

Abstract

Post-translational modifications that do not result in a change in mass are particularly difficult to detect by mass spectrometry. For example, isomerization of aspartic acid or epimerization of any chiral residue within a peptide do not lead to mass shifts but can be identified by examination of independently acquired tandem mass spectra or by combination with another technique. For analysis of a biological sample, this means that liquid chromatography or some other type of separation must be used to first separate the isomers from one another. Furthermore, each specific m/z of interest must be sampled repeatedly to allow for comparison of the tandem mass spectra from each separated isomer, which contrasts with the traditional approach in proteomics where the goal is typically to avoid resampling the same m/z. We illustrate that isomerization and epimerization of peptides can be identified in this fashion by examination of long-lived crystallin proteins extracted from a sheep eye lens. Tandem mass spectrometry relying on a combination of radical directed dissociation (RDD) and collision induced dissociation (CID) following separation by liquid chromatography was used to identify modified peptides. Numerous sites of isomerization and epimerization, including several that have not been previously identified, were determined with peptide specificity. It is demonstrated that the specific sites of amino acid isomerization within each peptide can be identified by comparison with synthetic peptides. For α-crystallin proteins, the sites that undergo the greatest degree of isomerization correspond to disordered regions, which may have important implications on chaperone functionality within the context of aging.
The eye lens is a very peculiar and interesting construct.1,2 It is composed of 90% crystallin proteins.3 Among the crystallins, alpha crystallins are most abundant and serve as structural elements and as chaperones.4,5 To achieve the desired index of refraction required for an optic, the concentration of proteins within the lens must be high.6 At the same time, aggregation of proteins into particles capable of scattering light must also be avoided. The alpha crystallins assemble into large oligomeric species of between 20 and 40 monomers which are highly dynamic in nature. Numerous studies have indicated a preference for even numbered oligomers, suggesting that the assemblies are comprised of dimer building blocks.7 Additionally, within each monomer several structural regions have been identified that each serve distinct roles. The central alpha crystallin domain folds into a well-defined structure that is conserved across many of the small heat shock proteins. In contrast, the N-terminal region and the C-terminal extension do not form well-defined structures at the monomer level; however, these regions help regulate higher order oligomer assembly and are necessary for chaperone activity.
As fiber cells in the eye lens mature, all organelles are ejected. The mature fiber cells perform very few metabolic functions, have low oxygen and energy demands, and are avascular. Perhaps most interestingly, the crystallin proteins that were present in the lens of a person when born are still present when they die. In other words, there is no turnover of proteins within lens fiber cells. Given these properties, it is not surprising that many studies have focused on examination of the changes that occur to lens proteins upon aging. It has been established that numerous post-translational modifications occur to crystallin proteins as a function of aging. Deamidation, truncation, glycation, phosphorylation, disulfide bond formation, oxidation, acetylation, and methylation are among the most commonly studied modifications.8−12 However, there is also evidence that epimerization, which occurs when a single amino acid undergoes stereoinversion, is also an important modification that occurs as a function of aging.13−15 Epimerization leads to no change in mass and it is significantly more difficult to detect than the other post-translational modifications (PTMs) listed above, which may explain why epimerization has received significantly less attention.16
Aspartic acid is a special amino acid in the context of PTMs that are not accompanied by changes in mass. Backbone attack of the side chain yields a succinimide ring that can lead to the formation of several isomeric forms. Simple ring opening will yield a mixture of aspartic acid and isoaspartic acid, where the side chain has essentially inserted into the peptide backbone.17 In addition, the chiral alpha hydrogen atom in the succinimide ring can undergo stereoinversion, which can lead to the formation of d-aspartic acid and d-isoaspartic acid. Since these two pathways can occur in conjunction, the end result is that aspartic acid is frequently converted into four isomeric states, none of which are distinguished by a shift in mass. For asparagine, these states can also be populated, though only when accompanied by deamidation. Both isomerization and epimerization significantly perturb the local structure of the molecule at the affected residue. For example, it has been demonstrated that substitution of d-residues can significantly reduce the propensity of peptides to adopt alpha helical structures.18 Similar structural changes may also explain why elevated epimerization is also associated unfavorably with numerous diseases. For example, racemization of serine residues in β-amyloid increases the rate of aggregation and accelerates degeneration of neuronal cells, which may be connected to the cause of Alzheimer’s disease.19 Epimerized residues are also more frequently detected in crystallin proteins from cataract sufferers than in age controlled healthy individuals.20,21 It is clear from these initial findings that characterization of isomeric PTMs, though difficult, is warranted.
