

Abstract
The enzyme soybean lipoxygenase (SLO) has served as a prototype for hydrogen-tunneling reactions, as a result of its unusual kinetic isotope effects (KIEs) and their temperature dependencies. Using a synergy of kinetic, structural, and theoretical studies, we show how the interplay between donor–acceptor distance and active-site flexibility leads to catalytic behavior previously predicted by quantum tunneling theory. Modification of the size of two hydrophobic residues by site-specific mutagenesis in SLO reduces the reaction rate 104-fold and is accompanied by an enormous and unprecedented room-temperature KIE. Fitting of the kinetic data to a non-adiabatic model implicates an expansion of the active site that cannot be compensated by donor–acceptor distance sampling. A 1.7 Å resolution X-ray structure of the double mutant further indicates an unaltered backbone conformation, almost identical side-chain conformations, and a significantly enlarged active-site cavity. These findings show the compelling property of room-temperature hydrogen tunneling within a biological context and demonstrate the very high sensitivity of such tunneling to barrier width.
Despite over a century of experimental and theoretical inquiry, the physical basis for enzymatic rate accelerations continues to be debated and challenged. The early focus of researchers on a reduction of barrier height as the origin of catalysis1 was later modified by the recognition of an important role for reduced barrier width in atom- and group-transfer reactions.2−8 Following the first definitive report of the involvement of room-temperature barrier penetration (tunneling) in the hydride-transfer reaction catalyzed by mammalian alcohol dehydrogenase,2 the widespread importance of quantum mechanical tunneling in enzymatic C–H activation has become apparent.9,10 One of the unique aspects of nuclear tunneling reactions is their ability to provide insight into the role for reduced barrier width that is coupled to active-site motions in enzymatic reactions. However, while theoretical treatments predict a dependence of the tunneling rate and kinetic isotope effect (KIE) on barrier width,11−15 the complexity of biological systems makes the demonstration of such correlations very challenging.5,6
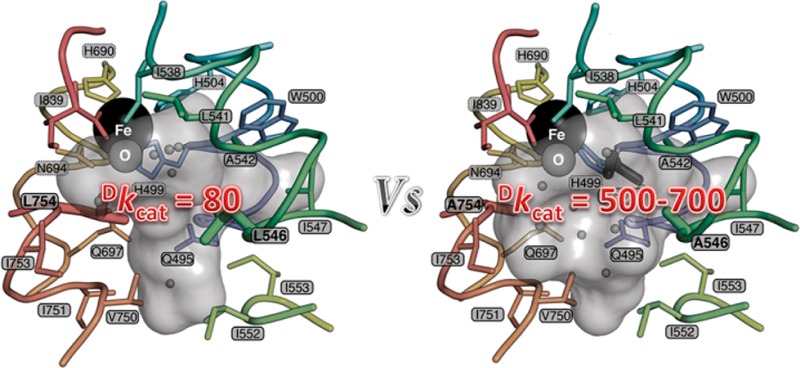
The enzyme soybean lipoxygenase (SLO) has served as a prototype for hydrogen-tunneling reactions since the initial detection of a very large, nearly temperature-independent KIE (kH/kD = 80) in native enzyme.13 By virtue of its tunneling mechanism in both native and single-site mutant enzymes, SLO offers a paradigmatic system in which to pursue the impact of barrier shape on tunneling efficiency.16,17 In the present study of a double mutant (DM) SLO, we show a subtle yet fatal protein disruption that both increases the hydrogen donor–acceptor distance and disables the ability of the protein to recover to a suitable tunneling distance. The kinetic behavior, with a room-temperature deuterium isotope effect of 540–730, is unprecedented in enzyme reactions. Very large KIEs have recently been implicated in several solution reactions.18,19
Studies of the DM SLO L546A/L754A were initiated by the measurement of steady-state kinetic properties using all-protio-linoleic acid as substrate and a spectroscopic assay that allows continuous monitoring of the dienoic hydroperoxide product at 234 nm. Collection of kinetic data from 15 to 50 °C [Table S1 in the Supporting Information (SI)] reveals the following properties in relation to wild-type (WT): an overall rate of catalysis [kcat(H)] that is decreased 104-fold together with a substantial elevation of the Arrhenius parameters AH and Ea (Table 1). While a large increase in Ea may not be unexpected, given the enormous reduction in kcat, the origin of an AH that is also significantly larger than the WT value is less clear. Several control experiments, including circular dichroism (Figure S1, SI) and analytical size-exclusion chromatography under the conditions of the kinetic assays (Figure S2, SI), eliminated extensive protein unfolding, structural rearrangement, or aggregation as the cause of the dramatic decrease in activity.
