

Abstract
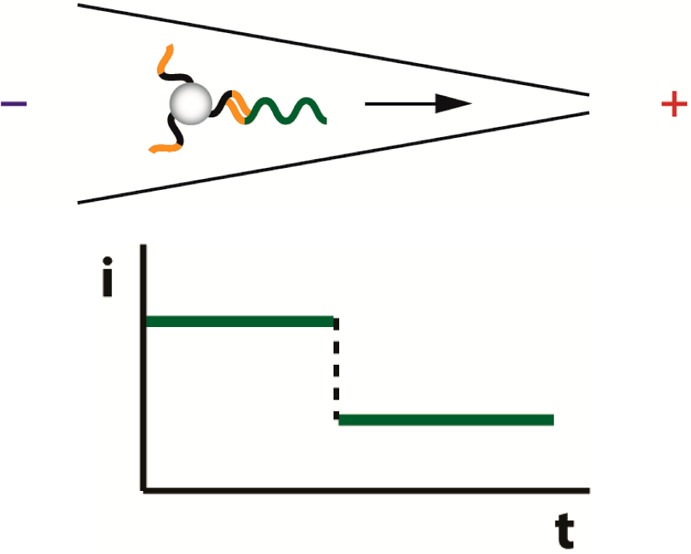
Target DNA fragments at 10 fM concentration (approximately 6 × 105 molecules) were detected against a DNA background simulating the noncomplementary genomic DNA present in real samples using a simple, PCR-free, optics-free approach based on electromechanical signal transduction. The development of a rapid, sensitive, and cost-effective nucleic acid detection platform is highly desired for a range of diverse applications. We previously described a potentially low-cost device for sequence-specific nucleic acid detection based on conductance change measurement of a pore blocked by electrophoretically mobilized bead-(peptide nucleic acid probe) conjugates upon hybridization with target nucleic acid. Here, we demonstrate the operation of our device with longer DNA targets, and we describe the resulting improvement in the limit of detection (LOD). We investigated the detection of DNA oligomers of 110, 235, 419, and 1613 nucleotides at 1 pM to 1 fM and found that the LOD decreased as DNA length increased, with 419 and 1613 nucleotide oligomers detectable down to 10 fM. In addition, no false positive responses were obtained with noncomplementary, control DNA fragments of similar length. The 1613-base DNA oligomer is similar in size to 16S rRNA, which suggests that our device may be useful for detection of pathogenic bacteria at clinically relevant concentrations based on recognition of species-specific 16S rRNA sequences.
Ultrasensitive, rapid, and cost-effective sequence-specific detection of nucleic acids (NAs) is of great significance for pathogen detection,1 medical diagnostics,2 drug discovery,3 and forensic investigations.4 Many strategies and technologies with high sensitivity and specificity have been developed for NA detection using optical,5 mechanical,6 and electrochemical signal transduction.7 For instance, an electrochemical strategy with dual amplification by polymerase reaction and hybridization chain reaction (HCR) enabled an 8 fM concentration limit of detection (LOD)8 of target NA. In a study by Zeng and co-workers, a lateral flow biosensor based on isothermal strand-displacement polymerase reaction and gold nanoparticles was designed for visual detection of NAs with a LOD of 0.01 fM.9 The same group developed a hairpin DNA probe and gold nanoparticle assay for multiplex DNA detection with a 0.1 fM LOD.10 In another approach, an electrochemiluminescence DNA sensor combined with isothermal circular amplification exhibited a 5 aM detection limit.11 A sensor based on a conducting nanowire with a LOD of 0.1 fM was fabricated for detection of a 19 bp breast cancer gene by the Mulchandani group.12 Giri et al. adapted a quantum dot (QD) barcode platform to detect infectious agents with a 10 fM concentration limit.13 Also, magnetic particle-mediated aggregation combined with rolling circle amplification was used to detect specific NAs with a 124 fM detection limit.14 In another approach, a PCR based magnetic assembled sensor was developed for DNA detection at 4.26 aM.15 However, the majority of these technologies rely on sample amplification using PCR, HCR, or rolling circle amplification to reach low detection limits and require expensive instrumentation and/or labels other than a complementary oligonucleotide probe. Therefore, the need remains for development of technologies for fast, sensitive, and cost-effective detection of specific NAs.
