

Abstract
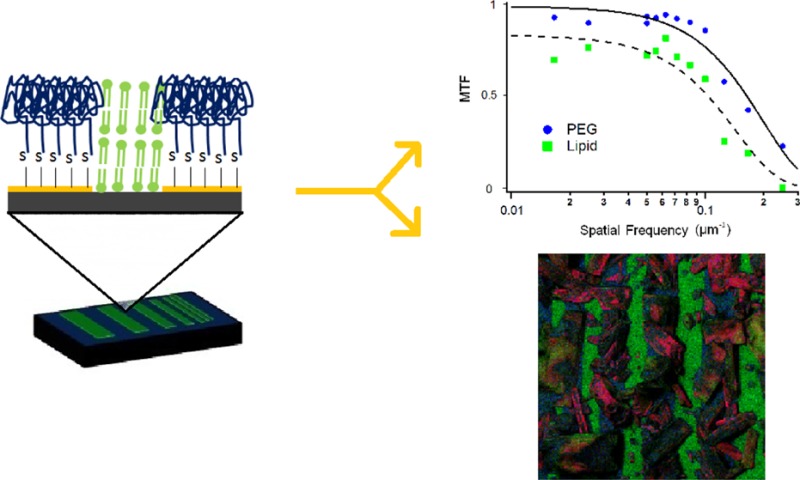
An organic lateral resolution test device has been developed to measure the performance of imaging mass spectrometry (IMS) systems. The device contains periodic gratings of polyethylene glycol (PEG) and lipid bars covering a wide range of spatial frequencies. Microfabrication technologies were employed to produce well-defined chemical interfaces, which allow lateral resolution to be assessed using the edge-spread function (ESF). In addition, the design of the device allows for the direct measurement of the modulation transfer function (MTF) to assess image quality. Scanning electron microscopy (SEM) and time-of-flight secondary ion mass spectrometry (TOF-SIMS) were used to characterize the device. TOF-SIMS imaging was used to measure the chemical displacement of biomolecules in matrix-assisted laser desorption/ionization (MALDI) matrix crystals. In a proof-of-concept experiment, the platform was also used to evaluate MALDI matrix application methods, specifically aerosol spray and sublimation methods.
Imaging mass spectrometry (IMS) is a powerful method used to map the spatial distributions of chemicals in biological materials.1 The method encompasses a number of ionization techniques, including matrix-assisted laser desorption/ionization (MALDI),2 desorption electrospray ionization (DESI),3 and secondary ion mass spectrometry (SIMS),4 each with unique advantages and challenges in the imaging modality.3 All imaging systems contain components that blur the resulting image and limit the lateral resolution of the technique. In IMS, the beam size and shape, detection scheme, secondary ion optics, stage or rastering alignment, step size/pixel size, ion signal-to-noise intensity, and chromatic aberration of the beam are all potential sources of image degradation. Spatial resolution limits down to 7 μm for MALDI,5 200 μm for DESI,3 and 50 nm for SIMS6 have recently been reported, and efforts to improve these values are the subject of ongoing research.7−9
Sensitivity is one of the largest factors limiting the lateral resolution in imaging mass spectrometry, especially for single cell analyses.9−11 The useful lateral resolution, ΔL, can be described giving the minimum pixel size that yields a selected signal-to-noise ratio (S/N). Thus, in the case of Poisson counting statistics,
| 1 |
where I is the intensity of the signal and S/N is the signal-to-noise ratio.12,13 Simply, the useful lateral resolution, ΔL, is the length of a pixel needed to produce a detectable signal, typically S/N > 3. This fact emphasizes the relationship between sensitivity and image quality and, hence, the need to assess both parameters when evaluating the performances of an imaging system.
In this context, in order to identify losses in image quality and assess their effect on IMS-based image quality, a lateral resolution reference material is required. Reference materials are commonly employed in the SIMS community to characterize spatial resolution.14−17 The BAM-L200 nanopatterned certified reference material is composed of metallic species (i.e., indium, gallium, arsenic, and aluminum),15 which makes it an ideal target for SIMS but incompatible with MALDI and DESI analyses. Since MALDI often requires cocrystallization of the matrix with the anayte and DESI requires the dissolution of analyte in desorbed droplets, the chemical nature of this reference material makes it an unsuitable target for evaluating MALDI and DESI imaging. Patterns printed with an inkjet printer have been successfully employed to evaluate spatial lateral resolution in DESI3 and electrostatic spray imaging.18 However, to date, this method has not been employed to evaluate image quality in MALDI imaging systems. There is an obvious need for fundamental MALDI imaging investigations. For example, without a reliable reference material, it is difficult to gauge improvements to lateral resolution and image quality afforded by emerging technologies.7,19−21
In this paper, the development of an organic lateral resolution test device to measure the performance of IMS systems is presented. The device is specifically designed for the quick assessment of the lateral resolution for various IMS technologies. Microfabrication techniques have been used to pattern the surface of the device.22 A device with periodic gratings of polyethylene glycol (PEG) and untethered lipid features ranging from a few micrometers to a millimeter and sharp “chemical edges” (i.e., no diffusive chemical mixing) between the PEG and lipids regions was produced. The test device was used as a platform to assess the blurring of the measured image, taking advantages of its known characteristics. In addition, the chemical displacement of untethered lipids during MALDI matrix application has been observed with this new device. We show that, in some cases, lipids can migrate as far as 16 ± 15 μm away from their initial position after matrix application.