The majority of work in this area involving mass spectrometry has focused on examination of isolated molecules. For example, it has been demonstrated that differences in MS2 spectra can be used to distinguish aspartic acid from isoaspartic acid.22,23 Similarly, differences in MS2 spectra can be used to distinguish peptide epimers.24−27 Quantitative analysis is typically carried out by calculation of an R value28 that corresponds to the degree of difference between the two fragments that change the most in the MS2 spectra obtained from each isomer (additional details about R values are provided below). R values of 1 correspond to identical spectra, whereas larger values reflect differences between the spectra being compared. Importantly, this method requires that both the all l peptide and the epimer with a single d residue be independently evaluated. Furthermore, not all fragmentation methods are equivalent in epimer disambiguation. For example, although collision induced dissociation (CID) can yield acceptable results, it generally offers less structural sensitivity than electron or radical based dissociation methods. Recent work has demonstrated that radical directed dissociation (RDD) yields the highest R values for epimer detection and has the advantage of the greatest flexibility in terms of charge state selection.29 For identification of isoaspartic acid, electron capture dissociation (ECD) is advantageous because it yields a characteristic fragment that can facilitate identification.30,31
Implementation of mass spectrometry in conjunction with liquid chromatography for the analysis of more complicated isomer containing samples requires additional considerations. Fortunately, analysis of biological samples within the context of aging simplifies the experiment in one important way: some of the original isomer will always be present. All processes by which spontaneous epimerization/isomerization occur are incomplete, therefore some of the original peptide or protein will always remain. Given this information, the challenge can be broken down into two components: separation and characterization. Separation is typically carried out with standard liquid chromatography (LC), which is capable of baseline separating many isomers (including epimers) using typical C18 columns (i.e., columns packed with chiral media are not required).16,32 Therefore, separation can be carried out in a similar fashion to other proteomics experiments. However, characterization requires that the same m/z be examined multiple times, which is typically avoided in most proteomics experiments. For evaluation of isomeric species, multiple tandem MS spectra at the same m/z must be acquired to confirm epimerization and for calculation of relevant R values. The need to acquire multiple spectra for the same m/z values must be then balanced against the traditional goal of also simultaneously examining as many unique peptides as possible. These requirements place limits on the complexity of samples that can be evaluated in an online fashion for isomer focused proteomics experiments.
In the present work, we describe characterization of isomeric PTMs in sheep crystallins extracted from the eye lens. The results from both RDD and CID on LC separated peptides were combined to improve isomer identification. A short exclusion time and a target peptide mass list were used to ensure that each peptide was examined multiple times to allow for comparison of the relevant tandem mass spectra. Three crystallin proteins (αA-, αB-, and βB3-crystallin) were identified from the ovine database with excellent sequence coverage. Several additional proteins that are predicted to be associated with crystallin are also identified from the newly released sheep genome. Many previously uncharacterized sites of isomerization for crystallins were identified. The results illustrate that the greatest degree of isomerization and epimerization occurs in the disordered N-terminal and C-terminal regions of αA- and αB-crystallin, which are abundant and important proteins in the lens that function as chaperones and also serve as structural elements.
Experimental Methods
Materials
Organic solvents and reagents were purchased from Sigma-Aldrich (St. Louis, MO) or Acros Organics (Geel, Belgium) and used without further purification. Water was purified to 18.2 MΩ by a Millipore 147 (Billerica, MA) Direct-Q system. Amino acids and resin were purchased from Ana Spec (Fremont, CA). Trypsin was purchased from Sigma-Aldrich (St. Louis, MO).
Peptide and Radical Precursor Synthesis
All synthetic peptides were synthesized manually using standard fmoc procedures with Rink Amide Resin or Wang Resin for the solid support.33N-Hydroxysuccinimide (NHS) activated iodo-benzoyl esters were synthesized by a previously reported procedure.34
Protein Extraction and Digestion
Sheep eyes were obtained from discarded tissue from Corona Cattle Inc. (Corona, CA). The lenses were separated, washed with distilled water, and then homogenized in 50 mM Tris/HCl, pH 7.8 buffer. The homogenate was centrifuged at 10,000 rpm for 20 min at 4 °C. The supernatant was purified by dialysis against water and lyophilized. The lyophilized protein was dissolved in 50 mM NH4HCO3 buffer, pH 7.8, and the disulfide bonds were reduced in 100 mM DTT at 95 °C for 5 min. After returning to room temperature, 100 mM iodoacetamide solution was added and the mixture was incubated in the dark for 20 min. Then proteins were digested with trypsin overnight at 37 °C, with the protein-enzyme ratio at 50:1. For the iodo-benzoic modification, the digestion mixture was first purified with a peptide trap (Michrom Bioresource Inc.). Approximately 5 nmol protein digestion mixture, 15 μL of 15 mM NHS ester dioxane solution, and 5 μL borate buffer (pH 8.6) were combined and incubated for 1 h at 40 °C. Important: Note that dimethyl sulfoxide (DMSO) should not be used for this step because it can easily cause aspartic acid isomerization. The modification side products at arginine and tyrosine side chain were removed by incubating the reaction mixture in 1 M hydroxylamine (pH 8.5 adjusted by NaOH) for 4 h. The exact same procedure was performed on control peptides. Since the deamidation and racemization of asparaginyl and aspartyl residues are nonenzymatic spontaneous reactions that can occur under physiological conditions,35 control experiments with a synthetic peptide (TVLDSGISEVR) were performed to ensure that our sample preparation procedure does not induce any isomerization. After reduction by DTT in 95 °C and incubation with trypsin overnight, no isomerization was detected by LC-MS (data not shown).
Mass Spectrometry and Radical Directed Dissociation
Solutions were analyzed by an LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific, San Jose, CA) with a standard electrospray ionization (ESI) source. The back plate of the mass spectrometer was modified with a quartz window to transmit fourth harmonic (266 nm) laser pulses from a Nd:YAG laser (Continuum, Santa Clara,CA). Photodissociation of the labeled peptide homolytically cleaves the C–I bond in the chromophore and produces a radical peptide. Further MS3 experiments were performed by reisolation and CID of the radical species.
