

Abstract
Peripheral myelin protein 22 (PMP22) is a tetraspan membrane protein strongly expressed in myelinating Schwann cells of the peripheral nervous system. Myriad missense mutations in PMP22 result in varying degrees of peripheral neuropathy. We used Rosetta 3.5 to generate a homology model of PMP22 based on the recently published crystal structure of claudin-15. The model suggests that several mutations known to result in neuropathy act by disrupting transmembrane helix packing interactions. Our model also supports suggestions from previous studies that the first transmembrane helix is not tightly associated with the rest of the helical bundle.
Peripheral myelin protein 22 (PMP22) is a member of the claudin/EMP/PMP22 tetraspan membrane protein family and is strongly expressed in the myelinating Schwann cells of the peripheral nervous system.1,2 Among its functions, PMP22 is critical to the formation and maintenance of the myelin ultrastructure,1−3 including possible roles in the tight junctionlike assemblies therein.4,5 A number of genetic aberrations, including more than 40 different missense mutations that encode single-amino acid changes in PMP22 distributed throughout its sequence,2 result in mild to severe peripheral neuropathy and disability (Figure S1 of the Supporting Information).
These peripheral neuropathies include heritable neuropathy with liability to pressure palsies (HNPP, mild neuropathy), Dejerine Sottas syndrome (DSS, severe), and Charcot-Marie-Tooth disease (CMTD, moderate to severe).2 It is believed that most disease mutant forms of PMP22 induce misfolding of the protein, leading to loss of function and possible toxicity from accumulated misfolded protein.4−11
Previous work indicates at least some PMP22 disease mutants are considerably destabilized; even wild-type (WT) PMP22 is only marginally stable,12−14 being transported to cell plasma membranes with an efficiency of only ∼20%.11 This inherent instability is among the reasons an experimental high-resolution structure of PMP22 has thus far proved elusive.
In this study, we utilized the recently published 2.4 Å crystal structure of claudin-15 (Protein Data Bank entry 4P79),15 the first high-resolution structure of a claudin/EMP/PMP22 family member, as a template for building a homology model of PMP22. The model presented here provides a step toward the goal of discriminating mechanisms of disease-inducing mutations.
Briefly, we employed BCL::Align, an alignment program that accounts for sequence identity and similarity as well as secondary structure and transmembrane region predictions,16 to generate an alignment of PMP22 (NP_696997.1) with claudin-15 (NP_068365.1). The alignment was truncated to cover only portions of the protein present in the crystal structure (Figure 1; see the Supporting Information for details), and the confidence of this alignment was evaluated (Figure S2 of the Supporting Information). In the final alignment, sequences were 25% identical and ∼60% similar. Interestingly, TM1 was much more divergent (only 13% identical) than the other transmembrane helices (TM2–TM4 being 36, 50, and 38% identical, respectively). Extracellular loop 1 (ECL1) was relatively well-conserved (30% identity), while there was limited conservation in the intracellular loop (ICL, 7%) and ECL2 (14%).
Figure 1.
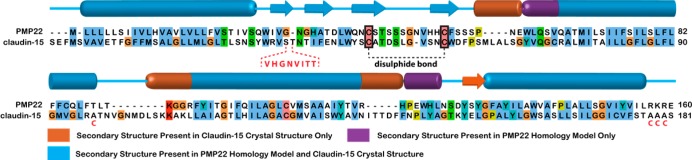
Final alignment of human PMP22 with murine claudin-15 utilized for homology modeling, with secondary structure indicated. Orange secondary structure elements are observed in the claudin-15 crystal structure, but not in the final top-scoring models; purple elements are observed in the final model but not in claudin-15. The sequence in ECL2 that was unresolved in the crystal structure and was removed in the final alignment is colored red within the dashed lines; the claudin-15 disulfide bond is denoted in black, and the C-to-A mutations in the claudin-15 crystal construct are depicted below the sequence in red.
Using the loop rebuilding utility within Rosetta 3.5, a starting set of homology models of PMP22 was constructed (see the Supporting Information for details). Knowledge-based potentials included within the calculation utilized secondary structure predictions as well as transmembrane residue lipid-facing propensity (so-called “lipophilicity”) generated within the Rosetta membrane ab initio utility.17−19 These models were scored by Rosetta,20 and the top models were relaxed iteratively (see Figure 2 and Figures S3 and S4 of the Supporting Information).