We previously demonstrated a binary-mode, label-free, optics-free, potentially low-cost sequence-specific NA detection device with a detection limit of 10 pM.16 For many applications, a binary-mode device that provides a rapid, accurate yes/no response regarding the presence/absence of a specific NA sequence can address the question of primary concern. For example: Is the mutation present? Does the sample contain NA identifying an individual? Is a pathogenic species present in a sample beyond some threshold concentration? It is important to note that questions regarding threshold concentrations can be addressed through sample processing given a limit of detection for the binary-mode device. Our system utilizes polystyrene beads conjugated with peptide nucleic acid (PNA) oligonucleotide probes. In PNA, the negatively charged phosphodiester backbone of DNA/RNA is replaced with repetitive uncharged units of N-(2-aminoethyl) glycine to which the bases are attached with a methyl carbonyl linker, thus enabling the construction of charge-neutral probe–bead conjugates.17 Introduction of complementary 20-mer target DNA to these neutrally charged PNA–bead conjugates resulted in sequence-specific binding of single-stranded DNA to the PNA, imparting negative charge to the assembly thereby making it electrophoretically mobile. Following placement of these bead conjugates with hybridized target in a conical capillary with tip diameter smaller than the bead diameter, the beads were electrophoretically driven toward the capillary tip, blocking it and producing a large, easily measurable, and persistent change in electrical current (Figure 1).
Figure 1.
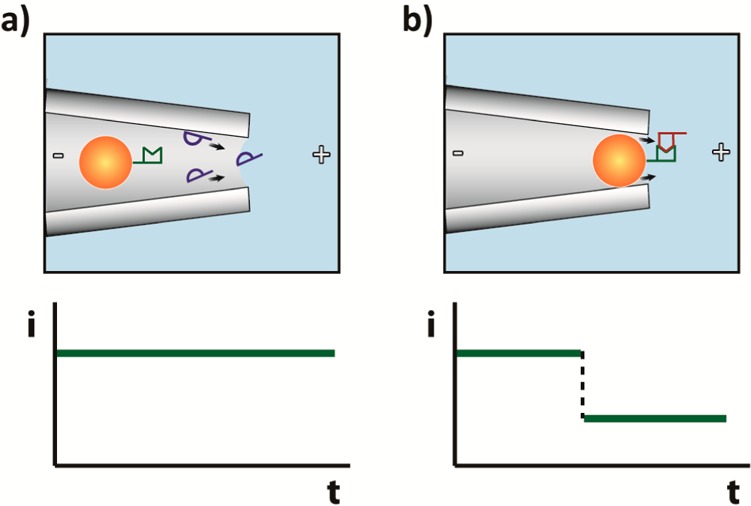
(a) In an applied electric field (+ and – symbols), charge neutral PNA–beads in a conical capillary are electrophoretically immobile in the presence of noncomplementary NA, which passes through the capillary pore without significantly altering the measured current. (b) Complementary NA binds to the PNA–bead, making the complex negatively charged and electrophoretically mobile, thereby resulting in the NA–PNA–bead blocking the capillary, which results in a large and persistent reduction in conductance.
In the work described here, we explored the ability of our device to detect longer DNA fragments. Because our detection concept is dependent upon charge imparted to the PNA–beads by the bound target NA, we hypothesized that the increased charge added to the bead conjugate per target DNA molecule would result in a reduced limit of detection.
PROCEDURE
Materials
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Carboxylic acid-functionalized, 3 μm diameter polystyrene microspheres were purchased from Polysciences, Inc. (Warrington, PA). Peptide nucleic acid (PNA) was purchased from Bio-Synthesis, Inc. (Lewisville, TX) as high performance liquid chromatography (HPLC)-purified and lyophilized powders. The PNA capture probe used was NH2-(CH2CH2O)12-GC AA CA GT CT TC. Methoxypolyethylene glycol amine, CH3O-(CH2CH2O)3-NH2, MW 350, was obtained from Nanocs, Inc. (New York, NY). Prepulled borosilicate micropipettes with a 2 μm inside tip diameter were bought from World Precision Instruments, Inc. (Sarasota, FL). NovaBlue competent Escherichia coli K-12 cells and pET-21b (+) plasmid vector were purchased from EMD Millipore, Inc. (Billerica, MA). Restriction endonucleases ScaI, PvuI, PstI, BsaI, and EcoNI were obtained from New England BioLabs, Inc. (Ipswich, MA). Finally, QIAprep Spin Miniprep, QIAquickGel Extraction, and MinElute Reaction Cleanup Kits were purchased from QIAGEN (Valencia, CA).
