

Abstract
Subambient pressure ionization with nanoelectrospray (SPIN) has proven to be effective in producing ions with high efficiency and transmitting them to low pressures for increased sensitivity in mass spectrometry (MS) analysis. Here we present evidence that the SPIN source not only improves MS sensitivity but also facilitates the detection of more labile compounds. The gentleness of conventional heated capillary electrospray ionization (ESI) and the SPIN designs was compared in conjunction with the liquid chromatography mass spectrometry (LC–MS) analysis of colominic acid and N-glycans containing sialic acid. Prior experiments conducted with the SPIN interface demonstrated the ability to detect labile glycans such as heavily sialylated and polysialic acid N-glycans, which are difficult to detect with a conventional ESI-MS interface. Colominic acid is a mixture of sialic acid polymers of different lengths containing labile glycosidic linkages between monomer units necessitating a gentle ion source. These labile covalent bonds may display similar behavior to sialic acid chains in N-glycans during MS analysis. By coupling the SPIN source with high-resolution mass spectrometry and using advanced data processing tools, we demonstrate much extended coverage of sialic acid polymer chains as compared to conventional ESI-MS and the ability to detect sialic acid containing N-glycans without the need of sample derivatization. In addition, we show that SPIN-LC–MS is effective in elucidating polymer features with high efficiency and high sensitivity previously unattainable by the conventional ESI-LC–MS methods.
Electrospray ionization (ESI) is generally regarded as a soft ionization technique. However, in-source fragmentation is typically observed for extremely labile compounds, potentially limiting their identification and structural characterization.1,2 Although it is possible to “soften” the ESI source by careful tuning of the ion optics’ voltages3−5 and lowering the temperature (e.g., of the heated capillary inlet) of the ESI mass spectrometry (ESI-MS) interface,6−12 these changes generally result in significant losses of sensitivity due to less effective desolvation. By altering these ion energy sensitive parameters, any ESI source can also be tuned in either a soft condition to reduce ion in-source fragmentation1 or to provide harsher conditions to induce ion in-source fragmentation, a preferable alternative in some applications to collision-induced dissociation commonly obtained in an ion trap or a collision cell.13,14 For effective ESI-MS analysis of extremely labile compounds, a more gentle ionization condition could prevent undesired in-source fragmentation which can lead to misidentifications and losses in coverage of intact labile compounds.2 A key challenge is doing this while maintaining sufficient sensitivity for the characterization of these compounds.
Subambient pressure ionization with nanoelectrospray (SPIN) source was developed in an attempt to eliminate the major ion losses in the ESI-MS interface.15 In the SPIN source, the ESI emitter is moved from the atmospheric pressure environment, as used in any conventional ESI source design, into the first vacuum stage of the mass spectrometer with its operating pressures typically of 15–30 Torr. The ESI emitter in the SPIN source is positioned at the entrance of an electrodynamic ion funnel allowing the entire electrospray plume to be collected by the ion funnel. Significant improvements in MS sensitivity using the SPIN source have been demonstrated in our early studies as compared to using the standard atmospheric pressure ESI interface which requires a small orifice or heated capillary inlet.15,16 A parallel experimental evaluation has demonstrated the ability of the SPIN source to transfer 50% of analyte molecules in solution into the low-pressure regions of the mass spectrometer as gas-phase ions.17 In addition to the major improvement in MS sensitivity, glycan profiling studies using the SPIN source also showed coverage increased by 25% for glycan families in human serum when compared to using the conventional ESI with a heated capillary/dual ion funnel interface.18 This significant increase in glycan coverage mostly came from the detection of large heavily sialylated glycans, both extremely labile and with relatively low ionization efficiency, that are not typically detected by using the conventional heated capillary inlet ESI-MS interface configuration.19−21 Many analytes were also detected with higher charge states using the SPIN source.18 These studies strongly hinted that the primary reason for the increase in identified structural features was due to the decrease in undesirable in-source fragmentation in the SPIN source.
To further probe this hypothesis, we compared the structural features identified from the analysis of known labile compounds using a conventional ESI-MS interface and a SPIN-MS interface. As noted in a prior study, heavily sialylated glycans and polysialic acid N-glycans were observed in human serum with the SPIN-MS interface most of which were completely absent with a ESI-dual ion funnel interface.18 These previous findings inspired our current study to perform a systematic evaluation of the ionization conditions with different MS interfaces operating at different conditions by using extremely labile compounds of sialylated glycans and colominic acid. Colominic acid is a polymer of particular interest because it is a mixture of homopolymers of sialic acid with varying degrees of polymerizations (DP) and the glycosidic bond being in proximity to the carboxylic acid on sialic acid renders these compounds very labile and difficult to analyze.22,23 For this reason, colominic acid was first used as a model compound in this study to gain understanding of the in-source fragmentation at different MS interface configurations. Specifically, we compared the liquid chromatography–mass spectrometry (LC–MS) analysis of colominic acid and sialylated glycans between a standard heated capillary ESI-MS interface operated at varying conditions and the SPIN-MS interface while keeping the sample and LC conditions constant. Conditions of the ESI-MS interface were initially tuned for optimum sensitivity, and subsequently adjusted for a gentler ionization condition by lowering the inlet capillary temperature and reducing the tube lens voltage. High-resolution MS allowed comprehensive analysis of polysialic acid and the unambiguous identification of polymer lengths and structures. The SPIN source enabled the detection of colominic acid compounds with higher DPs and intact sialylated glycans with significantly reduced in-source fragmentation without any sacrifice in MS sensitivity. In harmony with previous results, we also observed a shift toward higher charge states, and the greatest gains in sensitivity were observed for the compounds with the highest DP. The experimental data presented in this report positively confirms for the first time that the SPIN-MS interface provides gentler ionization conditions than a conventional ESI-MS interface.