Figure 2.
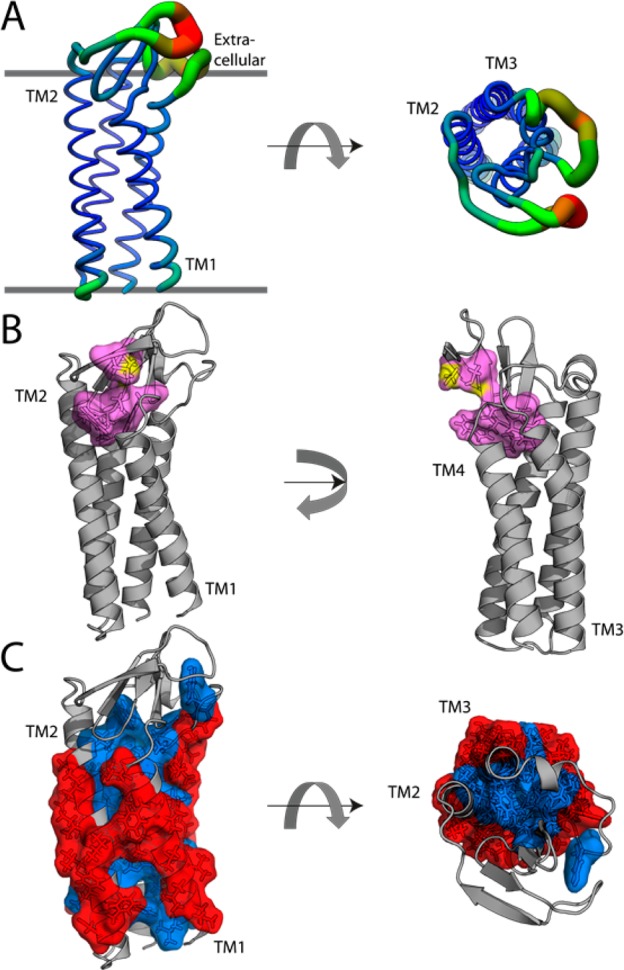
(A) Top-scoring PMP22 model color-coded according to the average chain root-mean-square deviation (rmsd) in the top 10 scoring models. The rmsd ranges from 0.6 Å (blue, thin backbone trace) to >10 Å (red, thick backbone trace). (B) Top-scoring model with the claudin motif residues highlighted in cyan as stick and surface view. Sulfur atoms are colored yellow. (C) Top-scoring model showing the most (red) and least (blue) “lipophilic” sites as determined by the LIPS algorithm.23 The Protein Data Bank format coordinates of this model are available in the Supporting Information. The extracellular face of the protein is at the top in the left panels.
The top-scoring PMP22 model (Protein Data Bank format coordinates in the Supporting Information) was evaluated with MolProbity21 (see the Supporting Information for details). After energy minimization, only the first four of five extracellular β-strands present in the claudin-15 template were retained (Figure 2A); these strands are all in ECL1. On the basis of the root-mean-square deviation (rmsd) from the top 10 models (Figure 2A), Rosetta most confidently predicts the TM1–TM4 region with slight uncertainty at the TM1 N-terminus. The predictions for ECL1 appear to be relatively uniform within the β-strands but have very weak convergence in the loop of the first β-hairpin.
Additionally, there is conformational heterogeneity among high scoring models present in both ECL2 and the ICL. It is observed that a portion (W-DLW) of the conserved claudin motif (W-[N/G/D]LW-C-C)22 dips back into the membrane to stabilize the helical packing on the extracellular side of the helical bundle (Figure 2B). While claudins have an extracellular disulfide bond, it is unclear whether a bond forms between the corresponding Cys pair in PMP22. This bond was not therefore not enforced in the generation of this model (3.6 Å between sulfur atoms). Repeating model generation with a forced disulfide bond did not require gross alterations in the structure (overall rmsd to the reduced form structure of 1.96 Å), suggesting that this model may be accurate in either case (Figure S5 of the Supporting Information). We also note that the computed “lipophilicity”23 predicts transmembrane helix–helix contacting faces that are fully consistent with what is seen in the model (Figure 2C).
