

Transition-metal catalyzed cross-coupling reactions encompass highly versatile methods for the construction of complex molecules from simple building blocks.[1] Efficient catalytic systems for a wide range of substrates are utilized in both research laboratories and industry. The most efficient and commonly employed cross-coupling catalysts feature 2nd- and 3rd-row transition metals, most notably palladium, to achieve high turnover numbers.[1a,2] Despite the mature state of this synthetic methodology, cross-coupling catalysis continues to attract significant interest, especially with respect to development of more sustainable catalysts based on abundant 1st-row transition metals.[1a,j,k,3] Various cross-coupling catalysts with 1st-row metals have been reported, and those based on nickel are particularly promising.[1g,4]
While nickel catalysts are effective in many of the same transformations known for palladium, recent reports indicate that nickel also promotes couplings of more challenging substrates, including Grignard reagents and unactivated alkyl halides.[5] These catalysts are thought to employ mechanisms that are distinct from those of palladium, involving one-electron redox processes that feature Ni(III)-alkyl or -aryl species as key intermediates.[6] While a few Ni(III) species of this type have been isolated and characterized, none have been rigorously provento be an intermediate in a catalytic cross-coupling.[7] Thus, a primary objective of research in this area is the acquisition of mechanistic information, and defining the role of nickel(III) species in an operative catalytic cycle. In this context, it was of interest to investigate the recently reported nickel(III) alkyl complex (Me)Ni[N(SiMe3)DIPP]2 (1)[7a] and explore its potential relevance as a catalytic intermediate. This investigation led to the discovery and mechanistic elaboration of C-C cross-couplings catalyzed by Ni[N(SiMe3)DIPP]2 (2). The latter complex appears to represent a new class of coupling catalysts, based on a two-coordinate bis(amido) ligand framework.
Reaction of 1 with one equivalent of PhI or C12H25I in THF did not lead to any observed products within the time that 1 decomposed to ethane and Ni[N(SiMe3)DIPP]2 (2, ca. 24 h). However, in light of a mechanism for nickel-catalyzed alkyl-alkyl cross-couplings recently proposed by Breitenfeld and Hu,[6a] in which the alkyl halide is initially activated by a nickel(II) alkyl complex, the reduction of 1 to a Ni(II) analogue was examined. Cyclic voltammetry revealed that 1 undergoes a reversible Ni(II/III) reduction at -1.30 V (E1/2 vs. Fc/Fc+, ipa/ipc = 0.98), suggesting that 1 might be readily reduced to the corresponding, anionic Ni(II) methyl complex. Indeed, reduction of 1 with KC8 at -30 °C in toluene over 25 minutes, followed by precipitation from toluene/pentane, provided the blue, anionic nickel(II) methyl complex K{(Me)Ni[N(SiMe3)DIPP]2} (3) in 64% yield (Scheme 1).
Scheme 1.
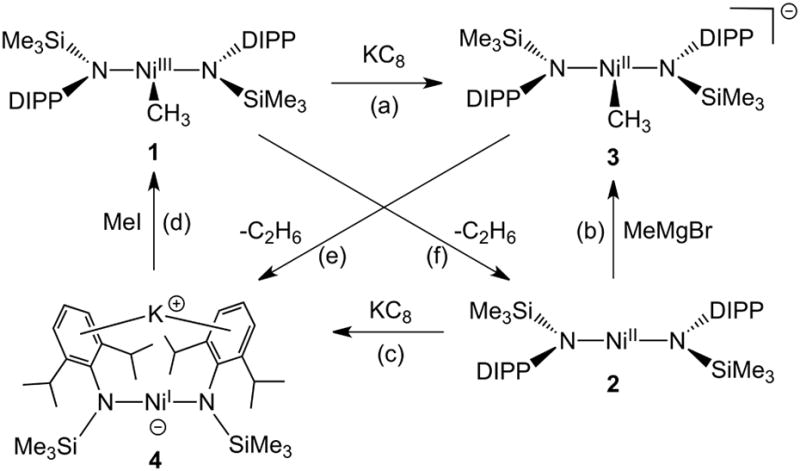
Reactions of related Ni-bis(amido) species. Conditions: (a) toluene, 25 minutes. (b) THF, seconds (c) toluene, 15 minutes. (d) THF, 30 minutes. (e) benzene, 7 days. (f) benzene, 1 day.
