

Abstract
Epithelial permeability is often increased in inflammatory bowel diseases. We hypothesized that perturbed mitochondrial function would cause barrier dysfunction and hence epithelial mitochondria could be targeted to treat intestinal inflammation. Mitochondrial dysfunction was induced in human colon-derived epithelial cell lines or colonic biopsy specimens using dinitrophenol, and barrier function was assessed by transepithelial flux of Escherichia coli with or without mitochondria-targeted antioxidant (MTA) cotreatment. The impact of mitochondria-targeted antioxidants on gut permeability and dextran sodium sulfate (DSS)–induced colitis in mice was tested. Mitochondrial superoxide evoked by dinitrophenol elicited significant internalization and translocation of E. coli across epithelia and control colonic biopsy specimens, which was more striking in Crohn’s disease biopsy specimens; the mitochondria-targeted antioxidant, MitoTEMPO, inhibited these barrier defects. Increased gut permeability and reduced epithelial mitochondrial voltage-dependent anion channel expression were observed 3 days after DSS. These changes and the severity of DSS-colitis were reduced by MitoTEMPO treatment. In vitro DSS-stimulated IL-8 production by epithelia was reduced by MitoTEMPO. Metabolic stress evokes significant penetration of commensal bacteria across the epithelium, which is mediated by mitochondria-derived superoxide acting as a signaling, not a cytotoxic, molecule. MitoTEMPO inhibited this barrier dysfunction and suppressed colitis in DSS-colitis, likely via enhancing barrier function and inhibiting proinflammatory cytokine production. These novel findings support consideration of MTAs in the maintenance of epithelial barrier function and the management of inflammatory bowel diseases.
The mammalian gut harbors an immense and diverse microbiota, and host-bacteria interactions are key determinants of digestive health and general well-being.1,2 While providing distinct benefits to the host (eg, vitamin synthesis), commensal bacteria that cross the epithelium and enter the mucosa have the potential to provoke inflammation, and movement into the circulation can result in sepsis and death. Consequently, the barrier function of the epithelium, both intrinsic (eg, epithelial tight junctions and cell membranes) and extrinsic (eg, mucus) elements, is a critical component of innate defense. Thus, the commensal bacteria-host interaction pivots on the gut epithelium as a point of first contact and knowledge of the dynamic nature of this interface is important to understanding normal intestinal function and disease.3
The epithelial barrier is not static; rather, it is highly dynamic and tightly regulated to maintain gut homeostasis. However, uncontrolled or prolonged increases in gut permeability have the potential to initiate or exaggerate enteric inflammatory disease. For example, the consensus on the etiology of inflammatory bowel disease (IBD; Crohn’s disease or ulcerative colitis) is that disease develops because of an inappropriate immune response to the gut microbiota in a genetically susceptible individual.4 This suggests a barrier defect because microbes, or their products, in the gut lumen must access the mucosal immune system by crossing the epithelial cell layer. Although increased epithelial permeability as a primary cause of IBD remains unproved,3 the leaky gut hypothesis5 is supported by observations in experimental models of colitis3,6,7 and increased epithelial permeability has been repeatedly demonstrated in active IBD.8,9 Therefore, the ability to enhance the barrier property of the epithelium would be of value in ameliorating enteric inflammatory disease.
Given that control of epithelial permeability (apical junction complex formation and transcellular permeation) is energy dependent, mitochondria should be essential for appropriate regulation of barrier function. Indeed, factors such as infection, nonsteroidal anti-inflammatory drugs, and smoking, which can contribute to the pathophysiological characteristics of Crohn’s disease, also perturb mitochondrial function.10–12 Furthermore, structurally abnormal mitochondria have been observed in tissue from patients with gut inflammation,13 in animal models of gut disease,14 and in epithelial monolayers treated with bacterial toxins or low-grade pathogens.15 However, despite these findings and the keen interest in mitochondria in the pathophysiological characteristics of neuromuscular disease, diabetes, and obesity,16–18 there are limited data on the role of mitochondria in colitis.
We have previously shown that model epithelia treated with dinitrophenol (DNP) to uncouple oxidative phosphorylation display decreased barrier function, as characterized by lower transepithelial resistance (TER) indicative of increased paracellular permeability, and the internalization and translocation of noninvasive, nontoxigenic commensal E. coli.13,19 The latter is particularly intriguing for two reasons: entry of commensal bacteria into the enterocyte likely represents a threat and the enterocytes’ response (eg, IL-8 synthesis) could promote inflammation; and the contribution of transcellular permeability to a barrier defect is not well understood and needs to be comprehensively assessed and fully integrated to any consideration of the epithelial barrier and innate defense. Thus, the current study was designed to uncover the mechanism(s) underlying the increased internalization and translocation of commensal Escherichia coli across gut epithelia, which occurs as a consequence of metabolic stress evoked by targeted perturbation of mitochondrial function.
Materials and Methods
Human Tissue
Human tissue was obtained and used with patient consent following the guidelines on use of human tissue at the University of Calgary (Calgary, AB, Canada) and the Regional Ethics Committee, Linköping. Animal studies were conducted at the University of Calgary and conformed to national guidelines, as overseen by the Health Sciences Animal Care Committee.
Reagents
DNP, rotenone, oligomycin A,20 hydrogen peroxide, N-acetylcysteine, and fluorescein isothiocyanate (FITC)–4-kDa dextran were from Sigma Chemical Company (St. Louis, MO). MitoQ (mQ) was generated by Dr. Michael Murphy (Cambridge, UK). Killed FITC–E. coli (K12), MitoSox, Mitotracker, and DAPI were from Molecular Probes (Eugene, OR). Dextran sodium sulfate (DSS) was from MP Biomedicals, LLC (Solon, OH). MitoTEMPO (MitoT or mT) was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).13,19–25 The E. coli strains HB101 and F18 were provided by Dr. Philip M. Sherman (Hospital for Sick Children, University of Toronto, Toronto, ON, Canada).
Cell Culture and Bacterial Translocation
The T84 and HT-29 human colonic epithelial cell lines were maintained as described.13 A total of 106 cells/mL were seeded into 12-well plates and used at approximately 70% confluence on the basis of phase-contrast microscopy or onto 3.0-μm porous filter supports and cultured for 7 days until electrically confluent (T84 >1000 Ω·cm2; HT-29 >300 Ω·cm2).13 E. coli (strains HB101 and F18) were grown overnight in Luria-Bertani broth, and 106 colony-forming units (CFU) were added to the epithelium (luminal aspect of filter-grown polarized preparations). Bacterial internalization was assessed by killing extracellular bacteria with 200 μg/mL gentamicin (1 to 2 hours; confirmed by a lack of colony growth on LB plates), followed by enterocyte lysis (0.1% Triton X-100, Sigma Chemical Company), and culturing serial dilutions of the cell extract. In other experiments, gentamicin was followed by extensive washing, fresh medium was added, samples were collected 6 and 16 hours later and cultured for 24 hours on LB agar, and CFUs were counted. Bacterial translocation was assessed by sampling the medium from the basolateral compartment of filter-grown epithelia.21 All drugs were added 30 minutes before or simultaneously with the E. coli, with or without metabolic stressor, and were tested (at the doses used) for bacteriostatic and bacteriocidal effects.