LC-MS Data Acquisition and Analysis
An Agilent 1100 series HPLC system (Agilent, Santa Clara, CA) with a BetaBasic-18 column (150 × 2.1 mm, particle size 5 μm) was coupled to the LTQ mass spectrometer. Peptides were separated using 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in acetonitrile (mobile phase B) with a flow rate of 0.2 mL/min. The digestion mixture was separated by the following gradient: 5% B to 20% B over 25 min, 20% B to 30% B over the next 35 min, 30–50% B over the next 15 min, and 50% B to 95% B over the final 10 min. The MS instrument was operated in the data-dependent mode. In a CID-only LC-MS run, the first scan event is full MS from m/z 300 to 2000 Da, followed by ultrazoom (scan event 2) and CID-MS2 (scan event 3). In a RDD LC-MS run, the laser pulses were triggered during the MS2 (scan event 3) and the CID was performed as a pseudo-MS3 step (scan event 4).36 Since the photodissociation of 4-iodo-benzoic labeled peptide will always produce the radical peptide as the major product, the precursor ion of CID in the MS3 step is the radical species rather than the original peptide. The exclusion time was 60 s for the identification of peptides and 16 s for the isomer discrimination.
MS data were acquired with Xcalibur software (Thermo Fisher Scientific). The raw files were converted to mgf files by MM File Conversion. The mgf files were searched with X! Tandem (version 2013.02.01.1) against the ovis aries database UniProt 2014 06, 26,849 entries). The cleavage sites were set as lysine and arginine (semi cleavage was turned on), allowing up to two missed cleavages and one point mutation. Carbamidomethylation (+57.02 Da at Cys) was set as fixed modification, and N-acetylation was considered a variable modification. For the modified digestion mixture, the 4-iodobenzoic acid modification (+230.01 Da) was considered a variable modification at either the N-terminus or lysine side chain. The parent monoisotopic mass error was set to ±1 Da and the fragment monoisotopic mass error was set to ±0.4 Da. The minimum parent ion mass was set to 400 Da. The criteria used for accepting peptides identification is e < 0.005 for peptides. The false discovery rate is 1%, calculated by searching the data against the reversed database. Given that the content of the eye lens is ∼90% crystallin proteins, the data was also searched against a smaller database that contains primarily crystallin proteins. A few additional peptides were identified in this fashion and their identities were confirmed by manual assignment of the MS/MS data.
Calculation of R Values
To quantify the isomer discrimination sensitivity, an R value originally reported by Tao et al.28 for chiral selectivity is used. In the present work, the R is calculated by eq 1. RA and RB represent ratios of the relative intensities of a pair of fragment ions which varies the most between two isomers. Therefore, Risomer = 1 indicates that the two tandem MS spectra are exactly identical and no isomerization occurs. If Risomer > 1, a larger number reflects a higher probability of isomerization. The statistical significance of the results is addressed in the Results and Discussion. In addition, we use an S value calculated in the same fashion to provide a quantitative measure of the similarity of experimental spectra to those obtained from synthetic standards. In the case of the S value, the number should be smaller than the threshold. Although the same formula is used in both cases, since the value should be higher in one case and lower in the other, we have given them different designations to avoid confusion.
| 1 |
While comparing the tandem mass spectra between synthetic standards and experimental peptides, the experiments for unknown peptides and the standard peptides have to be conducted in two separate LC-MS runs. More error arises from the different ionization efficiency or other random uncertainties between different LC-MS runs. Hence, to establish the S value threshold for positive identification of peptide isomers by comparing the MS/MS spectra with standard peptides, another set of standard LC-MS runs were performed. Six S values were obtained by comparing the MS/MS spectra acquired during different LC-MS runs of the same peptides. The threshold for positive identification is 1.9 for CID calculated from a standard t test (six S values are 1.63, 1.13, 1.58, 1.62, 1.72, 1.22) and 3.2 for RDD (six S values are 2.38, 2.02, 2.22, 3.21, 2.07, 2.70). See Supporting Information for additional details.
Results and Discussion
General Approach
Our general experimental procedure for identifying peptide epimers in a mixture of proteins is shown in Figure 1. Following protein isolation and digestion with trypsin, the sample is split into two pools and half of it is covalently modified. There are two advantages to covalent modification. First, the covalent modification allows for incorporation of a chromophore suitable for converting the peptides into radical species and analysis by RDD. Previous work has demonstrated that RDD is the most sensitive method for epimer discrimination.29 Second, the covalent modification frequently enhances the separation of epimers as is described in greater detail below. There are also obvious disadvantages to covalent modification, including loss of sample and additional experimental complexity. Therefore, the unmodified samples are also analyzed using standard CID. Although CID provides less ability to distinguish epimers, more peptides and particularly those in low abundance are able to be analyzed. Given that distinguishing epimers is a difficult task, it is also beneficial to carry out the analysis using two independent methods. Both the modified and unmodified samples are then subjected to a typical proteomics LC-MS run using CID for the purpose of peptide identification.
Figure 1.

Workflow to identify peptide isomers in a protein digestion mixture by LC-MS/MS.
Using this information, a target peptide list is generated and all charge states of these peptides are then examined in a second LC-MS run by both CID and RDD with a 16 s exclusion window. Multiple charge states are examined because R-values frequently vary significantly for different charge states. The 16 s exclusion window is used because it enables the examination and re-examination of up to four coeluting species within typical LC peak widths. Therefore, every peptide on the target list will be examined multiple times, even if it is only at the leading and trailing edges of a single LC peak (see Discussion below for the significance of re-examining the same LC peak). Although this strategy will limit the complexity of sample that can be analyzed in a single run, it should be possible to mitigate this problem by carrying out additional runs (if needed).