Theory
In this section, we detail the different methods typically used to analyze the resolution of an imaging system. Resolution, the ability to distinguish two adjacent features, is an indicator of image quality. The ability of the imaging system to adequately transfer features from the object to an image is limited by several blur-inducing parameters. Briefly, as detailed in the Supporting Information, SI-1, an image can be described as the convolution of the object with the impulse function of the imaging system (typically, a Gaussian-shaped curve).23 The images obtained from a point, line, or edge (i.e., the point spread function (PSF), line spread function (LSF), and edge spread function (ESF)) can be used to evaluate blurring induced by the imaging system and hence assess the limit resolution of the system (details are given in the Supporting Information). Once the amount of blurring is determined, the minimum distance required to distinguish two features in an image, or lateral resolution, can be evaluated.
The de facto standard methods used to measure spatial resolution in imaging technologies are typically derived from the LSF or ESF. For IMS, particularly SIMS, the width of the incident beam is often used to approximate lateral resolution. For practical purposes, the beam profile is often approximated by a model function, typically a Gaussian profile, characterized by its standard deviation, σ, or its full width half-maximum (fwhm) which is approximated by 2(2 ln(2))1/2σ.24 The ESF (see details in the Supporting Information, SI-2) conveniently gives access to these parameters. Indeed, the ESF is obtained by the convolution of an edge profile with a Gaussian beam function, resulting in an image profile described by the error function, erf. As shown below in the Results and Discussion section, a linescan can be obtained across the edge of a chemical feature, thus leading to the recording of the ESF, which is then fitted with an error function,
| 2 |
where Imax and Imin are the maximum and minimum ion intensities and σ is a parameter describing the width of the ESF. In this case, the fwhm is equal to the Δ88–12%, (i.e., the distance separating the points of the scan where the signal is 12% and 88% of its original intensity).24
| 3 |
Unfortunately, the method detailed above does not consider the effect sensitivity has on image resolution. Near the limit of detection, for a particular analyte, lateral resolution is no longer limited by the width of the beam but by the intensity of signal. The inability of an imaging systems or mass spectrometer to detect variation in the signal intensities is reflected in the contrast of the resulting image. The contrast, C, of an ion image is defined as
| 4 |
where Imax and Imin are the maximum and minimum intensities measured in an image. The contrast of the image relative to the object is known as the modulation transfer function (MTF),25−27
| 5 |
where Cimage(f) and Cobject(f) are the spatial frequency of the image and object, respectively. The object contrast, Cobject(f), is assumed to be 1 because of the discrete repartition of the different chemical components. As detailed in the Supporting Information, the MTF can be calculated from the ESF and used to evaluate beam width.
Experimental Section
Microfabrication
The mask, a 4 in. quartz crystal coated with a layer of chromium, was produced at the Nanofabrication Laboratory, Department of Microtechnology and Nanoscience at Chalmers University of Technology. The mask contained five copies of the pattern, presented in Figure 1A.
Figure 1.

(A) Three-tier design of the IMS lateral resolution test device. (B) Schematic of the lithography process. (C) Schematic depicting the functionalization of the gold–silicon pattern with organic molecules, PEG, and lipid.
The silicon wafers (3 in. diameter) were washed in Milli-Q deionized water (Millipore AB, Solna, Sweden, >18 MΩ cm). Organic residues were removed with a base piranha etch (3:1 mixture of ammonium hydroxide and hydrogen peroxide). (CAUTION: “Piranha” solution reacts violently with organic materials; it must be handled with extreme care.) The silicon wafers were then washed 3 times in deionized water, dried with nitrogen, and spin-coated with UV5 positive DUV photo resist (3000 rpm for 60 s). The photoresist coated silicon wafer was prebaked on a hot plate at 135 °C for 3 min. The DUV photolithography mask aligner (KS MJB3-DUV, Karl Suss) was operated at 220 nm with an intensity of 2.2 mW/cm2. The sample was irradiated for 4 s and then baked at 130 °C for 90 s (Figure 1B, step 1). The developer (0.26 N) was used to remove all the non-cross-linked material (Figure 1B, step 2). Again, the silicon wafer was washed in deionized water and dried with nitrogen. The wafer was sputter coated with a thin layer of titanium followed by a thin layer of gold (Figure 1B, step 3). A standard lift off technique was used to remove the photoresist (Figure 1B, step 4), leaving a gold and silicon patterned surface.
Functionalization
Before functionalization, the surface of the IMS test device was cleaned in a solution containing deionized water, ammonia, and hydrogen peroxide (3:1:1 v/v) for 30 min at 60 °C. The IMS test device was washed with deionized water and dried with nitrogen. The IMS test device was incubated overnight (an excess of 18 h) in a 10 mg/mL solution of methoxy-PEG thiol 2000 (Sigma-Aldrich) in ethanol (99.5%), as shown in Figure 1C, step 1. The substrate was washed with ethanol, then washed with deionized water, and dried under nitrogen gas. The troughs were filled with lipids by immersing the test device in a solution of liposomes, 1,2-dihexadecanoyl-sn-glycero-3-phosphocholine [PC(16:0/16:0)], and cholesterol (Avanti Polar Lipids, USA), followed by multiple washes to remove lipids deposited on the PEG (Figure 1C, step 2). Details for the preparation of the liposome solution are provided in the Supporting Information, SI-3.