ROS Detection and Quantification
A total of 2 × 105 T84 cells were seeded onto glass coverslips, and mitochondria-produced superoxide was detected using 5 μmol/L Mitosox Red (Molecular Probes) and mitochondria were detected with 200 nmol/L Mitotracker-Green (Molecular Probes).22 Random images on the basis of 2 mg/mL DAPI (to identify nuclei) staining were captured on an Olympus BX41 microscope (Olympus Canada, Markham, ON, Canada) (60× objective) with Qcapture software (Olympus Canada).22 Identical conditions and exposure times were used in each experiment and set to avoid overexposure of the Mitosox, Mitotracker Green, and DAPI fluorescence.
Fluorometric Assessment of Phagosomal pH
Carboxyfluorescein succinimidyl ester–labeled, IgG-coupled, 3-μm silica particles were prepared for assessment of phagosomal pH in live murine bone marrow–derived macrophages.26 Measurements were performed using a FLUOstar Optima fluorescent plate reader (BMG Labtech, Ortenberg, Germany) in assay buffer consisting of PBS supplemented with 1 mmol/L CaCl2, 2.7 mmol/L KCl, 0.5 mmol/L MgCl2, 5 mmol/L dextrose, and 0.25% gelatin. Cells were pretreated with rotenone, oligomycin A, or DNP for 30 minutes before addition of the particles. Phagosomal pH was calculated by recording the ratio of the fluorescent emission at 520 nm of carboxyfluorescein succinimidyl ester conjugated to experimental particles excited at 490 and 450 nm, followed by polynomial regression to a standard curve using an adapted nigericin/high K+ method.27
Ussing Chamber Studies
Human colonic biopsy specimens, surgical specimens (exposed surface area of 1.8 and 9.2 mm2, respectively),24 or 0.6-cm2 segments of murine midcolon28 were mounted in Ussing chambers, and the spontaneous potential difference was clamped at 0 V by the automated and continuous injection of current, the short-circuit current (ISC). After a 20-minute (murine) or 40-minute (human) equilibration, baseline ISC and potential difference were used to calculate TER. 51Cr-EDTA (34 μCi/mL; Perkin Elmer, Boston, MA) or dead FITC-labeled E. coli K12 (108 particles; Molecular Probes) and 0.1 mmol/L DNP were then added to the mucosal side of the tissues, with or without a 20-minute pretreatment with 10 μmol/L MitoTEMPO; samples were collected over a 2-hour period for flux calculations.24 At the end of the experiment, a final TER recording was made and 10−5 mol/L cAMP-dependent Cl− secretagogue, forskolin, was added to the serosal side of the tissue and ΔISC was recorded as a measure of tissue viability.28,29
qPCR Analysis
Expression of ATP synthase, peroxiredoxin 1, and NADH CoQ reductase mRNAs in colonic biopsy specimens was assessed by real-time PCR. Total RNA was extracted using the PureLink RNA Mini Kit (Invitrogen, Burlington, ON, Canada), cDNA was reverse transcribed from 0.5 μg of RNA with the iScript cDNA Synthesis kit (Bio-Rad, Mississauga, ON, Canada), and real-time PCRs were performed using reagent concentrations and a standard protocol for iQTM SYBR Green Supermix (Biorad, Toronto, ON, Canada). Briefly, cDNA was incubated in a 20-μL reaction, containing 1 μL cDNA, 300 μmol/L of nucleotide primers, and 10 μL of 2× iQTM SYBR Green Supermix (Biorad). The PCR conditions were an initial activation step at 95°C for 2 minutes, followed by a 15-second denaturing step at 95°C, an annealing step at 55°C for 15 seconds, and an extension step at 68°C for 20 seconds. Forty cycles were completed and followed by a melting curve analysis. Expression is presented relative to the housekeeping gene, 18s rRNA. Primers were designed using the primer3 program (http://bioinfo.ut.ee/primer3-0.4.0/primer3, last accessed October 20, 2010) and are available at http://www.ncbi.nlm.nih.gov/genbank: i) ATP synthase (GenBank accession number NM_001686.3), forward primer, 5′-GCAGATTTTGGCAGGTGAAT-3′, and reverse primer, 5′-AGGGGCAAGGAGAGAGACA-3′; ii) peroxiredoxin 1 (GenBank accession number NM_006793), forward primer, 5′-TGGGGTCTTAAAGGCTGATG-3′, and reverse primer, 5′-TCCCCATGTTTGTCAGTGAA-3′; iii) NADH CoQ reductase (GenBank accession number NM_002496.3), forward primer, 5′-CCACCATCAACTACCCGTTC-3′, and reverse primer, 5′-AAGCCGCAGTAGATGCACTT-3′; and iv) 18s rRNA (GenBank accession number NR_003286.2), forward primer, 5′-ATACATGCCGAAGGGCGCTG-3′, and reverse primer, 5′-AGGGGCTGACCGGGTTGGTT-3′.30
Colitis
Colitis was induced in 7- to 9-week-old male Balb/C mice (Charles River Laboratories, Senneville, QC, Canada) by administering 5% (w/v) DSS (approximately 40,000 kDa) in their drinking water for 5 days. Animals underwent necropsy after 3 or 5 days of DSS, or after 5 days of DSS and then 3 days of normal drinking water, with or without 100 μg MitoTEMPO (i.p.) on days 0 to 4 (prophylactic regimens). Alternatively, mice received 5% DSS for 5 days, followed by 3 days of normal drinking water and a once-daily treatment of MitoTempo (days 5 to 7), and underwent necropsy on day 8 (Figure 1). Colitis was assessed by a disease activity score on the basis of body weight, colon length, and macroscopic appearance of the animal, its colon, and stool.31 Hematoxylin and eosin (H&E)–stained paraffin sections of mid-distal colon were scored for histopathological features using a validated scoring system in a blinded manner (D.M.M., A.W.).31 In some experiments, epithelial barrier function was assessed by either gavaging mice with 100 μL of 50 mg of FITC-dextran (3 to 5 kDa; Molecular Probes) and measuring fluorescence in the serum 4 hours later; or excising small pieces of colon, weighing, rinsing in gentamicin, and then serially diluting homogenates, plating onto blood agar, culturing at 37°C for 24 hours, and counting bacterial colonies.
Figure 1.

Schematic of the experimental protocols used to assess the impact of systemic MitoTEMPO on DSS-induced colitis in BALB/c mice. MitoTEMPO was used in a prophylactic regimen in which mice underwent necropsy 3 (I) or 5 (II) days after DSS or after 5 days of DSS and then 3 days of normal water (III). When used in a treatment regimen, MitoTEMPO was delivered after removal of the DSS water and animals underwent necropsy on day 8 (IV).