Table 1. Kinetic Parameters of Wild-Type and Double Mutant Soybean Lipoxygenase.
Low-temperature X-ray crystallographic data for the DM SLO variant at close to atomic resolution (1.7 Å) reveal a protein backbone that is practically superimposable (0.30 Å Cα-RMSD) on the WT enzyme model (Figure 1A). Of significance to the mechanism, an expanded active site is detected (Figure 1B; Figure S3, SI). In addition to the reduction in side-chain volume, the α-carbons of the mutant residues move apart by 0.26 Å. In contrast, the active site contracts by up to 0.46 Å in the orthogonal direction; i.e., the Fe-coordinating shell moves toward the mutant positions (Figure S3, SI). The net volume increase is associated with a change in solvation, allowing entry of four additional water molecules as well as a molecule of ethylene glycol from the cryoprotectant which displaces two waters in the WT structure (Figure 1B). Although the activated oxygen of the iron center occupies a similar position in both WT and DM structures, the iron–oxygen distance shrinks by 0.05 Å, and the coordinating H499 χ2 rotates 56° in the DM (Figure 1C; Figure S4, SI). In addition, analysis of anisotropic thermal displacements indicates that the principal vector of vibration for the iron turns by ∼90° but is orthogonal to the iron–oxygen bond in both structures (Figure 1C; Figure S5, SI). By and large, the populations of side-chain conformations are similar between the two proteins. Analysis of the electron density maps using Ringer20 reveals that a minor conformation of I553 in the WT structure becomes the major form with the Cδ pointing into the cavity (Figure 1C; Figure S5, SI). Conversely, structures of I553 truncations reveal increased flexibility of L546, showing that these residues are sterically coupled.17 Otherwise, the increase in active-site volume does not significantly change the populations of side-chain conformations in the substrate cavity or throughout the protein (Figure 1C; Figure S5, SI). In summary, the mutations expand the volume accessible to substrate and subtly alter the coordination and displacements of the iron atom while otherwise maintaining the topography of the active site. The observed change in shape and volume of the cavity may be expected to alter the conformation of bound substrate and/or the resulting hydrogen vibrational wave function overlap from the substrate to the Fe(III)–OH acceptor.
Figure 1.
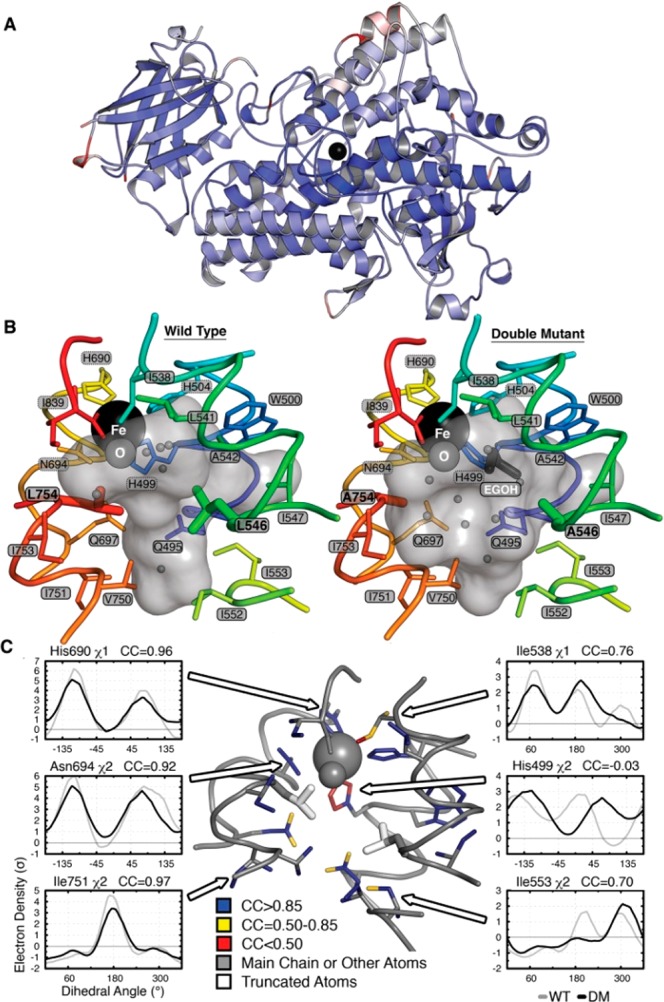
Crystal structure of DM SLO. (A) Superposition of the ribbon diagrams of WT and DM SLO with the Fe (black sphere) in the center. Colors represent Cα-RMS displacements from 0 Å (blue) to 0.5 Å (white) to 1 Å (red). (B) Solvent-accessible surface of the active-site cavity in the WT and DM SLO structures. The mutations make the cavity larger, allowing binding of ethylene glycol (dark gray sticks) and altering the water (small gray spheres) distribution. (C) Side-chain populations are similar in the DM and WT enzymes. Side chains lining the cavity are colored according to the correlation coefficient (CC) of the electron density (σ) sampled around each dihedral angle using the program Ringer. CC values above 0.85 (blue) reflect nearly identical conformational distributions, while CC values of 0.85–0.5 (yellow) or below 0.5 (red) reflect increasingly larger population shift. Representative plots of electron density vs dihedral angle show similar distributions (left) and the three largest differences (right).