Matrix Deposition
The matrix solution contained 30 mg/mL 2,5-dihydroxybenzoic acid (DHB) in deionized water/methanol (1:1, v/v) solvent with 0.2% trifluoroacetic acid (TFA). The matrix was applied using the ImagePrep (Bruker, Germany) automated spray device (see details in the Supporting Information, SI-4).
Instrumentation
A TOF.SIMS 5 mass spectrometer (ION-TOF, Münster, Germany) was used for the time-of-flight secondary ion mass spectrometry (TOF-SIMS) analyses. A pulsed 25 keV Bi3+ primary ion source was used as the analysis beam (pulse width = 23 ns, mass resolution m/Δm = 5000). For the high lateral resolution imaging, pulse width = 100 ns and mass resolution m/Δm = 500 were used. The primary ion dose was kept below the static limit (1013 primary ions/cm2). Data was collected in the positive ion mode.
MALDI data was acquired on an UltrafleXtreme MALDI instrument (Bruker Daltonik GmbH, Germany) equipped with a Nd:YAG smartbeam II laser (355 nm). The instrument was operated in the reflectron mode. Positive ions were detected from 300 Da to 3 kDa. The laser power was optimized on the sample before the data acquisition. For the MTF analysis, the “small” laser focus setting (20 μm spot size) was used at a 50 μm step size. 1000 laser shots were acquired at each shot at a 1 kHz repetition rate.
Data Processing
MATLAB, OriginPro, and ImageJ software were used in the analysis of the images and the linescans. Where applicable, numerical results are reported as average ± standard deviation (SD). All of the ion images and spectra were normalized to the most intense pixel and peak, respectively. Additional image processing protocols are provided in the Supporting Information.
Results and Discussion
Device Design
The IMS lateral resolution prototype test device contains alternating bars of PEG and lipid at various spatial frequencies (see Figure 1A). The device covers a large functional range to accommodate DESI-, MALDI-, and SIMS-based imaging. The pattern consists of three tiers; the period grating (1 PEG bar + 1 lipid bar) increases by 2n micrometers, where n is a natural number, for bars below 10 μm, by 20n micrometers for bars between 10 and 100 μm, and by 200n micrometers for bars between 100 μm and 1 mm. The pattern is reversible, producing the same pattern with both positive and negative photoresists. The transitions between the three tiers are easily located and are useful in determining a position on the device. The total size of the pattern is ∼11 × 10 mm.
Characterization of the Unfunctionalized Test Device
Scanning electron microscopy (SEM) was used to characterize the test device substrate before it was functionalized with organic compounds. As shown in the SEM images, Figure 2A,B, the microfabrication process successfully produced sharp interfaces between the gold and silicon surface regions. The smallest feature on the test device was a 3.0 μm silicon bar followed by a 4.0 μm gold bar. Artifacts from the lift off procedure can be seen on the SEM images, Figure 2C,D. In some areas, excess gold was present causing the gold layer to curl back at the interface. These features were relatively small, less than 250 nm in width, and had a negligible impact on the TOF-SIMS images. Linescans across the Au/SiO2 interface were obtained to assess the sharpness of the microfabricated bars. To do so, the section of the linescan at the interface was fitted to a sigmoidal curve (see eq 2 and Figure 2E). The Δ88%–12% was measured to be 53 nm, a value sufficiently below achievable lateral resolution of most IMS techniques. Therefore, the edges of the device were sharp enough to conclude that any additional loss in resolution would be the result of the imaging technique, not the device.
Figure 2.
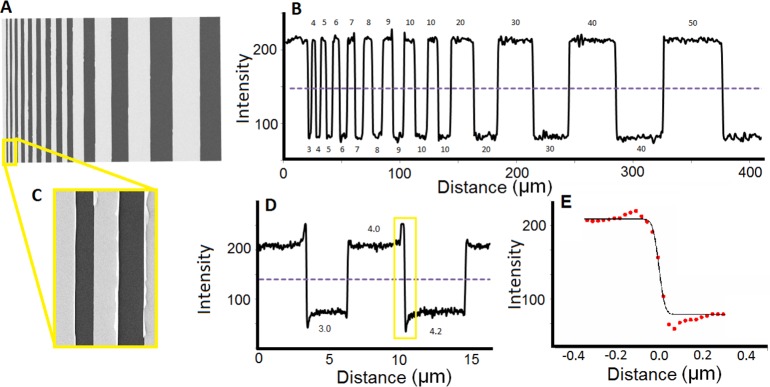
(A) SEM image of the unfunctionalized test device and (B) the corresponding linescan. (C) A zoomed-in SEM image of the 3 and 4 μm bars and (D) the corresponding linescan. (E) The sharp interface between the gold and silicon surface was fitted to the error function; the Δ88%–12% lateral resolution was measured to be 53 nm.
TOF-SIMS Analyses of the Functionalized Test Device
TOF-SIMS analyses were performed on the functionalized test device, and the resulting mass spectra, corresponding to two chemical regions, are shown in Figure 3A. The most intense peaks in the PEG region correspond to the protonated monomer at m/z 45 [C2H4O + H]+, polymer fragments at m/z 107 [(C2H4O)2 + H2O + H]+, and unknown species at m/z 109. The most intense peaks in the lipid regions correspond to the PC headgroup fragments at m/z 86 [C5H12N]+ and m/z 184 [C5H15NO4P]+. TOF-SIMS images of the device show alternating bars of PEG and lipid. As expected, the gold regions were successfully covered with PEG and the SiO2 regions were filled with lipid (see Figure 3B). The intensity of a selected ion was summed along the axis parallel to the bars to produce a linescan (see Figure 3C). The full width at half-maximum (fwhm) of the peaks in the linescan was used to estimate the width of the bars (see Table S1 in the Supporting Information, SI-5).