Isomer Separation
Comparison of potentially distinguishing MS/MS spectra first requires that the isomers of interest be evaluated independently of one another. Although separations can be carried out with chiral media,37 isomers (including epimers) can also be separated on traditional columns, which are more frequently used.32 Incomplete separations complicate analysis and make quantitation significantly more challenging. Fortunately, the covalent modification that we use to install labile bonds for RDD also changes the chromatographic behavior of the modified molecules. An example of this is shown in Figure 2 for the peptide DAEFR, which is a small tryptic peptide from β-amyloid. All of the four forms of aspartic acid (l-Asp,d-Asp, l-isoAsp, d-isoAsp) have been detected in the human brain and the isomerization may be related to the pathology of Alzheimer’s disease.38 Complete separation of these four isomers is difficult to achieve because of their structural similarity.39 Figure 2a shows the LC chromatogram of mixture of DAEFR, DAEFR, DAEFR, DAEFR (underlined residues corresponds to d-amino acid and bold residues correspond to isoaspartic acid). Only three LC peaks are observed because the l-isoAsp and d-Asp containing peptides coelute. 4-iodobenzoic acid (structure shown in the inset of Figure 2b) is one of the chromophores that can be used for generating radicals for RDD (IX will be used to represent the 4-iodobenzoic acid modification of X, where X is any amino acid).34 Figure 2b shows the LC chromatogram of the same four peptide isomers after covalent modification by 4-iodobenzoic acid. All four DAEFR isomers are nearly baseline separated. In addition, the elution times are shifted and the elution order is changed. It is clear that the addition of the hydrophobic tag significantly impacts the elution properties of DAEFR, leading to improved separation.
Figure 2.

LC chromatogram of the four isomers of DAEFR (a) before and (b) after modification by 4-iodo-benzoic acid. The four isomers are easily separated after modification. (c) LC chromatogram of a peptide mixture containing IDVGSNK-NH2, IDVGSNK-NH2, and ILDLAGR. The IDVGSNK epimers cannot be completely separated. (d) MS3 (RDD) spectra extracted from points marked by blue asterisks in panel c. The two spectra are significantly different, indicating partial separation of the two peptides. (e) LC chromatogram of a single peptide from the same run at a later elution time. (f) MS3 (RDD) spectra corresponding to the blue asterisks from panel e. The two spectra are almost identical. IX represents the 4-iodo-benzoic acid modification of X where X is any amino acid. Underlined residues correspond to d-amino acids, and bold residues correspond to isoaspartic acid.
Some peptide isomers do not separate even after modification with 4-iodobenzoic acid. For example, in Figure 2c a single peak is detected for the peptide DVGSNK-NH2 despite the fact that two epimers (both d- and l-Ser) are present in the solution. Fortunately in this case the two fragmentation spectra for the epimers are sufficiently distinct that the presence of two species can still be detected. This is achieved by examination of the leading and trailing edge of the LC peak, which yields the corresponding MS/MS spectra shown in Figure 2d and an R value of 3.1. Analysis of these same epimers when injected individually into the instrument yields an R value of 13.29 It is clear from this data that the epimers are still partially resolved in the LC even though a single peak is apparent on the chromatogram. For comparison, the LC chromatogram and MS/MS spectra for the leading and trailing edge of a peak containing a single synthetic peptide that was added to the sample are shown in Figures 2e and 2f. In this case, the two spectra are virtually identical as expected.
Data Analysis
When attempting to identify potentially isomerized peptides in unknown samples, candidate peptides from different LC peaks are selected if they have the same mass and exhibit similar fragmentation patterns. In addition, the leading and trailing edge of each individual LC peak is examined for differing MS/MS spectra. In each situation, the R value is calculated from the relevant tandem MS spectra. Theoretically, any Risomer value which is bigger than 1 should indicate the presence of isomers. However, in reality, MS/MS spectra are not perfectly reproducible (especially on the limited LC-MS time scale) and the relative fragmentation abundances of the same peptide in two different spectra acquired at different times are always slightly different. Previous studies have reported that values of R > 1.2 are sufficient to indicate statistically significant differences in spectra, based on the reproducibility of ion intensity measurements from direct injection experiments.26 However, in an LC-MS run, the error is higher because only a few scans are averaged to obtain the MS/MS spectra for one peptide. In contrast, spectra are usually averaged over 50–100 scans in direct infusion experiments, which significantly reduces variation of the mean intensities. To establish the relevant threshold for R values reported herein, a standard LC-MS run was performed on a mixture of synthetic peptides without any isomers using both CID and RDD under identical conditions to those employed on actual samples. The R values for each peptide were calculated from two dissociation spectra at the beginning and the end of each LC peak. Six R values were obtained this way for CID: 1.13, 1.20, 1.88, 1.55, 1.27, and 1.35. A standard t test was performed and the 99% confidence interval corresponds to R values from 0.94 to 1.85. On the basis of these numbers, we have set the threshold to identify peptide isomers from CID fragmentation in this work to R values >1.9. For RDD, the ion intensities vary slightly more and the threshold to identify peptide isomers corresponds to R values >2.4 (See Supporting Information). Finally, to eliminate interference from chemical noise, the relative intensities of the fragment pairs for the calculation of R values must be higher than 10% of the base peak in at least one of the two spectra. Furthermore, peaks assigned as sequential fragments and 13C isotopes tend to have higher errors and are not used to determine R values.