Experimental Section
Sample Preparation
The sodium salt of colominic acid from Escherichia coli was purchased from Sigma-Aldrich (St. Louis, MO). Colominic acid samples were diluted to a concentration of 660 μM, and 5 μL aliquots were loaded onto a graphite column. Samples were desalted with nanopure water (Barnstead Nanopure Infinity System, Dubuque, IA) on-column for 60 min prior to LC–MS analysis. A pooled reference human blood serum sample was obtained from Sigma-Aldrich (male, blood type AB, not heat-inactivated) and prepared with a previously described method to produce reduced nonderivatized glycans.18
High-Performance Liquid Chromatography
Separations were performed on graphitized carbon columns packed in house with 3 μm Hypercarb particles (Thermo Fisher, Madison, WI) using a fused-silica capillary, 75 μm i.d., 360 μm o.d., and 75 cm long (Polymicro Technologies, Phoenix, AZ). Solvents A and B consisted of 0.1% formic acid (Sigma-Aldrich, St. Louis, MO) in nanopure water and 0.2% trifluoroacetic acid (Sigma-Aldrich) in acetonitrile (Fisher Scientific, Pittsburgh, PA), respectively. The LC separation was performed with a linear gradient from 30% to 60% solvent B provided by an Agilent 1260 Series LC pump (Santa Clara, CA) over a period of 100 min. Serum samples were loaded with 1% B solvent mixture, and a 90 min gradient was used consisting of (time in minutes: % mobile phase B) 0:1%, 1:4%, 2:6%, 91:30%, 95:95%, 96:95%, 100:1%.
Mass Spectrometry
Mass spectra were acquired on a Thermo Orbitrap Exactive mass spectrometer (Thermo Scientific, San Jose, CA). In the conventional atmospheric ESI source configuration a standard heated capillary interface was used as illustrated in Figure 1a. The temperature of the heated capillary inlet was adjusted from 150 to 300 °C, and the dc voltages on the tube lens and skimmer were adjusted to achieve either optimal sensitivity or ionization gentleness. The instrument was tuned using the LTQ Velos ESI positive ion calibration solution (Thermo Scientific, San Jose, CA). In the SPIN source configuration the ESI emitter was moved to the first vacuum region of the mass spectrometer and positioned at the entrance of the ion funnel.15,18 As depicted in Figure 1b, in this configuration the standard ion optics up to lens L0 were replaced by two ion funnels operating at different vacuum pressures. Independent of the source configuration the instrument was always operated in ultrahigh-resolution mode with an m/z range of 200–3000 with a 3 microscan average per spectrum. The AGC was set for high dynamic range with a maximum ion injection time of 100 ms. Electrospray emitters were fabricated by chemically etching fused-silica capillary tubing with a 10 μm i.d. and 150 μm o.d. (Polymicro Technologies, Phoenix, AZ) as described previously.24
Figure 1.
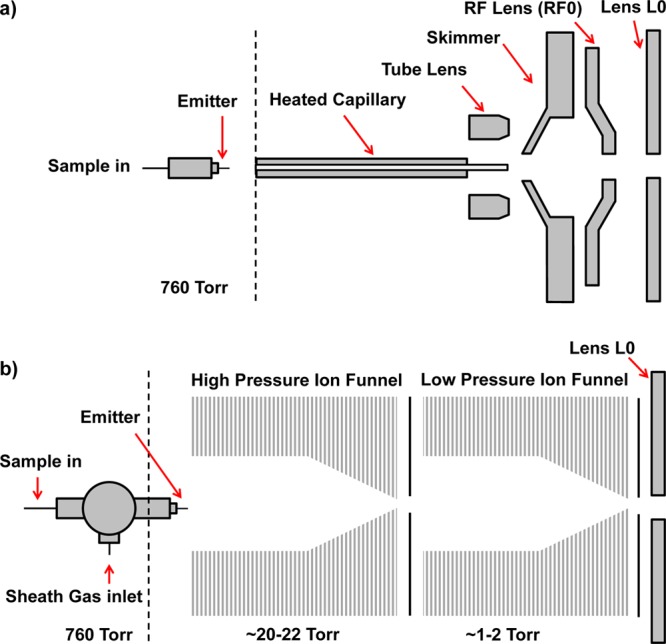
Diagram of the standard ESI (a) and SPIN (b) interfaces used with the Exactive MS. The dual ion funnel interface replaces the ion optics in front of lens L0, and the electrospray emitter tip is placed inside the first vacuum region of the MS.
Data Processing
The colominic acid and human serum LC–MS data sets were processed with the Glycomics Quintavariate Informed Quantification (GlyQ-IQ) software.25 Briefly, GlyQ-IQ is a targeted software package designed for searching and identifying glycan features in LC–MS data sets. GlyQ-IQ annotates glycans by using exact mass, modeled and fit isotope profiles, modeled and fit extracted ion chromatograms, glycan family relationships, and, if detected, in-source fragmentation information. A list of polysialic acid masses was used to populate the glycan target list which includes polymers with different degrees of polymerization extending from 2 to 50. Lactonized polysialic acid polymers were also included by including lactone modifications on each glycosidic bond allowing for the condensation reaction conversion to a lactone bond. For example, a polymer with four sialic acids monosaccharides could contain zero, one, two, or three lactone bonds. The formation of each lactone bond decreases the molecular mass by 18.01 Da from a loss of water molecule. The data was processed on a 48 node, 1504 core Microsoft Windows 2012 R2 HPC cluster utilizing AMD Interlagos CPUs (2.1Ghz) and an EMC Isilon running the OneFS file system.