Figure 3.
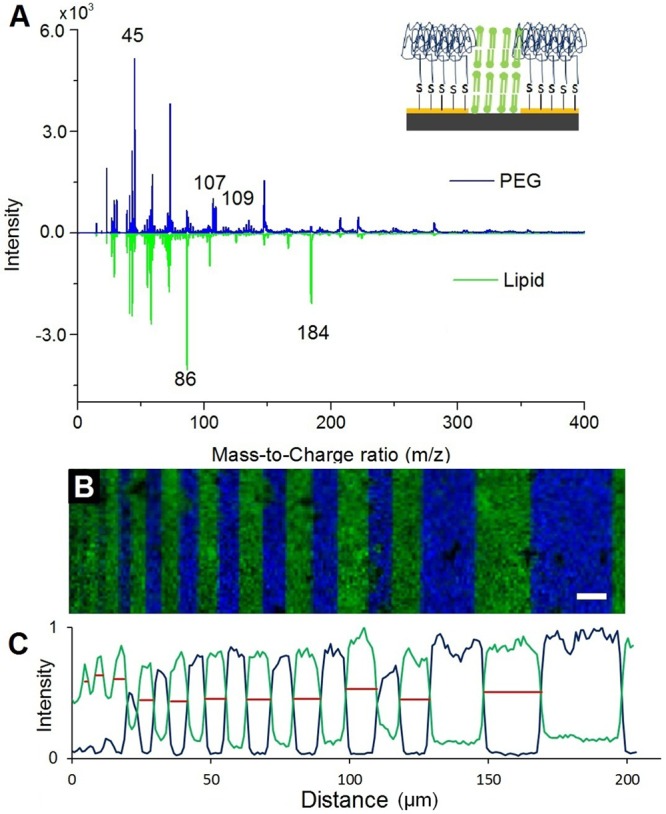
(A) Mass spectra obtained from the two distinct chemical regions of the device; the PEG (blue, top) and lipid (green, bottom) regions. (B) TOF-SIMS image showing the distribution of the PEG polymer fragments at m/z 45, m/z 107, and m/z 109 (blue) and lipid headgroup fragments at m/z 184 and m/z 124.9 (green). The scale bar is 10 μm. (C) The ion intensities for the PEG fragment (m/z 107) and the lipid fragment (m/z 184) were summed along the bar orientation and plotted as a function of lateral space. The fwhm for both the lipid (red) and PEG were measured and reported in Table S1 (see Supporting Information).
The de facto method for evaluating lateral resolution, as described in the Theory section, was applied to the SIMS image of the device. The chemical edges in the PEG linescan, Figure 3C, were fitted to the function described in eq 2. The Δ88–12% or fwhm of the beam, here used as an indicator of lateral resolution, was measured for each PEG bar. These values are reported in Table S2 (see Supporting Information, SI-5). The average Δ88–12% was 1.45 ± 0.64 μm, corresponding to an average σ of 0.62 ± 0.27 μm.
To demonstrate the issue with reporting fwhm, we examine the two signals from the PC(16:0/16:0) molecule with very different signal strengths; the protonated molecular ion of PC(16:0/16:0) at m/z 734.5 and fragment ion at m/z 184. The molecular ion peak at m/z 734 does not have enough signal-to-noise (Imax = 3.2 counts and Imin= 1.8 counts) to produce a useful image. However, when the signal intensity for m/z 734.5 and m/z 184 across a chemical edge are plotted in Figure 4A and fitted to eq 2, the measured Δ88–12% resolutions were 2.28 and 2.26 μm, respectively. The de facto method did not factor in signal intensity relative to the noise or contrast and therefore provided a lateral resolution measurement that did not reflect the image quality.
Figure 4.

(A) Intensity of the molecular ion (m/z 734 dark green) and fragment ion (m/z 184, light green) signal of lipid, PC(16:0/16:0), at the chemical interface. Both traces were fit to the error function (solid line). (B) The normalized ion intensity of PEG (m/z 107) plotted as a function of bar width (blue) and the linear relationship between the expected bar width and the measured bar width (red). (C) The MTF for PEG (m/z 107, blue) and lipid (m/z 184, green). Both traces were fit to a Gaussian function (solid line).
The effects of sensitivity on lateral resolution can be observed directly with the device. The average ion intensity for PEG is plotted as a function of bar width in Figure 4B. At low spatial frequencies (i.e., for large bars), the chemical contrast for PEG and the lipid are constant; however, as spatial frequency increases and the bars get closer and closer together, the chemical contrast for PEG decays. The extrapolated data point for zero intensity, y(0), is approximately 3.7 μm. Therefore, the narrowest detectable bar for this particular image acquisition is 4 μm in width. In the same plot, the linear relationship between the expected bar width and the measured bar width is shown. The plot shows that, even though the IMS system can resolve high spatial frequencies, the resulting image suffers from a significant loss in contrast as the bars get narrower (below ∼7 μm). This result demonstrates that the lateral resolution limit of the imaging system results from both the ability to resolve contrast and topology.