Localization of the isomerized residue in a peptide requires comparison of data obtained from biological sources with standard peptides. In these comparisons, the data for sample peptides and standard peptides are acquired in separate LC-MS runs. Hence, more error arises from different ionization efficiency or other random uncertainties between different LC-MS runs. Similar metrics to those used to distinguish dissimilar peptides from each other can be used to identify whether two peptides are likely to be the same. Although the R value equation is used for the comparison, we will refer to similarity scores as S values to avoid confusion. The criteria in this case will be that the S value should be lower than the threshold, which will indicate that the two peptides cannot be distinguished from each other and are likely identical. To establish the S value threshold for positive identification of peptide isomers by comparing the MS/MS spectra with standard peptides, a set of standard LC-MS runs were performed as described in the Experimental Methods section. The threshold for positive identification is 1.9 for CID and 3.2 for RDD. S values below these numbers will indicate that the peptides are likely the same.
In addition to the types of amino acid isomerization discussed up to this point, there are a few other modifications that do not result in any mass change that are worth mentioning. For example, cis/trans-proline isomerization can have significant structural implications. However, this type of isomerization is typically dynamic and it is unlikely that cis/trans-proline isomers of small peptides can be chromatographically separated at room temperature.40 Furthermore, our results do not suggest a bias toward identification of isomerization for peptides that contain proline, as proline is present in both isomerized and unmodified peptides. Another type of modification that could occur in some rare cases involves two amino acids that are inverted in sequence,41 which could lead to separation of the isomers by liquid chromatography. It is not anticipated that such isomers will occur frequently enough to significantly impact our results, and these isomers would be identified by analysis of the MS/MS data if fragmentation between the relevant residues was observed.
Sheep Crystallins
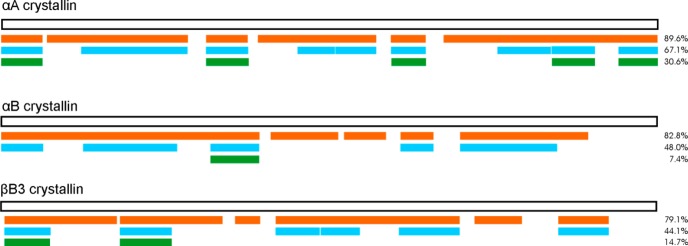
We applied the protocol described above to examine crystallin proteins extracted from the eye lens of an approximately one year old sheep. Three variants of crystallin identified from the ovine database were examined in significant detail (αA-crystallin, αB-crystallin, βB3-crystallin).42 A comprehensive list of identified peptide isomers from these three proteins is provided in Table 1, which includes many isomers that have never been previously identified (see Supporting Information for representative mass spectra of some of the peptides). The sequence coverage, degree of isomerization, and epimerization for these proteins are summarized in graphical format in Figure 3. The sequence coverage (orange bar in Figure 3) is excellent for all three proteins. Areas that are missing are primarily because of very short peptide fragments. The degree of isomerization is shown for each protein in the blue bars of Figure 3. These bars represent the presence of any isomer that was detected (presumably most are due to the presence of isoaspartic acid or aspartic acid/serine epimerization). The degree of isomerization is highest for αA-crystallin (62%), indicating that the most abundant protein is also subject to the greatest amount of modification. αB-crystallin and βB3-crystallin exhibit a similar degree of isomerization, just below 50% of the total sequence. In the green bars in Figure 3, the extent of epimerization is shown. We consider a peptide to be epimerized if the number of detected isomers is >4 for peptides containing two aspartic acids, >2 for peptides containing one aspartic acid, or >1 for peptides that lack aspartic acid. The degree of suspected epimerization is again greatest for αA-crystallin. Although it is possible that some of the isomers identified in the blue but not green bars of Figure 3 could also be epimers, this possibility is probably less likely given that aspartic acid is the most likely residue to isomerize and the rate of isoaspartic acid formation is greater than that of epimerization. Nevertheless, it is possible for exceptions to exist. Additionally, a detailed list of other identified proteins predicted to be crystallins in the newly released sheep database is shown in Supporting Information Table S2.
Table 1. Identified Peptide Isomers from Sheep Eye Lens Digesta,
| peptide sequence | crystallin | number of peaks in LC separation | relative abundance (%)f | number of peptide isomers confirmed by MS/MSb |
|---|---|---|---|---|
| Ac-1MDIAIQHPWF K11c | αA | 4 | 3.7%, 1.2%, 93.9%, 1.2% | 4 |
| 22LFDQFFGEGL FEYDLLPFLSSTISPYYR49 | αA | 3 | 7.8%, 32.4%, 59.8% | 3 |
| 55TVLDSGISEV R65 | αA | 3 | 47.4%, 50.8%, 1.8% | 4g,i |
| 79HFSPEDLTVK88 | αA | 2 | 10.4%, 89.6% | 2 |
| 89VQEDFVEIHG K99 | αA | 1 | 2 | |
| 104QDDHGYISR112d,e | αA | 5 | 4.7%, 21.1%, 15.1%, 3.1%, 56.0% | 4 |
| 132SLSADGMLTF SGPK145 | αA | 2 | 13.7%, 86.3% | 2 |
| 146VPSGVDAGHS ER157 | αA | 3 | 8.7%, 53.3%, 38.0% | 3 |
| 164EEKPSSAPSS173e | αA | 3 | 11.8%, 82.3%, 5.9% | 3 |
| Ac-1MDIAIHHPWI R11 | αB | 2 | 9.1%, 91.9% | 2 |
| 23LFDQFFGEHL LESDLFPAST SLSPF47 | αB | 2 | 9.3%, 90.7% | 2 |
| 57APSWIDTGLS E MR69 | αB | 6 | 1.6%, 34.0%, 1.0%, 1.8%, 59.7%, 1.9% | 4 |
| 108QDEHGFISR116d,e | αB | 2 | 4.2%, 95.8% | 2 |
| 124IPADVDPLTI TSSLSSDGVL TVNGPR149 | αB | 1 | 2 | |
| Ac-2AEQHSAPEQA AAGK15c | βB3 | 1 | 2i | |
| 39C**ELTAEC**PNL TESLLEK55h | βB3 | 2 | 14.2%, 85.8% | 2 |
| 89WDAWSNSHHSDSLL102 | βB3 | 1 | 2 | |
| 103SLRPLHIDGP DHK115 | βB3 | 1 | 2 | |
| 129MEIVDDDVPS LWAHGFQDR147 | βB3 | 1 | 2 | |
| 180HWNEWDANQP QLQSVR195 | βB3 | 1 | 2 |
Single letter codes are used for amino acids. Underlined residues correspond to most likely sites of epimerization. Bold residues are likely sites of isomerization. The UniprotKB Accession Numbers for the identified proteins are αA crystallin, Q5ENZ0, αB crystallin, W5Q0R4, and βB3 crystallin, Q52NW3.