Probe Coupling to Microspheres
50 μL of 3 μm diameter, carboxylic acid-functionalized polystyrene microspheres at 1.69 × 109/mL were washed three times with MES buffer (60 mM 2-(N-morpholino) ethanesulfonic acid, pH 5.5). The diameter (determined by dynamic light scattering (DLS)) and ζ-potential (see below) of the beads before conjugation was measured with a Zetasizer Nano-ZS (Malvern Instruments) and found to be 3716 nm and −87 mV, respectively. After each wash, the microspheres were centrifuged at 14000 rpm for 15 min; after the third wash, the beads were resuspended in 0.6 mL of coupling buffer (100 mM 1-[3-(dimethylamine)propyl]-3-ethylcarbodiimide (EDC) in MES buffer) and incubated at 50 °C for 45 min. 10 nM amine-functionalized PNA probes was added to the coupling buffer and incubated with the beads at 50 °C for 2 h. mPEG-amine (100 mM) was added to the reaction mixture and incubated at 50 °C for 1 h to reduce nonspecific binding of nucleic acids to the beads. 100 mM ethanolamine was added to the beads to cap residual carboxyl groups and incubated at 50 °C for an additional hour. Finally, the beads were washed four times in 0.4× SSC buffer (60 mM NaCl, 6 mM trisodium citrate, 0.1% Triton X-100, pH 8) and stored in PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) at 4 °C. Prior to hybridization, the zeta potentials of PNA–bead batches were measured in 1 mM KCl, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, pH 7.0) to ensure the near electroneutrality of the beads. The ζ-potential after four washes typically was ∼−2.4 mV. At this low potential, the beads tended to aggregate, which prevented meaningful diameter measurement by DLS.
Sample Preparation
Plasmid Preparation
Competent E. coli K-12 bacteria (NovaBlue) were transformed with the pET-21b(+) vector following the Novagen protocol. NovaBlue cells were removed from freezer and thawed on ice for 2–5 min. 20 μL cell aliquots were placed in prechilled polypropylene tubes to which 1 μL of purified plasmid DNA was added and incubated on ice for 5 min. Then the tubes were heated for 30 s in a 42 °C water bath and placed on ice for 2 min. 80 μL of room temperature SOC medium (2% tryptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, and 20 mM glucose) was added to each tube. An agar plate containing Luria Broth (LB) (10 g/L tryptone, 10 g/L NaCl, and 5 g/L yeast extract in DI water, pH 7.5) was coated with 60 μL SOC medium and 100 mg/L ampicillin. 25 μL of the induced cells was spread over this plate and incubated inverted at 37 °C for 15 h. After incubation, a single colony of plasmid-induced NovaBlue cells was selected from the agar plate and incubated in 10 mL LB media supplemented with 100 mg/L ampicillin at 37 °C for 8 h to provide a starter culture. Subsequently, 1 mL of starter culture was diluted into 500 mL of LB medium and the solution was incubated at 37 °C for 14 h. The bacterial culture was aliquoted into 10 50 mL Falcon tubes and centrifuged at 25000g for 15 min at 4 °C. The supernatant was removed and the pellets were stored at −80 °C.
For plasmid extraction, one bacterial pellet was thawed in a 50 °C DI water bath. Plasmid extraction and purification was performed following the QIAprep Spin Miniprep Kit protocol. The concentration of extracted plasmids was measured using a NanoDrop2000 (ThermoScientific) spectrophotometer at 260 nm wavelength.