Analysis of 3 by single-crystal X-ray diffraction revealed molecular units containing a T-shaped, three-coordinate nickel center, linked into a polymeric chain via coordination of the potassium ions to the aryl rings of adjacent units (Figure 1). Comparison of this structure to that of the nickel(III) species 1 reveals that the nickel(II) complex 3 possesses a longer average Ni-N bond length (av. 1.891(2) Å Ni(II); 1.813(2) Å Ni(III)), as expected based on a larger covalent radius for the more reduced nickel center. However, the NiII–C bond length in 3 (av. 1.891(2) Å) is unexpectedly shorter than the corresponding NiIII–C distance in 1 (1.923(4) Å), presumably due to population of a Ni–σ* orbital in the latter (see Supporting Information, page S10). Despite being three-coordinate, 3 is low-spin and diamagnetic, with the methyl resonance appearing as a singlet in the 1H NMR spectrum at -0.95 ppm. Like complex 1, 3 is thermally unstable and decomposes over a period of 7 days in C6D6via reductive homolysis to the previously reported anionic nickel(I) complex K{Ni[N(SiMe3)DIPP]2} (4),[7a] with elimination of ethane (1H and 13C NMR spectroscopy). Compound 3 exhibits no signs of decomposition after several months as a solid stored at -30 °C.
Figure 1.
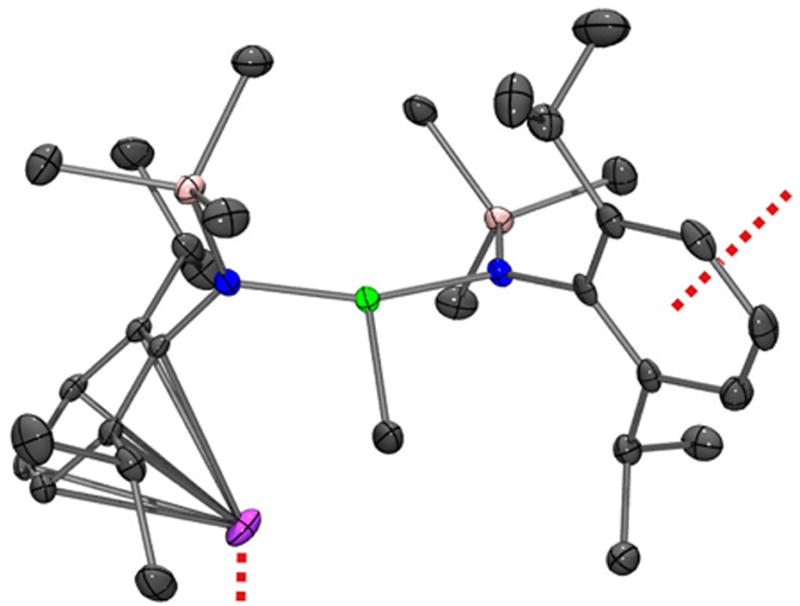
ORTEP diagram of 3. Thermal ellipsoids shown at 50% probability. Dashed red lines indicate intermolecular connectivity in the solid state. Selected bond lengths and angles: Ni-C: 1.879(2) Å; Ni-N: 1.8914(2) Å (av.); N-Ni-N: 161.42(7)°.
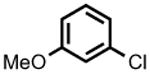
The accessibility of analogous Ni(II) and Ni(III) alkyls in this system, and the possible roles for such species as intermediates in catalytic C-C cross-coupling reactions, prompted an examination of 2 as a catalyst for the cross-coupling of Grignard reagents with alkyl and aryl halides. As shown in Table 1, 2 is a competent catalyst for the cross coupling of aryl halides with both aryl and methyl Grignard reagents at ambient temperature. Electron-poor substrates appear to be preferred (e.g., 8-bromoquinoline (93%) vs. 3-bromoanisole (<10%); entries 4 and 6 respectively), though electron rich aryl iodide substrates also performed well. This catalytic system is also effective for more challenging aryl chloride substrates as well as aryl bromides and iodides. Heterocyclic compounds, including coordinating pyridine units, are also well tolerated despite concern that strong coordination of pyridines to 2 could inhibit catalysis.[8] Attempts to couple aryl and alkyl Grignards with unactivated alkyl iodides resulted in only trace amounts of the cross-coupled product with the balance of products going to C12 alkene isomers and dodecane (entry 10).
