J Comp Neurol. 1996;364:254–266. doi: 10.1002/(SICI)1096-9861(19960108)364:2<254::AID-CNE5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 61.Fort P., Luppi P.H., Jouver M. Glycine immunoreactive neurons in the cat brain stem reticular formation. Neuroreport. 1993;4:1123–1126. [PubMed] [Google Scholar]
- 62.Mineff E.M., Popratiloff A., Romansky R., Kazakos V., Kaimaktschieff V., Usunoff K.G., Ovtscharoff W., Marani E. Evidence for a possible glycinergic inhibitory neurotransmission in the midbrain and rostral pons of the rat studied by gethyrin. Arch Physiol Biochem. 1998;106:210–220. doi: 10.1076/apab.106.3.210.4377. [DOI] [PubMed] [Google Scholar]
- 63.Ye M., Hayar A., Strotman B., Garcia-Rill E. Cholinergic modulation of fast inhibitory and excitatory transmission to pedunculopontine thalamic projecting neurons. J Neurophysiol. 2010;103:2417–2432. doi: 10.1152/jn.01143.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saitoh K., Hattori S., Song W.J., Isa T., Takakusaki K. Nigral GABAergic inhibition upon cholinergic neurons in the rat pedunculopontine tegmental nucleus. Eur J Neurosci. 2003;18:879–886. doi: 10.1046/j.1460-9568.2003.02825.x. [DOI] [PubMed] [Google Scholar]
- 65.Gai W.P., Halliday G.M., Blumbergs P.C., Geffen L.B., Blessing W.W. Substance containing neurons in the mesopontine tegmentum are severely affected in Parkinson's disease. Brain. 1991;114:2253–2267. doi: 10.1093/brain/114.5.2253. [DOI] [PubMed] [Google Scholar]
- 66.Brooks P.L., Peever J.H. Impaired GABA and glycine transmission triggers cardinal features of rapid eye movement sleep behavioral disorder in mice. J Neurosci. 2011;31:7111–7121. doi: 10.1523/JNEUROSCI.0347-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Olson E.J., Boeve B.F., Solber M.H. Rapid eye movement sleep behaviour disorder: demographic, clinical and laboratory findings in 93 cases. Brain. 2000;123:331–339. doi: 10.1093/brain/123.2.331. [DOI] [PubMed] [Google Scholar]
- 68.Schenck C.H., Mahowald M.W. REM sleep behavior disorder: clinical, developmental, and neuroscience perspectives 16 years after its formal identification in SLEEP. Sleep. 2002;25:120–138. doi: 10.1093/sleep/25.2.120. [DOI] [PubMed] [Google Scholar]
- 69.Nandi D., Aziz T.Z., Giladi N., Winter J., Stein J.F. Reversal of akinesia in experimental parkinsonism by GABA antagonist microinjections in the pedunculopontine nucleus. Brain. 2002;125:2418–2430. doi: 10.1093/brain/awf259. [DOI] [PubMed] [Google Scholar]
- 70.Shink E., Sidibé M., Smith Y. Efferent connections of the internal globus pallidus in the squirrel monkey: II. Topography and synaptic organization of pallidal efferents to the pedunculopontine nucleus. J Comp Neurol. 1997;382:348–363. [PubMed] [Google Scholar]
- 71.Lynch J.W. Native glycine receptor subtypes and their physiological roles. Neuropharmacology. 2009;56:303–309. doi: 10.1016/j.neuropharm.2008.07.034. [DOI] [PubMed] [Google Scholar]
- 72.Chase M.H. Confirmation of the consensus that glycinergic postsynaptic inhibition is responsible for the atonia of REM sleep. Sleep. 2008;31:1487–1491. doi: 10.1093/sleep/31.11.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Krenzer M., Anaclet C., Vetrivelan R., Wang N., Vong L., Lowell B.B., Fuller P.M., Lu J. Brainstem and spinal cord circuitry regulating REM sleep and muscle atonia. PLoS One. 2011;6:e24998. doi: 10.1371/journal.pone.0024998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takakusaki K., Obara K., Nozu T., Okumura T. Modulatory effects of the GABAergic basal ganglia neurons on the PPN and the muscle tone inhibitory system in cats. Arch Ital Biol. 2011;149:385–405. doi: 10.4449/aib.v149i4.1383. [DOI] [PubMed] [Google Scholar]