Cytokine and ATP Production
A total of 2.5 × 105 T84 epithelial cells were seeded into 12-well plates and treated with 1% to 4% DSS or 10 ng/mL lipopolysaccharide, culture medium was collected 4 to 48 hours later, and IL-8 was measured by ELISA (R&D Systems Inc.; assay detection limit, 9 pg/mL). ATP levels in epithelia 16 hours after exposure to DNP, rotenone, or oligomycin A were measured by commercial assay, following the manufacturer’s instructions (Cell Viability Assay, catalogue number G7573; Promega, Madison, WI) and normalized to 100 μg protein.
Protein Immunodetection
Standard immunocytochemistry and immunoblotting protocols were used to detect nuclear factor, erythoid 2–related factor (Nrf2), a sensor of oxidative stress, in glass coverslip or plastic-grown T84 epithelium, respectively, treated with E. coli, DNP, or both agents.32 For immunostaining, cells were fixed for 10 minutes with 100% methanol and the primary anti-Nrf2 antibody (sc-722; Santa Cruz Biotechnology, Inc.), used at 1:100 dilution. For immunoblotting, the anti-Nrf2 antibody was used at 1:1000 dilution. Whole epithelial cell extracts were assessed for expression of the mitochondrial-specific voltage-dependent anion channel (VDAC; 1:1000, number 4866; Cell Signaling Technology, Beverly, MA).
Statistical Analysis
Data are given as means ± SEM. Multiple group comparisons were done by one-way analysis of variance, followed by pairwise post hoc statistical tests, with P < 0.05 as a level of statistical significant difference.
Results
Perturbed Mitochondrial Function Increases Epithelial Permeability
Disruption of T84 epithelial cell mitochondria by DNP (H+ ionophore), oligomycin A (inhibits ATP synthase), or rotenone (blocks electron transport chain complex I) at the doses used had marginal effects on TER (a measure of passive ion flux) over a 16-hour time frame; however, in the presence of noninvasive, nonpathogenic E. coli (HB101), a significant decrease in TER occurred (Figure 2A). Moreover, bacterial translocation was substantially increased (Figure 2B), highlighting the impact of metabolic stress in the context of commensal bacteria on gut permeability. The drugs, at the doses used, did not cause epithelial apoptosis and were neither bacteriostatic nor bacteriocidal (data not shown); however, they did lower epithelial ATP levels by approximately 25 to 30% (Figure 2A), consistent with previous findings.19
Figure 2.

Perturbed epithelial mitochondrial activity reduces barrier function. Confluent filter-grown T84 epithelial cell monolayers display reduced TER 24 hours after exposure to E. coli (Ec; strain HB101, 106 CFU inoculum) that is enhanced by 0.1 mmol/L DNP, 1 μmol/L rotenone (Rot), or 1 μmol/L oligomycin A (OmA) cotreatment (n = 3 experiments, nine epithelial preparations) (A, top panel). Epithelial ATP levels 16 hours after treatment (n = 9 to 12 monolayers from two experiments) (A, bottom panel). E. coli translocation (B) and internalization (C) across filter-grown T84 cell monolayers are significantly increased by DNP, Rot, or OmA cotreatment, as is E. coli internalization into plastic-grown T84 epithelia (n = 3 to 4 experiments, 8 to 13 epithelial preparations) (D). E: After killing extracellular bacteria, significant numbers of E. coli are recovered in the culture medium from DNP-treated epithelial monolayers, which is reduced by treatment with the mitochondria-targeted antioxidants, 10 μmol/L MitoQ (mQ) or 10 μmol/L MitoTEMPO (mT) (n = 4 epithelial monolayers from one representative experiment of four experiments). F: Application of 0.5 mmol/L H2O2 directly to plastic-grown T84 cell monolayers does not evoke increased internalization of E. coli (n = 3 experiments, 9 monolayers/condition). Data are given as means ± SEM. ∗P < 0.05 versus Ec, †P < 0.05 versus control (con), and ‡P < 0.05 versus Ec + DNP.
Seeking to differentiate between paracellular and transcellular bacterial translocation,10 treatment with gentamicin revealed a significant increase in E. coli internalization into epithelia treated with the metabolic stressors (Figure 2, C and D), and a portion of the bacteria escaped death and re-entered the culture medium (Figure 2E) [findings reproduced with the HT-29 epithelial cell line (Figure 2E) and E. coli (F18; data not shown)]. Macrophages treated with DNP retained their ability to acidify the phagosome (data not shown), suggesting that reduced phagosomal killing did not underlie the escape of E. coli from DNP-treated epithelia. Although many stimuli can perturb mitochondrial function,13 indomethacin-induced enteropathy has been associated with mitochondrial dysfunction and, corroborating the findings herein, we have provided data implicating reactive oxygen species (ROS) in indomethacin-induced increased bacterial translocation across T84 epithelial cell monolayers.22
More important, H2O2 added to T84-epithelia (ie, extracellular ROS20) did not significantly increase the internalization of E. coli (Figure 2F), suggesting that the DNP-evoked increased bacterial internalization could be via intracellular ROS signaling.
Blocking Mitochondrial ROS Reduces Bacteria Internalization and Transcytosis
Perturbation of mitochondrial function causes ROS generation.23,33 Although DNP might be predicted to elicit mitochondrial ROS, the possibility of phagosome- or endoplasmic reticulum (ER)-derived ROS, perhaps because of communication with mitochondria,34 was addressed by experiments with MitoSOX to fluorescently detect mitochondria-derived superoxide, and MitoTEMPO and mQ, antioxidants whose physicochemical properties direct them to mitochondria.35 There was marked up-regulation of mitochondrial superoxide in DNP, with or without E. coli–treated epithelia (Figure 3A), compared with controls or DNP only. Use of MTAs partially inhibited the decrease in TER (Figure 3B) and, more important, substantially reduced bacterial internalization, release (Figure 2E), and translocation (Figure 3, C and D) across T84-epithelia.
Figure 3.

Targeting mitochondria-derived superoxide reduces bacterial transcytosis across model epithelia. A: Representative epifluorescence images showing mitochondria (Mitotracker) and that mitochondria-derived superoxide (MitoSox) is a rapid (30 minutes) and sustained (16 hours) response to DNP (0.1 mmol/L); and this response is greatest after treatment with DNP and E. coli (Ec; inoculum at 106 CFU). B: The decrease in TER elicited by DNP + E. coli is partially corrected by cotreatment with the mitochondria-targeted antioxidants, 10 μmol/L MitoQ (mQ), or 10 μmol/L MitoTEMPO (mT) (starting TER range = 984 to 1974 Ω cm2). MitoTEMPO completely blocks the increased bacterial internalization and translocation of E. coli across filter-grown T84 monolayers (n = 12 epithelia from four experiments) (C) and internalization into plastic-grown epithelia induced by DNP (n = 9 epithelia from three experiments) (D). Barrier studies were conducted at 16 hours after treatment; effect of 5 mmol/L N-acetylcysteine (NAC) as a general antioxidant is shown in B. Data are given as means ± SEM. ∗P < 0.05 versus Ec, †P < 0.05 versus control (con), and ‡P < 0.05 versus DNP + Ec.