Table 1.
Substrate table for Grignard/Ar-X cross-coupling reactions catalysed by 5 mol % Ni[N(SiMe3)DIPP]2 (2) with 1.1 eq. R'MgBr.
| Entry | R-X | R'MgBr | Time | Yield (Et2O) | Yield (THF) |
|---|---|---|---|---|---|
| 1 |
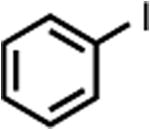
|
PhMgBr | 30 m | 98% | 92% |
| 2 |
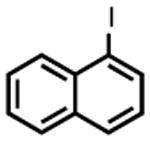
|
PhMgBr[a] | 30 m | 100% | 67% |
| MeMgBr | 1.5 h | 74% | 87% | ||
| EtMgBr | 40 m | 30% | - | ||
| 3 |
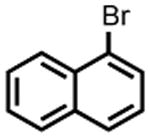
|
PhMgBr | 1 h | 68% | 95% |
| 4 |
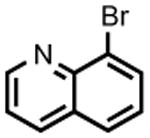
|
PhMgBr | 1.5 h | 93% | 18% |
| 5 |
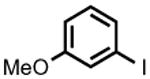
|
PhMgBr | 1.5 h | 99% | 33% |
| MeMgBr | 3.5 h | 93%[b] | - | ||
| 6 |
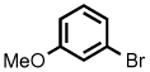
|
PhMgBr | 5.5 h | <10% | 56% |
| 7 |
|
PhMgBr | 66 h | <10% | 59% |
| 8 |
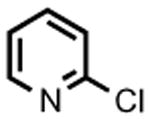
|
PhMgBr | 2 h | 78% | 47% |
| 9 |
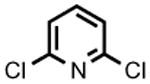
|
PhMgBr[c] | 6 h | 92% | - |
| 10 | C12H25I | MeMgBr | 45 m | <1% | <10% |
| PhMgBr | 45 m | 0% | 0% | ||
| 11 |
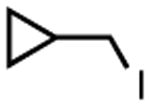
|
PhMgBr | 40 m | - | 79%[d] |
Catalysis performed using 0.5 mol % 2.
Yield determined by calibrated GC/FID instead of NMR due to the volatility of the product.
2.1 eq. of PhMgBr were used.
Yield is of the ring-opened product 4-phenyl-1-butene.
Use of EtMgBr as the Grignard reagent resulted in a substantially reduced yield compared to MeMgBr (30% vs. 74% in the coupling with 1-iodonaphthalene; Table 1, entry 2). To evaluate the stability of the anionic Ni(II) ethyl complex, the stoichiometric reaction between 2 and EtMgBr in d8-THF was monitored by 1H NMR spectroscopy. Addition of EtMgBr resulted in an immediate color change and complete consumption of 2. Stoichiometric quantities of ethylene were detected in solution, suggesting that β-hydride elimination had occurred. Similar results have been observed for other Ni(II) alkyl complexes,[9] and β-hydride elimination is thought to represent a significant hurdle for Ni-catalyzed carbon-carbon couplings involving alkyl groups.[10]
Analysis of the isolated, post-catalytic reaction mixture from catalytic coupling of MeMgBr with PhI revealed that the primary nickel-containing product was anionic 3 (presumably as the BrMg+ salt), accounting for 89% of the initial catalyst loading (1H NMR spectroscopy in d8-THF vs. internal standard). Small quantities of 4 also present in the post-catalytic reaction mixture may result from slight thermal decomposition of 3. The relatively large proportion of molecular Ni complexes present after catalysis suggests that these species may be directly involved in the catalytic cycle. Additionally, the yield and qualitative rate of the coupling reactions were found to be unaffected by the presence of mercury, further supporting the involvement of a homogeneous catalyst.