As a probe of the impact of an enlarged active-site cleft on hydrogen vibrational wave function overlap, turnover rates for reaction of the isotopically labeled substrate, 11,11-2H2-linoleic acid (d2-LA), were pursued initially using the continuous UV–vis spectroscopic assay of protio-substrate. Steady-state assays of SLO routinely show a short lag, reflecting the need to generate trace amount of product hydroperoxide that binds to the enzyme and converts the precursor Fe(II)–OH2 form to the active Fe(III)–OH state.21 However, spectroscopic monitoring of SLO with d2-LA failed to show any change in absorbance at all temperatures monitored (from 10 to 50 °C) after several hours of incubation. As an alternative, a discontinuous HPLC assay was used to directly detect the product (see SI). Incubation of d2-LA with DM SLO showed a long lag phase (∼3 h) followed by a linear rate for product formation (Figure 2A). Comparison of the linear portion of this curve to a similar assay with protio-LA (with the more typical lag time of ∼3 min) gives an estimated value Dkcat = 729 ± 26 (Table 1). Controls conducted to test for any time-dependent inactivation of DM SLO during the long lag phase indicate full maintenance of enzyme activity during the 4 h of incubation (Figure S6, SI).
Figure 2.
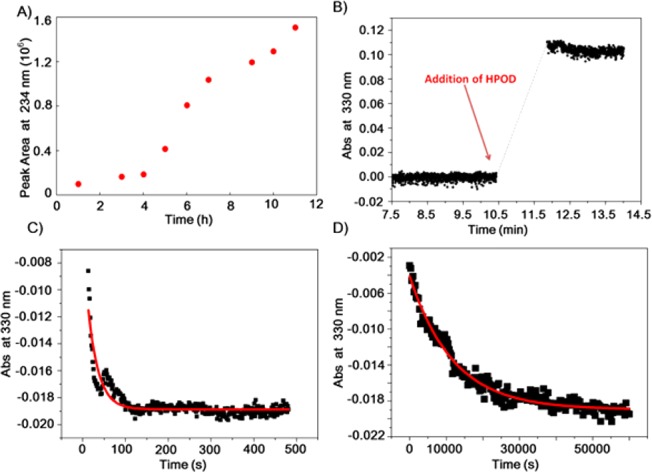
Kinetic traces of DM SLO. (A) Time course of reaction of DM SLO (1 μM in the reaction mixture) with 33 μM d2-LA at 30 °C in 0.1 M borate, pH 9. At specific times, a 3 mL aliquot was acidified, methylene chloride was extracted, and the solution was evaporated and HPLC-separated. The points were obtained from the area of the product peak at 234 nm. (B) Typical absorbance change at 330 nm during preactivation of DM SLO by adding 2 equiv of 13-(S)-HPOD. (C,D) Pre-steady-state kinetic traces for absorbance at 330 nm vs time for the anaerobic reaction between ferric SLO and linoleic acid. Reaction of protio-LA at 35 °C is shown in C, and that of d2-LA is shown in D. The UV–vis spectrophotometer was reset to zero when substrate was added, leading to negative values of absorbance in C,D.