The device was used to measure the MTF of the image. The MTF is a commonly used method to evaluate the spatial frequency response in a number of imaging systems, including optical microscopy techniques and medical imaging techniques.25−27 This method has been previously applied to SIMS imaging of inorganic material; however, it is not routinely applied to IMS.28 The analysis of the MTF is more informative than the mere measurements of the fwhm.29 Since contrast depends on the intensity of the signal and noise level, parameters that affect these variables, such as secondary ion yields, ionization efficiency, and matrix suppression effects, influence the lateral resolution measurement. As a consequence, the modulation of the contrast is a critical factor for the quality of chemical imaging, as the resolution of the topology is not sufficient to produce a good image.
The MTF for PEG (m/z 107) and lipid (m/z 184) was plotted as a function of spatial frequency (see Figure 4C). The MTF can be directly obtained from the intensities of the linescans shown in Figure 3C. Both the PEG and lipid signals reach a steady state at spatial frequency = 0.08 μm–1 (6 μm bar width); below this frequency, the ion contrast is no longer influenced by the resolution of the imaging system. The MTF shown in Figure 4 was fit with Gaussians. From the trace, we can use the MTF50%, the point where the MTF has decreased to half its maximal value, to assess lateral resolution. The corresponding frequency, f50%, is related to σ following (see Supporting Information, SI-6, Equation S-12)
| 6 |
The computed values for σ were 1.56 and 1.13 μm for the lipid and PEG traces, respectively. This corresponds to fwhm of 3.67 and 2.67 μm for the lipid and PEG traces, respectively.
The fwhm and σ values obtained from the MTF are 2-times higher than the values obtained from the de facto method reported above. Unfortunately, the de facto methods overestimate the resolving capabilities of the system in the case of sensitivity-limited analytes. This result strongly advocates the use of the MTF in place of the ESF to assess the resolution of IMS, as the complexity of the processes resulting in the final image cannot be solely accounted for by merely analyzing the resolution of the sample topology. Overall, the device offers several advantages for evaluating image resolution: the device allows the user (i) to record simultaneously the MTF and ESF, (ii) to obtain a direct measurement of the MTF via the test device and image contrast, and (iii) to avoid additional processing steps (i.e., derivation and Fourier transforms) which are more sensitive to noise levels, thus improving the reliability of the analysis.
Characterizing MALDI Matrix Application Methods
The device was used to characterize MALDI matrix application methods and evaluate their effects on chemical lateral displacement. A matrix is an essential component in MALDI imaging; it absorbs the laser radiation and acts as a proton source during ionization. Unfortunately, the size of the matrix crystals and the method used to apply the matrix affect the image quality. In this experiment, MALDI matrix was sprayed on the device and SIMS imaging was used to characterize crystal size, as well as the incorporation and chemical displacement of the analyte into the matrix crystals.
An ion image showing the distribution of matrix (m/z 136, 137, 154, and 155), PEG (m/z 107 and 109), and lipid (m/z 86, 124, and 184) is shown in Figure 5A. The SIMS images reveal a preferential deposition of the matrix on the PEG-functionalized bars. It is important to note that the chemical composition of the substrate influences the localization of matrix molecules. In order to avoid potential artifacts in MALDI imaging experiments, it is critical to understand the interactions between the substrate and the matrix, as well as between the matrix and the sample. The image also shows matrix crystals spanning across multiple bars of PEG. Such features are mainly observed in regions where the bar width is 10 μm and below. The ion intensities for these chemical components have been summed along the vertical axis to form a linescan, Figure 5B. The linescan reveals the level of chemical mixing and image blurring caused by the deposition of matrix.
Figure 5.
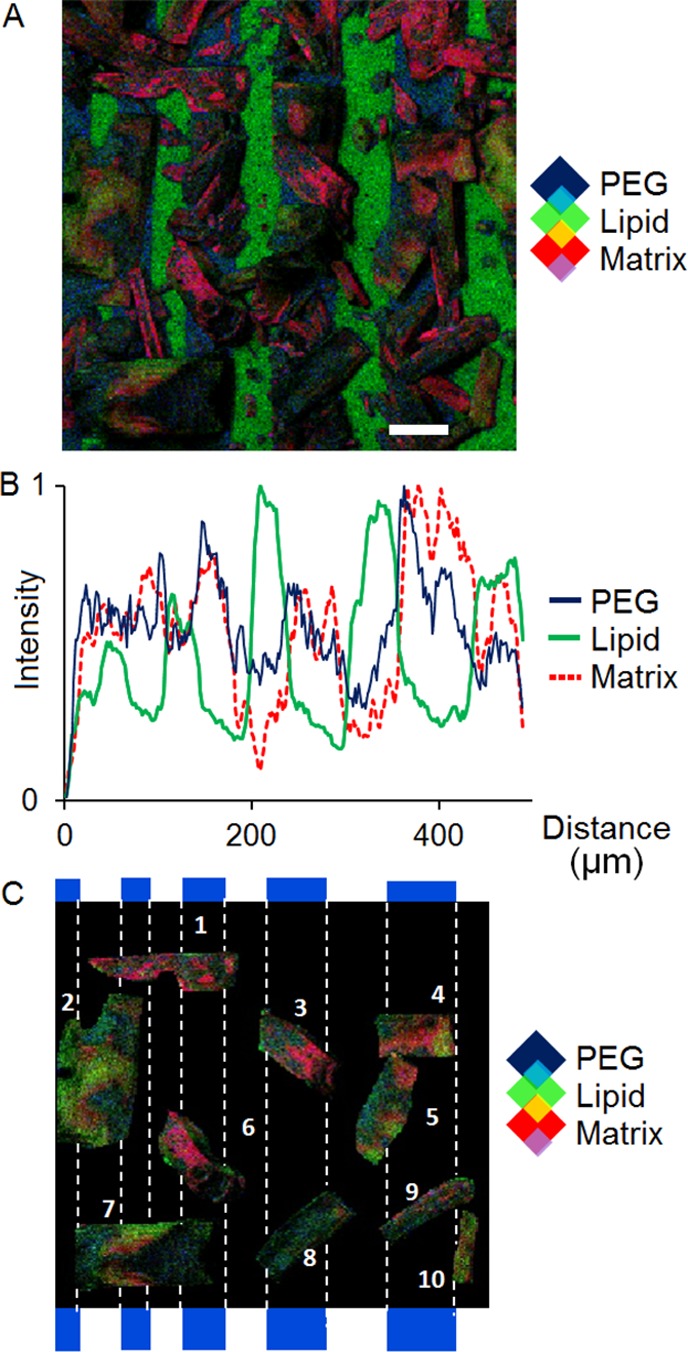
(A) TOF-SIMS image showing the distribution of matrix (red), PEG (blue), and lipid (green) after solvent-based matrix application (scale bar: 50 μm). (B) The linescan of the three components demonstrates the mixing that occurs as a result of matrix application. (C) Ten individual matrix crystals were isolated from the ion image in order to emphasize the degree of chemical mixing and show the distribution of analyte molecules in matrix crystals.