Given the efficiency of carbon-carbon coupling catalysis by 2, the relatively broad substrate scope, and the isolation of potential intermediates, it was of interest to explore the catalytic mechanism. Mechanistic studies involved observation of stoichiometric transformations for candidate intermediates 1-4, and led to the proposed mechanism illustrated in Scheme 2. Initial addition of the Grignard reagent to the neutral nickel(II) bis(amido) complex 2 occurs to form an anionic nickel(II) alkyl or aryl complex (3′) analogous to 3. This anionic complex then reduces the organic halide, with formation of an organic radical species and a nickel(III) alkyl or aryl complex (1′). The nature of this radical is currently undefined, and especially given the complexity of the reaction mixture, such radical species may not be “free”. The organic radical then combines with a second equivalent of 3′ to form the cross-coupled product. This step may involve an anionic nickel(III) complex 5′, which rapidly undergoes C–C reductive elimination to give the anionic nickel(I) complex 4; alternatively, the radical may directly attack the metal-bound alkyl group of 3′. The nickel(I) anion then reduces the nickel(III) complex resulting from reduction of the alkyl or aryl halide, to reform one equivalent each of 2 and 3′. Based on the measured reduction potentials of 1 (-1.30 V vs. Fc/Fc+, vide supra) and 2 (-1.28 V vs. Fc/Fc+),[7a] this final redox step is expected to result in an equilibrium mixture of reactants and products.
Scheme 2.

Proposed mechanism of catalytic coupling of Grignard reagents with aryl halides by 2. All species have been directly observed except 5′ and Ar•.
The feasibility of each step of the proposed catalytic cycle was tested by investigating relevant stoichiometric reactions of 1-4 and the catalytic substrates. The first step of this mechanism, the addition of a carbanion to 2, was easily demonstrated by the reaction of 2 with 1.1 equivalents of MeMgBr at -30 °C in THF over 30 seconds, which resulted in formation of a solution having the characteristic blue color of 3. Analysis of the products by 1H NMR and UV-vis spectroscopies revealed the presence of a diamagnetic species with spectra indistinguishable from those of 3 (see Supporting Information, page S8). The methylation of 2 with MeMgBr to form 3, the resting state of the catalysis, proceeds well within the timeframe of catalysis and in nearly quantitative yield (>98% by UV-vis).
The stoichiometric (1:1) reaction of 3 with 1-iodonaphthalene was observed to proceed sluggishly over 45 minutes to give the cross-coupled product 1-methylnaphthalene in only 13% yield. This reaction also produces significant quantities of the nickel(III) methyl complex 1, along with 1,1′-binaphthalene (64%) and unreacted 1-iodonaphthalene (23%). Addition of one equivalent of 1 to the reaction mixture (1, 3 and 1-iodonaphthalene in a 1:1:1 ratio) did not result in improved yield of the cross-coupled product (10%), indicating that 1 is not an effective trap for the species produced by reduction of the aryl iodide. In fact, two equivalents of 3 are required for efficient conversion of the aryl halide to product, as indicated by reaction of 1-iodonaphthalene with two equivalents of 3 in THF, which proceeded to completion within minutes to cleanly produce 1-methylnaphthalene in 98% yield. Thus, the cross-coupling of aryl halide requires two equivalents of 3, which appears to be the species that traps the radical rather than 1. Analysis of the post-coupling reaction mixture indicated the presence of all four species 1-4, as expected given the rapid equilibrium that converts 1 and 4 to 2 and 3 (vide infra).
The nature of the reduced aryl or alkyl halide was probed with use of the radical clock substrate (iodomethyl)cyclopropane. The cross-coupling of (iodomethyl)cyclopropane with 1.1 equivalents of PhMgBr catalyzed by 5 mol % of 2 resulted in a 79% yield of 4-phenyl-1-butene, the product resulting from rearrangement of the (cyclopropyl)methyl radical (Table 1, entry 11). This suggests that a radical intermediate results from reduction of the alkyl halide, as shown in the proposed mechanism of Scheme 2. No unrearranged product was observed, and no coupling products of any kind were observed in the absence of 2.