Results and Discussion
In order to evaluate the ionization gentleness of the SPIN source and interface, a comparison of the polymer coverage from the LC–MS analyses of colominic acid was conducted between the SPIN-MS and the conventional heated capillary ESI-MS interfaces. All the sample prep procedures, LC separation conditions, and MS parameters were held constant during the experiments so that any change could be prescribed solely to the differences in the interface. For the convenience of the comparison, all the experimental data representing different colominic acid degrees of polymerization under different source configurations are presented in a grid format shown in Figure 2. The horizontal index represents the observed degree of colominic acid polymerization (DP), and the vertical index represents the observed number of lactones for each DP. The lactonization of polysialic acid occurs through the internal esterification of the carboxyl groups of adjacent hydroxyl groups and is well-known to stabilize the labile bonds.26,27 Each individual square in the grid correlates to a given DP with a given degree of lactonization. Under acidic conditions, colominic acid will lactonize in solution and form a cluster of polymers at each given DP with different degrees of lactonization.26,28 To illustrate this, the inset in Figure 2 shows the mass spectrum of the DP 3 polymer cluster acquired using the conventional heated capillary ESI source. Different degrees of DP 3 polymer lactonization are evident by the different peaks with a mass difference of 18 Da indicating consecutive water losses from each additional DP 3 lactonization. Of particular interest is the overall coverage of the colominic acid polymers under different ion source configurations. To make a comprehensive comparison between the conventional ESI-MS and the SPIN-MS interfaces, the inlet heated capillary temperature of the conventional interface was tuned separately at 300 °C for optimal sensitivity and at 150 °C for gentler ionization conditions using the tuning solution. The highest observed DP, as shown by the blue squares in Figure 2, was 30 monomer units at the heated capillary temperature of 300 °C. This detection limit was further increased to 38 monomer units (the green squares in Figure 2) at the heated capillary temperature of 150 °C while the sensitivity of the instrument decreased significantly. Repeating the same experiment with the SPIN source, the maximum detected DP further increased to 47, as shown by the red squares in Figure 2, with still better sensitivity than ESI source at 300 °C heated capillary temperature. Every DP identified with the ESI-MS interface was also identified with the SPIN-MS, and DPs between 8 and 20 were observed in the 300 °C configuration to have a higher number of lactones than 150 °C and the SPIN configurations. Many additional structures of colominic acid polymers were positively identified by SPIN-MS which were completely absent in data acquired with the conventional ESI-MS at both heated capillary temperatures.
Figure 2.

(a) Colominic acid coverage by ESI at 300 °C (blue), 150 °C (green), and SPIN (red) with each square relating to a given DP (horizontal index) with a fixed number of lactones (vertical index). Vertical divisions within a given square demonstrate that a given polymer was observed under all three conditions, and horizontal divisions indicate observation at two conditions.
In addition to tuning the temperature of the heated capillary inlet, the attempt was made to tune the conventional ESI-MS interface to even gentler conditions by adjusting the declustering dc field between the tube lens and the skimmer. When the instrument was tuned for optimal sensitivity, as was the case in the 300 °C temperature condition, the voltage on the tube lens was set to 190 V and the skimmer at 46 V with the pressure in the skimmer interface region being roughly 1.30 Torr. The tube lens voltage was subsequently decreased to 55 V to minimize in-source fragmentation at both heated capillary temperatures of 150 and 300 °C, respectively. The experimental evaluation indicated that reducing the declustering field showed essentially no gain in polymer coverage and resulted in further losses of instrument sensitivity, confirming that the heated capillary temperature effectively defined the gentleness of the interface under such conditions.
To further explore the mechanism behind the different polymer coverage under the different ion source configurations, Figure SI1 in the Supporting Information shows the DP versus elution time for all the polymers detected via LC–MS analyses of colominic acid at 300 °C ESI and 150 °C ESI interfaces and SPIN-MS interface, respectively. For all three configurations the detected DP number initially increased as the elution time increased. However, for the 300 °C ESI interface (Supporting Information Figure SI1a), a noticeable DP detection threshold appears at ∼60 min into the LC gradient with a maximum DP of 30. After this time point, only a mixture of DPs smaller than 30 was detected. Since the larger the DP number corresponds to more fragile polymers, the later eluting longer length DPs beyond ∼60 min were most likely broken into shorter DP polymers by the in-source fragmentation of glycosidic bonds for the 300 °C heated capillary inlet temperature.27 This effect was partially mitigated by lowering the heated capillary temperature to 150 °C (Supporting Information Figure SI1b). The maximum DP threshold in 150 °C ESI interface was observed later in the LC gradient at ∼70 min and increased to DP 38, beyond which the later eluting larger DP polymers were again “disassociated” by in-source fragmentation. In contrast, the maximum detected DP in the SPIN source configuration (Supporting Information Figure SI1c) extended across most of the LC separation. A less pronounced DP threshold (DP 47) only appeared at close to the end of the LC separation. Shorter length DPs were still observed later in the LC gradient for the SPIN source, indicating DP number larger than 47 may still experience in-source fragmentation even at the SPIN operating conditions. However, data acquired with the SPIN-MS interface exhibited better polymer coverage as compared to using the conventional ESI-MS interface at both heated capillary temperatures (i.e., the most gentle ion source among three tested in this study).