Ten crystals from the SIMS image were isolated and characterized (see the Supporting Information, SI-7, Table S3). The average length and width of the DHB crystals were 124.7 ± 31.9 μm and 49.4 ± 20.5 μm, respectively. The TOF-SIMS image shows the incorporation and mixing of lipid (green) and PEG (blue) in the matrix (red). The average counts per square micrometer and the percent coverage for various PEG, lipid, and matrix chemical peaks were calculated for each crystal, and the average is displayed in Table S4, in the Supporting Information. Despite the preferential nucleation of matrix crystals on the PEG bars, lipids molecules were more mobile and readily incorporated into the matrix crystal compared to the PEG molecules.
The distribution of analytes in MALDI matrix crystal has previously been measured with TOF-SIMS,30,31 MALDI,32 and confocal fluorescence microscopy;33 however, without a fixed origin, the chemical displacement cannot be assessed. Since the device provides the known fixed origins of the lipid and PEG before matrix was applied, the diffusion length of PEG into the lipid domain and vice versa can be accurately measured, as shown in Figure 6. Details on the data processing procedures are provided in the Supporting Information, SI-8. In this case, for a single matrix crystal spanning across the 70 μm PEG bar, the average lipid signal was detected approximately 16 ± 15 μm away from the interface (see Table 1). Unlike the PEG, the lipids are not tethered to the surface of the device and are allowed to migrate freely. The growth of the matrix crystal on the PEG bar also displaced PEG molecules, originally localized to the PEG region, into the lipid region. Hence, the distance between the PEG containing pixel detected in the lipid region and the known interface was also measured. The average PEG signal was detected 5.5 ± 5 μm away from the interface. Overall, the measured displacement of the lipids exceeded the lateral resolution measured for the device before matrix was applied. As a result, the delocalization of the chemical on the device can only be attributed to diffusion during the matrix application.
Figure 6.
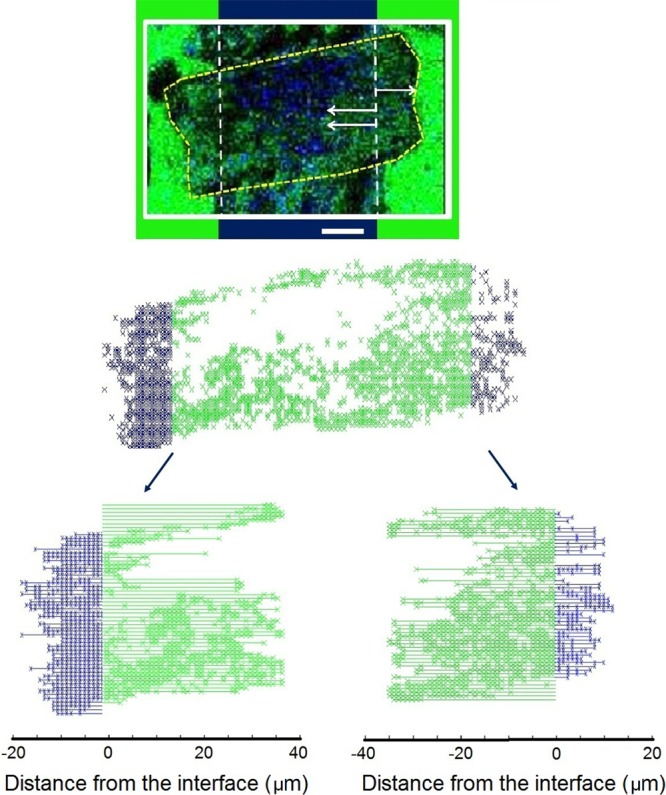
Migration distance of PEG and lipid molecules in a single matrix crystal was measured using TOF-SIMS imaging and the device. TOF-SIMS image of a single matrix crystal is isolated (top); pixels in the PEG region that contained lipid peaks with S/N > 3 (middle, green) and pixels in the lipid region of the device that contained lipid peaks with S/N > 3 (middle, blue) are isolated, and the distance between these pixels and the PEG-lipid interface is measured (bottom). The scale bar shows 20 μm.