The enormous magnitude of the KIE of the DM, together with the inherent complexity of steady-state measurements, led us to pursue single-turnover kinetics as a means of corroboration. The method employed involves monitoring the time course for reduction of the active-site Fe(III)–OH to Fe(II)–OH2 by either the protio- or d2-LA.22 This single-turnover time course represents the first half-reaction of the SLO reaction, which is followed by an O2-dependent oxidation during steady-state turnover.23 There are two important differences between the present work and previous analysis of the WT enzyme. First, the DM SLO is sufficiently slow that hand mixing can be used in place of the earlier stopped-flow protocol. Second, the extremely slow reaction of the DM SLO with deuterated substrate made leakage of molecular O2 into the reaction chamber a serious complicating feature. For this reason, comparative single-turnover studies to obtain the KIE were carried out under conditions of strict anaerobiosis within an airtight glovebox.
Ferric SLO, the active enzyme species, was freshly prepared by treatment of ferrous SLO with the enzymatic product, 13-(S)-HPOD. This pre-activation process, monitored by UV–vis spectroscopy, leads to an increase of a 330 nm species (active, ferric form, Figure 2B).24 Oxygen plays a critical role in pre-activation by facilitating the dissociation of a ferric enzyme–radical intermediate complex, and must be rigorously scrubbed from the reaction vessel before initiation of reaction with substrate.21 Repeated measurements of the reduction of absorbance vs time for the reaction of protio-LA at 30 °C indicated a single-exponential decay process with a rate constant of 0.0250 ± 0.0004 s–1 (Figure S7, SI). This rate constant is close to, but slightly faster than, the earlier steady-state value of kcat(H) (30 °C, Table 1), and is well within the range of the variability in rate among different SLO preparations.
The anaerobic reduction of activated DM enzyme by protio- vs d2-LA was monitored by UV–vis spectroscopy within the anaerobic glovebox (35 °C, Figure 2C,D). The kinetics trace with d2-LA continued out to nearly 15 h. A control reaction using pre-activated enzyme was run to demonstrate enzyme stability under the anaerobic condition to 10 h (Figure S8, SI). The rate constant for the reaction of DM SLO with d2-LA, obtained by fitting to a single-exponential decay, is (7.68 ± 0.67) × 10–5 s–1. This corresponds to Dkcat = 537 ± 55 in relation to the rate of protio-LA obtained in a similar assay (Table 1). The fact that single-turnover kinetics is inherently independent of the concentration of catalyst eliminates the possibility of non-productive inhibitory binding of substrate as the origin of the KIE. We further note almost identical amplitudes for kinetic traces monitoring protio and deuterio substrates. The slightly smaller value for Dkcat in the pre-steady-state compared to the steady-state condition (Table 1) is consistent with the higher experimental temperature in the former case.25
The enormous size of the room-temperature KIEs with DM SLO provides a rigorous experimental benchmark for theoretical discrimination. These KIEs approach, or even exceed, values that have been traditionally attributed to reactions at very low temperatures, ∼100 K,26,27 and show the compelling role for tunneling in SLO. The previously developed non-adiabatic hydrogen-tunneling analysis of both the rate constant and KIE was undertaken to understand the experimental results.13,28−30 In a first approximation, the slow rate and the temperature dependence of the rate for protio-LA with DM SLO were well fit (Figure S9B, SI) using a value for the driving force similar to that of the WT, together with a greatly elevated reorganization energy (λ = 45.6 kcal/mol vs 13.4 kcal/mol used to refit the WT data). Parsing of the contributions to the free energy barrier into a term arising from a large-scale stochastic conformational search (defined as the work term, W)31−33 and a term associated with electrostatic reorganization (represented by λ) provided an equally good fit for the DM SLO (W = 7.72 kcal/mol and λ = 13.4 kcal/mol; Figure S9C, SI). Fitting of the enormous KIE in DM SLO was possible by co-variation of the equilibrium distance between the donor carbon of the substrate and the acceptor oxygen at the iron center (Req) and the frequency for the donor–acceptor distance sampling (Ω),13,29,30,33 as shown in Figure 3. These two fitted parameters are independent of the choice of λ within the physically reasonable regime (SI). As a frame of reference, the fitted parameters obtained from the WT KIE and its temperature dependence are Ω = 175 cm–1 and Req = 2.71 Å.29 In comparison, the possible values of Req for DM SLO are in the range 2.79–2.91 Å, elongated in relation to those of the WT enzyme by ∼0.1–0.2 Å.