Plasmid Digestion and DNA Isolation and Purification
The isolated plasmid was double digested by selected restriction enzyme pairs from ScaI, PvuI, PstI, BsaI, and EcoNI (Figure 2a). The enzyme pairs were selected such that target fragments of different lengths were produced containing the sequence, 3′-GA AG AC TG TT GC-5′; and control fragments of different lengths were produced not containing the target sequence. Specifically, ScaI and PvuI, acting at bases 4537 and 4427 on the plasmid respectively, produced a 110-base, target-containing fragment, T1 (Figure 2), with the remainder of the plasmid forming a 5332-base, non-target-containing fragment. Similarly, ScaI (4537) and PstI (4302) produced target fragment T2 (235 bases); while ScaI (4537) and BsaI (4118) produced target fragment T3 (419 bases), and PvuI (4427) and EcoNI (598) produced target fragment T4 (1613 bases). Control fragments not containing the target sequence were produced using PvuI and PstI (C1, 125 bases), PstI and BsaI (C2, 184 bases), PvuI and BsaI (C3, 309 bases), and ScaI and EcoNI (C4, 1503 bases).
Figure 2.

Schematic of DNA oligomer preparation. (a) Purified pET-21b plasmids were enzymatically digested by selected pairs of ScaI, PvuI, PstI, BsaI, and EcoNI restriction enzymes, producing fragments of different lengths. The target DNA sequence complementary to the PNA probe is located beginning at plasmid position 4427 (orange band). Plasmid digestion by ScaI and PvuI produced a 110-base, target-containing fragment, T1. Plasmid digestion by PvuI and PstI produced a 125-base, target-free control fragment, C1. Other fragments were produced similarly: T2 (235 bases) using ScaI and PstI, T3 (419 bases) using ScaI and BsaI, T4 (1613 bases) using by PvuI and EcoNI), C2 (184 bases) using PstI and BsaI, C3 (309 bases) using PvuI and BsaI, and C4 (1503 bases) using ScaI and EcoNI. (b) Following digestion, the DNA was isolated by gel electrophoresis, extracted, and purified. (c) Purified double-stranded DNA was denatured and hybridized with bead–PNA probe conjugates. (d) DNA–PNA–bead mixture was injected into the micropipette for electrical detection.
After plasmid digestion, target and control DNA fragments were isolated by 1% agarose gel electrophoresis, were excised from the gel, and were purified using QIAquick gel extraction and MinElute cleanup kits (Figure 2b) and eluted into 10 μL of buffer EB (10 mM Tris–Cl, pH 8.5). The DNA concentration of the fragment preparations was measured using the NanoDrop spectrophotometer (260 nm wavelength) prior to hybridization experiments with bead conjugates.
Hybridization
Double-stranded DNA samples were denatured into single strands in a 90 °C water bath for 20 min. Prior to hybridization, 2.1 × 106 PNA–beads were washed twice with 0.4× SSC buffer and once with hybridization buffer (750 mM NaCl, 10 mM Tris–HCl, pH 7.0). DNA samples and PNA–beads were resuspended in hybridization buffer and incubated at 37 °C overnight (Figure 2c). Target and control DNA of each strand length was hybridized with PNA–beads in a 100 μL reaction volume at different concentrations, following a serial dilution scheme.
ζ-Potential and Electrical Measurements
The ζ-potential and electrical measurements were performed as previously described.16 Following incubation with single-stranded target and/or control DNA, PNA–beads were injected into the micropipette and drawn toward the pore (i.e., pipette tip) under 25 V applied potential difference (Figure 2d). The apparatus containing the capillary and electrodes was mounted on the stage of a Leica DMIRB inverted microscope for simultaneous microscopic observation. ζ-Potentials and measured currents for each experiment are listed in the Supporting Information. With the exception of the runs at 10 fM, one capillary was used for each DNA concentration at each size of target or controlled DNA, and three capillaries were employed for the runs at 10 fM. Thus, a total of 48 capillaries were used to obtain the data described below (see the Supporting Information). In addition, control runs were performed in which PNA–beads not exposed to DNA and DNA alone were added to the capillaries.
Results and Discussion
Previously, we used our platform for sequence specific detection of 20-mer DNA containing a portion of the anthrax LF gene with a demonstrated detection limit of 10 pM.16 Many samples of biological origin prepared by shearing or other methods contain DNA of 1000 bases or more.18−20 Additionally, as our method relies on the electrophoretic blockade of a pipette tip by beads that have acquired their electrophoretic mobility through the binding of negatively charged target DNA, use of longer DNA strands would impart more negative charge per bound DNA molecule. Notably, 16S rRNA, commonly used to identify the presence of specific microbial species in a sample, measures ∼1500 bases, which is of similar length to our largest target DNA, T4 (1613 bases). We hypothesized that increasing the length of the target DNA would enable detection at lower DNA concentrations.