Table 1. Displacements of PEG and Lipid Molecules Incorporated into a Single Matrix Crystala.
| left (μm) | right (μm) | total (μm) | ||
|---|---|---|---|---|
| LIPID | (m/z 184) | 18 ± 11 | 14 ± 10 | 16 ± 15 |
| PEG | (m/z 107) | 6 ± 4 | 5 ± 3 | 5.5 ± 5 |
Pixels with S/N > 3 of lipid were isolated, and the distances between these pixels and the interface were measured. Distance measurements were collected for both the right and left sides of the crystal.
The device presented here is therefore a potential platform for the precise measurement of chemical displacement for various sample preparation methods. In this case, the matrix was applied by spraying the surface of the device with a fine mist of matrix solution. The size of the mist droplets, the ratio of organic solvent to aqueous solvent in the matrix solution, the concentration of the matrix, drying time between spray cycles, and the hydrophobicity of the substrate/sample are all operational parameters that influence the diffusion of material and limit the potential lateral resolution of the technique.
The device can be used to evaluate the quality of MS images obtained with MALDI. The MTF of the lipid signal obtained from the device sprayed with matrix is plotted in Figure 7A. The f50% of the MTF plot is approximately 0.0024 μm–1. On the basis of eqs 3 and 6, the fwhm resolution is estimated to be approximately 184 μm. The large spatial resolution is expected on the basis of large laser step size, 50 μm, and the mobility of the untethered lipids. The device can also be used to evaluate differences in sample preparation. In a proof-of-concept experiment, two different matrix application methods, a spray method and a solvent-free sublimation method, were applied to the device and analyzed with MALDI. The mass spectra obtained for the two sample preparation methods reveal two distinct chemical signatures (see Figure 7B). A large PEG-related peak at m/z 1830 was observed with the spray-based method but not within the solvent-free method. The lack of solvent in the sublimation method most likely prevented the extraction and, therefore, the detection of this particular PEG molecule. Overall, the proof-of-concept experiment shows that the device is a viable platform for evaluating MALDI imaging, as well as evaluating the effect sample preparation has on MALDI analyses.
Figure 7.
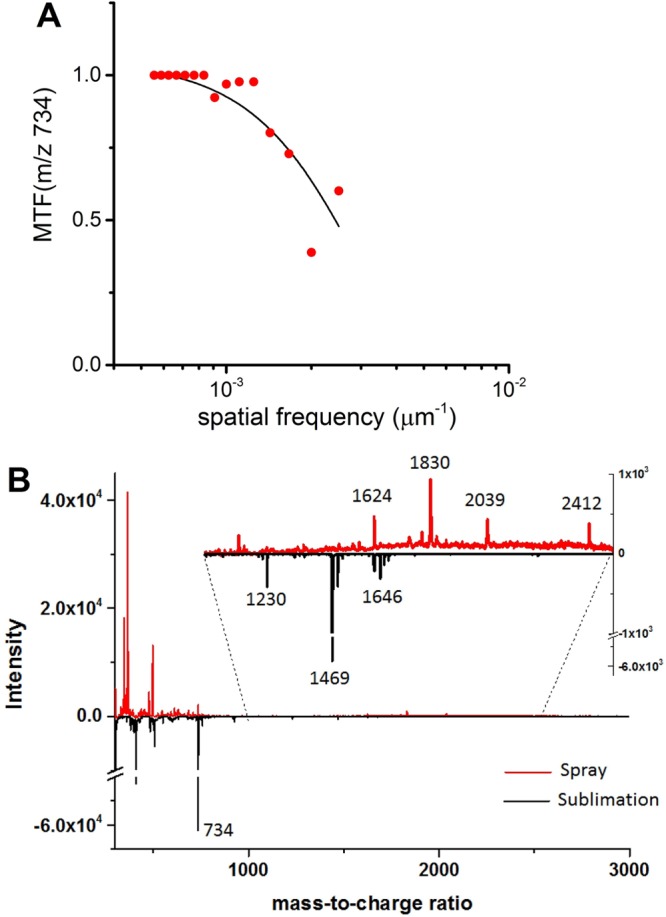
(A) The MTF for the protonated molecular ion of PC(16:0/16:0) at m/z 734.5 obtained after imaging the device using a solvent-based spray matrix application method (image step size: 50 μm). (B) MALDI spectra from the device using the spray-based matrix application (top) and solvent-free sublimation method (bottom).
Conclusions
A prototype lateral resolution test device for evaluating image quality in IMS techniques was developed and characterized. The device was designed for the quick assessment of the modulation transfer function (MTF). The device, in combination with SIMS imaging, was used to measure matrix crystal size, analyte incorporation to the matrix, and the chemical displacement of the analyte after matrix deposition. This flexible platform was also used to evaluate MALDI-based sample preparation methods.
Acknowledgments
The European Research Council (Advanced Grant), Knut and Alice Wallenberg Foundation, the Swedish Research Council (VR), and the National Institutes of Health are acknowledged for financial support. This work was partially supported by the National Physical Laboratory’s Strategic Programme (project SR116301-NiCE-MSI).