The last step in the catalytic cycle, the reduction of 1 by 4, to form 2 and 3, was demonstrated by addition of one equivalent of 1 to the NnBu4+ salt of 4 (4a)[7a] in THF, resulting in an immediate color change from dark green to dark grey/black. Due to extensive paramagnetic broadening in d8-THF solutions of paramagnetic species 1, 2 and 4, it was not possible to accurately measure the equilibrium constant by 1H NMR. However, all four species 1-4 were detected and the NnBu4+ salt of 3 was isolated from the other Ni-containing species in 29% yield (1H NMR spectroscopy in d8-THF vs. an internal standard, see Supporting Information, page S4), consistent with the equilibrium mixture predicted by the observed redox potentials.
This redox equilibrium step plays an important role in determining the efficiency of catalysis. This is indicated, for example, by stoichiometric reactions of 3 with 1-iodonaphthalene. This reaction produces a low yield of cross-coupled product (13%); however, addition of one equivalent of MeMgBr to the reaction mixture led to a substantially better yield (77%). This is attributed to the Grignard reagent's effect on the redox equilibrium step of the cycle (Scheme 2). Apparently, the rapid reaction of MeMgBr with 2 to form 3 pulls the redox equilibrium to the right (to 2 and 3 and ultimately, to only 3). Consistently, addition of one equivalent of MeMgBr to an equilibrium mixture of 1-4 (produced by reaction of 4a with 1) resulted in an immediate color change from dark grey/black to the characteristic blue color of 3. Analysis of the resulting products by 1H NMR spectroscopy indicated that all Ni-containing species had been converted to 3. By shifting the equilibrium in this way, the Grignard reagent ensures efficient recycling of 1 and 4 to 3, such that every radical in the cycle has a coupling partner. In the absence of Grignard, 3 is consumed in the reduction of the aryl halide and because the redox reaction produces an equilibrium mixture, the Ni-byproducts are not efficiently recycled. Additionally, 4 was found to react with aryl halides to produce undesired homocoupling products: reaction of 4 with 1-iodonaphthalene in THF over 1 hour produced 1,1′-binaphthalene in 80% yield. This unproductive process is also prevented by the Grignard's effect on the equilibrium. Notably, this reaction of 4 with aryl halide is substantially slower than the oxidation of 4 by 1, a result predicted by Breitenfeld and Hu for an analogous nickel(I) intermediate in their system.[6a]
The stoichiometric reaction chemistry described above provides compelling support for the proposed mechanism of Scheme 2. Significantly, this mechanism involves several intermediates (1-4) that have been isolated, completely characterized, and observed to participate in catalytically competent reaction steps. These results lend substantial support to proposed mechanisms for nickel-mediated cross-couplings that feature one-electron redox events for the nickel species,[4d] and especially corroborate the bimetallic oxidative addition mechanism (featuring comparable but inner sphere redox events) suggested by Breitenfeld and Hu.[6a] This study also provides experimental mechanistic evidence for the roles of nickel(I) and nickel(III) intermediates in this catalysis. Of particular interest is demonstration of a function for the nickel(III) alkyl complex (1), especially given previous speculation about such species in cross-coupling chemistry. In catalysis initiated by the bis(amido) complex 2, the neutral nickel(III) alkyl species appear to participate only in redox reactions, and not directly in any of the key bond-forming or bond-breaking steps. This nickel(III) species is formed as a byproduct of the C–X bond activation, and plays a key role in oxidizing nickel(I) in the catalytic mixture back to nickel(II). The latter redox process not only provides nickel(II) species required for the coupling steps, but removes nickel(I) which is active for the nonproductive homo-coupling of the halide substrate.
A somewhat more speculative aspect of this reaction mechanism concerns detail for reaction of the radical species with the anionic nickel(II) complex. This step may involve attack of the radical on the nickel center, to generate the nickel(III) complex 5′ (Scheme 2), from which the product is formed by reductive elimination. Alternatively, the organic radical might directly attack the alkyl or aryl ligand of 3′ to give 4 and the coupled product. The pathway involving 5′ is currently assumed, given spectroscopic observation of related intermediates by Vicic[11] as well as the observation of reductive elimination from Ni(III) alkyl species by Kochi[12] and Mirica.[7d] Also, the two-electron Ni(III/I) reductive elimination from 5′ seems reasonable given that the reverse process, a two-electron Ni(I/III) oxidative addition, has been experimentally demonstrated in the independent synthesis of 1 (Scheme 1).