To assess the capability of our system to detect DNA of various longer lengths and to investigate the impact of target DNA length on the concentration LOD, we produced DNA of 110, 235, 419, and 1613 nucleotides in length all containing the same 12 base target sequence. Control studies were performed with DNA of similar lengths (125, 184, 309, and 1503 nucleotides) in which the target sequence was not present. For efficient production of target and control DNA21 we performed controlled restriction enzyme digests of pET21b(+) plasmid DNA produced by a bacterial expression system. The 12-mer target sequence complementary to our PNA probe was located at the 5′- end of the 110-mer and 1613-mer target strands, but was in the middle of the 235-mer and 419-mer targets (Figure 2a).
Following digestion, gel electrophoresis, excision, purification, and characterization, each of the target and control DNA samples were separately diluted to 1 pM, 100 fM, 10 fM, and 1 fM. The DNA in each of these solutions was denatured and hybridized with the PNA–beads. Beads incubated with each target and control concentration (1 pM–10 fM) were injected into a fresh capillary and the resultant current measured during voltage application (Figure 3). The open pore currents consistently ranged from 500 to 700 nA due to variations in capillary diameters (see the Supporting Information).16 When a block was observed, the potential was maintained for at least 60 s following the block. Blocks that persisted for ≥60 s were deemed to be permanent. Blocked currents were 10 to 30% less than open pore currents due to variations both in capillary and bead diameters (see the Supporting Information).16 To show that permanent blocks were a result of electrophoresis and not an adhesion of the bead to the capillary wall, we repeatedly reversed the voltage and reapplied it to open the pore and obtain another block (Figure 3b). Some bead blocks were observed to be transient, where the bead traveled back down the capillary after a short time (average time of 10.8 s) (Figure 3c). This was also observed in our previous work, where we hypothesized that removal of weakly attached, nonspecifically bound DNA from the bead reduced its charge, reducing the electrophoretic force on the bead and enabling its removal from the tip by the opposing electroosmotic16 flow.
Figure 3.
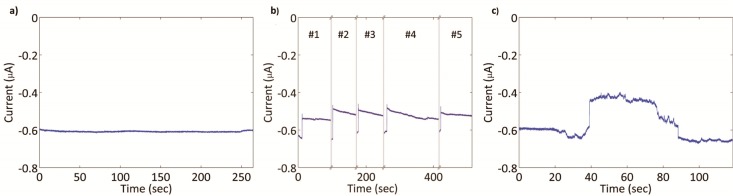
(a) Sample current trace measured with an unblocked capillary tip (pore) at −25 V. (b) Sample data showing five bead blockages (numbered) of at least 60 s and four intermittent, brief field reversals (+25 V) to remove beads from the pore (gray bars). The measured current exhibits a large step reduction (to a less negative value) when a bead immobilizes at the capillary tip. (c) Sample transient block of the pore showing brief current reduction and return to its preblock level under constant applied voltage of −25 V.
The measured currents and ζ-potentials for beads incubated with DNA oligomers at each concentration are listed in the Supporting Information. Interestingly, the measured ζ-potentials did not consistently correlate with length of bound DNA or its concentration, although it appears that a ζ-potential of at least −35 to −40 mV is required for a preparation to give a permanent block. This inconsistent correlation could be due to the heterogeneity of the population, the conformation of bound DNA, and the existence of nonspecifically bound DNA.22
Beads incubated with the 110-mer target DNA were unable to block the capillaries at any concentration tested (1 pM–1 fM), except for one observation at 10 fM. To verify that the 110-mer target was functional, we successfully detected it at 10 pM concentration (the lower limit of detection found in our previous work with 20-mer target DNA16). Permanent, reversible blocks were successfully detected for the 235-mer targets down to 100 fM and for the 419-mer and 1613-mer targets down to 10 fM. At 1 fM, no blockades were obtained for target or control DNA of any length. Table 1 shows the results of the capillary blockade detection for the different target and control DNA lengths as a function of DNA concentration. In each of the three experiments shown, the 1613-mer target DNA was detected at 10 fM.