- 75.Clements J.R., Grant S. Glutamate-like immunoreactivity in neurons of the laterodorsal tegmental and pedunculopontine nuclei in the rat. Neurosci Lett. 1990;120:70–73. doi: 10.1016/0304-3940(90)90170-e. [DOI] [PubMed] [Google Scholar]
- 76.Parent A., Parent M., Charara A. Glutamatergic inputs to midbrain dopaminergic neurons in primates. Parkinsonism Relat Disord. 1999;5:193–201. doi: 10.1016/s1353-8020(99)00037-1. [DOI] [PubMed] [Google Scholar]
- 77.Ames A., Nesbett F.B. Pathophysiology of ischemic cell death: II. Changes in plasma membrane permeability and cell volume. Stroke. 1983;14:227–233. doi: 10.1161/01.str.14.2.227. [DOI] [PubMed] [Google Scholar]
- 78.McBain C.J., Traynelis S.F., Dingledine R. Regional variation of extracellular space in the hippocampus. Science. 1990;249:674–677. doi: 10.1126/science.2382142. [DOI] [PubMed] [Google Scholar]
- 79.Hoogendijk W.J., Pool C.W., Troost D., van Zwieten A., Swaab D.F. Image analyser-assisted morphometry of the locus coeruleus in Alzheimer disease, Parkinson's disease and amyotrophic lateral sclerosis. Brain. 1995;118:131–143. doi: 10.1093/brain/118.1.131. [DOI] [PubMed] [Google Scholar]
- 80.Menke R.A., Scholz J., Miller K.L., Deoni S., Jbabdi S., Matthews P.M., Zarei M. MRI characteristics of the substantia nigra in Parkinson's disease: a combined quantitative T1 and DTI study. Neuroimage. 2009;47:435–441. doi: 10.1016/j.neuroimage.2009.05.017. [DOI] [PubMed] [Google Scholar]
- 81.Halliday G.M. Thalamic changes in Parkinson's disease. Parkinsonism Relat Disord. 2009;15:S152–S155. doi: 10.1016/S1353-8020(09)70804-1. [DOI] [PubMed] [Google Scholar]
- 82.Lim A.S., Moro E., Lozano A.M., Hamani C., Dostrovsky J.O., Hutchison W.D., Lang A.E., Wennberg R.A., Murray B.J. Selective enhancement of rapid eye movement sleep by deep brain stimulation of the human pons. Ann Neurol. 2009;66:110–114. doi: 10.1002/ana.21631. [DOI] [PubMed] [Google Scholar]
- 83.Lax N.Z., Pienaar I.S., Reeve A.K., Hepplewhite P.D., Jaros E., Taylor R.W., Kalaria R.N., Turnbull D.M. Microangiopathy in the cerebellum of patients with mitochondrial DNA disease. Brain. 2012;135:1736–1750. doi: 10.1093/brain/aws110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Elson J.L., Samuels D.C., Turnbull D.M., Chinnery P.F. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am J Hum Genet. 2001;68:802–806. doi: 10.1086/318801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tuppen H.A., Blakely E.L., Turnbull D.M., Taylor R.W. Mitochondrial DNA mutations and human disease. Biochim Biophys Acta. 2010;1797:113–128. doi: 10.1016/j.bbabio.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 86.Kraytsberg Y., Kudryavtseva E., McKee A.C., Geula C., Kowall N.W., Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 87.Sciacco M., Bonilla E. Cytochemistry and immunocytochemistry of mitochondria in tissue sections. Methods Enzymol. 1996;264:509–521. doi: 10.1016/s0076-6879(96)64045-2. [DOI] [PubMed] [Google Scholar]
- 88.Bua E., Johnson J., Herbst A., Delong B., McKenzie D., Salamat S., Aiken J.M. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. 2006;79:469–480. doi: 10.1086/507132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.