For peptides observed in multiple HPLC peaks, MS/MS spectra are compared carefully to confirm the number of peptide isomers.
Ac- represents N-terminal acetylation.
N-terminal glutamine cyclization.43
These peptides were identified by searching the data against a smaller database as detailed in the Experimental Methods section above.
Calculated from peak area in the extracted ion chromatogram.
Determined from the combination of RDD and CID results.
The double star represents iodoacetamide modified cysteine (+57 Da).
Isomers were confirmed by comparison with synthetic peptides.
Figure 3.
Sequence coverage (orange), degree of isomerization (blue) and degree of epimerization (green) for αA-crystallin, αB-crystallin, and βB3-crystallin. The white bar represents the full protein sequence.
Identification of peptide isomers as outlined in Table 1, and Figure 3 does not reveal the amino acid specific sites of isomerization. Previous work has revealed that aspartic acid is the most likely site for both isomerization and epimerization because of aging.14 Therefore, peptides containing aspartic acid are likely modified at this residue. Serine is the second most likely site to undergo spontaneous epimerization.20 All peptides that we have identified contain either aspartic acid or serine or both. It is possible to positively identify the site of isomerization by synthesizing synthetic standards and comparing the respective MS/MS spectra, as detailed below. Importantly, recent work has also demonstrated that ion mobility is able to pinpoint sites of epimerization for some peptides, which may simplify site identification in future experiments on crystallins.44 For the present study, we synthesized a small number of authentic standards to compare with our results. TVLDSGISEVR is a tryptic peptide from α-crystallin which separates into 3 peaks by LC (Supporting Information Figure S1). By comparing the MS/MS CID spectra with those obtained from synthetic peptides where all four different forms of aspartic acid were incorporated, only the peak at 24.76 min is immediately identified as an L-isoAsp variant with an SCID value of 1.3 (SCID <1.9 indicates high similarity, see description above). The peak at 28.53 min is closest to the d-Asp variant with an SCID value of 2.1 (which may be above threshold due to the very low intensity of this peptide). The peak at 26.59 min does not match any of the isomers well, with SCID values of 2.7, 13, 3.5, and 3.9 for l-Asp, l-isoAsp, d-Asp, and d-isoAsp, indicating that the remaining two Asp isomers are coeluting or that potentially the serine20 residue is also epimerized. After iodo-benzoic acid modification, three peaks are again detected by LC (Supporting Information Figure S2); however, comparison with the synthetic peptides by RDD reveals that the three peaks represent l-isoAsp, d-isoAsp, and l-Asp with corresponding SRDD values of 2.6, 2.4, and 3.1 (SRDD < 3.2 indicates high similarity, see description above). The greater structural sensitivity of RDD allows for more confident assignment of the data (all relevant S values are shown in Supporting Information Figure S2). Taken together, the combination of RDD and CID confirms the presence of all four Asp isomers and suggests that the most likely explanation for the unidentified peak in Supporting Information Figure S1 is not serine epimerization but rather coelution of d-isoAsp and l-Asp.
The importance of using MS/MS data in conjunction with LC separation is further illustrated by examples extracted from the data in Table 1. For example, Ac-2AEQHSAPEQAAAGK15 is detected in a single LC peak, but the leading and trailing edges of the peak are sufficiently distinct to confirm the presence of two isomers. Furthermore, this peptide does not contain aspartic acid, which suggests that epimerization at serine is the most likely cause for the presence of two isomers. This hypothesis was confirmed by comparison with synthetic standards containing d-Ser as shown Supporting Information Figure S4. Similarly, a total of seven peptides are detected as single LC peaks in Table 1, yet the MS/MS data confirm the presence of two isomers for all of these peptides. If separation by LC alone were used for isomer identification, modification of all of these peptides would be missed. There are also situations where the number of peaks that are separated by LC exceed the number of isomers that can be confirmed by examination of the MS/MS data (for example 57APSWIDTGLSEMR69). It is possible that CID may not be able to distinguish these isomers (although it should be noted that failure to distinguish epimers has never been observed in RDD experiments on synthetic standards). Another explanation could be that coelution with another molecule may lead to observation of the same peptide in two LC peaks. In such cases, closer scrutiny is warranted to determine the true number of isomers.