Although the same LC separation conditions were used for all the LC–MS analyses of colominic acid at the three tested ion source configurations, the observed mass spectra from each configuration differed significantly. As an example, Figure 3 shows the averaged mass spectra for the LC elution time window between 70 and 80 min for 300 °C ESI source (Figure 3a), 150 °C ESI source(Figure 3b), and SPIN source (Figure 3c) configurations, respectively. This LC elution time window was chosen based in Figure 2 with the expectation that higher DP polymers should be detected by MS at later elution times. The predominant ions detected using the 300 °C ESI source, as shown in Figure 3a, were singly charged short polymers DP 2, DP 3, DP 4, and DP 5. Polymer clusters were shifted by mass differences of 273.09 or 291.10 Da indicating a sialic acid monomer monosaccharide difference.26 Multiply charged large polymers, such as 3+ DP 19, 2+ DP 13, and 4+ DP 26 (Figure 3a inset), were detected at more than an order of magnitude lower intensity. In fact, the singly charged short polymers DP 2 to DP 5 were observed to be the dominant MS peaks throughout the entire LC–MS analysis under the 300 °C ESI source configuration indicating extensive in-source fragmentation in the interface. The ionization conditions were shown to be significantly gentler using the 150 °C capillary inlet. While the singly charged shorter polymers DP 2 to DP 5 were still the most abundant peaks in Figure 3b under the 150 °C inlet configuration, many multiply charged large polymers, such as 4+ DP 25, 4+ DP 26, 5+ DP 32 were detected (Figure 3b inset) as compared to the 300 °C inlet configuration. However, the intensity of the most abundant peaks for 150 °C interface decreased roughly by a factor of 10 as compared to the 300 °C interface, implying a practical sensitivity limit preventing further reducing the heated capillary temperature for even gentler ionization condition. The most striking differences in the mass spectrum were observed with the SPIN configuration. In concert with previous results obtained with the SPIN source,18 we observed a notable shift in the charge state distribution toward higher charge states. A representative example of this phenomenon is illustrated by the mass spectrum in Figure 3c. Compared to the data obtained at the same LC elution window with the ESI interface, the observed MS peak envelope for individual DPs is pushed to higher charge states and the most abundant peak became 5+ DP 25. A more comprehensive plot of the polymer distribution versus the observed charge state for different interface configurations is shown in Figure SI2 of the Supporting Information. The predominant singly charged short polymer peaks observed in high abundance under both 300 °C interface and 150 °C interface configurations were completely absent for the SPIN source. The inset in Figure 3c also shows that the significantly larger 6+ DP 37 and 6+ DP 38 ions were observed in the SPIN source configuration which were not detected in ESI source configurations during the selected LC elution time window. For the 5+ DP 32 polymer, observed only in 150 °C ESI and SPIN sources, the detected ion intensity for SPIN is over an order of magnitude higher than what was detected in the 150 °C ESI configuration. A similar increase in sensitivity was observed for all DPs commonly detected in the same charge state with both ion sources.
Figure 3.
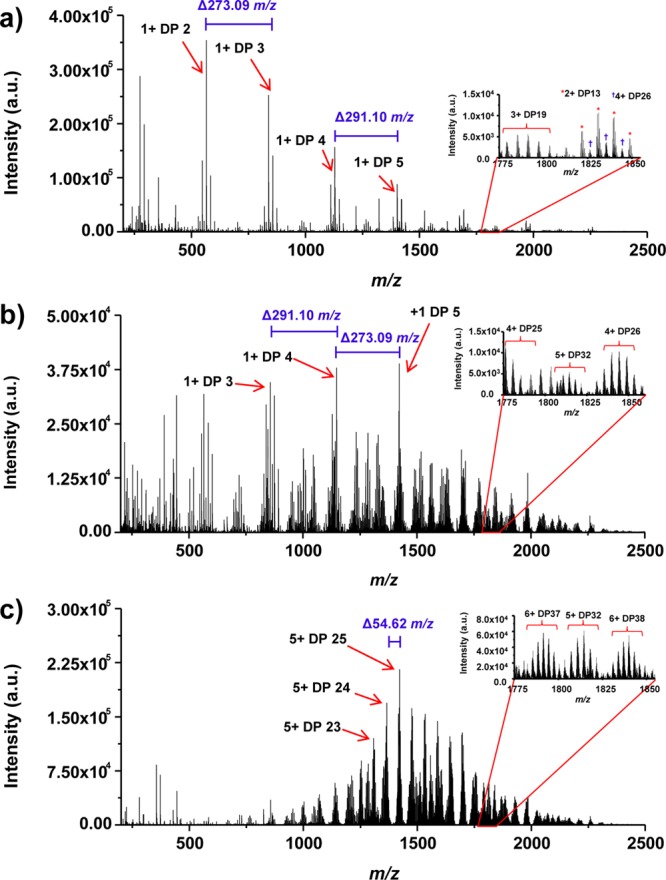
Averaged mass spectra acquired from elution times 70–80 min from the LC–MS analysis of colominic acid for the conventional ESI interface operated with capillary inlet temperatures of 300 °C (a), and 150 °C (b), and for the SPIN interface (c). The numbers in blue indicate a sialic acid monomer mass difference between polymer units.
Figure 4 summarizes all DPs identified using the GlyQ-IQ software tool for ESI 300 °C (Figure 4a), ESI 150 °C (Figure 4b), and SPIN (Figure 4c) configurations, respectively, within the same selected LC elution time window as shown in Figure 3 (i.e., from 70 to 80 min). Ion intensities in each figure were normalized to the most abundant peak detected in the respective ion source configuration. The most abundant ions were DP 2 and DP 5 for the 300 °C ESI source and 150 °C ESI source. Larger DP polymers (10 < DP < 40) were identified in both ESI source configurations with mostly less than 10% and 25% of the base peak intensity for ESI 300 °C and ESI 150 °C source configurations, respectively. Conversely, as shown in Figure 4c, the most abundant peak in SPIN interface configuration shifted to DP 25 and larger DPs were observed in much higher relative abundances.