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ M.K.P.: National Centre of Excellence in Mass Spectrometry Imaging (NiCE-MSI), National Physical Laboratory (NPL), Teddington, Middlesex, TW11 0LW, U.K.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- McDonnell L. A.; Heeren R. Mass Spectrom. Rev. 2007, 26, 606–643. [DOI] [PubMed] [Google Scholar]
- Cornett D. S.; Reyzer M. L.; Chaurand P.; Caprioli R. M. Nat. Methods 2007, 4, 828–833. [DOI] [PubMed] [Google Scholar]
- Ifa D. R.; Wiseman J. M.; Song Q.; Cooks R. G. Int. J. Mass Spectrom. 2007, 259, 8–15. [Google Scholar]
- Winograd N. Anal. Chem. 2005, 77, 142 A–149 A. [Google Scholar]
- Schober Y.; Guenther S.; Spengler B.; Römpp A. Anal. Chem. 2012, 84, 6293–6297. [DOI] [PubMed] [Google Scholar]
- Boxer S. G.; Kraft M. L.; Weber P. K. Annu. Rev. Biophys. 2009, 38, 53–74. [DOI] [PubMed] [Google Scholar]
- Boggio K. J.; Obasuyi E.; Sugino K.; Nelson S. B.; Agar N. Y.; Agar J. N. Expert Rev. Proteomics 2011, 8, 591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillion F.; Horreard F.; Stadermann F.. Recent results and developments on the CAMECA NanoSIMS 50. Proceedings of the 12th International Conference on Secondary Ion Mass Spectrometry, Brussels, Belgium, September 5–10, 1999. [Google Scholar]
- Passarelli M. K.; Ewing A. G. Curr. Opin. Chem. Biol. 2013, 17, 854–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trouillon R.; Passarelli M. K.; Wang J.; Kurczy M. E.; Ewing A. G. Anal. Chem. 2012, 85, 522–542. [DOI] [PubMed] [Google Scholar]
- Passarelli M. K.; Ewing A. G.; Winograd N. Anal. Chem. 2013, 85, 2231–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollmer F. Appl. Surf. Sci. 2004, 231, 153–158. [Google Scholar]
- Gilmore I. S. J. Vac. Sci. Technol., A 2013, 31, 050819–050819. [Google Scholar]
- Lee J.; Ninomiya S.; Matsuo J.; Gilmore I.; Seah M.; Shard A. Anal. Chem. 2009, 82, 98–105. [DOI] [PubMed] [Google Scholar]
- Senoner M.; Wirth T.; Unger W.; Österle W.; Kaiander I.; Sellin R.; Bimberg D. Surf. Interface Anal. 2004, 36, 1423–1426. [Google Scholar]
- Senoner M.; Wirth T.; Unger W.; Escher M.; Weber N.; Funnemann D.; Kromker B. J. Surf. Anal. 2005, 12, 78. [Google Scholar]
- Senoner M.; Unger W. Surf. Interface Anal. 2007, 39, 16–25. [Google Scholar]
- Qiao L.; Tobolkina E.; Lesch A.; Bondarenko A.; Zhong X.; Liu B.; Pick H. M.; Vogel H.; Girault H. H. Anal. Chem. 2014, 86, 2033–2041. [DOI] [PubMed] [Google Scholar]
- Jurchen J. C.; Rubakhin S. S.; Sweedler J. V. J. Am. Soc. Mass Spectrom. 2005, 16, 1654–1659. [DOI] [PubMed] [Google Scholar]
- Hankin J. A.; Barkley R. M.; Murphy R. C. J. Am. Soc. Mass Spectrom. 2007, 18, 1646–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavalin A.; Todd E. M.; Rawhouser P. D.; Yang J.; Norris J. L.; Caprioli R. M. J. Mass Spectrom. 2012, 47, 1473–1481. [DOI] [PubMed] [Google Scholar]
- Gunnarsson A.; Kollmer F.; Sohn S.; Höök F.; Sjövall P. Anal. Chem. 2010, 82, 2426–2433. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Kashti T.; Kella D.; Frank T.; Shaked D.; Ulichney R.; Fischer M.; Allebach J. P. Proc. SPIE 2012, 10.1117/12.912989. [DOI] [Google Scholar]
- Seah M. Surf. Interface Anal. 2002, 33, 950–953. [Google Scholar]
- Fujita H.; et al. IEEE Trans. Med. Imaging 1992, 11, 34–39. [DOI] [PubMed] [Google Scholar]
- Tzannes A. P.; Mooney J. M. Opt. Eng. 1995, 34, 1808–1817. [Google Scholar]
- Boreman G. D.Modulation Transfer Function in Optical and Electro-optical Systems; SPIE Press: Bellingham, WA, 2001; Vol. 4. [Google Scholar]
- Senoner M.; Wirth T.; Unger W. E. J. Anal. At. Spectrom. 2010, 25, 1440–1452. [Google Scholar]
- Smith S. W.The Scientist and Engineer’s Guide to Digital Signal Processing; California Technical Publishing: San Diego, 1997. [Google Scholar]
- Luxembourg S. L.; McDonnell L. A.; Duursma M. C.; Guo X.; Heeren R. M. Anal. Chem. 2003, 75, 2333–2341. [DOI] [PubMed] [Google Scholar]
- Hanton S. D.; Cornelio Clark P. A.; Owens K. G. J. Am. Soc. Mass Spectrom. 1999, 10, 104–111. [Google Scholar]
- Bouschen W.; Spengler B. Int. J. Mass Spectrom. 2007, 266, 129–137. [Google Scholar]
- Dai Y.; Whittal R. M.; Li L. Anal. Chem. 1996, 68, 2494–2500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.