Figure 3.
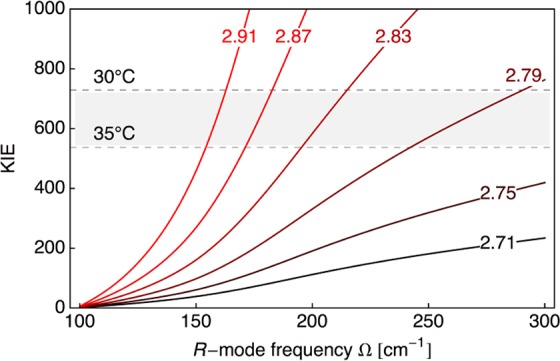
Predicted KIE for DM SLO as a function of the donor–acceptor equilibrium distance (Req) and the frequency of the donor–acceptor distance sampling (Ω). The curves are labeled according to Req given in Å. In this model, the reorganization energy is λ = 45.616 kcal/mol, the reaction free energy is ΔG° = −5.4 kcal/mol, and no additional work term is included. The shaded area of the plot corresponds to the experimental range of the observed KIE between 30 and 35 °C. The corresponding plot with λ = 13.408 kcal/mol and an additional work term of W = 7.723 kcal/mol is given in Figure S10 in the SI.
Equally important for the present study, the computed frequencies for the donor–acceptor distance sampling mode in the DM SLO range from 150 to 300 cm–1 and are elevated in relation to frequencies previously estimated for a series of single-site mutants at I553 (Table S2, SI).17,29 Often, active-site mutations are accompanied by decreases in the force constant for the donor–acceptor distance sampling mode.11,15 The latter facilitates sampling of the shorter donor–acceptor distances characteristic of WT enzyme.13,17,34 DM SLO is the first protein variant that appears unable to undergo a dynamic recovery from initially expanded donor–acceptor distances to the tunneling configurations characteristic of the native enzyme. From an average donor–acceptor extension of 0.15 Å for DM SLO in conjunction with the frequencies shown in Figure 3, we estimate a rate reduction of ∼102-fold. The additional reduction in rate (104-fold overall) likely results from a combination of non-optimal protein conformational subspecies and increased λ. Significantly, reduced sampling that is accompanied by extended donor–acceptor distances consigns protium, and especially deuterium, to active-site configurations of poor hydrogen vibrational wave function overlap, leading to the unusually large KIE. These combined properties have allowed a direct test of the predictions from non-adiabatic quantum theory11−15 regarding the size of the KIE as a function of donor–acceptor distance.
To conclude, the combined observations of an enlarged active site for the DM SLO (Figure 1) with conspicuously inflated KIE values (Figure 2) provide experimental verification of the critical role of hydrogen tunneling and the associated barrier width in enzyme-supported C–H activation. Further, the distinctive features between DM and other SLO mutants13,17,30 show how protein flexibility and the ensuing generation of close donor–acceptor distances can contribute to the enormous rate enhancements brought about by enzymes. The study of tunneling in C–H activation reactions affords a unique and sensitive window into the geometric and dynamic contributions that determine enzymatic fitness.
Acknowledgments
We thank Dr. Santosh Sivaramakrishnan at the University of California, San Francisco, for assistance with pre-steady-state kinetics of d2-LA. This work was supported by the National Institutes of Health (GM025765 to J.P.K.; P50GM082250 to T.A.; GM056207 to S.H.-S.; and T32GM008295 to C.A.M.C.).