Figure 4.
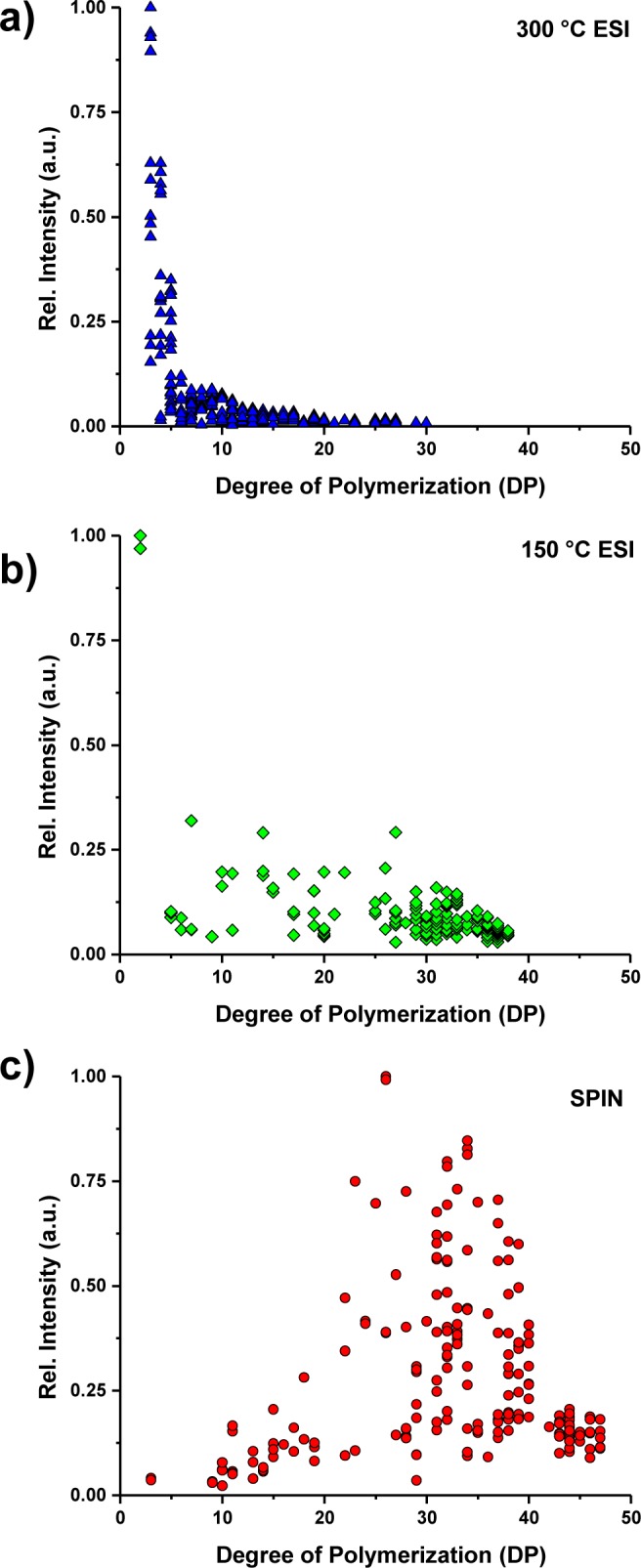
Polymer structures were extracted from the mass spectra shown in Figure 3 via the GlyQ-IQ software, and a plot of degree of polymerization vs relative intensity (normalized to the most abundant peak) is shown for ESI 300 °C (a), ESI 150 °C (b), and the SPIN interface (c).
For each observed colominic acid polymer, the mass spectrum contained a distribution of peaks corresponding to different degrees of lactonization. Lactone profiles for various DPs observed in multiple charge states from the SPIN source configuration are further shown in Figure 5a. In these plots, the intensity of each observed lactone was normalized to the most abundant lactone and plotted as a function of percent lactonization (number of lactones divided by the DP). For all polymers shown in the plot, the most abundant peak was always observed at ∼50% lactonization, independent of charge state and length of the polymer. This phenomenon was observed across all DPs detected, and Figure 5b shows a plot of the degree of polymerization against the degree of lactonization for all DPs observed with the SPIN source. For each polymer identified, only the most abundant peak from the lactone profile was included in the plot. The slope of the best fit line is roughly 0.50, which indicates that the most abundant peak for each observed DP correlated to roughly 50% lactonization. Similar trends were observed from the analyses conducted in both conventional ESI configurations indicating that the degree of lactonization was also independent of the interface configuration. Additional experiments are being conducted to probe whether the observed lactone profile has biological significance or is merely the consequence of sample preparation or LC conditions.
Figure 5.