Table 1. Summary of Detection Results for Target and Control Samplesa.
| target |
control |
|||
|---|---|---|---|---|
| [DNA] | length | detection? | length | detection? |
| 1 pM | 110 | no | 125 | no |
| 235 | yes | 184 | nob | |
| 419 | yes | 309 | no | |
| 1613 | yes | 1503 | yesc | |
| 100 fM | 110 | nob | 125 | no |
| 235 | yes | 184 | nob | |
| 419 | yes | 309 | no | |
| 1613 | yes | 1503 | no | |
| 10 fM expt 1 | 110 | no | 125 | no |
| 235 | no | 184 | no | |
| 419 | yes/nod | 309 | no | |
| 1613 | yes | 1503 | nob | |
| 10 fM expt 2 | 110 | yesd | 125 | nob |
| 235 | no | 184 | no | |
| 419 | no | 309 | no | |
| 1613 | yes | 1503 | nob | |
| 10 fM expt 3 | 110 | no | 125 | no |
| 235 | no | 184 | nob | |
| 419 | yes | 309 | no | |
| 1613 | yes | 1503 | nob | |
| 1 fM (2 expts) | 110 | no | 125 | no |
| 235 | no | 184 | no | |
| 419 | no | 309 | no | |
| 1613 | no | 1503 | no | |
A positive result indicates a blockade measured for >60 s that was reversible. A negative result is indicated when no block or a transient block (<60 s) was observed. Measurement details are provided in the Supporting Information.
Transient block observed.
Permanent block then transient block after reversal.
Transient block observed, followed by permanent block.
All control experiments showed either transient or no blockade of the pore for all DNA lengths and concentrations measured, with one exception. At 1 pM, the beads incubated with the 1503-mer control initially showed a permanent block, which was not repeated following voltage reversal; instead the previously blocking bead gave a transient block. Thus, the devices yielded essentially no false positive results.
To simulate the detection of our target sequence against a background of genomic DNA, we incubated PNA–beads with a solution containing 10 fM of the 1613-mer target sequence and 30 pM of the 1503-mer control sequence. 30 pM 1503-mer DNA approximates the 4.6 Mb E. coli genome at 10 fM after shearing. Repeatable permanent blocks of the capillary pore were observed (Table S-22, Supporting Information). As a control experiment, we incubated the PNA–beads with 30 pM of 1503-mer DNA without 10 fM of the 1613-mer target sequence and observed no permanent or transient blocks.
The successful detection of 1613-mer DNA at 10 fM concentration is consistent with our hypothesis that the concentration limit of detection could be lowered from 10 pM found in our previous work by increasing the length of the DNA bound to the bead and therefore the amount of charge imparted to the bead. By increasing the charge per bound strand, we may be compensating for a decrease in electrophoretic mobility resulting from a decreased number of bound DNA oligomers per bead at lower DNA concentrations.
We may estimate the number of bound DNA oligomers per bead using Poisson statistics:
where μ is the average number of DNA molecules per bead and Pμ(v) is the probability of finding ν molecules on one bead. Our 100 μL reaction volume contained 2.1 × 106 beads; and 10 fM DNA in this volume is approximately 6.0 × 105 molecules, which gives a bead-to-DNA ratio of 3.5:1 and a μ of 0.287 DNA/bead. With this μ, we obtain the probability of a bead having zero bound DNA of P(0) = 0.751, probability of one bound DNA, P(1) = 0.215, P(2) = 0.031, etc. Multiplication of these probabilities by the total bead count yields the number of beads having a number of bound DNA (Supporting Information, Table S-23): 1.6 × 106 beads with no DNA, 4.5 × 105 beads with 1 bound DNA, 6.5 × 104 beads with 2 bound DNA, 6190 beads with 3 bound DNA, 443 beads with 4 bound DNA, 25 beads with 5 bound DNA, 1 with 6 bound DNA, and less than 1 with more than 6 bound DNA. This analysis assumes that all DNA molecules are bound to beads (and remain bound) and ignores the binding of the noncomplementary DNA to the beads (the DNA after purification is double-stranded and denatured before incubation with the beads, giving a complementary and noncomplementary population of DNA). As such, it represents an estimate of the maximum of the number of bound target DNA per bead.