Supporting Information Available
Materials and methods, Tables S1–S4, Figures S1–S10, and supporting references. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ Kiverdi, Inc., 2929 Seventh St., Berkeley, CA 94710
Author Contributions
∥ S.H. and S.C.S. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Wolfenden R. Acc. Chem. Res. 1972, 5, 10. [DOI] [PubMed] [Google Scholar]
- Cha Y.; Murray C. J.; Kliman J. P. Science 1989, 243, 1325. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Klinman J. P. J. Am. Chem. Soc. 2011, 133, 17134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S. D.; Schramm V. L. Nat. Chem. Biol. 2009, 5, 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay S.; Scrutton N. S. Nat. Chem. 2012, 29, 161. [DOI] [PubMed] [Google Scholar]
- Glowacki D. R.; Harvey J. N.; Mulholland A. J. Nat. Chem. 2012, 29, 169. [DOI] [PubMed] [Google Scholar]
- Major D. T.; Heroux A.; Orville A. M.; Valley M. P.; Fitzpatrick P. F.; Gao J. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 20734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson M. H. M.; Parson W. W.; Warshel A. Chem. Rev. 2006, 106, 1737. [DOI] [PubMed] [Google Scholar]
- Allemann R. K., Scrutton N. S.. Quantum Tunneling in Enzyme-Catalyzed Reactions; Royal Society of Chemistry: London, 2009. [Google Scholar]
- Klinman J. P.; Kohen A. Annu. Rev. Biochem. 2013, 82, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov A. M.; Ulstrup J. Can. J. Chem. 1999, 110, 1085. [Google Scholar]
- Hammes-Schiffer S. Acc. Chem. Res. 2001, 34, 273–281. [DOI] [PubMed] [Google Scholar]
- Knapp M. J.; Rickert K.; Klinman J. P. J. Am. Chem. Soc. 2002, 124, 3865. [DOI] [PubMed] [Google Scholar]
- Kiefer P. M.; Hynes J. T. J. Phys. Chem. A 2004, 108, 11793. [Google Scholar]
- Hammes-Schiffer S.; Stuchebrukhov A. A. Chem. Rev. 2010, 110, 6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman M. H.; Wiseman J. S.; Klinman J. P. J. Am. Chem. Soc. 1994, 116, 793. [Google Scholar]
- Meyer M. P.; Tomchick D. R.; Klinman J. P. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh M. H. V.; Meyer T. J. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh M. H. V.; Meyer T. J. Angew. Chem., Int. Ed. 2002, 41, 1395. [DOI] [PubMed] [Google Scholar]
- Lang P. T.; Ng H.-L.; Fraser J. S.; Corn J. E.; Echols N.; Sales M.; Hoton M. J.; Alber T. Protein Sci. 2010, 19, 1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.; Brash A. R. J. Biol. Chem. 2010, 285, 39876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T.; Glickman M. H.; Sun S.; Klinman J. P. J. Am. Chem. Soc. 1996, 118, 10319. [Google Scholar]
- Glickman M. H.; Klinman J. P. Biochemistry 1996, 35, 12882. [DOI] [PubMed] [Google Scholar]
- Feiters M. C.; Assa R.; Malmström B. G.; Veldink G. A.; Vliegenthart J. F. G. Biochim Biophys. Acta 1986, 873, 182. [Google Scholar]
- It is currently unclear how the temperature dependence of the KIE for the DM SLO will compare to the results for single-site mutants characterized previously.11,15 The extremely slow rate for DM SLO necessitates modification of existing methodologies before KIE measurements will become available over a wide temperature range.
- Le Roy R. J.; Murai H.; Williams F. J. Am. Chem. Soc. 1980, 102, 2325. [Google Scholar]
- Brunton G.; Griller D.; Barclay L. R. C.; Ingold K. U. J. Am. Chem. Soc. 1976, 98, 6803. [Google Scholar]
- Hatcher E.; Soudackov A. V.; Hammes-Schiffer S. J. Am. Chem. Soc. 2004, 126, 5763. [DOI] [PubMed] [Google Scholar]
- Hatcher E.; Soudackov A. V.; Hammes-Schiffer S. J. Am. Chem. Soc. 2007, 129, 187. [DOI] [PubMed] [Google Scholar]
- Edwards S. J.; Soudackov A. V.; Hammes-Schiffer S. J. Phys. Chem. B 2010, 114, 6653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus R. A. J. Phys. Chem. 1968, 72, 891. [Google Scholar]
- Marcus R. A. J. Am. Chem. Soc. 1969, 91, 7224–7225. [Google Scholar]
- The equilibrium distance (Req) can be viewed as the average distance within an ensemble of ground-state enzyme–substrate complexes that are in a region of conformational space amenable to reaction. These configurations are sampled among a large number of accessible protein conformational ground states and are typically associated with short donor–acceptor distances. A common property of mutants is the presence of elongated values for Req relative to WT, requiring increased donor–acceptor distance sampling to reach the shorter distances with higher tunneling probabilities.
- Pudney C. R.; Johannissen L. O.; Sutcliffe M. J.; Hay S.; Scrutton N. S. J. Am. Chem. Soc. 2010, 132, 11329. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.