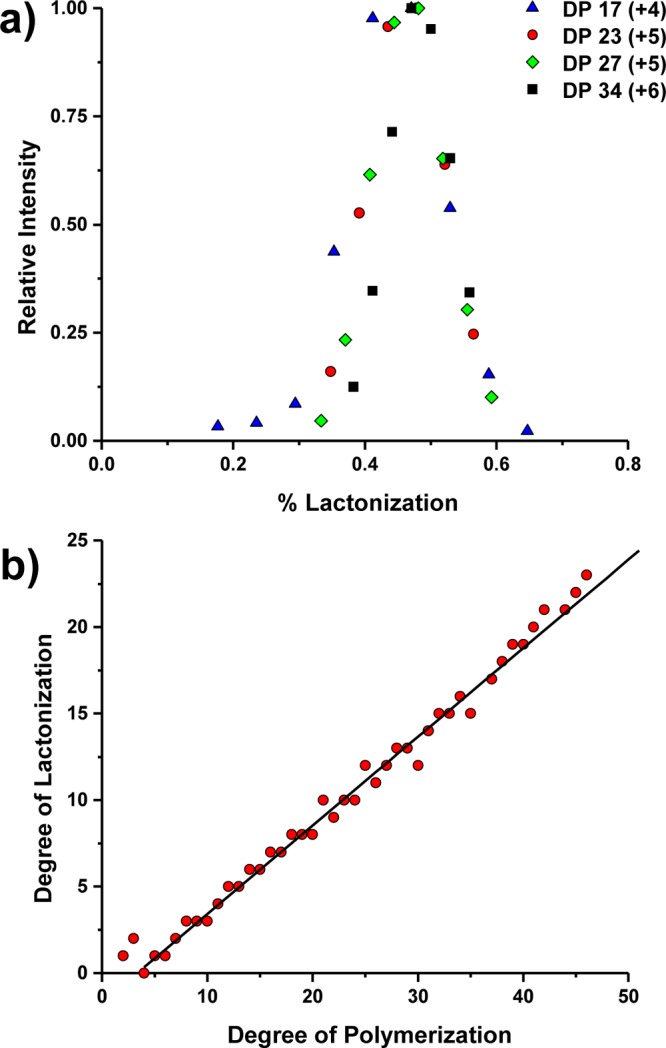
(a) Plot of the percent lactonization vs relative intensity for the polymers DP 17 (blue triangles), DP 23 (red circles), DP 27 (green diamonds), and DP 34 (black squares). The intensity was normalized to the most abundant lactone for each polymer length. The charge state of the peaks is indicated in parentheses. (b) Plot of the degree of lactonization vs the degree of polymerization for the most abundant degree of lactonization for a given polymer length.
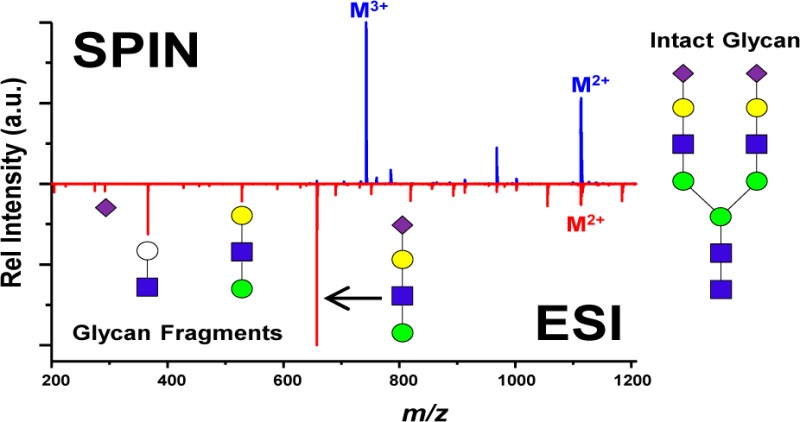
In addition to the studies conducted with colominic acid, the structural features observed from the LC–MS analysis of N-glycans containing sialic acid were further compared between the ESI-MS and the SPIN-MS interfaces. These glycans are relevant because of the labile nature of the sialic acid residue making the MS detection of the intact glycans very challenging.29,30 Similar to the colominic acid experiment, the LC–MS analyses of these sialic acid containing glycans were performed with the ESI-MS interface operated at both high heated capillary temperature (300 °C) for optimal sensitivity and low temperature (150 °C) for reduced in-source fragmentation and with the SPIN-MS interface. Figure 6 shows the mass spectra of a representative sialic acid containing N-glycan, Hex5HexNAc4NeuAc2 (2224.01 Da), extracted from the LC–MS analysis of human serum with different source configurations. The intact glycan in Figure 6 was first positively identified by using the GlyQ-IQ informatics software. The corresponding fragments in the spectra were then manually identified from the structure of the glycan. The spectra shown in the figure were obtained by averaging 60 scans across the chromatographic peak eluted at the same time in all instrument configurations. With the ESI-MS interface at the capillary temperature of 300 °C, the intact glycan, highlighted in red in Figure 6, was observed only at the 2+ charge state (1113.39 m/z) and in low abundance with an observed ratio of the extracted ion current (EIC) to total ion current (TIC) of 6.2%, as shown in Figure 6a. The predominant peaks in the spectrum were from glycan fragment ions, and their respective putative structures are annotated in the figure. Concurrent with the colominic acid analysis, the sialic acid residue fragment ions were present in the spectrum at 292.10 m/z indicating the loss of a sialic acid. By lowering the capillary temperature to 150 °C, as shown in Figure 6b, the intensity of the fragment ions decreased significantly, whereas the abundance of the intact glycan peak was greatly increased. In addition, the 3+ charge state of the intact glycan ions at 742.60 m/z was clearly visible in the spectrum. By summing the EICs for both charge states, the ratio of analyte EIC to the TIC increased to 29.8%. The mass spectrum observed with the SPIN-MS interface is shown in Figure 6c. The sialic acid containing fragment ions present in the ESI spectra (at 819.29 m/z, 657.24 m/z, and 292.10 m/z) are noticeably absent from the spectrum while the intensity of the intact glycan increased substantially. The ratio of analyte EIC to TIC improved to 42.6%. When compared to the signal obtained with the ESI-MS interface operated at 150 °C, the intensity of the 2+ and 3+ intact glycan ions using SPIN-MS interface increased by factors of roughly 4.4 and 46.5, respectively. Similar results were observed for other glycans identified in the human serum samples with an additional example of the N-glycan, Hex6HexNAc5NeuAc3 (2881.03 Da), shown in the Supporting Information as Figure SI3. These results confirm again that the SPIN-MS interface not only improves analyte sensitivity but also greatly reduces in-source fragmentation preserving intact sialic acid containing glycans.