Repeating this analysis with the same number of beads, but for DNA detection at 1 fM gives μ = 0.0287, corresponding to 97.2% of the beads bound to zero DNA molecules, 58 498 beads bound to one DNA, 838 beads bound to 2 DNA, 8 beads bound to 3 DNA, and less than 1 on average bound to more than 3 DNA (Supporting Information, Table S-24). On the basis of this analysis, we suggest two possible reasons why the detection was successful at 10 fM but not 1 fM. (1) Only beads containing 4, 5, or 6 DNA (present at 10 fM but not 1 fM) have electrophoretic mobility sufficient to block the capillary; (2) beads containing fewer bound DNA (e.g., 3) can produce blockades; but less than 100% yield in bead preparation, combined with the smaller numbers of these beads present at 1 fM (8 out of 2.1 × 106), combine to give a low probability of blockage by such beads.
The assumption that almost all DNA is bound to the beads is supported by equilibrium calculations of DNA–PNA binding on the bead surface (Supporting Information). A conservative estimate of the number of carboxylic acid groups per bead is ∼107,23 and successful conjugation of just 10% of these would result in 106 PNA/bead. Using a PNA–DNA equilibrium constant of 109 M–1,24 we find that 97.2% of 10 fM DNA is bound.
The results presented here suggest that it may be possible to obtain a lower concentration LOD with this device by further lengthening the DNA target, which would impart more charge per bead. Also, an increase in the length of the PNA probe, increasing the PNA–DNA binding constant, would result in a larger DNA bound fraction at low concentrations. For shorter DNA lengths, it may also be possible to obtain a lower concentration LOD by decreasing the number of beads used. According to our Poisson calculations, a 10-fold to 100-fold decrease in bead count per experiment would increase μ and therefore increase the number of beads bound to several DNA. For example, 1 fM DNA measured with 2.1 × 105 beads would result in 619 beads with 3 DNA, 44 with 4 DNA, and 2 with 5 DNA. At 1 fM DNA measured and 2.1 × 104 beads thousands of beads bound to 4 or more DNA would result (Supporting Information, Tables S-25 and S-26). However, in our 100 μL reaction volume, 2.1 × 106 beads was the minimum usable aliquot per experiment, given the limitations of pipetting, centrifugation, and zeta potential measurement during bead preparation. The volumetric limitation of our current hybridization procedure motivates a future microfluidic approach to reduce our bead-to-DNA ratio and potentially improve our detection limit beyond 10 fM.
Conclusions
As we hypothesized, the detection limit of our device improved from 10 pM to 10 fM with increased DNA length. At DNA concentrations of 1 pM and 100 fM, the 110-mer target was not detectable, while longer 235-mer, 419-mer and 1613-mer targets were detectable. At 10 fM concentration, only 419-mer and 1613-mer targets generated permanent blocks. Also, these results suggest that the position of the complementary target sequence does not impair detection in our platform. Both 419- and 1613-mers, with the target sequence positioned at the middle and end of the strands, respectively, were detectable by our device at 10 fM concentration and above with no false positives. Thus, the complementary probe sequence could be chosen from any position within the nucleic acid target to obtain the highest specificity and capture efficiency. In addition to detecting target sequences in an isolated environment, we demonstrated detection in a simulated real sample with background noncomplementary DNA, again with no false positives. These results suggest the potential clinical usefulness of our device to detect larger nucleic acids of length similar to 1613 bases, such as bacterial 16S rRNA. For example, given our ability to detect 1613-mer target NA at ∼6 × 105 molecules/100 μL and an average of 104 16S rRNA per bacterial cell, our device should be capable of detecting ∼60 viable bacterial cells/100 μL (∼600 cells/mL), which is well below the clinically important threshold level of 105 viable cells/mL in urine that is indicative of infection.25 However, the successful testing of our device with samples from complex media such as urine has not yet been demonstrated and is the subject of our current research.
Acknowledgments
Research reported in this publication was supported by NHGRI of the National Institutes of Health under award number R21HG006157 and National Science Foundation award #1265061. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or National Science Foundation. The authors thank UCLA-DOE facility personnel for assistance with the NanoDrop measurements. The authors also thank Prof. Tatiana Segura and her research group for assistance with zeta potential measurements.