This catalytic system is noteworthy as one of the only examples of an intact two-coordinate metal complex that competently catalyzes an organic transformation. Previously, Fe[N(SiMe3)2]2 has been described as an active precatalyst for the hydrosilylation of organic carbonyls,[13] and 2 catalyzes the hydrosilylation of alkenes.[8] The catalytic mechanism of Scheme 2 is unusual in several respects that appear to leverage the very low coordination number of 2. Direct alkylation of 2 by the Grignard without halogen displacement exploits the inherent electron deficiency of two-coordination, to stabilize a strongly reducing, anionic intermediate (3). The various redox processes of the mechanism proceed without ligand exchange, which is facilitated by the ability of this stable two-coordinate framework to support multiple oxidation states. These results suggest that two-coordinate complexes are a promising new class of potential catalysts whose unique chemical properties enable novel metal-mediated transformations.
In conclusion, a well-defined catalytic system for C-C coupling of aryl halides with Grignard reagents catalyzed by a two-coordinate nickel complex has been identified and mechanistically evaluated. Isolation and identification of the catalytic intermediates and stoichiometric reactions among those intermediates provides strong support for activation of the aryl halide by two metal centers and implicates a nickel(II) alkyl species as the coupling partner of the generated aryl radical rather than a nickel(III) alkyl species.
Supplementary Material
Footnotes
We gratefully acknowledge funding from the National Science Foundation for this work under grant no. CHE-1265674 and for the Molecular Graphics and Computational facility (College of Chemistry, University of California, Berkeley) under grant no. CHE-0840505. We also acknowledge the National Institutes of Health for funding of the ChexRay X-ray crystallographic facility (College of Chemistry, University of California, Berkeley) under grant no. S10-RR027172.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Jana R, Pathak TP, Sigman MS. Chem Rev. 2011;111:1417–1492. doi: 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cahiez G, Moyeux A. Chem Rev. 2010;110:1435–1462. doi: 10.1021/cr9000786. [DOI] [PubMed] [Google Scholar]; c) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457–2483. [Google Scholar]; d) Liu C, Zhang H, Shi W, Lei A. Chem Rev. 2011;111:1780–1824. doi: 10.1021/cr100379j. [DOI] [PubMed] [Google Scholar]; e) Beletskaya IP, Ananikov VP. Chem Rev. 2011;111:1596–1636. doi: 10.1021/cr100347k. [DOI] [PubMed] [Google Scholar]; f) Roglans A, Pla-Quintana A, Moreno-Mañas M. Chem Rev. 2006;106:4622–4643. doi: 10.1021/cr0509861. [DOI] [PubMed] [Google Scholar]; g) Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita AM, Garg NK, Percec V. Chem Rev. 2011;111:1346–1416. doi: 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Suzuki A. J Organomet Chem. 1999;576:147–168. [Google Scholar]; i) Chinchilla R, Nájera C. Chem Rev. 2007;107:874–922. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]; j) Bolm C, Legros J, Paih JL, Zani L. Chem Rev. 2004;104:6217–6254. doi: 10.1021/cr040664h. [DOI] [PubMed] [Google Scholar]; k) Sherry BD, Fürstner A. Acc Chem Res. 2008;41:1500–1511. doi: 10.1021/ar800039x. [DOI] [PubMed] [Google Scholar]