Isomerization and Functionality
The crystallin proteins are primarily responsible for maintaining transparency in the eye lens and are therefore critically important for eyesight. Crystallins function as both structural and chaperone proteins, indicating that modifications such as isomerization or epimerization may significantly influence their effectiveness. The degree of isomerization for the two most abundant proteins, αA-crystallin and αB-crystallin, is shown in Figure 4 as a function of the structural region of the protein. Both α-crystallins form dynamic assemblies of larger oligomers where each monomer is comprised of three domains, an N-terminal region, an alpha crystallin domain, and a C-terminal extension. The terminal domains are largely disordered and thought to mediate oligomer assembly, which is vital for chaperone functionality. In Figure 4, the sequence of each protein is represented by the color coded bar with aspartic acid and serine residues indicated by black and white asterisks, respectively. Below each bar, the degree of isomerization detected within each peptide is shown (as determined by the total fractional abundance of isomers). It is clear that αA-crystallin undergoes the greatest extent of isomerization, with significant amounts of isomerization being detected in both the N-terminal domain and the C-terminal extension. The alpha crystallin domain, which corresponds to the ordered, globular part of the structure, is isomerized to a lesser extent although some modification is noted. αB-crystallin is less abundant, and appears to be modified primarily in the N-terminal region. It is certainly possible that epimerization or formation of isoaspartic acid could significantly perturb the delicate structural interactions that regulate the overall oligomerization state of the crystallins and significantly impact their ability to function properly.
Figure 4.

Isomerization ratio of αA and αB crystallins. Different colors indicate the three structural regions of crystallin, with the N-terminal region in orange, the α-crystallin domain in blue, and the C-terminal extension in purple.45 The black asterisks represent aspartic acid residues and the white asterisks represent serine residues. The stars (peptide 89–99 in αA and peptide 124–149 in αB) indicate regions where isomerization was detected but not quantified because of incomplete separation by HPLC.
Conclusions
Although direct comparison between different species and age groups is difficult, the results outlined in Table 1 reveal numerous previously undetected sites of isomerization relative to examination of human crystallin proteins from significantly more aged samples.16 Given that isomerization is known to increase as a function of age, it is likely that the methods outlined herein would lead to identification of many additional sites in samples of greater age. The results obtained for the crystallin proteins illustrate that it is possible to identify nonmass shifting PTMs by LC-MS/MS analysis in samples of significant complexity. Instances of partial separation of isomers in these experiments serve to highlight the importance of using MS/MS for isomer identification, otherwise numerous isomers that fail to chromatographically resolve will be missed. In addition, we chose conservative threshold values for epimer/isomer identification, suggesting that there may be some additional epimers/isomers that were not identified.
Results obtained on eye lens sheep crystallins reveal that a significant amount of isomerization can be observed even for a young animal. Both epimerization of aspartic acid and serine containing peptides was observed. The most abundant protein, αA-crystallin was isomerized and epimerized to the greatest extent. Examination of the isomerization in relation to the structural regions of αA-crystallin reveals that modification is more prevalent in regions of the protein that are not structurally well ordered. These modifications may impact in the functionality of αA-crystallin and may be one of the causes of age-related cataract. The greater isomerization of unstructured regions may also suggest that natively disordered proteins in general are more susceptible to isomerization, although this idea will have to be examined in future studies.
Acknowledgments
The authors gratefully acknowledge funding from the National Center for Research Resources (5R21RR032391-02) and the National Institute of General Medical Sciences (8 R21 GM103531-02) from the National Institutes of Health.
Supporting Information Available
Additional material as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sharma K. K.; Santhoshkumar P. Biochim. Biophys. Acta 2009, 1790, 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemendal H.; de Jong W.; Jaenicke R.; Lubsen N. H.; Slingsby C.; Tardieu A. Prog. Biophys. Mol. Biol. 2004, 86, 407–485. [DOI] [PubMed] [Google Scholar]
- Bloemendal H. Science 1977, 197, 127–138. [DOI] [PubMed] [Google Scholar]