Supporting Information Available
Measured ζ-potentials, blockade currents, and Poisson distribution calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Palchetti I.; Mascini M. Anal. Bioanal. Chem. 2008, 391, 455–471. [DOI] [PubMed] [Google Scholar]
- Gubala V.; Harris L. F.; Ricco A. J.; Tan M. X.; Williams D. E. Anal. Chem. 2012, 84, 487–515. [DOI] [PubMed] [Google Scholar]
- Seidel M.; Niessner R. Anal. Bioanal. Chem. 2008, 391, 1521–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey L.; Mitnik L. Electrophoresis 2002, 23, 1386–1397. [DOI] [PubMed] [Google Scholar]
- Freeman R.; Girsh J.; Willner I. ACS Appl. Mater. Interfaces 2013, 5, 2815–2834. [DOI] [PubMed] [Google Scholar]
- Prakrankamanant P.; Leelayuwat C.; Promptmas C.; Limpaiboon T.; Wanram S.; Prasongdee P.; Pientong C.; Daduang J.; Jearanaikoon P. Biosens. Bioelectron. 2013, 40, 252–257. [DOI] [PubMed] [Google Scholar]
- Lazerges M.; Bedioui F. Anal. Bioanal. Chem. 2013, 405, 3705–3714. [DOI] [PubMed] [Google Scholar]
- Wang C.; Zhou H.; Zhu W.; Li H.; Jiang J.; Shen G.; Yu R. Biosens. Bioelectron. 2013, 47, 324–328. [DOI] [PubMed] [Google Scholar]
- Lie P.; Liu J.; Fang Z.; Dun B.; Zeng L. Chem. Commun. (Cambridge, U. K.) 2012, 48, 236–238. [DOI] [PubMed] [Google Scholar]
- Liu J.; Chen L.; Lie P.; Dun B.; Zeng L. Chem. Commun. (Cambridge, U. K.) 2013, 49, 5165–5167. [DOI] [PubMed] [Google Scholar]
- Zhou H.; Liu J.; Xu J.-J.; Chen H.-Y. Chem. Commun. (Cambridge, U. K.) 2011, 47, 8358–8360. [DOI] [PubMed] [Google Scholar]
- Bangar M. a.; Shirale D. J.; Purohit H. J.; Chen W.; Myung N. V.; Mulchandani A. Electroanalysis 2011, 23, 371–379. [Google Scholar]
- Giri S.; Sykes E. a; Jennings T. L.; Chan W. C. W. ACS Nano 2011, 5, 1580–1587. [DOI] [PubMed] [Google Scholar]
- Lin C.; Zhang Y.; Zhou X.; Yao B.; Fang Q. Biosens. Bioelectron. 2013, 47, 515–519. [DOI] [PubMed] [Google Scholar]
- Ma W.; Yin H.; Xu L.; Wang L.; Kuang H.; Xu C. Chem. Commun. (Cambridge, U. K.) 2013, 49, 5369–5371. [DOI] [PubMed] [Google Scholar]
- Esfandiari L.; Monbouquette H. G.; Schmidt J. J. J. Am. Chem. Soc. 2012, 134, 15880–15886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen P. E.; Egholm M.; Berg R. H.; Buchardt O. Sci. Res. (N. Y., NY, U. S.) 1991, 254, 1497–1500. [DOI] [PubMed] [Google Scholar]
- Martins S. a M.; Prazeres D. M. F.; Fonseca L. P.; Monteiro G. a Anal. Bioanal. Chem. 2008, 391, 2179–2187. [DOI] [PubMed] [Google Scholar]
- Xin Z.; Chen J. Plant Methods 2012, 8, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gansauge M.-T.; Meyer M. Nature Protocols 2013, 8, 737–748. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Stanford W. L.; Chan W. C. W. Small 2011, 7, 137–146. [DOI] [PubMed] [Google Scholar]
- Gagnon Z.; Senapati S.; Gordon J.; Chang H.-C. Electrophoresis 2008, 29, 4808–4812. [DOI] [PubMed] [Google Scholar]
- Thomson D.; Dimitrov K.; Cooper M. Analyst 2011, 136, 1599–1607. [DOI] [PubMed] [Google Scholar]
- Park H.; Germini A.; Sforza S.; Corradini R.; Marchelli R.; Knoll W. Biointerphases 2006, 1, 113–122. [DOI] [PubMed] [Google Scholar]
- Kwon J. H.; Fausone M. K.; Du H.; Robicsek A.; Peterson L. R. Am. J. Clin. Pathol. 2012, 137, 778–784. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.