Figure 6.

Mass spectra of an N-glycan containing sialic acid, Hex5HexNAc4NeuAc2 (2224.01 Da), from the LC–MS analysis of human serum obtained from the conventional ESI-MS interface operated with capillary inlet temperatures of 300 °C (a) and 150 °C (b), and for the SPIN interface (c). The red arrows represent the 3+ and 2+ charge states of the observed intact glycan. Consortium for functional glycomics (CFG) nomenclature was used to illustrate putative glycan structure with each sugar type denoted by a shape and isomers differentiated by color. Yellow and green circles represent hexoses with galactose and mannose stereochemistries, respectively, blue squares represent N-acetylhexosamine with glucose stereochemistry, and purple diamonds represent N-acetylneuraminic acid (sialic acid). White circles were used as a modification to represent a generic hexose.
The gentler ionization conditions and the shift in charge state envelope toward higher charge state observed in the SPIN-MS as compared to the conventional heated capillary ESI-MS interfaces are most likely due the differences in the droplet desolvation process between these interfaces. Although the initial formation of charged droplets in electrospray follows the same mechanism in both SPIN and conventional ESI sources,31 the regions and rates in which droplet desolvation occurs differ significantly. In the heated capillary ESI-MS interface, desolvation occurs dominantly in the higher pressure region inside the heated capillary as the charged droplets are transmitted from ambient pressures to the first vacuum stage of the instrument. The extended period of charged droplet desolvation in a narrow bore heated inlet capillary in the conventional ESI-MS interface design results in significant gas-phase ion/molecule or ion/ion reactions decreasing the analyte charge state,32,33 and the fully desolvated analyte ions may fragment following proton transfer reactions from the carboxylic acid to the glycosidic bond. In addition, transport through the heated capillary can result in preferential loss of higher mobility species, i.e., higher charge state ions, biasing data toward lower charge states.34 In contrast, droplet desolvation in the SPIN source occurs inside the ion funnel in the cooler and subambient pressure region, a much gentler environment reducing both charge state bias from our previous studies and ion in-source fragmentation as evident in this study using labile colominic acid polymers and N-glycans. These results suggest that the greater speed of the transfer process plays a role in the distinctive properties of SPIN observed in this work.
Conclusions
Through the LC–MS analyses of labile colominic acid polymers and N-glycans obtained from human serum, we compared the ionization conditions between using the conventional heated capillary ESI-MS interface and the SPIN-MS interface. We demonstrated that for SPIN-MS the coverage of colominic acid increased by ∼25% compared to the conventional ESI-MS interface. Tuning the conventional ESI interface to provide gentler conditions increased polymer coverage but at the cost of significant loss of sensitivity. In the SPIN-MS interface, both gentle ionization conditions and high sensitivity can be achieved simultaneously. For polymers observed in both interface configurations, the SPIN source demonstrated on average more than an order of magnitude enhancement in sensitivity. The extended colominic acid DP coverage and increase in observed charge states in SPIN are indicative of gentler ionization conditions, with each polymer observed in the mass spectrum having a lactone profile with the most abundant peak correlating to roughly 50% lactonization. The SPIN source also demonstrated a shift toward higher charge states allowing for extended coverage by reducing the m/z of the detected polymers. Because the colominic acid polymer is composed of sialic acid residues containing similar glycosidic linkages and structural features to sialic acid containing N-glycans, it can be used to tune ion source conditions to reduce in-source fragmentation for more effective analysis. Analysis of N-glycans containing sialic acid with the SPIN-MS interface showed a notable decrease in in-source fragmentation and greater than an order of magnitude enhancement in sensitivity as compared to using the conventional heated capillary ESI-MS interface. These studies indicate that analyses of labile compounds, including colominic acid and glycan, are greatly facilitated by SPIN-MS.
Acknowledgments
Portions of this research were supported by the NIH National Cancer Institute (1R33CA155252), as well as General Medical Sciences (GM103493-12), and by the Department of Energy Office of Biological and Environmental Research Genome Sciences Program under the Pan-omics project. All the experiments were performed in the Environmental Molecular Sciences Laboratory, a U.S. Department of Energy (DOE) national scientific user facility located at PNNL in Richland, Washington. High-performance computing research was performed using PNNL Institutional Computing. PNNL is a multiprogramming national laboratory operated by Battelle for the DOE under contract DE-AC05-76RLO01830.