Previous studies indicate that even WT PMP22 is only marginally stable,12−14 and nuclear magnetic resonance (NMR) studies indicate that under micellar conditions at 45 °C WT PMP22 occupies a folding intermediate in which TM1 dissociates from the rest of the transmembrane domain, with TM2–TM4 forming a molten globule-like bundle.13 The TrJ disease mutant (L16P in TM1) increases the propensity of this helix to dissociate. Interestingly, Rosetta found the initial conformation of TM1 in the WT protein to be unfavorable; consequently, the loop rebuilding and side chain repacking algorithms readjusted the position of the packing of the bundle in nearly every case. In our final WT model, TM1 of PMP22 is packed much less tightly to TM2–TM4 than the corresponding helices of claudin-15 (Figure S6 of the Supporting Information). Additionally, the L16 residue, along with several other disease mutation sites, appears to be involved in TM1 packing with the helical bundle (Figure 3A and Figure S7 of the Supporting Information).
Figure 3.
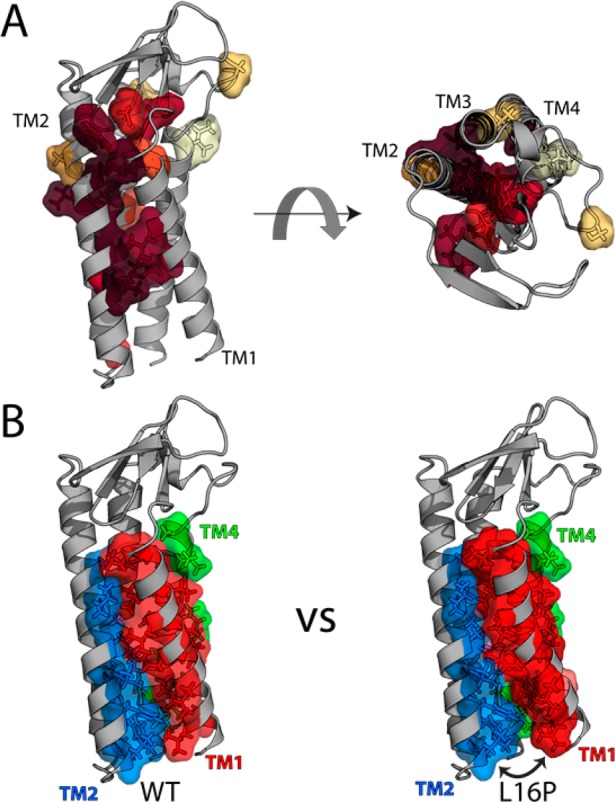
Assessment of disease mutation locations in the PMP22 model. (A) PMP22 homology model with color coding of wild-type residues mutated in neuropathies according to patient motor nerve conduction velocities (NCVs), with maroon having the lowest NCVs and cream representing a benign polymorphism (see Table S1 of the Supporting Information). Note that for a number of known disease mutations, patient nerve conduction velocities have not been reported, such that the associated sites are not highlighted in this figure. Note also that the lone site of a severe mutation facing the lipid environment is a proline substitution (L71P) in the middle of a TM2, which is expected also to disrupt helical packing. (B) Comparison of the packing interface between the WT model and the top two L16P models, showing a reduced interface for L16P between TM1 and the rest of the bundle: red for TM1, marine for TM2, violet for TM3, green for TM4, and salmon for the additional contacting residue on L16P TM1.
A number of the most severe disease mutations (associated with patients presenting nerve conduction velocities of <10 m/s), including L16P, are at residues located along the helix-packing interface between TM1 and TM2/TM4, while less severe mutation sites tend to either face the lipid or “cap” the helices (Figure 3A and Table S1 of the Supporting Information). Modeling of the L16P mutation with Rosetta generates structures with a significantly higher Rosetta energy (p < 0.0001). These models conform to the predictions made by the NMR data; the size of the TM1 interface with TM2–TM4 is reduced, with predicted structures sharing an interface with either the N- or C-terminal side of L16P TM1, but not both (Figure 3B and Figure S8 of the Supporting Information).
This study provides the first high-resolution working model for PMP22 and will be used as a springboard for future work through its potential predictive power. Future studies will focus on verifying which disease mutations are indeed destabilizing as well as providing experimental restraints for refinement of this computational model.