MTAs Block Epithelial Barrier Dysfunction in Crohn’s Disease
Analysis of control human colonic tissue (Table 1) in Ussing chambers revealed DNP-evoked decreases in TER, and increased transepithelial fluxes of 51Cr-EDTA (paracellular marker) and E. coli (Figure 4). MitoTEMPO cotreatment did not affect the decrease in TER but significantly reduced the fluxes of 51Cr-EDTA and E. coli.
Table 1.
Characteristics of Patients from Whom Colonic Resected Tissues or Biopsy Specimens Were Obtained for Use in Ussing Chamber Analysis or qPCR
| Patient no. | Diagnosis | Procedure | Sex | Age (years) | Anti-inflammatory medications |
|---|---|---|---|---|---|
| 1 | Colon cancer | Surgery | M | 96 | — |
| 2 | Colon cancer | Surgery | M | 75 | — |
| 3 | Polyp control | Endoscopy | M | 52 | — |
| 4 | Polyp control | Endoscopy | F | 40 | — |
| 5 | Polyp control | Surgery | M | 32 | — |
| 6 | Colon cancer | Surgery | F | 62 | — |
| 7 | Colon cancer | Surgery | F | 52 | — |
| 8 | Colon cancer | Surgery | F | 79 | — |
| 9 | Colon cancer | Surgery | M | 64 | — |
| 10 | Colon cancer | Surgery | M | 82 | — |
| 11 | Colon cancer | Surgery | F | 66 | — |
| 12 | Colon cancer | Surgery | F | 83 | — |
| 13 | Crohn’s disease | Surgery | M | 22 | — |
| 14 | Crohn’s disease | Surgery | F | 37 | Prednisone |
| 15 | Crohn’s disease | Endoscopy | F | 49 | — |
| 16 | Crohn’s disease | Endoscopy | M | 50 | — |
| 17 | Crohn’s disease | Surgery | F | 36 | — |
| 18 | Crohn’s disease | Surgery | M | 36 | — |
| 19 | Colon cancer screening | Biopsy | M | 36 | — |
| 20 | Colon cancer screening | Biopsy | M | 52 | — |
| 21 | Colon cancer screening | Biopsy | M | 32 | — |
| 22 | Colon cancer screening | Biopsy | F | 42 | — |
| 23 | Colon cancer screening | Biopsy | F | 44 | — |
| 24 | Colon cancer screening | Biopsy | F | 45 | — |
| 25 | Crohn’s disease, inactive | Biopsy | M | 48 | Prednisone, azathioprine |
| 26 | Crohn’s disease, inactive | Biopsy | M | 55 | Methotrexate |
| 27 | Crohn’s disease, inactive | Biopsy | M | 57 | Infliximab |
| 28 | Crohn’s disease, inactive | Biopsy | F | 19 | Azathioprine, infliximab |
| 29 | Crohn’s disease, inactive | Biopsy | M | 34 | Prednisone, methotrexate |
| 30 | Crohn’s disease, inactive | Biopsy | M | 24 | Azathioprine, infliximab |
| 31 | Crohn’s disease, inactive | Biopsy | M | 76 | Adalimumab |
| 32 | Crohn’s disease | Biopsy | F | 32 | Azathioprine, adalimumab |
| 33 | Crohn’s disease | Biopsy | M | 30 | Azathioprine |
| 34 | Crohn’s disease | Biopsy | M | 48 | Salafalk |
| 34 | Crohn’s disease | Biopsy | M | 32 | Azathioprine, adalimumab |
| 36 | Crohn’s disease | Biopsy | M | 42 | Methotrexate |
| 37 | Crohn’s disease | Biopsy | F | 36 | Adalimumab |
For patients 1 to 18, Ussing chamber analysis was used (University of Linköping, Linköping, Sweden); and for patients 19 to 37, qPCR was used (University of Calgary, Calgary, AB, Canada).
—, No data.
Figure 4.

Metabolic stress–induced decreases in epithelial barrier function in human colon are inhibited by MitoTEMPO (MitoT). Biopsy specimens from normal human colon mounted in Ussing chambers and treated with 0.1 mmol/L DNP display a decrease in TER (A) and increased flux of 51Cr-EDTA (B), with the latter being significantly reduced by 10 μmol/L MitoT cotreatment (n = 6 to 11). C: DNP-induced increases in FITC-labeled dead E. coli (K12) flux is suppressed by MitoT (n = 6 to 11). One fluorescent unit equals 3.2 × 103E. coli CFU (2-hour fluxes). Data are given as means ± SEM. ∗P < 0.05 versus control (con) and DNP.
Quantitative PCR (qPCR) revealed up-regulation of peroxiredoxin-1 (a cytosolic antioxidant) and ATP-synthase mRNA in inflamed biopsy specimens from patients with Crohn’s disease, consistent with a compensatory response to oxidative stress (Figure 5A), adding additional support for assessing the impact of metabolic stress in this disorder (NADH CoQ reductase mRNA was unaffected). Also, 50% of biopsy specimens from patients with inactive disease displayed increased peroxiredoxin-1 and ATP-synthase mRNA, begging the questions if this is indicative of low-grade inflammation and/or a propensity for relapse and if markers of mitochondrial function could be predictive for relapses in IBD.
Figure 5.

Tissues from patients with Crohn’s disease display increased baseline E. coli flux that is significantly enhanced by metabolic stress and reduced by MitoTEMPO. A: qPCR on biopsy specimens from patients with Crohn’s disease (each bar indicates a patient) reveals increased expression of peroxiredoxin-1 and ATP synthase mRNA, indicative of a response to metabolic stress. B:E. coli flux across biopsy specimens from patients with Crohn’s disease mounted in Ussing chambers is significantly increased by 0.1 mmol/L DNP, and this barrier defect is inhibited by 10 μmol/L MitoTEMPO (MitoT). One fluorescent unit equals 3.2 × 103E. coli CFU (2-hour fluxes). ∗P < 0.05, ∗∗P < 0.01 versus the indicated group. Con, control.
Tissue from patients with Crohn’s disease displayed a significant increase in baseline E. coli flux, consistent with a barrier defect,8,9 that was increased approximately sevenfold by DNP, indicating enhanced susceptibility to oxidative stress (Figure 5).9 Remarkably, MitoTEMPO reduced the DNP-induced transepithelial flux of E. coli in all six patients by 7% to 71% (Figure 5). However, the increased baseline translocation of E. coli was unaffected by MitoTEMPO cotreatment (n = 4).
Systemic Delivery of MitoTEMPO Reduces Murine Colitis
Four different protocols were used to assess the impact of systemic delivery of MitoTEMPO on murine colitis (Figure 1). Analysis of mice after 3 days of exposure to DSS revealed, as expected, only minor macroscopic disease (compare Figures 6A and 7A), no loss of body weight, and mild derangement of colonic architecture (Figure 6, A and B). However, there was a significant increase in the lumen-to-blood flux of FITC-dextran (4 kDa) (Figure 6C) and a subtle reduction in epithelial mitochondrial VDAC protein expression (Figure 6D). These early epithelial defects could be important in the manifestation of DSS-induced colitis.
Figure 6.