- 2.Seechurn CCCJ, Kitching MO, Colacot TJ, Snieckus V. Angew Chem Int Ed. 2001;51:5062–5085. doi: 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- 3.a) Vechorkin O, Hu X. Angew Chem Int Ed. 2009;48:2937–2940. doi: 10.1002/anie.200806138. [DOI] [PubMed] [Google Scholar]; b) Steib AK, Kuzmina OM, Fernandez S, Flubacher D, Knochel P. J Am Chem Soc. 2013;135:15346–15349. doi: 10.1021/ja409076z. [DOI] [PubMed] [Google Scholar]; c) Ge S, Green RA, Hartwig JF. J Am Chem Soc. 2014;136:1617–1627. doi: 10.1021/ja411911s. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bullock RM. Catalysts Without Precious Metals. Wiley-VCH; Weinheim: 2010. [Google Scholar]
- 4.a) Csok Z, Vechorkin O, Harkins SB, Scopelliti R, Hu X. J Am Chem Soc. 2008;130:8156–8157. doi: 10.1021/ja8025938. [DOI] [PubMed] [Google Scholar]; b) Jones GD, Martin JL, McFarland C, Allen OR, Hall RE, Haley AD, Brandon RJ, Konovalova T, Desrochers PJ, Pulay P, Vicic DA. J Am Chem Soc. 2006;128:13175–13183. doi: 10.1021/ja063334i. [DOI] [PubMed] [Google Scholar]; c) Breitenfeld J, Scopelliti R, Hu X. Organometallics. 2012;31:2128–2136. [Google Scholar]; d) Hu X. Chem Sci. 2011;2:1867–1886. [Google Scholar]
- 5.a) Böhm VPW, Weskamp T, Gstöttmayr CWK, Herrmann WA. Angew Chem Int Ed. 2000;39:1602–1604. doi: 10.1002/(sici)1521-3773(20000502)39:9<1602::aid-anie1602>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]; b) Xi Z, Liu B, Chen W. J Org Chem. 2008;73:3954–3957. doi: 10.1021/jo800197u. [DOI] [PubMed] [Google Scholar]; c) Xu YC, Zhang J, Sun HM, Shen Q, Zhang Y. Dalton Trans. 2013;42:8437–8445. doi: 10.1039/c3dt00086a. [DOI] [PubMed] [Google Scholar]
- 6.a) Breitenfeld J, Ruiz J, Wodrich MD, Hu X. J Am Chem Soc. 2013;135:12004–12012. doi: 10.1021/ja4051923. [DOI] [PubMed] [Google Scholar]; b) Tsou TT, Kochi JK. J Am Chem Soc. 1979;101:7547–7560. [Google Scholar]; c) Biswas S, Weix DJ. J Am Chem Soc. 2013;135:16192–16197. doi: 10.1021/ja407589e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Lipschutz MI, Yang X, Chatterjee R, Tilley TD. J Am Chem Soc. 2013;135:15298–15301. doi: 10.1021/ja408151h. [DOI] [PubMed] [Google Scholar]; b) Lee CM, Chen CH, Liao FX, Hu CH, Lee GH. J Am Chem Soc. 2010;132:9256–9258. doi: 10.1021/ja102430d. [DOI] [PubMed] [Google Scholar]; c) Grove DM, van Koten G, Zoet R, Murrall NW, Welch A. J Am Chem Soc. 1983;105:1379–1380. [Google Scholar]; d) Zhang B, Tang F, Luo J, Schultz JW, Rath NP, Mirica LM. J Am Chem Soc. 2014 doi: 10.1021/ja5024749. Article ASAP. [DOI] [PubMed] [Google Scholar]
- 8.Lipschutz MI, Tilley TD. Chem Commun. 2012;48:7146–7148. doi: 10.1039/c2cc32974c. [DOI] [PubMed] [Google Scholar]
- 9.Liang LC, Chien PS, Lin JM, Huang MH, Huang YL, Liao JH. Organometallics. 2006;25:1399–1411. [Google Scholar]
- 10.Breitenfeld J, Vechorkin O, Corminboeuf C, Scopelliti R, Hu X. Organometallics. 2010;29:3686–3689. [Google Scholar]
- 11.Zhang CP, Wang H, Klein A, Biewer C, Stirnat K, Yamaguchi Y, Xu L, Gomez-Benitez V, Vicic DA. J Am Chem Soc. 2013;135:8141–8144. doi: 10.1021/ja4030462. [DOI] [PubMed] [Google Scholar]
- 12.Tsou TT, Kochi JK. J Am Chem Soc. 1978;100:1634–1635. [Google Scholar]
- 13.Yang J, Tilley TD. Angew Chem Int Ed. 2010;49:10186–10188. doi: 10.1002/anie.201005055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.