Acknowledgments
We thank Dr. Jonathan Schlebach, Amanda Duran, and Stephanie DeLuca for their critical input regarding this work.
Supporting Information Available
Materials and methods, table, references, figures and captions, and atomic coordinates. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ These authors contributed equally to this work.
This work was supported by National Institutes of Health (NIH) Grants U54 GM094608, R01 HL122010, and R01 DC007416 to C.R.S. and NIH Grants R01 GM099842 and R01 DK097376 and National Science Foundation (NSF) Grant CHE 1305874 to J.M. K.F.M. was supported by NSF Predoctoral Fellowship DGE090966. B.M.K. was supported by NIH Grant T32 NS007491-13.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Jetten A. M.; Suter U. (2000) Prog. Nucleic Acid Res. Mol. Biol. 64, 97–129. [DOI] [PubMed] [Google Scholar]
- Li J.; Parker B.; Martyn C.; Natarajan C.; Guo J. (2013) Mol. Neurobiol. 47, 673–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amici S. A.; Dunn W. A. Jr.; Notterpek L. (2007) J. Neurosci. Res. 85, 238–249. [DOI] [PubMed] [Google Scholar]
- Guo J.; Wang L.; Zhang Y.; Wu J.; Arpag S.; Hu B.; Imhof B. A.; Tian X.; Carter B. D.; Suter U.; Li J. (2014) Ann. Neurol. 75, 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notterpek L.; Roux K. J.; Amici S. A.; Yazdanpour A.; Rahner C.; Fletcher B. S. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 14404–14409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colby J.; Nicholson R.; Dickson K. M.; Orfali W.; Naef R.; Suter U.; Snipes G. J. (2000) Neurobiol. Dis. 7, 561–573. [DOI] [PubMed] [Google Scholar]
- Liu N.; Yamauchi J.; Shooter E. M. (2004) Neurobiol. Dis. 17, 300–309. [DOI] [PubMed] [Google Scholar]
- Naef R.; Adlkofer K.; Lescher B.; Suter U. (1997) Mol. Cell. Neurosci. 9, 13–25. [DOI] [PubMed] [Google Scholar]
- Naef R.; Suter U. (1999) Neurobiol. Dis. 6, 1–14. [DOI] [PubMed] [Google Scholar]
- Sanders C. R.; Ismail-Beigi F.; McEnery M. W. (2001) Biochemistry 40, 9453–9459. [DOI] [PubMed] [Google Scholar]
- Pareek S.; Notterpek L.; Snipes G. J.; Naef R.; Sossin W.; Laliberte J.; Iacampo S.; Suter U.; Shooter E. M.; Murphy R. A. (1997) J. Neurosci. 17, 7754–7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers J. K.; Mobley C. K.; Sanders C. R. (2008) Biochemistry 47, 10620–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakura M.; Hadziselimovic A.; Wang Z.; Schey K. L.; Sanders C. R. (2011) Structure 19, 1160–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlebach J. P.; Peng D.; Kroncke B. M.; Mittendorf K. F.; Narayan M.; Carter B. D.; Sanders C. R. (2013) Biochemistry 52, 3229–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H.; Nishizawa T.; Tani K.; Yamazaki Y.; Tamura A.; Ishitani R.; Dohmae N.; Tsukita S.; Nureki O.; Fujiyoshi Y. (2014) Science 344, 304–307. [DOI] [PubMed] [Google Scholar]
- Dong E.; Smith J.; Heinze S.; Alexander N.; Meiler J. (2008) Gene 422, 41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth P.; Schonbrun J.; Baker D. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 15682–15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth P.; Wallner B.; Baker D. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy V.; Schonbrun J.; Baker D. (2006) Proteins 62, 1010–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R.; Baker D. (2008) Annu. Rev. Biochem. 77, 363–382. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. III; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. (2010) Acta Crystallogr. D66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause G.; Winkler L.; Mueller S. L.; Haseloff R. F.; Piontek J.; Blasig I. E. (2008) Biochim. Biophys. Acta 1778, 631–645. [DOI] [PubMed] [Google Scholar]
- Adamian L.; Liang J. (2006) BMC Struct. Biol. 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.