Increased intestinal permeability is an early feature of DSS-induced colitis. BALB/c mice were exposed to 5% DSS for 3 days, with or without 100 μg MitoTEMPO (MitoT; i.p., daily), and colitis was assessed (Figure 1). A and B: DSS-treated mice appear normal, but on autopsy show signs of mild macroscopic disease (n = 11 to 12, three experiments) and histological damage (n = 3 to 4) (compare to Figure 7) that is reduced in Mito + DSS-treated mice. C: Lumen-to-blood flux of FITC-dextran (4 kDa) increases in the DSS-treated mice and is inhibited by MitoT (n = 7 to 8, two experiments). D: Immunoblotting of whole cell extracts of isolated colonic epithelial cells reveals a modest reduction in the mitochondria-specific VDAC protein. Data are given as means ± SEM. ∗P < 0.05 versus control (cont), †P < 0.05 versus DSS. Original magnification, ×200 (H&E images). L, lumen of colon; m, external muscle layers.
Figure 7.

A: MitoTEMPO (MitoT) suppresses colitis (n = 14) that is accompanied by enhanced barrier function measured by FITC-dextran flux (n = 3) and translocation of bacteria into the mucosa (n = 5 to 10). B–D: The anticolitic benefit of MitoTEMPO in the prolonged DSS model (Figure 1); MitoTEMPO used as either a prophylactic or a treatment significantly reduces macroscopic and histological disease. Data are given as means ± SEM [n = 15 to 19 mice from four experiments and five mice from one experiment in the prophylactic (III) and treatment (IV) regimens, respectively]. ∗P < 0.05 versus control (con), †P < 0.05 versus DSS. Original magnification, ×200 (H&E images). L, lumen of colon; m, external muscle layers.
Severe colitis was obvious in mice exposed to DSS for 5 days (Figure 7, A–D) (±3 days normal water) (Figure 7B). Mice receiving MitoTEMPO as a prophylactic (Figure 7, A and B) or a treatment (Figure 7C) developed significantly less severe disease. Epithelial barrier function, as assessed by FITC-dextran fluxes and culture of bacteria from gut segments, was substantially reduced in MitoTEMPO-treated mice (Figure 7A). Segments of colon from DSS-treated mice mounted in Ussing chambers revealed characteristic reductions in forskolin-stimulated short-circuit current,36 indicative of altered epithelial electrogenic ion transport, which were unaffected by MitoTEMPO (data not shown).
MitoTEMPO Reduces DSS-Induced ProInflammatory Cytokine Protection in Vitro
In contrast to lipopolysaccharide, DSS-evoked increases in IL-8 production by T84 epithelia were inhibited by MitoTEMPO (Figure 8).
Figure 8.
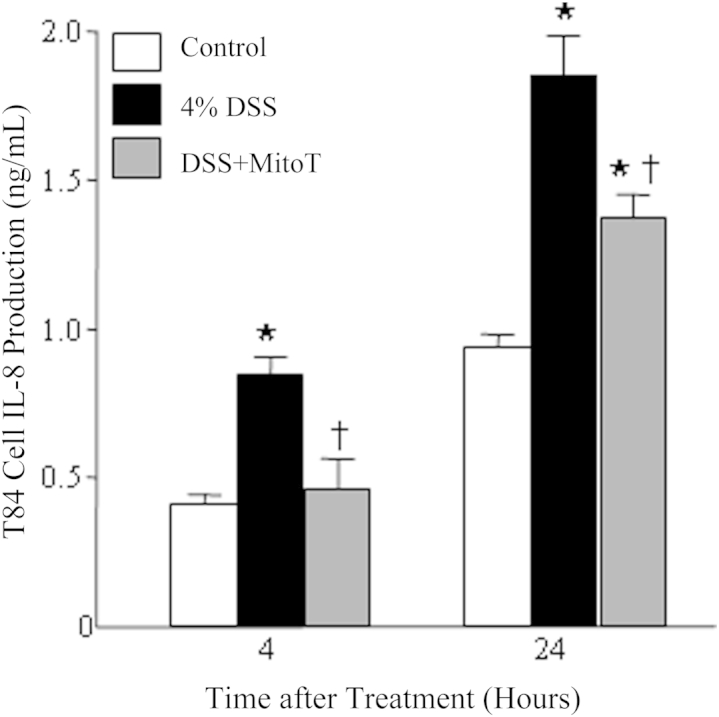
IL-8 production by T84 epithelia exposed to DSS is suppressed by cotreatment with 10 μmol/L Provided reagents MitoTEMPO (MitoT). Data are given as means ± SEM (n = 9 cell preparations from three separate experiments). ∗P < 0.05 versus control, †P < 0.05 versus DSS.
Discussion
Maintenance of the epithelial barrier or restoration of a defective barrier is an important component of intestinal homeostasis, and a laudable goal in treating intestinal inflammatory diseases, such as IBD. Yet, despite the fact that the control of paracellular and transcellular permeation is critically energy dependent, the potential to target the epithelial mitochondria to extrinsically regulate gut function has not been addressed. This is surprising given that many of the stimuli that derange mitochondrial activity (eg, toxins and psychological stress10) are implicated in IBD pathogenesis or as triggers of disease relapse, and in the light of the putative roles of autophagy37 and ER stress38 in IBD, both of which impinge on mitochondrial biology.20 Indeed, in many respects, mitochondria and the ER can be considered a functional unit.34
Herein, we extend our previous findings13,21 showing that perturbed mitochondrial activity increases epithelial permeability by demonstrating, for the first time to our knowledge, that mitochondria-derived superoxide is crucial for the increased transcellular permeation of commensal bacteria and that this can be countered by MTAs. In addition, perturbed epithelial mitochondria and increased gut permeability are identified as early events in DSS-induced colitis and MitoTEMPO reduced the severity of DSS-induced colitis, correlating with improved barrier function and an ability to suppress proinflammatory cytokine output. These mechanistic studies highlight the importance of mitochondria, specifically mitochondria-derived superoxide, as a signaling molecule, in the control of epithelial barrier function and host-bacteria interactions in the gut.
Although increases in paracellular permeability are readily demonstrable in models of colitis and IBD,3 a contribution from increased transcellular permeation to a barrier defect is poorly understood. Certainly, mitochondrial dysfunction increases paracellular permeability, but herein we opted to focus on transcellular permeability because of the paucity of data in this area. Juxtaposing our analyses of metabolic stress13 with assessment of bacterial pathogens39 and proinflammatory cytokines40 on model human colon-derived epithelial cells, a body of evidence is emerging that underlines the need to incorporate consideration of transcellular passage of microbes/antigens into any discussion of epithelial barrier function. Additional emphasis is given to this perspective because internalization of bacteria would be expected to activate the enterocyte, with putative consequences for mucosal immunity.
The mechanism by which perturbed epithelial mitochondrial activity (DNP, rotenone, and oligomycin A evoked) resulted in internalization and translocation of two noninvasive commensal E. coli strains remained an open question. Earlier pharmacological and molecular analyses had indicated a role for NF-κB in the DNP-induced epithelial barrier dysfunction,21 and we hypothesized that ROS participated in this event, noting that some inhibitors of NF-κB (eg, pyrrolidine dithiocarbamate) can have antioxidant activity.