Supporting Information Available
Additional information as noted in the text, including a plot of the elution time versus the degree of polymerization, a comprehensive plot of the polymer distribution versus the observed charge state for the different interface configurations, and additional mass spectra from the N-glycan Hex6HexNAc5NeuAc3 (2881.03 Da). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Gabelica V.; De Pauw E. Mass Spectrom. Rev. 2005, 24, 566–587. [DOI] [PubMed] [Google Scholar]
- Kim J. S.; Monroe M. E.; Camp D. G.; Smith R. D.; Qian W. J. J. Proteome Res. 2013, 12, 910–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo J. A.; Udseth H. R.; Smith R. D.; Futrell J. H. Rapid Commun. Mass Spectrom. 1988, 2, 207–210. [Google Scholar]
- Yan Z. Y.; Caldwell G. W.; Jones W. J.; Masucci J. A. Rapid Commun. Mass Spectrom. 2003, 17, 1433–1442. [DOI] [PubMed] [Google Scholar]
- Naban-Maillet J.; Lesage D.; Bossee A.; Gimbert Y.; Sztaray J.; Vekey K.; Tabet J. C. J. Mass Spectrom. 2005, 40, 1–8. [DOI] [PubMed] [Google Scholar]
- Rockwood A. L.; Busman M.; Udseth H. R.; Smith R. D. Rapid Commun. Mass Spectrom. 1991, 5, 582–585. [Google Scholar]
- Busman M.; Rockwood A. L.; Smith R. D. J. Phys. Chem. 1992, 96, 2397–2400. [Google Scholar]
- Penn S.; He F.; Green M. K.; Lebrilla C. J. Am. Soc. Mass Spectrom. 1997, 8, 244–252. [Google Scholar]
- Sakamoto S.; Fujita M.; Kim K.; Yamaguchi K. Tetrahedron 2000, 56, 955–964. [Google Scholar]
- Gabelica V.; De Pauw E. J. Am. Soc. Mass Spectrom. 2002, 13, 91–98. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K. J. Mass Spectrom. 2003, 38, 473–490. [DOI] [PubMed] [Google Scholar]
- Gabelica V.; De Pauw E.; Karas M. Int. J. Mass Spectrom. 2004, 231, 189–195. [Google Scholar]
- Rostom A. A.; Tame J. R. H.; Ladbury J. E.; Robinson C. V. J. Mol. Biol. 2000, 296, 269–279. [DOI] [PubMed] [Google Scholar]
- Hunter C. L.; Mauk A. G.; Douglas D. J. Biochemistry 1997, 36, 1018–1025. [DOI] [PubMed] [Google Scholar]
- Page J. S.; Tang K.; Kelly R. T.; Smith R. D. Anal. Chem. 2008, 80, 1800–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K.; Page J. S.; Marginean I.; Kelly R. T.; Smith R. D. J. Am. Soc. Mass Spectrom. 2011, 22, 1318–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marginean I.; Page J. S.; Tolmachev A. V.; Tang K.; Smith R. D. Anal. Chem. 2010, 82, 9344–9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marginean I.; Kronewitter S. R.; Moore R. J.; Slysz G. W.; Monroe M. E.; Anderson G.; Tang K.; Smith R. D. Anal. Chem. 2012, 84, 9208–9213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell A. K.; Harvey D. J. Rapid Commun. Mass Spectrom. 1996, 10, 1027–1032. [DOI] [PubMed] [Google Scholar]
- Zaia J. Mass Spectrom. Rev. 2004, 23, 161–227. [DOI] [PubMed] [Google Scholar]
- Isailovic D.; Plasencia M. D.; Gaye M. M.; Stokes S. T.; Kurulugama R. T.; Pungpapong V.; Zhang M.; Kyselova Z.; Goldman R.; Mechref Y.; Novotny M. V.; Clemmer D. E. J. Proteome Res. 2011, 11, 576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzi A. E.; Higa H. H.; Diaz S.; Varki A. J. Biol. Chem. 1994, 269, 23617–23624. [PubMed] [Google Scholar]
- Nakata D.; Troy F. A. J. Biol. Chem. 2005, 280, 38305–38316. [DOI] [PubMed] [Google Scholar]
- Kelly R. T.; Page J. S.; Luo Q.; Moore R. J.; Orton D. J.; Tang K.; Smith R. D. Anal. Chem. 2006, 78, 7796–7801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronewitter S. R.; Slysz G. W.; Marginean I.; Hagler C. D.; Lamarche B. L.; Zhao R.; Harris M. Y.; Monroe M. E.; Polyukh C. A.; Crowell K. L.; Fillmore T. L.; Carlson T. S.; Camp D. G.; Moore R. J.; Payne S. H.; Anderson G. A.; Smith R. D. Anal. Chem. 2014, 86, 6268–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galuska S. P.; Geyer R.; Muhlenhoff M.; Geyer H. Anal. Chem. 2007, 79, 7161–7169. [DOI] [PubMed] [Google Scholar]
- Galuska S. P.; Geyer H.; Bleckmann C.; Roehrich R. C.; Maass K.; Bergfeld A. K.; Muehlenhoff M.; Geyer R. Anal. Chem. 2010, 82, 2059–2066. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Lee Y. C. J. Biol. Chem. 1999, 274, 6183–6189. [DOI] [PubMed] [Google Scholar]
- Harvey D. J. Mass Spectrom. Rev. 1999, 18, 349–450. [DOI] [PubMed] [Google Scholar]
- Wheeler S. F.; Domann P.; Harvey D. J. Rapid Commun. Mass Spectrom. 2009, 23, 303–312. [DOI] [PubMed] [Google Scholar]
- Marginean I.; Page J. S.; Kelly R. T.; Tang K.; Smith R. D. Appl. Phys. Lett. 2009, 95, 184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLuckey S. A.; Stephenson J. L. Mass Spectrom. Rev. 1998, 17, 369–407. [DOI] [PubMed] [Google Scholar]
- Pitteri S. J.; McLuckey S. A. Mass Spectrom. Rev. 2005, 24, 931–958. [DOI] [PubMed] [Google Scholar]
- Page J. S.; Marginean I.; Baker E. S.; Kelly R. T.; Tang K.; Smith R. D. J. Am. Soc. Mass Spectrom. 2009, 20, 2265–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.