- Delbecq S. P.; Klevit R. E. FEBS Lett. 2013, 587, 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz J. Exp. Eye Res. 2009, 88, 190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A. R.; Lubsen N. H.; Slingsby C. Int. J. Biochem. Cell Biol. 2012, 44, 1687–1697. [DOI] [PubMed] [Google Scholar]
- Baldwin A. J.; Lioe H.; Robinson C. V.; Kay L. E.; Benesch J. L. P. J. Mol. Biol. 2011, 413, 297–309. [DOI] [PubMed] [Google Scholar]
- Hains P. G.; Truscott R. J. W. J. Proteome Res. 2007, 6, 3935–3943. [DOI] [PubMed] [Google Scholar]
- Hoehenwarter W.; Klose J.; Jungblut P. R. Amino Acids 2006, 30, 369–389. [DOI] [PubMed] [Google Scholar]
- Spector A.; Roy D. Proc. Natl. Acad. Sci. U. S. A. 1978, 75, 3244–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry R. E.; Swamy M. S.; Abraham E. C. Exp. Eye Res. 1987, 44, 269–282. [DOI] [PubMed] [Google Scholar]
- Wilmarth P. A.; Tanner S.; Dasari S.; Nagalla S. R.; Riviere M. A.; Bafna V.; Pevzner P. A.; David L. L. J. Proteome Res. 2006, 5, 2554–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz-Timme S.; Collins M. J. Ageing Res. Rev. 2002, 1, 43–59. [DOI] [PubMed] [Google Scholar]
- Fujii N. Biol. Pharm. Bull. 2005, 28, 1585–1589. [DOI] [PubMed] [Google Scholar]
- Cloos P. A. C.; Fledelius C. Biochem. J. 2000, 345, 473–480. [PMC free article] [PubMed] [Google Scholar]
- Fujii N.; Sakaue H.; Sasaki H.; Fujii N. J. Biol. Chem. 2012, 287, 39992–40002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger T.; Clarke S. J. Biol. Chem. 1987, 262, 785–794. [PubMed] [Google Scholar]
- Krause E.; Beyermann M.; Dathe M.; Rothemund S.; Bienert M. Anal. Chem. 1995, 67, 252–258. [DOI] [PubMed] [Google Scholar]
- Kaneko I.; Yamada N.; Sakuraba Y.; Kamenosono M.; Tutumi S. J. Neurochem. 1995, 65, 2585–2593. [DOI] [PubMed] [Google Scholar]
- Hooi M. Y. S.; Raftery M. J.; Truscott R. J. W. Protein Sci. 2013, 22, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooi M. Y. S.; Truscott R. J. W. Age 2010, 33, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargaeva N. P.; Lin C.; O’Connor P. B. Anal. Chem. 2009, 81, 9778–9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez L. J.; Shimizu T.; Satomi Y.; Betancourt L.; Besada V.; Padron G.; Orlando R.; Shirasawa T.; Shimonishi Y.; Takao T. Rapid Commun. Mass Spectrom. 2000, 14, 2092–2102. [DOI] [PubMed] [Google Scholar]
- Bai L.; Romanova E. V.; Sweedler J. V. Anal. Chem. 2011, 83, 2794–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams C. M.; Zubarev R. A. Anal. Chem. 2005, 77, 4571–4580. [DOI] [PubMed] [Google Scholar]
- Serafin S. V.; Maranan R.; Zhang K. L.; Morton T. H. Anal. Chem. 2005, 77, 5480–5487. [DOI] [PubMed] [Google Scholar]
- Sachon E.; Clodic G.; Galanth C.; Amiche M.; Ollivaux C.; Soyez D.; Bolbach G. Anal. Chem. 2009, 81, 4389–4396. [DOI] [PubMed] [Google Scholar]
- Tao W. A.; Zhang D. X.; Nikolaev E. N.; Cooks R. G. J. Am. Chem. Soc. 2000, 122, 10598–10609. [Google Scholar]
- Tao Y.; Quebbemann N. R.; Julian R. R. Anal. Chem. 2012, 84, 6814–6820. [DOI] [PubMed] [Google Scholar]
- Hurtado P. P.; O’Connor P. B. Mass Spectrom. Rev. 2012, 31, 609–625. [DOI] [PubMed] [Google Scholar]
- Yang H.; Fung E. Y. M.; Zubarev A. R. J. Proteome Res. 2009, 8, 4615–4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargaeva N. P.; Goloborodko A. A.; O’Connor P. B.; Moskovets E.; Gorshkov M. V. Electrophoresis 2011, 32, 1962–1969. [DOI] [PubMed] [Google Scholar]
- Chan W. C.; White P. D.. FMOC Solid Phase Peptide Synthesis, 1st ed.; Oxford University Press: New York, 2000. [Google Scholar]
- Ly T.; Zhang X.; Sun Q. Y.; Moore B.; Tao Y. Q.; Julian R. R. Chem. Commun. 2011, 47, 2835–2837. [DOI] [PubMed] [Google Scholar]
- McKerrow J. H. Mech. Ageing Dev. 1979, 10, 371–377. [DOI] [PubMed] [Google Scholar]
- Diedrich J. K.; Julian R. R. Anal. Bioanal. Chem. 2012, 403, 2269–2277. [DOI] [PubMed] [Google Scholar]
- Bai L.; Sheeley S.; Sweedler J. V. Bioanal. Rev. 2009, 1, 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda H.; Shimizu T.; Nakajima M.; Mori H.; Shirasawa T. Bioorg. Med. Chem. Lett. 1999, 9, 953–956. [DOI] [PubMed] [Google Scholar]
- Roher A. E.; Lowenson J. D.; Clarke S.; Wolkow C.; Wang R.; Cotter R. J.; Reardon I. M.; Zurcherneely H. A.; Heinrikson R. L.; Ball M. J.; Greenberg B. D. J. Biol. Chem. 1993, 268, 3072–3083. [PubMed] [Google Scholar]
- Kalman A.; Thunecke F.; Schmidt R.; Schiller P. W.; Horvath C. J. Chromatogr., A 1996, 729, 155–171. [DOI] [PubMed] [Google Scholar]
- Brüeckner C.; Bunz S.-C.; Neusüß C.; Scriba G. K. E. Chromatographia 2012, 75, 1205–1210. [Google Scholar]
- Robertson L. J. G.; David L. L.; Riviere M. A.; Wilmarth P. A.; Muir M. S.; Morton J. D. Invest. Ophthalmol. Vis. Sci. 2008, 49, 1016–1022. [DOI] [PubMed] [Google Scholar]
- Khandke K. M.; Fairwell T.; Chait B. T.; Manjula B. N. Int. J. Pept. Protein Res. 1989, 34, 118–123. [DOI] [PubMed] [Google Scholar]
- Jia C.; Lietz C. B.; Yu Q.; Li L. Anal. Chem. 2014, 86, 2972–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. K.; Kim R.; Kim S. H. Nature 1998, 394, 595–599. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.