The ROS are a natural by-product of the electron transport chain,35 and although a plethora of antioxidants exist to restrict ROS-driven cytotoxicity, a burgeoning literature attests to the signaling roles that ROS play.33,34 Increased T84 cell ROS production accompanied DNP and/or E. coli exposure, and the latter is in accordance with data from Neish and colleagues41 showing that commensal bacteria can elicit epithelial ROS production. This could be of particular relevance in gut disease, where mucus depletion allows direct bacteria-epithelial contact.42 However, only when combined with metabolic stress (eg, DNP) was significant internalization and passage of noninvasive, commensal E. coli across epithelial monolayers observed in our study; these are findings we have also recapitulated in normal human colon mounted in Ussing chambers. Given the prominent role of the commensal microbiota in enteric inflammatory disease43 and the relapsing-remitting nature of IBD, we speculate that perturbed host-bacteria interaction as a consequence of deranged mitochondrial activity and the concomitant increase in epithelial permeability, notably internalization and transcellular passage of microbes, contribute to the reactivation of enteric inflammation. Additional credence is given to this supposition by the increased sensitivity to uncoupling of oxidative phosphorylation in IBD, as demonstrated by the enhanced bacterial transcytosis across DNP-treated biopsy specimens from patients with Crohn’s disease.
Mitochondria are the major, but not the only, cellular source of ROS and so mechanistically it was important to assess the source of the ROS. Immunolocalization with Mitosox and use of MTAs indicated that mitochondrial superoxide predominantly drives the internalization and transcytosis of E. coli across stressed epithelium, in both cell lines in culture and human colonic tissue assessed ex vivo. Moreover, extracellular H2O2 (to mimic an inflammatory state) did not increase epithelial internalization of E. coli, suggesting that the ROS effect observed may be due to specific targeting of intracellular-generated ROS that cannot be recapitulated by extracellular ROS despite its ability to cross the cell membrane. Previous studies revealed no obvious apoptosis in DNP-treated epithelia within the parameters of the experiment.19,21,44 Collectively, these data are compatible with a scenario in which superoxide generated through perturbed mitochondrial function, along with a concomitant reduction in ATP,19 triggers the uptake of extracellular material for catabolism. We posit that this is a conserved cell survival mechanism45; having sensed metabolic stress, the cell seeks to internalize material for catabolism. The paradox therein is that, in the gut, excessive endocytosis of commensal bacteria could activate the enterocyte, provoking local inflammation that would be exaggerated further by the enhanced transcytosis of bacteria. Extrapolating from this, the hypothesis arises that MTAs, by virtue of their barrier-enhancing effect, could be used to treat colitis or prolong periods of disease remission. Protective effects of MTAs (ie, mQ and MitoE) have been shown in models of sepsis,46,47 but gut permeability was not assessed in those studies.
Increased gut permeability as a forerunner to colitis has been demonstrated in IL-10−/− and MDR1−/− mice,6,7 and penetration of bacteria deep into the mucus layer may occur in mice exposed to DSS before the development of inflammation.48 Herein, increased epithelial permeability and perturbed mitochondrial function (ie, mild reduction in VDAC expression) were noted early in the course of DSS: both irregularities were prevented by MitoTEMPO treatment. The prevention of the barrier defect defined by 4-KDa FITC-dextran is noteworthy, raising issues relating to the site of the barrier defect (eg, small versus large intestine), nature of the defect (ie, tight junction disruption or transcellular, altered epithelial apoptosis or proliferation), and target cell for the MitoTEMPO (ie, direct or indirect effects), all of which require a more extensive inquiry. Subsequently, it was determined that mice receiving MitoTEMPO had significantly less DSS-induced disease in all of the paradigms tested (Figure 1). Moreover, and in accordance with the in vitro analyses of human colon, the anticolitic effect of MitoTEMPO was associated with enhanced barrier function, as measured by transepithelial FITC-dextran and bacterial fluxes. Collectively, these data underline the link between mitochondria, epithelial permeability, and the development of inflammation.
Macrophage and epithelial activation (or death) have been implicated in DSS-induced colitis.49 In this context, MitoTEMPO suppression of DSS-induced tumor necrosis factor-α (a pivotal cytokine in IBD41; personal observation) and IL-8 (a potent neutrophil chemokine) synthesis by macrophages and epithelia, respectively, would be an important facet, in conjunction with enhanced epithelial barrier function, of MTA suppression of colitis. Indeed, these data complement the recent demonstration of suppression of inflammasome activation and IL-1β and IL-18 synthesis in the human THP-1 macrophage cell line by the MTA, mQ.50
In summary, metabolic stress compromises epithelial barrier function, promoting the transcytosis of bacteria across the epithelial layer that occurs, at least partially, via transcellular permeation. This oxidative stress–induced barrier defect may be particularly relevant because i) the commensal bacteria have been implicated in the etiology of IBD, ii) it would allow activation of the enterocyte, and iii) colonic tissue from patients with Crohn’s disease was sensitive to the uncoupling of oxidative phosphorylation, much more so than tissue from controls. These barrier defects in human tissues, and those observed in DSS-treated mice, and the severity of DSS-colitis were all ameliorated by MTA. We present these novel findings as proof-of-principle data to support the consideration of MTAs in IBD, a process that could be expedited as MTAs are undergoing assessment in a clinical trial.46
Acknowledgments
We thank the IBD Intestinal Tissue Bank at the University of Calgary for assistance with patient samples and ethics approval.
Author contributions were as follows: study design, J.D.S. and D.M.M.; generation of data, A.W., A.V.K., V.P., C.M.M., I.S., J.L., M.F., N.R., D.B., and M.D.; data analysis, A.W., A.V.K., V.P., C.M.M., I.S., J.L., M.F., P.L.B., J.D.S., and D.M.M.; provided reagents, M.P.M.; writing of manuscript, W.K.M., J.D.S., and D.M.M.; and review of manuscript, A.W., A.V.K., V.P., C.M.M., I.S., J.L., M.P.M., M.F., N.R., D.B., R.Y., M.D., P.L.B., W.K.M., J.D.S., and D.M.M.
Footnotes
Supported by a Canadian Institutes of Health Research grant MPO 126005 (D.M.M.) and a Swedish Medical Research Council grant (J.D.S.). D.M.M. holds a Tier 1 Canada Research Chair in Intestinal Immunophysiology.
Disclosures: M.P.M. is the inventor of MitoQ and has shares in a company commercializing MitoQ and related compounds. M.P.M. provided the MitoQ used in this study.
References
- 1.Hooper L.V., Gordon J.I. Commensal host-bacterial relationships in the gut. Science. 2001;292:1115–1118. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- 2.Lozupone C.A., Stombaugh J.I., Gordon J.I., Jansson J.K., Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turner J.R. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 4.Jostins L., Ripke S., Weersma R.K., Duerr R.H., McGovern D.P., Hui K.Y. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hollander D. Crohn’s disease: a permeability disorder of the tight junction? Gut. 1988;29:1621–1624. doi: 10.1136/gut.29.12.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madsen K.L., Malfair D., Gray D., Doyle J.S., Jewell L.D., Fedorak R.N. Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm Bowel Dis. 1999;5:262–270. doi: 10.1097/00054725-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Resta-Lenert S., Smitham J., Barrett K.E. Epithelial dysfunction associated with the development of colitis in conventionally housed mdr1a-/- mice. Am J Physiol Gastrointest Liver Physiol. 2005;289:G153–G162. doi: 10.1152/ajpgi.00395.2004. [DOI] [PubMed] [Google Scholar]
- 8.Zeissig S., Bürgel N., Günzel D., Richter J., Mankertz J., Wahnschaffe U., Kroesen A.J., Fromm M., Schlzke J.D. Changes in expression and distribution of claudins 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut. 2007;56:61–72. doi: 10.1136/gut.2006.094375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Söderholm J.D., Peterson K.H., Olaison G., Frazén L.E., Weström B., Magnusson K.E., Sjödahl R. Epithelial permeability to proteins in the non-inflamed ileum of Crohn’s disease. Gastroenterology. 1999;117:65–72. doi: 10.1016/s0016-5085(99)70551-2. [DOI] [PubMed] [Google Scholar]
- 10.Schoultz I., Söderholm J.D., McKay D.M. Is metabolic stress a common denominator in inflammatory bowel disease? Inflamm Bowel Dis. 2011;17:2008–2018. doi: 10.1002/ibd.21556. [DOI] [PubMed] [Google Scholar]
- 11.Rodiger W.E. The colonic epithelium in ulcerative colitis: an energy-deficiency disease? Lancet. 1980;2:712–715. doi: 10.1016/s0140-6736(80)91934-0. [DOI] [PubMed] [Google Scholar]
- 12.Singh S., Graff L.A., Bernstein C.N. Do NSAIDs, antibiotics, infections or stress trigger flares in IBD? Am J Gastroenterol. 2009;4:1298–1313. doi: 10.1038/ajg.2009.15. [DOI] [PubMed] [Google Scholar]
- 13.Nazli A., Yang P.C., Jury J., Howe K., Watson J.L., Söderholm J.D., Sherman P.M., Perdue M.H., McKay D.M. Epithelia under metabolic stress perceive commensal bacteria as a threat. Am J Pathol. 2004;164:947–957. doi: 10.1016/S0002-9440(10)63182-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodenburg W., Keijer J., Kramer E., Vink C., van der Meer R., Bovee-Oudenhoven I.M. Impaired barrier function by dietary fructo-oligosaccharides in rats is accompanied by increased colonic mitochondrial gene expression. BMC Genomics. 2008;9:1441–1451. doi: 10.1186/1471-2164-9-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma C., Wickham M.E., Guttman J.A., Deng W., Walker J., Madsen K.L., Jacobson K., Vogl W.A., Finlay B.B., Vallance B.A. Citrobacter rodentium infection causes both mitochondrial dysfunction and epithelial barrier disruption in vivo: role of mitochondrial associated protein (Map) Cellul Microbiol. 2006;6:1669–1686. doi: 10.1111/j.1462-5822.2006.00741.x. [DOI] [PubMed] [Google Scholar]
- 16.Chowdhury S.K., Smith D.R., Fernyhough R. The role of aberrant mitochondrial bioenergetics in diabetic neuropathy. Neurobiol Dis. 2013;51:56–65. doi: 10.1016/j.nbd.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 17.Rath E., Haller D. Inflammation and cellular stress: a mechanistic link between immune-mediated and metabolically-driven pathologies. Eur J Nutr. 2011;50:219–233. doi: 10.1007/s00394-011-0197-0. [DOI] [PubMed] [Google Scholar]
- 18.Tarnapolsky M.A., Raha S. Mitochondrial myopathies: diagnosis, exercise intolerance, and treatment options. Med Sci Sports Exerc. 2005;37:2086–2093. doi: 10.1249/01.mss.0000177341.89478.06. [DOI] [PubMed] [Google Scholar]
- 19.Nazli A., Wang A., Steen O., Prescott D., Lu J., Perdue M.H., Söderholm J.D., Sherman P.M., McKay D.M. Enterocyte cytoskeletal changes are crucial for enhanced translocation of nonpathogenic Escherichia coli across metabolically stressed gut epithelium. Infect Immun. 2006;74:192–201. doi: 10.1128/IAI.74.1.192-201.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tschopp J. Mitochondria: sovereign of inflammation? Eur J Immunol. 2011;41:1196–1202. doi: 10.1002/eji.201141436. [DOI] [PubMed] [Google Scholar]
- 21.Lewis K., Caldwell J., Phan V., Prescott D., Nazli A., Wang A., Söderholm J.D., Perdue M.H., Sherman P.M., McKay D.M. Decreased epithelial barrier function evoked by exposure to metabolic stress and nonpathogenic E. coli is enhanced by TNFα. Am J Physiol Gastrointest Liver Physiol. 2008;294:G669–G678. doi: 10.1152/ajpgi.00382.2007. [DOI] [PubMed] [Google Scholar]
- 22.Schoultz I., McKay C.M., Graepel R., Phan V.C., Wang A., Söderholm J.D., McKay D.M. Indomethacin-induced translocation of bacteria across enteric epithelia is reactive oxygen species-dependent and reduced by vitamin C. Am J Physiol Gastrointest Liver Physiol. 2012;303:G536–G545. doi: 10.1152/ajpgi.00125.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodriguez-Cuenca S., Cochemé H.M., Logan A., Abakumova I., Prime T.A., Rose C., Vidal-Puig A., Smith A.C., Rubinsztein D.C., Fearnley I.M., Jones B.A., Pope S., Heales S.J., Lam B.Y., Neogi S.G., McFarlane I., James A.M., Smith R.A., Murphy M.P. Consequences of long-term oral administration of the mitochondria-targeted antioxidant MitoQ to wild-type mice. Free Radic Biol Med. 2010;48:161–172. doi: 10.1016/j.freeradbiomed.2009.10.039. [DOI] [PubMed] [Google Scholar]
- 24.Keita Å.V., Salim S.Y., Jiang T., Yang P.C., Franzén L., Söderkvist P., Magnusson K.E., Söderholm J.D. Increased uptake of non-pathogenic E. coli via the follicle-associated epithelium in longstanding ileal Crohn’s disease. J Pathol. 2008;215:135–144. doi: 10.1002/path.2337. [DOI] [PubMed] [Google Scholar]
- 25.Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 26.Yates R.M., Russel D.G. Real-time spectrofluorometric for the lumen environment of the maturing phagosome. Methods Mol Biol. 2008;445:311–325. doi: 10.1007/978-1-59745-157-4_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rybicka J.M., Balce D.R., Chaudhuri S., Allan E.R., Yates R.M. Phagosomal proteolysis in dendritic cells is modulated by NADPH oxidase in a pH-independent manner. EMBO J. 2012;31:932–944. doi: 10.1038/emboj.2011.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu J., Wang A., Ansari S., Hersberg R.M., McKay D.M. Intra-colonic delivery of bacterial superantigens evokes an inflammatory response and exaggerates disease in mice recovering from colitis. Gastroenterology. 2003;125:1785–1795. doi: 10.1053/j.gastro.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 29.Asfaha S., Bell C.J., Wallace J.L., MacNaughton W.K. Prolonged colonic epithelial hyporesponsiveness after colitis: role of inducible nitric oxide synthase. Am J Physiol Gastrointest Liver Physiol. 1999;276:G703–G710. doi: 10.1152/ajpgi.1999.276.3.G703. [DOI] [PubMed] [Google Scholar]
- 30.Prescott D., McKay D.M. Aspirin-triggered lipoxin enhances macrophage phagocytosis of bacteria while inhibiting inflammatory cytokine production. Am J Physiol Gastrointest Liver Physiol. 2011;301:G487–G497. doi: 10.1152/ajpgi.00042.2011. [DOI] [PubMed] [Google Scholar]
- 31.Diaz-Granados N., Howe K., Lu J., McKay D.M. Dextran sulphate sodium-induced colonic histopathology, but not altered epithelial ion transport, is reduced by inhibition of phosphodiesterase activity. Am J Pathol. 2000;156:2169–2177. doi: 10.1016/S0002-9440(10)65087-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smyth D., McKay C.M., Gulbransen B.D., Phan V., Wang A., McKay D.M. Interferon-© signals via an ERK1/2-ARF6 pathway to promote bacterial internalization by gut epithelia. Cellul Microbiol. 2012;14:1257–1270. doi: 10.1111/j.1462-5822.2012.01796.x. [DOI] [PubMed] [Google Scholar]
- 33.Marchi S., Giorgi C., Suski J.M., Agnoletto C., Bononi A., Bonora M., De Marchi E., Missiroli S. Mitochondria-ROS crosstalk in the control of cell death and aging. J Signal Transduct. 2012;2012:329635. doi: 10.1155/2012/329635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bravo R., Vicencio J.M., Parra V., Troncoso R., Munoz J.P., Bui M., Quiroga C., Rodriguez A.E., Verdejo H.E., Ferreira J., Iglewski M., Chiong M., Simmen T., Zorzano A., Hill J.A., Rothermel B.A., Szabadkai G., Lavandero S. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci. 2011;124:2143–2152. doi: 10.1242/jcs.080762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reily C., Mitchell T., Chacko B.K., Benavides G., Murphy M.P., Darley-Usmar V. Mitochondrially-targeted compounds and their impact on cellular bioenergetics. Redox Biol. 2013;1:86–93. doi: 10.1016/j.redox.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirota C.L., McKay D.M. Loss of Ca2+-mediated ion transport during colitis correlates with reduced ion transport responses to a Ca-activated K channel opener. Br J Pharmacol. 2009;156:1085–1097. doi: 10.1111/j.1476-5381.2009.00122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Travassos L.H., Carneiro L.A., Ramjeet M., Hussey S., Kim Y.G., Magalhães J.G., Yuan L., Soares F., Chea E., Le Bourhis L., Boneca I.G., Allaoui A., Jones N.L., Nuñez G., Girardin S.E., Philpott D.J. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 38.Kaser A., Lee A.H., Franke A., Glickman J.N., Zeissig S., Tilg H., Nieuwenhuis E.E., Higgins D.E., Schreiber S., Glimcher L.H., Blumberg R.S. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalischuk L.D., Inglis G.D., Buret A.G. Campylobacter jejuni infection induces transcellular translocation of commensal bacteria via lipid rafts. Gut Pathog. 2009;1:2e. doi: 10.1186/1757-4749-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clark E., Hoare C., Tanianis-Hughes J., Carlson G.L., Warhurst G. Interferon-© induces translocation of commensal Escherichia coli across gut epithelial cells via a lipid raft-mediated process. Gastroenterology. 2005;128:1258–1267. doi: 10.1053/j.gastro.2005.01.046. [DOI] [PubMed] [Google Scholar]
- 41.Swanson P.A., II, Kumar A., Samarin S., Vijay-Kumar M., Kundu K., Murthy N., Hansen J., Nusrat A., Neish A.S. Enteric commensal bacteria potentiate epithelial restitution via reactive oxygen species-mediated inactivation of focal adhesion kinase phosphatases. Proc Natl Acad Sci U S A. 2011;108:8803–8808. doi: 10.1073/pnas.1010042108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vaishnava S., Yamamoto M., Severson K.M., Ruhn K.A., Yu X., Koren O., Ley R., Wakeland E.K., Hooper L.V. The antibacterial lectin RegIII© promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knights D., Lassen K.G., Xavier R.J. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut. 2013;62:1505–1510. doi: 10.1136/gutjnl-2012-303954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lewis K., Lutgendorff F., Phan V., Söderholm J.D., Sherman P.M., McKay D.M. Enhanced translocation of bacteria across metabolically-stressed epithelia is reduced by butyrate. Inflamm Bowel Dis. 2010;16:1138–1148. doi: 10.1002/ibd.21177. [DOI] [PubMed] [Google Scholar]
- 45.Ryder T.A., Bowen I.D. Endocytosis and aspects of autophagy in the foot epithelium of the slug Agriolimax reticulatus (Muller) Cell Tissue Res. 1977;181:129–142. doi: 10.1007/BF00222779. [DOI] [PubMed] [Google Scholar]
- 46.Zang Q.S., Sadek H., Maass D.L., Martinez B., Ma L., Kilgore J.A., Williams N.S., Frantz D.E., Wigginton J.G., Nwariaku F.E., Wolf S.E., Minei J.P. Specific inhibition of mitochondrial oxidative stress suppresses inflammation and improves cardiac function in a rat pneumonia-related sepsis model. Am J Physiol Heart Circ Physiol. 2012;302:H1847–H1859. doi: 10.1152/ajpheart.00203.2011. [DOI] [PubMed] [Google Scholar]
- 47.Lowes D.A., Webster N.R., Murphy M.P., Galley H.F. Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Br J Anaesth. 2013;110:472–480. doi: 10.1093/bja/aes577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johansson M.E., Gustafsson J.K., Sjöberg K.E., Petersson J., Holm L., Sjövall H., Hansson G.C. Bacteria penetrate the inner mucus layer before inflammation in the dextran sulphate colitis model. PLoS One. 2010;5:e12238. doi: 10.1371/journal.pone.0012238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mahler M., Bristol I.J., Leiter E.H., Workman A.E., Birkenmeier E.H., Elson C.O., Sundberg J.P. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. Am J Physiol Gastrointest Liver Physiol. 1988;274:G544–G551. doi: 10.1152/ajpgi.1998.274.3.G544. [DOI] [PubMed] [Google Scholar]
- 50.Dashdoji A., Jyothi K.R., Lim S., Jo A., Nguyen M.N., Ha J., Yoon K.S., Kim H.J., Park J.H., Murphy M.P., Kim S.S. Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing inflammasome-mediated inflammatory cytokines. BMC Med. 2013;11:178. doi: 10.1186/1741-7015-11-178. [DOI] [PMC free article] [PubMed] [Google Scholar]